Article Text

Abstract
Background Successful development of chimeric antigen receptor (CAR) T cell immunotherapy for children and adults with relapsed/refractory acute myeloid leukemia (AML) is highly desired given their poor clinical prognosis and frequent inability to achieve cure with conventional chemotherapy. Initial experiences with CD19 CAR T cell immunotherapy for patients with B-cell malignancies highlighted the critical impact of intracellular costimulatory domain selection (CD28 vs 4-1BB (CD137)) on CAR T cell expansion and in vivo persistence that may impact clinical outcomes. However, the impact of costimulatory domains on the efficacy of myeloid antigen-directed CAR T cell immunotherapy remains unknown.
Methods In this preclinical study, we developed six CAR constructs targeting CD33, a highly expressed and validated AML target, comprised of one of three single-chain variable fragments with CD3ζ and either CD28 or 4-1BB costimulatory domains. We systematically compared the preclinical in vitro and in vivo efficacy of T cells lentivirally transduced with CD33 CAR constructs (CD33CARTs) against human AML.
Results We observed potent in vitro cytokine production and cytotoxicity of CD33CARTs incubated with human CD33+ AML cell lines, as well as robust in vivo antileukemia activity in cell line and childhood AML patient-derived xenograft (PDX) models. Gemtuzumab-based CD33CARTs were unexpectedly toxic in vivo in animal models despite observed in vitro anti-leukemia activity. CD28-based CD33CARTs consistently induced more robust inhibition of leukemia proliferation in AML cell line and PDX models than did 4-1BB-based CD33CARTs. A ‘best-in-class’ lintuzumab-CD28/CD3ζ CAR construct was thus selected for clinical translation.
Conclusions CD33 is a critical antigen for potential immunotherapeutic targeting in patients with AML. Based on this rigorous preclinical evaluation, our validated clinical grade lintuzumab-CD28/CD3ζ CD33CART immunotherapy is now under evaluation in a first-in-child/first-in-human phase 1 clinical trial for children and adolescents/young adults with relapsed/refractory AML.
Trial registration number clinicaltrials.gov; NCT03971799.
- pediatrics
- receptors
- chimeric antigen
- immunotherapy
- adoptive
- translational medical research
- hematologic neoplasms
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information. All data relevant to the study are included in the article or uploaded as supplementary information. Data are available upon reasonable written request and with execution of appropriate institutional material transfer agreements.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- pediatrics
- receptors
- chimeric antigen
- immunotherapy
- adoptive
- translational medical research
- hematologic neoplasms
Introduction
Acute myeloid leukemia (AML) is the second most common leukemia occurring in children and adolescents and the most common acute leukemia in adults. Despite intensive multiagent chemotherapy and frequent consolidation with allogeneic hematopoietic stem cell transplantation (HSCT),1–3 only 60% of children and adolescents/young adults (AYAs) with AML achieve long-term remission, and outcomes are dismal for those patients who relapse. New therapeutic strategies are needed to increase remission rates, decrease relapse and improve overall survival of these high-risk patients.
Chimeric antigen receptor (CAR) T cell immunotherapy targeting CD19 or CD22 has achieved landmark clinical success with dramatic remission rates reported in patients with relapsed/refractory B-acute lymphoblastic leukemia (B-ALL) or diffuse large B cell lymphoma, leading to four FDA-approved or ‘fast track’-designated cell therapy products.4–8 Translation of this success to CAR T cell immunotherapy for AML has long been similarly desired. Several antibody-based immunotherapies have been developed to overcome AML-associated chemotherapy resistance, including monoclonal antibodies, radiolabeled antibody conjugates, antibody-drug conjugates (ADCs), and bispecific T cell-engaging antibodies.9 Many immunotherapeutic agents developed to date target CD33 (also known as siglec-3 (sialic acid-binding Ig-like lectin 3)), a cell surface glycoprotein involved in normal myelopoiesis that is expressed on >80% of childhood and adult AML cases and, thus, highly amenable to CAR T cell targeting.10–12 In pediatric patients, high CD33 surface protein expression has been associated with highest risk AML genetic alterations and inferior clinical outcomes.11 12 The largest clinical experience with immunotherapy for patients with AML has been with gemtuzumab ozogamicin (GO), a CD33 ADC incorporating a calicheamicin toxin that induces DNA strand breaks, apoptosis, and cell death of CD33+ cells. Effective single-agent activity and successful combination of GO with multiagent chemotherapy led to its approvals by the Food and Drug Administration and European Medicines Agency in adults and children with de novo or relapsed AML, although GO addition to chemotherapy has not improved outcomes for all patients.13–16 Subsequent clinical investigation of other CD33-targeting immunotherapy agents also demonstrated initial activity in adult patients, including the humanized monoclonal antibody lintuzumab (HuM195, SGN-33) and its successor ADC, vadastuximab talirine (SGN-33A), although clinical trials of both drugs were later halted due to toxicity concerns.17 18 Early phase studies of CD33xCD3 bispecific antibody immunotherapies are also ongoing with preliminary safety and tolerability reported recently by some studies.19 20 A recent small single-center phase 1 clinical trial of a CD33CART product with 4-1BB/CD3ζ endodomains in adults with relapsed/refractory AML-reported appreciable difficulty with autologous T cell apheresis, CAR T cell manufacturing, and/or infusion in this heavily pretreated patient population,21 highlighting the anticipated challenges of successful AML CAR T cell actualization.
Preclinical and clinical studies of CAR T cell immunotherapies have clearly demonstrated that variables in CAR construct design can appreciably affect T cell biology and antitumor efficacy.22 23 Best established is the impact of the T cell costimulatory domain included in the CAR construct. In the context of CD19-redirected CAR T cell immunotherapy (CD19CART) for B-cell malignancies, CD28-based products usually result in earlier expansion with decreased longer term persistence, while 4-1BB (CD137)-based CD19CART drives slower expansion and engenders prolonged T cell persistence.24–27 Based on the growing clinical experience with various CD19 CAR T cell products in patients with B-ALL or lymphoma, the importance of early T cell activation and rapid expansion versus persistence appears to be disease dependent. Recent experience with CD22 CAR T cell immunotherapy in patients with B-ALL has also highlighted the critical nature of the specific target as relates to both antigen characteristics, such as site density, and specific single-chain variable fragment (scFv) binder selection.28 29 Collectively, these experiences with clinical translation of CAR T cell immunotherapy indicate that optimal design depends on specific target antigen and disease characteristics.
In an effort to develop effective cellular immunotherapy for children and AYAs with AML,30 we systematically and rigorously evaluated optimal CAR construct designs. We selected CD33 as an ideal myeloid target antigen for first-in-child CAR T cell immunotherapy clinical development given its high surface expression on most AML cases, association between highest CD33 expression and inferior clinical outcomes in children with de novo AML11 and demonstrated improved relapse-free survival for a subset of children with de novo or relapsed AML treated with combined GO and chemotherapy.12 14 31 Importantly, increased veno-occlusive disease/sinusoidal obstruction syndrome potentially due to CD33 expression on normal hepatic Kupffer cells and/or calicheamicin itself was not observed with gemtuzumab addition to chemotherapy.14 32 Herein, we report the iterative preclinical development of second-generation CD33-targeted CAR T cells (CD33CART) containing CD3ζ and CD28 or 4-1BB costimulatory endodomains based on extensive in vitro and in vivo testing in human AML cell lines and childhood AML patient-derived xenograft (PDX) models. Data from these studies resulted in selection of an optimized second-generation CD28/CD3ζ CD33 CAR construct (CD33CARTs) containing a lintuzumab-derived single-chain variable fragment (scFv) and transduced T cell product that is now under clinical evaluation in children and AYAs with relapsed/refractory AML via a Pediatric Transplantation and Cellular Therapy Consortium-supported first-in-child/first-in-human phase 1 trial.
Methods
CD33CART design and T cell transduction
Six discrete second-generation CD33CARTs were developed using scFv sequences derived from published CD33 antibody sequences of GO (hP67.6),33 lintuzumab (SGN-33)34 and M195.34 These scFvs were linked with a CD8 hinge or CD28 transmembrane domain and paired with either 4-1BB (CD137) or CD28 costimulatory domains followed by a CD3ζ signaling domain. Constructs were cloned into a pELNS lentiviral vector backbone as described35 36 and as detailed in the online supplemental data. CD123 CAR constructs harboring a published 32 716 ScFv37 with 4-1BB or CD28 endodomains were used as positive controls in some experiments given previously demonstrated antileukemia activity in preclinical AML models.38–41 A green fluorescent protein (GFP)-encoding construct was used as a negative T cell transduction control (designated as (-)TC in the figures). Surface expression of CAR-transduced T cells was determined by flow cytometry using protein-L (ThermoFisher) as described42 43 or a biotinylated human siglec-3/CD33 protein (Acro Biosystems) followed by incubation with streptavidin-PE (BioLegend) for CD33CARTs and anti-FLAG for CD123-targeting CAR T cells (CD123CART).
Supplemental material
AML cell lines
Non-transduced parental and luciferase-transduced MOLM14, MV4;11, THP-1 and U937 human AML cell lines were used for in vitro and in vivo CD33CART testing as below and described.39 40 The CD33-negative human B-ALL cell line NALM-6 was used as a negative control in some studies. Cell lines were originally purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ) repository, and identity was confirmed via short tandem repeat analysis in our institutional core facilities. Cell lines were regularly tested to ensure that they were Mycoplasma-free.
In vitro analyses of CD33CART anti-leukemia activity
IL-2 and IFN-γ cytokine production and cytotoxicity assays were conducted as described39 44 with some modifications using human AML cell lines coincubated with vehicle, GFP-transduced T cells ((-)TC), one of six experimental CD33CARTs or CD123CARTs as a positive control. Complete assay methods are detailed in online supplemental data.
In vivo analyses of CD33CART anti-leukemia activity in xenograft models
CD33CART expansion and the ability of CD33CARTs to inhibit AML proliferation in vivo were assessed in bioluminescent human AML cell line xenograft models or primary PDX models using NSG or busulfan-conditioned NSGS mice as described39 40 and as detailed in online supplemental data. Animal experiments were conducted under protocols approved by the National Cancer Institute (NCI) and Children’s Hospital of Philadelphia (CHOP) Institutional Animal Care and Use Committees and in accordance with National Institutes of Health laboratory animal guidelines. Viably cryopreserved primary childhood AML specimens were banked and coded without patient identifiers at CHOP via institutional review board (IRB)-approved research protocols after obtainment of informed consent in accordance with the Declaration of Helsinki. Use of coded human leukemia specimens for PDX modeling was considered non-human subjects research by the CHOP IRB.
Statistical analyses
Data display and statistical analyses of in vitro and in vivo studies were performed using Prism (V.8.0) as described. Treatment groups were compared via Student’s t-test (two groups) or ANOVA (three or more groups) to assess for statistical significance. Post-tests for multiple comparisons following ANOVA was performed for several in vitro and in vivo studies. Specific statistical methods for relevant experiments are designated in the figure legends.
Results
Preclinical development of CD33CARTs
To develop the most effective CD33CART for future clinical translation, we created six second-generation CAR constructs using three different CD33 scFvs, 4-1BB or CD28 costimulatory domains, and a downstream CD3ζ stimulation domain (figure 1A). Protein L detection showed generally similar T cell transduction efficiency of CD33CARTs for each scFv irrespective of whether the construct was generated with a 4-1BB or a CD28 endodomain (figure 1B), although transduction efficiency varied among the three CD33 scFvs (online supplemental table 1). CD123CARTs were generated as positive controls for AML cell killing39 40 and also demonstrated high transduction efficiency. Activation markers expressed on CAR-transduced T cells varied by specific antigen ScFv and intracellular costimulatory domains (online supplemental figure 1).
Supplemental material
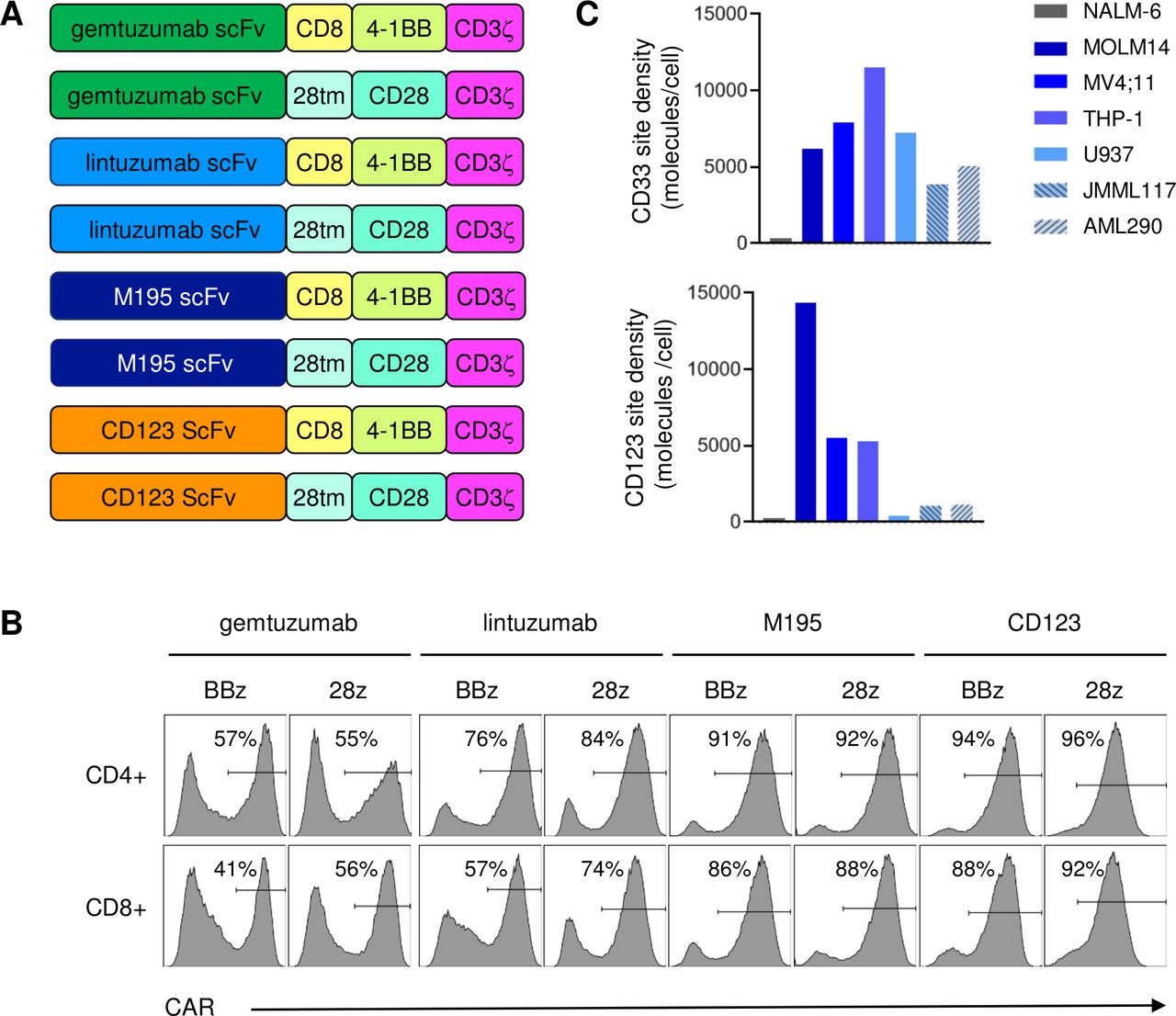
CD33 chimeric antigen receptor construct (CD33CART) design and validation. (A) Schema of second-generation CD33-redirectedCAR constructs. Single-chain variable fragments (ScFvs) were derived from commercially available CD33 antibodies gemtuzumab ozogamicin (hP67.6; green), lintuzumab (SGN-33; light blue), and M195 (dark blue) and paired with a CD8 hinge or CD28 transmembrane (28tm) domain linker, 4-1BB or CD28 costimulatory endodomain, and a CD3ζ endodomain (designated BBz and 28z, respectively). CD123 CAR T cells with a 32 716 ScFv were used as a positive control for some studies. (B) Flow cytometry analysis of protein L expression demonstrating transduction efficiency of T cells transduced with gemtuzumab-based (gem), lintuzumab-based (lin), and M195-based CD33 CAR constructs or CD123 CAR constructs. (C) Flow cytometry quantification of CD33 site density (molecules/cell) on AML cell lines (MOLM14, MV4;11, THP-1, U937) and childhood acute myeloid leukemia (AML) patient-derived xenograft (PDX) models (JMML117, AML290) used for in vitro and/or in vivo testing of CD33CARTs. CD123 site density is shown as a comparator. The B-ALL cell line NALM-6 was used as a CD33-negative control.
CD33 antigen site density of AML cell lines and PDX models
Assessment of human AML cell lines via PCR assay and Sanger sequencing for CD33 rs12459419 single-nucleotide polymorphisms associated with alternatively spliced isoforms and differential CD33 surface protein expression that may or may not influence response to gemtuzumab45–49 detected the CC genotype in MOLM14 and THP-1 and the CT genotype in MV4;11 and U937 (online supplemental table 2). Quantification of surface protein expression (‘site density’ measured as molecules per cell) via flow cytometric analysis of human AML cell lines and childhood AML PDX models JMML117 and AML29040 demonstrated a range of CD33 and CD123 expression, thereby allowing us to evaluate the potential anti-leukemia activity of CD33CARTs against lower and higher target antigen states in vitro and in vivo (figure 1C).
In vitro activity of CD33CARTs against AML
We then evaluated the potential of different AML cell lines to stimulate in vitro cytokine production by CD33CARTs in an in vitro coincubation assay. IL-2 and/or IFN-γ production was detected for all six scFv and costimulatory domain CD33CART configurations and with CD123CART; the highest cytokine production was induced by the MV4;11 and THP-1 cell lines (figure 2A). Importantly, we did not detect cytokine production following coculture of CD33CARTs with CD33-negative NALM-6 B-ALL cells, indicating minimal background activity in the absence of target antigen. Viability assays using the Cell-Titer Glo system (Promega) also showed potent inhibition of in vitro AML cell growth with all tested CD33CARTs and CD123CARTs using two different healthy T cell donors (figure 2B). Modest inhibition of NALM-6 cell growth with CD123CARTs was also observed, consistent with the known CD123 positivity of some B-ALL cells and with our and others’ prior studies,41 50 but not with CD33CARTs. However, no clear anti-AML superiority of one CD33CART product was identified in these in vitro assays.

Robust in vitro anti-leukemia activity of CD33 chimeric antigen receptor constructs (CD33CARTs). (A) CD33CART or CD123CART (positive acute myeloid leukemia (AML) killing control) cells were co-incubated with target-positive AML cells in vitro in a 1:1 ratio as indicated for 18 hours, and IL-2 and IFN-γ levels were quantified by ELISA in the culture supernatant. NALM-6 was again used as a negative control cell line. Experiments were performed in triplicate (technical replicates) using two normal T cell donors (biologic replicates). Data are displayed as mean±SD. (B) Cell-Titer Glo viability assays of human AML cell lines co-incubated with CD33CARTs or CD123CARTs in 1:1 effector: T cell show marked inhibition of cell growth over time with all tested CAR T cell products using two normal T cell donors. The B-ALL cell line NALM-6 was used as a CD33-negative control. T cell transduction efficiency ranged from 41% to 94% (mean 75%) depending on the CAR construct used. 28z=CD28/CD3ζ endodomains, BBz=4–1BB/CD3ζ endodomains. Cytokine and viability assays were performed in technical triplicates with biologic duplicates (T cell donor number 1 and number 2). Mean data are displayed for each condition with SD also shown for the viability assays. Data were analyzed by two-way ANOVA with multiple comparisons analysis of column means. All six CD33CARTs induced statistically significant killing of CD33 +AML cell lines at day 4 versus no T cell or GFP-transduced ((-)TC) negative controls (p<0.0001 for all comparisons), but were not statistically different from one another.
In vivo activity of CD33CARTs in AML cell line xenograft models
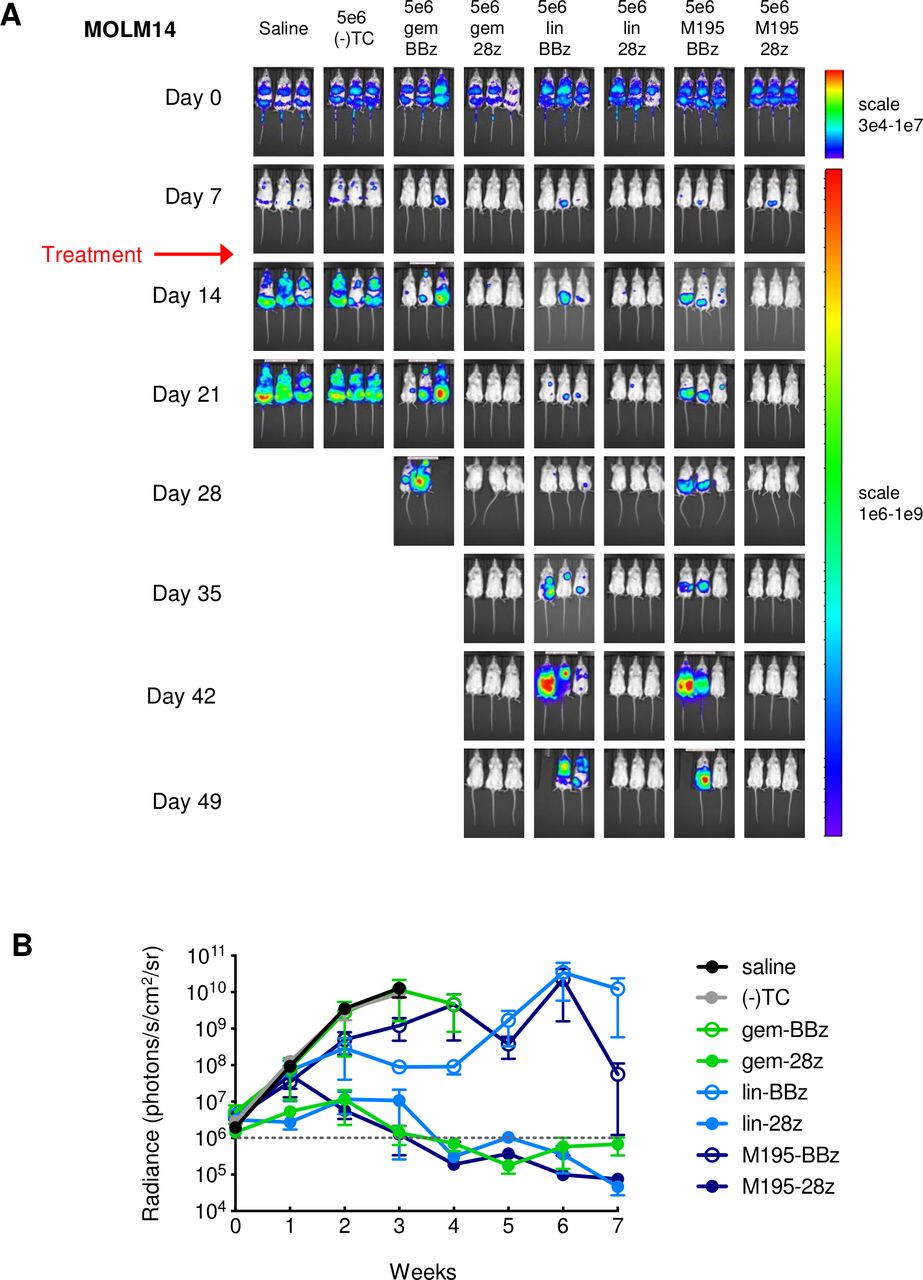
We next assessed the ability of CD33CARTs to inhibit leukemia proliferation in vivo using luciferase-transduced human AML cell lines engrafted in NSG mice. Treatment of CD33-bright MOLM14 xenograft mice with saline or (-)TC showed similar rates of leukemia progression by bioluminescent imaging (BLI) requiring animal euthanasia within 3 weeks of AML engraftment. Notably, potent anti-leukemia activity was observed following treatment with a single dose (5×106 cells) of five of the six CD33CARTs, facilitating long-term leukemia-free survival in many cohorts. Surprisingly, we observed minimal efficacy of gemtuzumab-4-1BB/CD3ζ CD33CART in this study, while CD33CART comprised of the same gemtuzumab ScFv with CD28/CD3ζ costimulatory domains efficiently eliminated leukemia. We further observed inhibition of MOLM14 proliferation with lintuzumab-4-1BB/CD3ζ and M195-4-1BB/CD3ζ CD33CARTs and curative effects of lintuzumab-CD28/CD3ζ and M195-CD28/CD3ζ CD33CARTs in several studies (representative data in figure 3). Long-term follow-up of lintuzumab-treated and M195-CD28/CD3ζ CD33CART-treated mice in this MOLM14 study until 6 months post-T cells demonstrated no evidence of BLI-detectable leukemia recurrence (data not shown).

CD33 chimeric antigen receptor construct (CD33CART) inhibits acute myeloid leukemia (AML) proliferation in vivo. (A) Luciferase-transduced MOLM14 cells (1×106) were injected intravenously via tail vein into NSG mice on day 0. Once engraftment was documented by bioluminescent imaging (BLI) on day 7, cohorts of three mice were randomized to intravenous treatment with saline, GFP-transduced T cells ((-)TC), or one of the CD33CARTs as designated (5×106 total cells/mouse; red arrow). Mice were followed by weekly BLI and euthanized when a predetermined maximal radiance level of 1×106 photons/s/cm2/sr was detected, indicative of leukemia progression. All three gemtuzumab-based (gem), lintuzumab-based (lin), and M195-based CD33CARTs demonstrated some anti-leukemia activity in vivo with complete inhibition of MOLM14 proliferation observed only in the 4-1BB/CD3ζ (BBz) construct-containing T cells versus partial activity with CD28/CD3ζ(28z) versions. (B) Graphic representation of summary BLI radiance data was performed in Prism. 28z, CD28/CD3ζ endodomains, BBz, 4–1BB/CD3ζ endodomains.
Dose titration experiments of CD28/CD3ζ-based CD33CARTs using a different healthy T cell donor in a subsequent MOLM14 xenograft experiment showed subcurative activity of 1×106 CAR T cell dosing, but effective leukemia eradication at both 5×106 and 1×107 total cell dosing of gemtuzumab-CD28/CD3ζ CD33CARTs and lintuzumab-CD28/CD3ζ CD33CARTs (online supplemental figure 2). The M195-CD28/CD3ζ CD33CART overall had less activity in eradicating MOLM14 in this study regardless of tested cell dose and was not ultimately curative. These data limited our further exploration of M195-CD33CARTs in later studies. We further confirmed greater activity of CD28/CD3ζ- over 4-1BB/CD3ζ-based CD33CARTs in bioluminescent MV4;11 and THP-1 AML cell line xenograft mice with superiority of lintuzumab-C33CARTs in these models, although some CD33CARTs only partially inhibited leukemia proliferation (online supplemental figures 3 and 4). We again observed less leukemia clearance and more toxicity with gemtuzumab-C33CARTs in both MV4;11 and THP-1 xenograft models with decreased murine weight, hypothermia, and lethargy that led to early death in many animals and was initially posited to be secondary to tumor lysis syndrome or cytokine release syndrome (CRS) given detectable CD33CART cells in murine peripheral blood. Flow cytometry analysis of bone marrow, spleen and liver tissues from gemtuzumab-CD33CART-treated cell line xenograft mice with residual leukemia showed that these sites were leukemia-free, while residual leukemia was detectable in (-)TC-treated cohorts (data not shown). However, additional studies of naïve NSG mice (without engrafted leukemia) treated with gemtuzumab-C33CART also resulted in frequent early animal death in the absence of known target antigen, suggesting possible murine cross-reactivity, xenogeneic graft-versus-host disease, or other non-specific toxicity (data not shown). These findings led us to conclude that while gemtuzumab-CD33CARTs harbor anti-AML activity, they may not be the best choice for further translation to the clinic.
In vivo activity of CD33CARTs against AML patient-derived xenograft models
Given our goals of early phase clinical trial testing of CD33CART in children with high-risk AML, we then sought to validate the preclinical in vivo activity observed in the AML cell line xenograft studies specifically in PDX models of CD33+ childhood AML. We observed potent inhibition of leukemia proliferation in an aggressive chemorefractory monosomy 7/PTPN11-mutant AML PDX model (JMML117) with superior activity of lintuzumab-CD33CARTs and M195-CD33CARTs versus gemtuzumab CD33CARTs (figure 4). CD28/CD3ζ-based CD33CARTs again induced greater anti-leukemia effects than did 4-1BB/CD3ζ-based CD33CARTs. Additional studies in a relapsed KMT2A-partial tandem duplication/FLT3-ITD AML PDX model (AML290) showed early gemtuzumab-CD33CART-induced toxicity of unclear etiology that required early animal euthanasia, similar to that observed in some AML cell line xenograft and naïve NSG mouse studies. Conversely, lintuzumab-CD33CARTs were well tolerated and effectively curative with enhanced activity again seen with CD28/CD3ζ-based vs 4-1BB CD3ζ-based T cells. Robust in vivo CD33CART expansion was also detectable by quantitative flow cytometry analysis of murine peripheral blood and end-study bone marrow and/or spleen tissues (figure 4).

CD33 chimeric antigen receptor construct (CD33CART) inhibits leukemia proliferation in vivo in childhood acute myeloid leukemia (AML) patient-derived xenograft models. Busulfan-conditioned NSGS mice engrafted with primary AML cells (tertiary PDX models (A) JMML117 and (B) AML290) were randomized (n=5 mice/cohort) to intravenous treatment with saline, irrelevant target GFP-transduced T cells ((-)TC), or one of the CD33CARTs as designated (5×106 total cells/mouse). CD33CART transduction efficiency ranged from 19.9% to 88.3% for these experiments with information detailed in online supplemental table 1. Mice were followed by weekly flow cytometric quantification of human CD33+/CD45 +AML and CD3+ CAR T cells in peripheral blood. Animals were euthanized at 2 or 4 weeks after treatment depending on rate of leukemia progression in control animals or >15% wt loss. AML cells and T cells were quantified in murine bone marrow and/or spleens as above. Superior leukemia clearance was again seen with the lintuzumab-based CD33CART with a CD28/CD3ζendodomain (lin-28z). *P<0.05, **p<0.01, ***p<0.001 by ANOVA with Dunnett post-test for multiple comparisons of treatment group versus saline control (black bars). Absence of symbol indicates lack of statistical significance.
Based on our collective in vitro and in vivo preclinical data, we selected the lintuzumab-CD28/CD3ζ CD33CART for clinical translation with the aim of best-achieving desirable on-target/on-tumor anti-AML efficacy and minimizing undesirable on-target/off-tumor toxicity risk due to long-term persistence.
Immunophenotype of CD33CARTs
Differences in the kinetics and accumulation of CD28-and 4-1BB-based CAR T cells have been reported in preclinical studies, and the majority of CD19CART clinical data in patients with B-cell malignancies have demonstrated greater long-term persistence of 4-1BB-based CAR T cells.51 Given potential for on-target/off-tumor targeting of non-malignant CD33+ myeloid cells, it may be clinically advantageous to minimize the long-term persistence of CD33CARTs. We first evaluated the phenotype of lintuzumab-CD28/CD3ζ and −4-1BB/CD3ζ CAR T cells and found that surface expression of the CD33CARTs increased following ex vivo co-culture with MOLM14 cells (online supplemental figure 5A), which was associated with complete leukemia eradication (online supplemental figure 5B). Moreover, CD33 antigen-induced stimulation of both CD33CARTs via MOLM14 coculture resulted in upregulation of T cell activation markers CD25 and CD71 on both CD4+ and CD8+ cells. However, CD25 upregulation was significantly higher for the CD28/CD3ζ as compared with the 4-1BB/CD3ζCD33CART at the studied time points (online supplemental figure 5C–E).
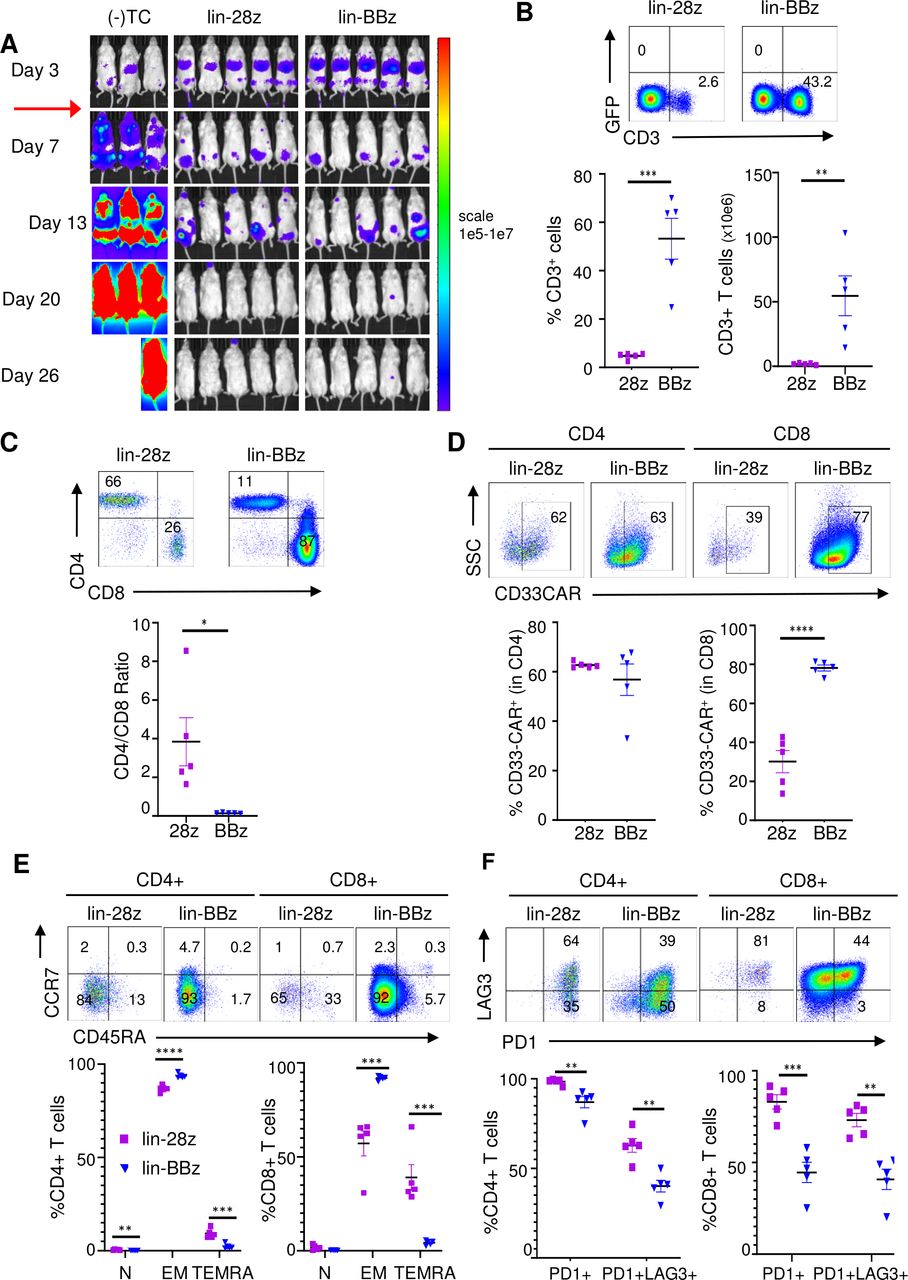
To assess whether or not these differences in the ex vivo activation status of lintuzumab-CD28/CD3ζ and −4-1BB/CD3ζ CAR T cells were associated with differential in vivo accumulation, we assessed CD33CART persistence in MOLM14-xenografted NSG mice at 4 weeks following adoptive CAR T cell transfer (figure 5A). Notably, we detected a significantly higher percentage of CD3+ T cells in the spleens of mice treated with CD33CARTs harboring 4-1BB/CD3ζ versus CD28/CD3ζ costimulatory domains at this relatively later experimental time point (figure 5B), suggesting preferential persistence of the former T cell products. Furthermore, the ratio of T cells transduced with the 4-1BB/CD3ζ was markedly skewed to the CD8 lineage (figure 5C) with the majority of these cells expressing surface CD33 CAR (figure 5D). Most notably, the phenotype of both CD4 and CD8 CAR T cells differed as a function of the included costimulatory domain with a significantly higher percentage of effector memory (EM) cells within the 4-1BB/CD3ζ-based as compared with the CD28/CD3ζ-based CD33CART population. Conversely, CD28/CD3ζ-based CD33CART exhibited a higher percentage of EM T cells re-expressing CD45RA (TEMRA; figure 5E), a subset that has been associated with defective long-term function.52 This phenotype could be of therapeutic interest in the context of a CD33CART where long-term anti-myeloid cell function would not be desirable. Furthermore, CD28/CD3ζ-based CD33CART showed markedly higher expression of the PD-1 and LAG3 exhaustion markers at the studied time points (figure 5F). Taken together, our preclinical data suggest that the lintuzumab-CD28/CD3ζ-based CD33CART could be specifically advantageous for future clinical translation due to its early potency and potential for less bystander toxicity given shorter T cell persistence in vivo, although potential for exhaustion may exist.

In vivo exhaustion potential of lintuzumab-based CD33 chimeric antigen receptor constructs (CD33CARTs) in an acute myeloid leukemia (AML) xenograft model. (A) Luciferase-expressing MOLM14 xenograft mice were created as in figure 3. Cohort of five mice were treated with one 4×106 dose of GFP-transduced T cells ((-)TC), lintuzumab-CD28/CD3ζ (lin-28z), or lintuzumab-4-1BB/CD3z (lin-BBz) CD33CART intravenous on day 4 (red arrow), then followed by bioluminescent imaging. CD33CART transduction efficiency ranged from 19.9% to 88.3% for these experiments with information detailed in online supplemental table 1. (B) Representative dot plots show flow cytometric detection of percentages of human CD3+ CD33 CART cells in harvested end-study murine spleens at day 26 following T cell treatment (upper panels). T cell quantification (lower panels) detected greater numbers in lin-BBz-treated versus lin-28z-treated animals. (C) Representative dot plots show flow cytometric analysis of CD4/CD8 T cell profiles (upper panels) with ratio calculation (lower panel) in end-study murine spleens. (D) Similar percentages of cell surface CD33 CAR positivity (bound to recombinant CD33-Fc fusion protein) were detected in CD4+ T cells, but not CD8+ T cells, when comparing lin-28z and lin-BBz CD33CARTs. (E) Naïve (N), effector memory (EM), and effector memory re-expressing CD45RA (TEMRA) human T cells in murine spleens at the day 26 end-study timepoint were further characterized by CCCR7+ CD45RA+, CCR7-CD45RA-, and CCR7-CD45RA+ flow cytometric profiling, respectively. Representative plots for CD4+ and CD8+CD33CARTs are shown (upper panels) and quantification of the different populations within the CD4+ (left) and CD8+ (right) subsets are shown. (F) Exhaustion phenotypes of CD33CARTs were evaluated as a function of PD1 and PD1/LAG3 expression profiles and representative dot plots are presented with %cells designated numerically for each gated quadrant (top). Quantification of PD1+ and PD1+/LAG3 +CD4+ and CD8+ T cells in murine spleens at the day 26 end-study timepoint are shown for lin-28z versus lin-4BBz CD33CART treatments. Paired data were analyzed by Student’s t-test with *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Evaluation of potential on-target/off-tumor toxicity of CD33CART
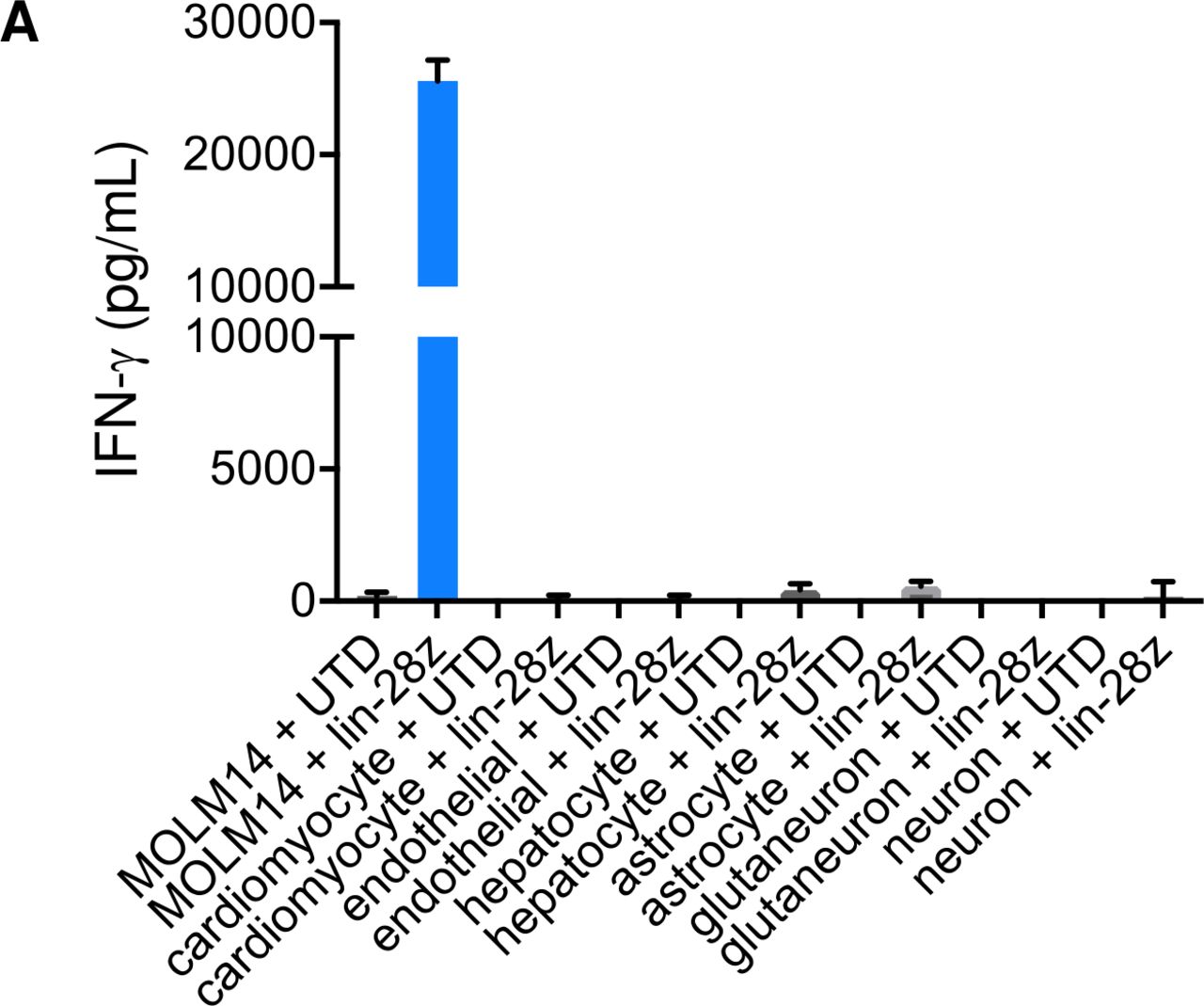
We and others previously demonstrated hematologic toxicity of CD123CART and CD33CART in preclinical AML models,39 40 53 which is an expected potential sequela of CAR T cells targeting CD33 or other myeloid antigens that could require allogeneic HSCT rescue of patients.53 54 To address potential on-target/off-tumor and off-target/off-tumor toxicity of CD33CART outside of the hematopoietic compartment that could also hinder successful clinical development, we co-incubated lintuzumab-CD28/CD3ζCD33CART with induced pluripotent stem cell (iPSC) lines representing normal human cardiac, endothelial, hepatic, and neural tissues and measured in vitro cytokine production as done in earlier AML cell line experiments. As expected, high levels of IFN-γ were detected with coculture of CD33CARTs with CD33+ MOLM14 cells (positive control). Notably, minimal or no cytokine production was detected with any of the normal tissue iPSC lines (figure 6). These data suggest lack of obvious toxicity of CD33CART against normal non-hematopoietic tissues.
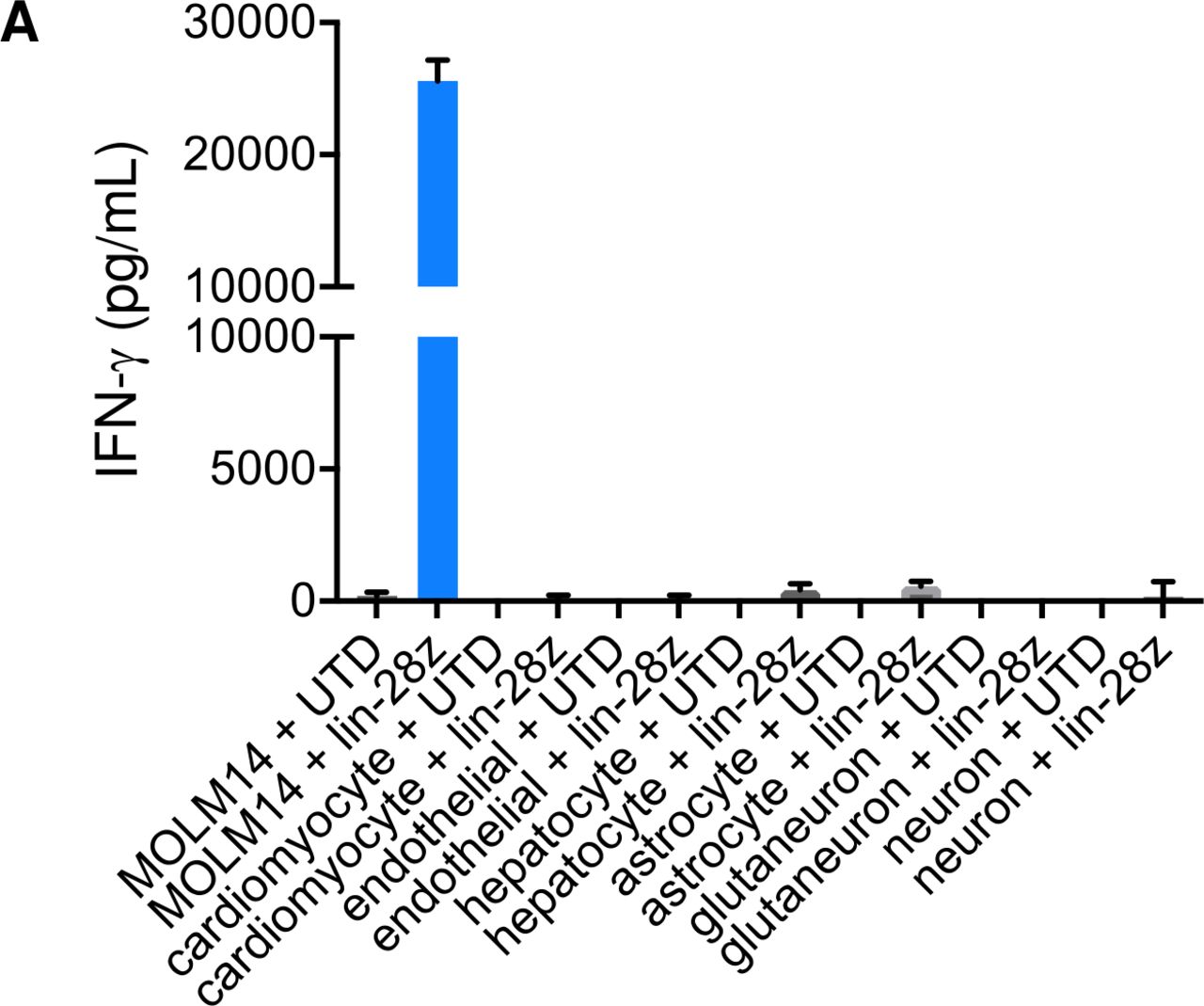
Minimal bystander toxicity of clinical CD33 chimeric antigen receptor construct (CD33CART) product. (A) 1×105 GFP-transduced T cells ((-)TC) or clinical-grade lintuzumab-CD28/CD3ζ (lin-28z) cells were co-incubated in a 1:1 ratio with induced pluripotent stem cell lines of the designated normal tissue types for 18 hours. IFN-γ levels were quantified by ELISA in the culture supernatant as in figure 2. MOLM14 cells were used as a CD33 +positive control. No appreciable cytokine production was detected in any of the normal tissue conditions.
Validation of clinical grade CD33CART activity
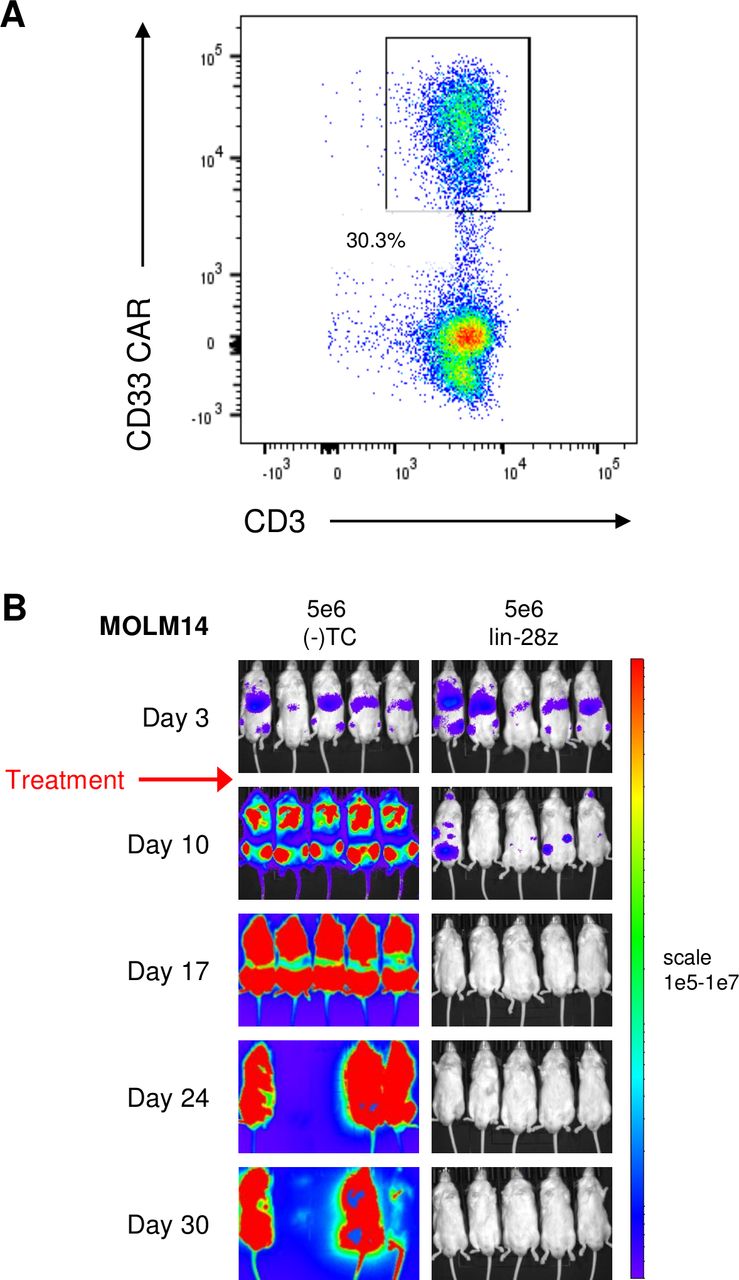
In preparation for phase 1 pediatric clinical trial evaluation, we manufactured clinical grade lintuzumab-CD28/CD3ζCD33CART from a healthy T cell donor using a Prodigy system (Miltenyi) at the Biopharmaceutical Development Program of the Frederick National Laboratory for Cancer Research under contract from the NCI. Confirmatory testing of the Prodigy-produced lintuzumab-CD28/CD3ζCD33CART in our bioluminescent MOLM14 xenograft model demonstrated swift leukemia eradication within 3 weeks of T cell treatment (figure 7), consistent with our preclinical research-grade studies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Preclinical validation of anti-leukemia activity of clinical-grade CD33 chimeric antigen receptor construct (CD33CART). The lintuzumab-based CD33CART with a CD28/CD3ζendodomain (lin-28z) was selected for further development and evaluation in a phase 1 clinical trial. (A) Transduction efficiency of clinical grade lin-28z CD33CART was 30.3% for this experiment. (B) Luciferase-transduced MOLM14 cells (1×106) were injected intravenous into NSG mice on day 0. Animals were assessed by bioluminescent imaging (BLI) and then randomized to treatment with GFP-transduced T cells ((-)TC) or clinical-grade lin-28z CD33CART cells (5×106 total cells/mouse) on day 3 (red arrow). Mice were followed by weekly BLI and euthanized when a predetermined maximal radiance level of 1×1010 photons/s/cm2/sr was detected Complete inhibition of leukemia proliferation was again observed with CD33CART.
Discussion
Developing effective CAR T cell immunotherapy for AML represents the next frontier in adoptive cellular therapy but has been challenging to date due to multiple factors. The lack of a universal AML antigen analogous to the ‘holy grail’ of CD19 in B-ALL highlights a pressing need for development of multiple approaches for different immunophenotypic subsets if appreciable cure of a broad range of high-risk chemorefractory patients is to be achieved. Most clinical immunotherapies to date have focused on targeting of CD33 and CD123 given their expression on the majority of AML cases,9 although both antigens are also expressed at lower levels on normal myeloid precursor cells. Desire for potent anti-AML efficacy, thus, must be equally balanced by careful attention to potential hematologic and non-hematologic toxicity, and HSCT rescue of patients may be required to mitigate risk of on-target/off-tumor CAR T cell immunotherapy-induced marrow aplasia. Early clinical experiences with cellular immunotherapy for adults with AML targeting CD123 have demonstrated safety and feasibility, although with limited remissions reported to date, as summarized in recent reviews.9 30 A single-case description of an adult with relapsed/refractory AML treated with a lentivirally transduced CD33CART comprised of an AM402974.1-derived CD33 ScFv and CD28/CD3ζ costimulatory domains also reported grade 3 CRS and achievement of initial leukemia remission, although the patient subsequently relapsed with CD33+ AML 9 weeks after receipt of CD33CART therapy and died of progressive disease.55 As aforementioned, a single-institution phase 1 trial (NCT03126864) of autologous CD33CART comprised of an unknown CD33 scFv and 4-1BB CD3ζ costimulatory domains with a truncated EGFR suicide switch reported successful infusion of 3 of 10 enrolled adult patients with relapsed/refractory AML at 0.3×10e6 CAR+ cells/kg dosing. Importantly, no dose-limiting toxicity was observed. Despite detection of CD33CART in peripheral blood, all three treated patients were removed from study by 31 days post-CD33CART due to disease progression and subsequently died of progressive AML.21
In this study, we report preclinical development of six second-generation CD33CARTs comprised of different ScFvs from clinically tested antibodies and intracellular costimulatory domains that differentially affect T cell potency and persistence in the context of CD19CART for B-cell malignancies. Rigorous in vitro and in vivo testing of these CD33CARTs against AML cell lines and pediatric AML PDX models facilitated critical biologic observations and enabled selection of an optimized T cell product that is now under clinical evaluation in a phase 1 trial in children and AYAs with relapsed/refractory AML. Importantly, we detected active cytokine production and cytotoxicity with most CD33CARTs coincubated with a range of CD33-expressing AML cell lines at levels analogous to those produced by our CD123CART positive controls, which provided enthusiasm for subsequent detailed in vivo assessment in preclinical leukemia models. We unexpectedly observed appreciable toxicity of gemtuzumab-based CD33CARTs, which was surprizing given demonstrated clinical efficacy of gemtuzumab in patients with AML.13 14 31 Conversely, lintuzumab-based and CD28/CD3ζ-based CD33CARTs consistently demonstrated superior inhibition of leukemia proliferation and long-term ‘cure’ in our AML xenograft models with seemingly minimal toxicity for up to 6 months of post-T cell observation in some models, although showed clear potential for exhaustion in our detailed T cell immunophenotyping analyses. It is plausible that differences in transduction efficiency of CD33CARTs influenced in vivo responses observed in xenograft model studies, although similar activity of all six CD33CARTs was detected in our in vitro assays. Finally, we validated potent anti-AML activity of the selected lintuzumab-CD28/CD3ζ CD33CART via clinical grade T cell manufacturing that is now under investigation in our pediatric phase 1 trial.
While our results refuted our initial hypothesis that gemtuzumab- and/or 4-1BB/CD3ζ CD33CARTs would have best efficacy against AML, as was shown by the groups of Kenderian et al,53 Schneider et al,49 Li et al,56 and Tambaro et al,21 our careful comparative preclinical investigation led to alternative selection of a lintuzumab-CD28/CD3ζ CD33CART as best-in-class in these studies for clinical translation. Choice of a shorter-acting T cell product with a CD28 co-stimulatory domain that is capable of inducing rapid remission may actually be advantageous in patients with relapsed/refractory AML by providing a safety mechanism against potentially life-threatening myelosuppression, although could risk occurrence of leukemia relapse if patients do not rapidly proceed to HSCT. While prolonged normal B-cell aplasia following CD19CART-4-1BB/CD3ζ immunotherapy has appeared clinically tolerable in patients with long-term supportive immunoglobulin replacement therapy, it is plausible that persisting myeloid antigen-directed CAR T cells could induce severe myelosuppression, a bystander toxicity that is highly undesirable in this high-risk and usually heavily chemotherapy-pretreated patient population. As such, an initially potent, but less persistent, CD28/CD3ζ-based CD33CART could both efficiently eradicate disease and allow for more optimal normal hematopoietic cell recovery.
Our studies of the lintuzumab-CD28/CD3ζ CD33CART identified potential for T cell exhaustion that has been reported with other CD28-based CAR T cell products,24 although sustained leukemia remission was achievable in several of our preclinical xenograft models. As a precaution, our current CD33CART phase 1 clinical trial requires identification of an allogeneic stem cell donor prior to study enrollment as a precaution in case patients experience CAR T cell-induced aplasia requiring stem cell rescue or achieve leukemia remission that should be swiftly consolidated with HSCT to optimize long-term cure. Other safety approaches to limit CAR T cell toxicity and facilitate hematologic recovery particularly prior to allogeneic transplantation include incorporation of genetic suicide switches and drug-mediated T cell depletion (eg, herpes simplex virus thymidine kinase, induced caspase-9 with chemical inducer of dimerization drugs, cetuximab against truncated epidermal growth factor receptor, rituximab against synthetic CD20, alemtuzumab against native CD52 and steroids or other lymphotoxic chemotherapy), as well as CRISPR/Cas9 editing of hematopoietic stem cells for transplantation. It is certainly possible that conditioning chemotherapy or total body irradiation transplant preparative regimens would be sufficient to eradicate persistent CAR T cells, as well. These considerations will be critical to successful actualization of cellular immunotherapy to the clinic for patients with AML.
Limitations of our studies included inability to evaluate potential hematologic toxicity of CD33CARTs simultaneously with in vivo anti-leukemia efficacy in our preclinical cell line xenograft and PDX models, which was also an issue in our earlier CD123CART studies that informed recent clinical trials (NCT02623582, NCT03766126).39 40 It is also plausible that the robust preclinical activity of our lintuzumab-CD28/CD3ζ CD33CART will not translate to clinical responses experienced by patients or that this CD33CART will indeed become exhausted and permit rapid AML relapse before consolidative HSCT can be performed.
In summary, our studies demonstrate careful and clinically pragmatic preclinical optimization of CD33CARTs that has enabled early phase clinical investigation of a lintuzumab-CD28/CD3ζ T cell product in children and AYAs with second or greater relapsed/refractory AML in our multi-institutional first-in-child/first-in-human CD33CART phase 1 trial supported by the Pediatric Transplant and Cellular Therapy Consortium. Other preclinical and early phase clinical studies are now exploring the therapeutic potential of CAR T cells targeting additional AML antigens, bispecific CAR T cells, allogeneic or donor-derived CAR T cells, natural killer (NK) cells, and CAR-NK cells, which will surely collectively lead to continued progress in our quest to develop successful immunotherapeutic approaches for children and adults with chemoresistant or relapsed AML.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information. All data relevant to the study are included in the article or uploaded as supplementary information. Data are available upon reasonable written request and with execution of appropriate institutional material transfer agreements.
Ethics statements
Patient consent for publication
Acknowledgments
We acknowledge the Frederick National Laboratory of Cancer Research Biopharmaceutical Development Program (BDP) Cell Therapy manufacturing and quality control groups for production of clinical-grade CD33CART validated in our preclinical studies. We are grateful to the Pediatric Transplant and Cellular Therapy Consortium and National Marrow Donor Registry program team, the St Baldrick’s Foundation, the Frederick National Laboratory of Cancer Research BDP team, and the Children’s Hospital of Philadelphia vector production core facility for their roles in and support of the CD33CART clinical trial that ensued from these preclinical studies. This work is dedicated in memory and celebration of Miss Nia Symone Stratton.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SKT and TJF contributed equally.
Presented at Presented in part at the 50th Annual Congress of the International Society of Paediatric Oncology in November 2018 (Kyoto, Japan).
Correction notice This article has been corrected since it was first published. Middle initials have been added to author names ‘Nirali N Shah’ and ‘Naomi A Taylor’.
Contributors HQ designed and performed research, analyzed and interpreted data, and assisted with manuscript writing. LY and JAC performed research, analyzed data, and assisted with manuscript writing. NNS interpreted data and assisted with manuscript writing. JAC, ST, MP, CDC, and LMN performed research and analyzed and interpreted data. ARW provided scientific oversight of clinical-grade CD33CART manufacturing for validation studies. NAT contributed key experimental reagents and scientific expertise, analyzed and interpreted data, and edited the manuscript. SKT and TJF designed and directed the study, analyzed and interpreted data, and wrote and/or edited the manuscript. All authors approved the final version of the manuscript.
Funding This study was supported by the National Institutes of Health (NIH) Intramural Research Program (HQ, NS, CDC, NST, TJF), NIH/National Cancer Institute (NCI) T32CA009615 (LMN), K08CA184418 (SKT), and 1U01CA232486 (SKT, TJF), Andrew McDonough B+Foundation (SKT), Gabrielle’s Angel Foundation for Cancer Research (SKT), Gerdin Charitable Foundation (SKT), Press On Fund of the Community Foundation for the Central Savannah River Area (SKT), Rally Foundation for Childhood Cancer Research (SKT), SchylerStrong Foundation, and St Baldrick’s Foundation/Stand Up to Cancer Pediatric Dream Team (SKT, TJF). Stand Up to Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. NNS is an NIH Lasker Clinical Research Scholar. TJF was the Robert and Kathleen Clark Endowed Chair in Pediatric Cancer Therapeutics at the Children’s Hospital Colorado. We thank Ms Tiffany Hylkema and Dr Soheil Meshinchi at Fred Hutchinson Cancer Research Center for assistance with CD33 genotyping of AML cell lines.
Disclaimer The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Competing interests HQ and TJF have filed United States patent number WO2019178382A1 ‘Anti-CD33 Chimeric Antigen Receptors and Their Uses.’ SKT is a scientific advisory board member for Aleta Biotherapeutics and Kura Oncology and has received research funding from Incyte Corporation and Gilead Sciences for unrelated studies. TJF has received research funding from Lentigen and Elevate Bio for unrelated studies and is a current employee of Sana Biotechnology. The remaining authors declare no relevant conflicts of interest.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.