Article Text

Abstract
Background The clinical benefits of antiprogrammed cell death protein 1 (PD-1) therapy are compromised by resistance in immunologically cold tumors. Convergence of immunotherapy and bioengineering is potential to overcome the resistance. Mesoporous silica nanoparticles (MSNs) are considered the most promising inorganic biological nanomaterials for clinical transformation, however, the fundamental influence of MSNs on immunotherapy is unclear. In this study, we aimed to investigate the role of MSNs in tumor resensitization and explore the feasibility of MSNs combined with anti-PD-1 in cancer therapy.
Methods Intrinsic and acquired resistant tumors, as well as spontaneous and secondary tumor recurrence models, were used to evaluate the influence of MSNs and the synergistical effect with anti-PD-1 therapy. The roles of CD8+ cytotoxic T-lymphocytes (CTLs) and macrophages were assessed in Rag-1-/- mice, ovalbumin/OT-1 TCR transgenic T-cell system, and other blocking mice models. Mechanistic studies were processed by transcriptomics analysis and conducted in primary cells, in vitro coculture systems, and Toll-like receptor 4 (TLR4) knockout mice.
Results Both granular and rod-shaped MSNs efficiently overcame tumor resistance with dependence on diameter and aspect ratio. Only once injection of MSNs in prior to anti-PD-1 markedly improved the treatment efficacy, protective immunity, and prognosis. MSNs per se boosted infiltration of CTLs as the early event (days 2–3); and synergistically with anti-PD-1 therapy, MSNs rapidly established a T cell-inflamed microenvironment with abundant high-activated (interferon-γ/tumor necrosis factor-α/Perforin/GranzymeB) and low-exhausted (PD-1/lymphocyte-activation gene 3 (LAG-3)/T-cell immunoglobulin and mucin-domain containing-3 (TIM-3)) CTLs. Chemokines Ccl5/Cxcl9/Cxcl10, which were produced predominantly by macrophages, promoted MSNs-induced CTLs infiltration. MSNs led to high Ccl5/Cxcl9/Cxcl10 production in vitro and in mice through regulating TLR4-NFκB axis. Blocking TLR4-NFκB axis in macrophages or CTLs infiltration abrogated MSNs-induced resensitization to anti-PD-1 therapy.
Conclusions MSNs efficiently and rapidly inflame immunologically cold tumors and resensitize them to anti-PD-1 therapy through TLR4-NFκB-Ccl5/Cxcl9/Cxcl10 axis. MSNs-based theranostic agents can serve as sensitizers for patients with resistant tumors to improve immunotherapy.
- immunotherapy
- combined modality therapy
- programmed cell death 1 receptor
- lymphocytes
- tumor-infiltrating
- macrophages
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunotherapy
- combined modality therapy
- programmed cell death 1 receptor
- lymphocytes
- tumor-infiltrating
- macrophages
Background
Immune checkpoints are important self-tolerance regulators in immune system. Under normal physiological conditions, immune checkpoint molecules turn down the activation signals from costimulatory molecules to maintain self-tolerance and prevent autoimmunity.1 However, tumor cells can use this mechanism to disrupt T cell activation and functional status, resulting in T cell exhaustion and tumor immune escape.2 Immune-checkpoint-pathway inhibitors (ICIs), notably antiprogrammed cell death protein 1 antibodies (αPD-1), show impressive clinical success for various types of cancer.3 However, fewer than 50% of patients derive objective responses to PD-1 blockade.4 A major factor to prompt initial ICIs resistance is the so-called ‘immunological cold’ tumors, characterizing lack of tumor infiltrating lymphocytes.5 To destruct cold tumors and further improve therapeutic outcomes and efficacy of ICIs, combination treatments using radiotherapy, chemotherapy, vaccines and other adjuvants have emerged, aimed at reversing resistance, imparting potent therapeutic effects and reducing side effects.6
Biomaterials at the nanoscale have attracted substantial attention in biomedicine field.7 Nanomaterials feature unique physicochemical properties and biological effects, including tunable shape and size, ease of synthesis, and versatility in surface functionalization.8 9 Since they can be engineered for unique composition and functionality, nanomaterials have been extensively used in medical diagnosis, monitoring, and treatment of cancer.10 In addition to functionalized materials, exploration of nanomaterials without specific functional enhancements can provide a fundamental regular pattern for the field of biomaterials. Among these, mesoporous silica nanoparticles (MSNs) are representative of a new generation of nanomaterials due to properties such as large surface areas (~1000 m2/g) and high pore volumes, controllable particle size, excellent biocompatibility/biodegradability, desirable chemical and thermal stabilities, and high loading efficiency of guest molecules.11–13 Based on the intrinsic mesoporous nanostructure and high porosity, MSNs have been extensively explored for a variety of biomedical applications, including drugs/gene delivery, bioimaging, and cancer therapy.12 Therefore, it is highly worthy for exploring the biological effects of MSNs themselves (without additional functional transformation) on cancer treatment and immunotherapy.
Herein, we report the resensitizing effect of MSNs with different crucial structural parameters on anti-PD-1 immunotherapy. Both granular and rod-shaped MSNs, even only injected once, are able to turn cold tumors hot and overcome resistance to anti-PD-1 therapy in diameter- and aspect ratio-dependent manners. Through targeting Toll-like receptor 4 (TLR4)/NF-κB pathway in macrophages, MSNs promote the tumorous infiltration of CD8+ cytotoxic T-lymphocytes (CTLs). With PD-1 blockade, MSNs synergically activate and reverse the exhaustion of CTLs to result in a long-lasting antitumor immunity. Our findings provide an intriguing insight into the biological impact of MSNs per se on immunotherapy and facilitate the broad applications of nanomedicines in PD-1-targeted cancer immunotherapy.
Materials and methods
Chemicals
Cetyltrimethylammonium chloride (CTAC) was purchased from Sigma-Aldrich. Hexadecyl cetyltrimethylammonium bromide (CTAB), triethanolamine (TEA), tetraethyl orthosilicate (TEOS), ethanol, ammonium hydroxide (NH3·H2O) and hydrochloric acid (HCl) were obtained from Sinopharm Chemical Reagent Co. Methoxy polyethylene glycol (PEG) silane, MW 2000 (Me-PEG2000-silane) was bought from JenKem Technology. All the mentioned chemicals and regents were used directly without further purification. Deionized water (DI water) was used in all experiments.
Synthesis of spherical MSNs with tunable sizes
Typically, the spherical MSNs were synthesized using a simple soft-templating method. Through adjustment and match of CTAC and TEA, spherical MSNs with varied sizes were synthesized. To synthesize spherical MSNs with diameters of 100, 65 and 45 nm, 2 g CTAC was mixed with 0.01 g, 0.02 g and 0.06 g TEA, respectively, and the mixed solutions were then diluted in 20 mL DI water by ultrasonic dispersion at room temperature. Then, the solutions were placed in the thermostatic water bath at 80°C, 300 r/min stirring for 20 min. Afterwards, 1.5 mL TEOS was added dropwise into the above-mentioned solutions under magnetic stirring (300 r/min) and the solutions were stirred for another 4 hours to conduct the condensation process in the thermostatic water bath at 80°C. After cooling down to room temperature, the product was collected by centrifugation (18 000 r/min, 20 min) and washed for several times with ethyl alcohol and DI water to remove the residual reactants. Finally, to dislodge the surfactants (CTAC), the prepared MSNs were extracted by circulation reflux with HCl alcohol solution (10% HCl) at 78°C for three rounds. To observe the size and morphology of the spherical MSNs, transmission electron microscopy (TEM, JEM-2100F) and scanning electron microscope (SEM, SU9000) were applied.
Synthesis of rod-like MSNs with tunable aspect ratios
The rod-like MSNs with varied aspect ratios were synthesized based on a single cationic surfactant CTAB and a simple silica source TEOS via a sol-gel reaction. Take rod-like MSNs with a 3:1 length-to-diameter ratio (L/D), for example, a mixture of CTAB (0.2 g, 8.0 mmol), DI water (70 mL), and ammonium hydroxide (0.7 mL, 85.5 mmol) was stirred at 30°C for 30 min at 450 rpm in a 125 mL wide-mouthed bottle. Then, 1.5 mL TEOS was added into the above solution dropwise and the condensation process was conducted under stirring for another 4 hours in a water bath at 30°C. After cooling down to room temperature, the product was gathered by centrifugation (18 000 r/min, 15 min) and washed for three times with alcohol, DI water, and alcohol to remove the residual reactants. Each washing round involved a sonication of 20 min at room temperature. Finally, to remove the surfactants (CTAB), the prepared MSNs were extracted by circulation reflux with HCl alcohol solution (10% HCl) at 78°C for three rounds. After washing with alcohol, the MSNs with non-CTAB were dispersed in alcohol for further modification.
To synthesize MSNs with controllable L/D, the molar quantities of CTAB and NH3·H2O was modulated. Keeping other parameters constant, when the molar quantities of CTAB and NH3·H2O were 8.0 mM:85.5 mM, 11.8 mM:171.0 mM and 23.6 mM:171.0 mM, respectively, the L/D have undergone a corresponding change of 3:1, 4:1 and 8.5:1. To determine the L/D and observe the nanostructure of the as-prepared MSNs intuitively, TEM and SEM were also used.
Surface PEGylation of MSNs
To ensure the stability and monodispersity of MSNs in physiological condition, the as-synthesized MSNs were surface-functionalized with PEG. Typically, 10 mg as-prepared MSNs were dispersed into Me-PEG2000-silane ethanol solution (100 mL, 0.5 mg/mL) under stirring at 60°C overnight. Then, PEG-MSNs was recollected by centrifugation (18 000 r/min, 10 min) and rinsed with ethyl alcohol and DI water for several times to remove the redundant Me-PEG2000-silane. The final obtained PEGylated MSNs were redispersed in PBS solutions and stored under 4°C for further use in cellular and animal experiments.
Mice and cell lines
C57BL/6 and BALB/C male mice, 4–6 weeks old, were purchased from Shanghai Slac Laboratory Animal and maintained in a pathogen-free vivarium under standard conditions. Rag1-/-, OT-1 and tumor necrosis factor (TNFR1)-/- mice, with a C57BL/6 background, were purchased from Shanghai Model Organisms Center. TLR4-/- mice with a C57BL/6 background were kindly provided by Professor Yingying Le.14 SIBS, CAS. β2-microglobulin (B2m)-sgRNA B16F10 tumor cell line was created by using the lentiCRISPRv2 system to deliver sgRNAs against the protein-coding genes, and deletion was confirmed via western blot (Control sgRNA: GCGAGGTATTCGGCTCCGCG, B2m sgRNA: AGTATACTCACGCCACCCAC). B16F10-ovalbumin (OVA) tumor cell line was created by transducing with OVA-expressing lentivirus, and expression was determined via flow cytometry using established anti-mouse SIINFEKL:H-2kb antibody. B2m-sgRNA B16F10 and B16F10-OVA tumor cells were cultured in DMEM medium. H22 and CT26 tumor cells were cultured in RPMI medium. All mediums were supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 µg/mL streptomycin. The cell lines used in our study were tested to be free of mycoplasma contamination.
In vivo tumor models
In the therapeutic experiments, B2m-sgRNA B16F10 (2×105) tumor cells were implanted subcutaneously into the left flank of C57BL/6 mice. H22 (1.5×106) or CT26 (1.5×106) tumor cells were implanted subcutaneously into the left flank of BALB/C mice. Tumor-bearing (about 0.5 cm in diameter) mice were randomized and injected intraperitoneally with PBS vehicle or MSNs prior to anti-PD-1 treatment. Anti-PD-1 antibody (5 mg/kg, BioXcell) was injected intraperitoneally every two or 3 days. Tumor volume was determined with the formula 1/2a×b2 (where a is the long diameter, and b is the short diameter). Mice were killed when the tumor volume reached 3000 mm3. For the tumor rechallenge model, B2m-sgRNA B16F10 tumor cells (2×105) were implanted subcutaneously into the left flank of C57BL/6 mice. The tumors were treated at the indicated time points and resected 4 days after MSNs administration. Then we injected the secondary tumor cells (2×105) contralateral to the primary tumor a week after surgery. Mice were observed for tumor growth and survival. To block T cell infiltration, mice were injected with 25 µg FTY720 (APExBIO) on day 3 and 5 µg on day 4–6 after tumor inoculation. To deplete macrophages, mice were injected with 150 µL clodronate liposomes (FormuMax) as the first dose and 100 µL clodronate liposomes every 3 days subsequently. To deplete CD8+ T cells, mice were injected with 200 µg neutralizing anti-CD8 antibody (BioXcell) every 4 days. For adoptive T cell therapy model, B16F10-OVA tumor cells were implanted subcutaneously into the left flank of Rag1-/- mice. OT-1 T cells were isolated from the spleens of OT-1 mice using the EasySep Mouse CD8+ T Cell Isolation Kit (Stemcell). When tumor volume was close to 500 mm3, we adoptively transferred OT-1 T cells (8×105) into Rag1-/- mice-bearing B16F10-OVA tumor. Then, the mice were treated with MSNs or PBS prior to anti-PD-1 treatment. Mice were monitored for tumor growth. For biodistribution of MSNs in mice, B2m-sgRNA B16-F10 tumor-bearing mice were intraperitoneally injected with MSNs, and kidney, liver, lung, spleen and tumor tissue were harvested at 24 hours post-injection and dissolved in aqua regia for ICP-MS measurement. The content of Si in each organ was expressed as percentage of the injected dose per gram of tissue (% ID/g).
Antibodies and reagents
Anti-PD-1 antibody (clone: RMP1-14) and isotype control were obtained from BioXcell. The fluorochrome-labeled antimouse antibodies used for flow cytometry measurements were Fixable Viability Stain 780 (565388; BD Biosciences); PE-CD45 (553081; BD Biosciences); FITC-CD45 (553080; BD Biosciences); BV510-CD45 (563891; BD Biosciences); FITC-CD3e (553061; BD Biosciences); BV421-CD8 (563898; BD Biosciences); BV510-CD8 (563068; BD Biosciences); APC-CD4 (553051; BD Biosciences); FITC-CD4 (553080; BD Biosciences); PE-Cy7-CD11b (552850; BD Biosciences); FITC-Ly6G (551460; BD Biosciences); APC-Ly6C (560595; BD Biosciences); BV421-F4/80 (123132; Biolegend); PerCP-Cy5.5-I-A/I-E (562363; BD Biosciences); PE-natural killer (NK)-1.1 (553165; BD Biosciences); PE-SIINFEKL:H-2kb (141603; BD Biosciences); APC-interferon-γ (IFN-γ) (554413; BD Biosciences); PE-PD-1 (109103; Biolegend); BV421-TIM3 (119723; Biolegend); PE-Cy7-LAG-3 (125226; Biolegend); PE-Cy7-Ccl5 (149106; Biolegend); PE-Cxcl9 (515604; Biolegend); CD16/32 (553141; BD Biosciences); APC-CD206 (141707; Biolegend). The anti-mouse antibodies used for immunobloting were anti-B2m antibody (ab75853; Abcam); anti-TLR4 antibody (14358; CST); anti-TRAF6 antibody (ab33915; Abacam); anti-MYD88 antibody (4283; CST); anti-TNFR1 antibody (ab19139; Abcam); anti-TRADD antibody (3694; CST); anti-RIP1 antibody (3493; CST); anti-TRAF2 antibody (4724; CST); anti-TAK1 antibody (5206; CST); anti-Phospho-TAK1 antibody (9339; CST); anti-IKKα/β antibody (2370; CST); anti-Phospho-IKKα/β antibody (2697; CST); anti-IκBα antibody (9242; CST); anti-Phospho- IκBα antibody (2859; CST); anti-Phospho-NF-κB P65 antibody (3033; CST); anti-NF-κB P65 antibody (4764; CST); anti-GAPDH antibody (2118; CST); Anti-rabbit IgG antibody (5127; CST).
Analysis of infiltrating immune cells by flow cytometry
Mice were sacrificed by cervical dislocation under anesthesia using 6% chloral hydrate. Tumor tissues were collected and cut into small pieces, then dissociated into single-cell suspensions with mouse tumor dissociation kits (Miltenyi Biotec) following the manufacturer’s instructions. After filtering through a 70 µm Falcon cell strainer, cells suspensions were blocked with antimouse CD16/32 antibody for 15 min and stained with Fixable Viability Stain (1:1000, BD Biosciences) for 10 min to exclude dead cells. After washing, cells were stained with indicated antibodies for 30 min in the dark. The cells were then washed twice with PBS and analyzed by BD Fluorescence Activated Cell Sorting (FACS) Aria II (BD Biosciences, San Jose, California, USA). For intracellular cytokine, tumor-infiltrating leukocytes were enriched using an Optiprep (Sigma) density gradient and restimulated at 37°C with Leukocyte Activation Cocktail plus BD GolgiPlug (BD Biosciences) for 4 hours, followed by fixation and permeabilization using the Cytofix/Cytoperm Fixation/Permeabilization kit (BD Biosciences). For intranuclear staining, cells were permeabilized with the Foxp3 Transcription Factor Staining Buffer Kits (eBioscience). Cell suspensions were stained as described above.
Quantitative real-time PCR
Total RNA was extracted with the TRIzol reagent (Invitrogen) and reverse-transcribed into cDNA. qRT-PCR was performed on an ABI7900HI (Applied Biosystems). Target gene expression was normalized to Actin. Primers were shown in online supplemental table 1.
Supplemental material
Protein extraction and immunoblotting
Cells were lysed with RIPA buffer supplemented with freshly added protease inhibitor cocktail (Roche). After incubation on ice for 30 min, cell lysates were centrifuged at 12 000 rpm for 20 min at 4°C. Subsequently, protein concentrations in the supernatant were determined by Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Supernatant was heated to 95°C in 6×Protein Loading Buffer (TransGen) for 10 min. Proteins were separated by SDS-PAGE and transferred to PVDF membrane (Millipore). Membranes were blocked in Tris-buffered saline plus 0.1% Tween 20 (TBST) containing 5% nonfat dry milk for 1 hour and probed with indicated antibodies overnight. Membranes were washed with TBST for three times and incubated with Horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 hours. The bands were detected with Tanon-5200 Chemiluminescent Imaging System (Tanon Science & Technology).
T cells migration assay
CD8+ T cells were isolated from C57BL/6 mice. Bone marrow-derived macrophages (BMDMs) were obtained by flushing the femurs of mice and cultured for 1 week in Iscove’s Modified Dulbecco’s Medium containing 20 ng/mL M-CSF. 1×106 CTLs in complete media were loaded into the top chamber of Transwell inserts (5.0 µm pore size, Corning). The lower chamber was filled with conditioned medium from Raw 264.7 cells or BMDMs after treated with MSNs for 12 hours. Plates were incubated for 3 hours, the contents of the bottom chamber were collected, and the CTLs were counted.
Microscale thermophoresis
Microscale thermophoresis (MST) measurements were performed on a Monolith NT.115 (NanoTemper). FITC-labeled MSNs were diluted with PBS to 0.2 mg/mL. Human TLR4 (1478-TR-050; R&D Systems) was diluted into a series of concentrations and mixed with the same volume of MSNs. Then samples were loaded into capillaries. The thermophoresis was monitored at 40% MST power. Data were analyzed and fit to the KD model using MO. Affinity software (V.2.3).
Transcriptomic analysis
Mice bearing B2m-sgRNA B16F10 tumor were treated with PBS vehicle or MSNs prior to anti-PD-1 treatment. After 11 days, mice were euthanized and the tumor tissues were collected for RNA extraction and transcriptome sequencing. Sequencing libraries were generated using NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA) following manufacturer’s recommendations. The library preparations were sequenced on an Illumina Novaseq platform (Novogene, Beijing, China) and 150 bp paired-end reads were generated. Reads were mapped to the reference genome using Hisat2 v2.0.5. FeatureCounts v1.5.0-p3 was used to count the reads numbers of each gene. And FPKM of each gene was calculated for subsequent expression analysis. Gene correlation was determined by Spearman’s correlation coefficients. Gene specific and transcriptional factor enrichment were analyzed through Metascape.15 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment and Gene Set Enrichment Analysis (GSEA) were performed by the R package ‘clusterProfiler’.
Statistical analysis
All values were presented as the means±SEM. The data were analyzed by using GraphPad Prism V.7 (GraphPad Software). Statistical significance was determined through unpaired Student’s t-test or log-rank (Mantel-Cox) test. The differences in tumor growth were analyzed at the end time point. A p<0.05 was considered significant.
Results
MSNs sensitize resistant tumors to anti-PD-1 therapy and improve survival
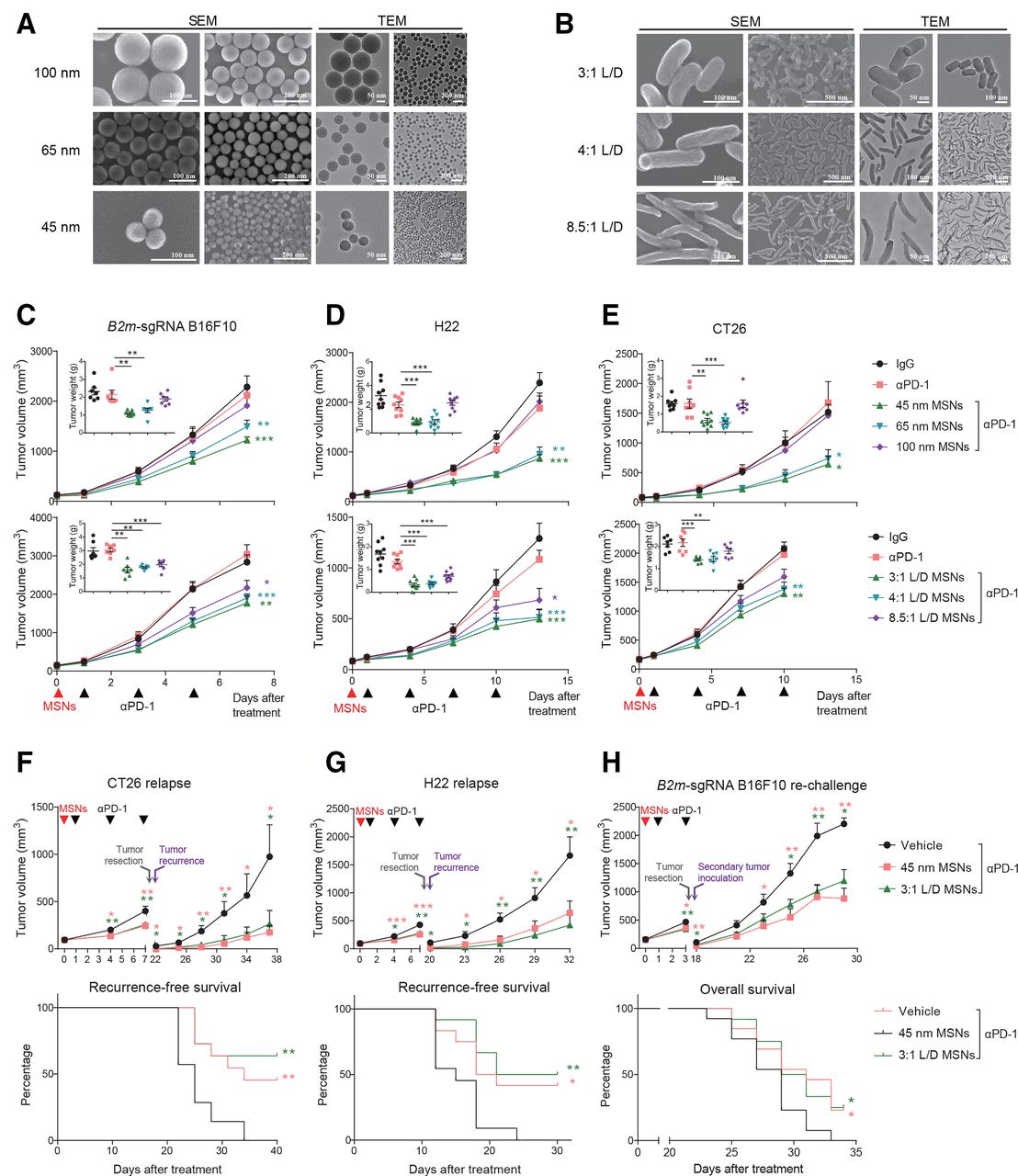
The properties such as size, shape, and surface functionalization of nanoparticles influence the biological effects in vivo. To assess the immunological effects and the influences of properties, MSNs with tunable sizes and shapes were synthesized. Spherical MSNs with diameters (ranging from ca. 45–100 nm) were facilely synthesized with excellent dispersity and evident mesoporous structure by regulating the mass ratio of TEA as the alkaline catalyst and CTAC as the structural-directing agent (figure 1A). Rod-like MSNs with varied L/D were synthesized by altering the dosage of surfactant (CTAC) during the synthesis process, which were directly observed as spirally mesoporous with homodispersed nanostructure (figure 1B). The preparation, nanostructure and composition made these MSNs suitable for evaluation of size-dependent and morphology-dependent effects.

MSNs overcome resistance and prevent relapse of αPD-1-resistant tumors. (A) SEM and TEM images of spherical MSNs with diameters of 100, 65 and 45 nm. (B) SEM and TEM images of rod-like MSNs with aspect ratios of 3:1, 4:1, 8.5:1. (C–E) Tumor growth and tumor weights of B2m-sgRNA B16F10 (C), H22 (D) and CT26 (E) tumor-bearing mice treated with different MSNs treatment (top: sizes; bottom: L/Ds) prior to αPD-1. (F, G) Growth of recurrent tumors (top) and recurrence-free survival (bottom) of tumor-bearing mice after indicated treatments. (H) Growth of rechallenged tumors (top) and overall survival (bottom) of B2m-sgRNA B16F10 tumor-bearing mice after indicated treatments. Error bars represent mean±SEM (n>6 per group in C–H). P value for tumor measurement was calculated by unpaired Student’s t-test. P value for survival curve was calculated by the log-rank (Mantel-Cox) test. (*P<0.05, **p<0.01, ***p<0.001). MSNs, mesoporous silica nanoparticles; PD-1, programmed cell death protein 1; TEM, tumor microenvironment.
To investigate the potential influence of MSNs on immunotherapy-resistant tumors, we employed syngeneic mouse models. First, we established acquired αPD-1-resistant tumors through impairing B2m in B16F10 melanoma (B2m-sgRNA B16F10, online supplemental figure 1A,B), and found that pretreatment with MSNs prior to αPD-1, even only once, significantly restored sensitivity to PD-1 blockade and eliminated resistant tumors in size- or L/D-dependent manner (figure 1C and online supplemental figure 1I). Smaller size and L/D MSNs yielded the most robust therapeutic effect across all groups (53% for 45 nm and 47% for 3:1 L/D). Second, we used two intrinsic resistant tumors, H22 hepatocellular carcinoma and CT26 colorectal carcinoma,16 17 for further validation. These tumors were also resensitized to αPD-1 by all MSNs except for spherical 100 nm particles (figure 1D,E and online supplemental figure 1I). Therefore, MSNs overcame resistance to PD-1 blockade in both acquired and intrinsic resistant tumors. Notably, MSNs without immunotherapy (plus IgG) did not elicit antitumor response (online supplemental figure 1C–I), suggesting that MSNs induced the anti-PD-1 sensitization rather than a direct therapeutic efficacy. Additionally, no obvious differences in body weight were observed among groups (online supplemental figure 2).
Supplemental material
Supplemental material
We next investigated whether MSNs could induce long-lasting antitumor immunity through two relapse models. In spontaneous relapse model, after MSNs and αPD-1 treatment, tumors were resected and the recurrence was monitored. Pretreatment with MSNs significantly reduced postoperative recurrence and the growth of recurrent tumors even if no longer therapy (figure 1F,G), suggesting that MSNs plus αPD-1 enhanced immune memory effect effectively. In rechallenged model, we inoculated secondary tumors contralaterally after MSNs/αPD-1 treatment and surgical removal of primary foci. The growth of secondary B2m-sgRNA B16F10 tumors were suppressed in MSNs plus αPD-1 groups, which achieved a significantly longer overall survival (figure 1H). These results demonstrate that MSNs enhanced the systemic antitumor immune response during PD-1 blockade therapy and improved survival.
MSNs overcome tumor resistance in a CTLs-dependent manner
To characterize the causality for MSNs-induced resensitization to anti-PD-1, we first measured biodistribution of MSNs in mice bodies. At 24 hours after injection, MSNs preferentially accumulated in tumors, however, there was no correlation between distribution amount and therapeutic efficiency (online supplemental figure 3). Then, we analyzed the immune composition in tumor microenvironment (TME) after treatment. In both intrinsic and acquired resistant tumors, MSNs (except 100 nm) plus αPD-1 treatment showed greater infiltration of immune cells (CD45+) compared with αPD-1 treatment alone (online supplemental figure 4A–C). The proportion of CD8+ CTLs increased significantly under treatment with both spherical and rod-like MSNs combined with αPD-1 in all types of tumor models (figure 2A and online supplemental figure 4D–F). Moreover, MSNs-induced elevation of CD45+ and CD8+ cells was positively correlated with the therapeutic effect, indicating that CD8+ CTLs played important roles in this process. Other immune cells, including CD4+ T cells, granulocytic myeloid-derived suppressor cells (MDSCs), monocytic MDSCs, macrophages, dendritic cells, and NK cells, did not always show a consistent trend under treatment with MSNs coupled with αPD-1 (figure 2A and online supplemental figure 4D–F).
Supplemental material
Supplemental material
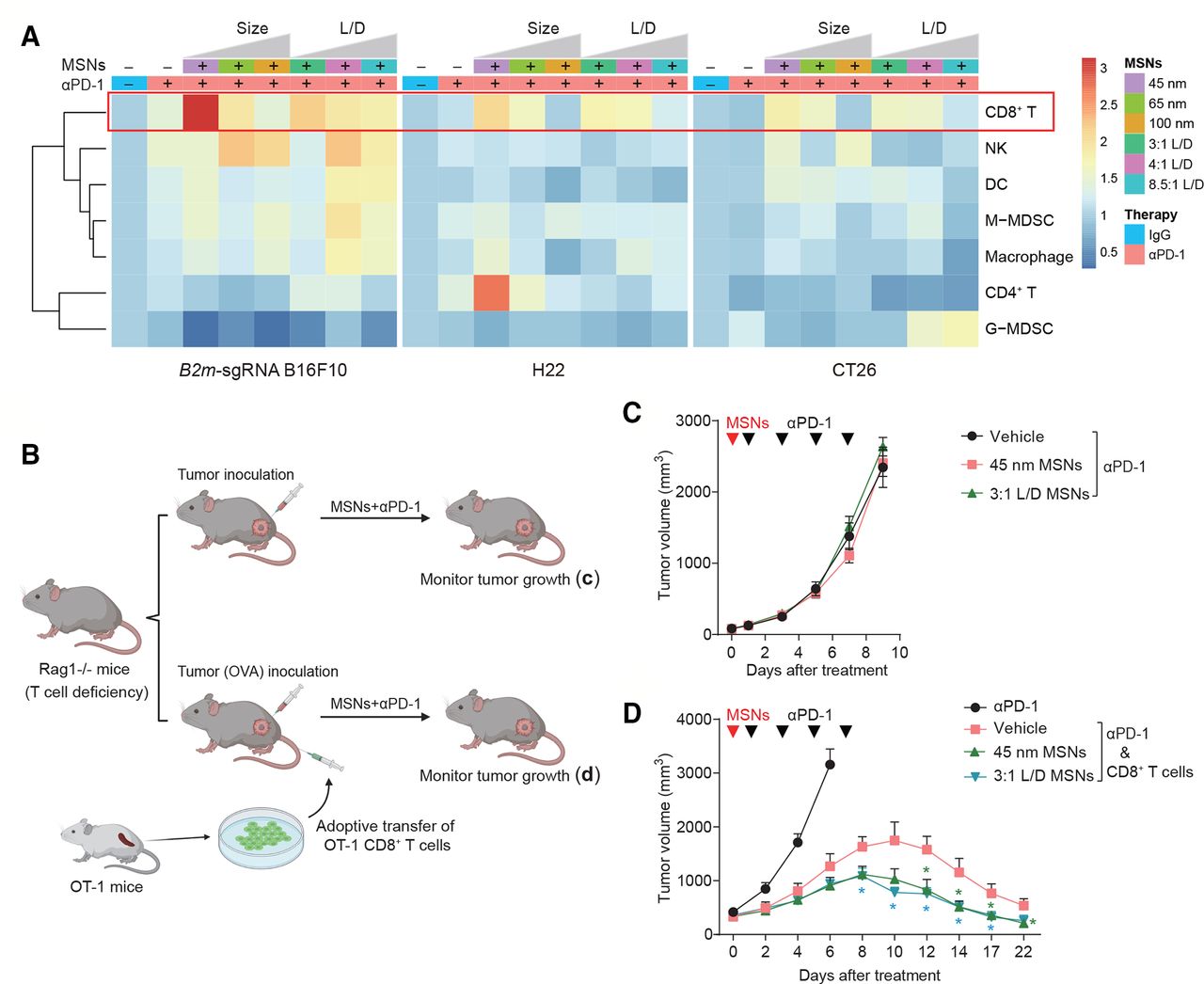
MSNs resensitize tumors to αPD-1 through CTLs. (A) Heatmap showing the proportion of tumor-infiltrating CD8+ T cells, NKs, DCs, M-MDSCs, macrophages, CD4+ T cells and G-MDSCs in the Tme after indicated treatments. (B) Scheme of tumor therapy in T cell-deficiency Rag1-/- mice and T cell-regained mice by transferring. (C) Growth of B2m-sgRNA B16F10 tumors in Rag1-/- mice after indicated treatments. (D) Growth of B16F10-OVA tumors in Rag1-/- mice with adoptive transfer of OT-1 CTLs. Error bars represent mean±SEM (n>5 per group in (C, D)). P value was calculated by unpaired Student’s t-test (*P<0.05, **p<0.01, ***p<0.001). CTLs, cytotoxic T-lymphocytes; DCs, dendritic cells; G-MDSCs, granulocytic myeloid-derived suppressor cells; M-MDSCs, monocytic MDSCs; MSNs, mesoporous silica nanoparticles; NKs, natural killer; OVA, ovalbumin; PD-1, programmed cell death protein 1.
In order to explore the involvement of CTLs in MSNs-induced antitumor effect, we adopted recombinase-activating gene 1-deficient (Rag1-/-) mice, which lack mature T and B cells,18 and set up an adoptive T cell therapy model with OVA-specific CTLs from OT-1 TCR transgenic mice (OT-1 CTLs) (figure 2B). B16F10 cells stably expressing chicken OVA, which are recognizable by T cells, were inoculated in this mice model. We found that, in Rag1-/- mice, pretreatment with MSNs can no longer overcome tumor resistance (figure 2C). However, adoptive transfer of OT-1 CTLs conferred Rag1-/- mice antitumor immunity and restored the resensitizing effect of MSNs (figure 2D). Consistently, the resensitizing effect of MSNs on CT26 tumor was abolished by CD8+ T cells depletion through anti-CD8 antibody (online supplemental figure 5). These results indicate that CTLs essentially mediated the antitumor immune responses induced by MSNs.
Supplemental material
MSNs and αPD-1 synergistically establish a T cell-inflamed TME at the beginning of treatment
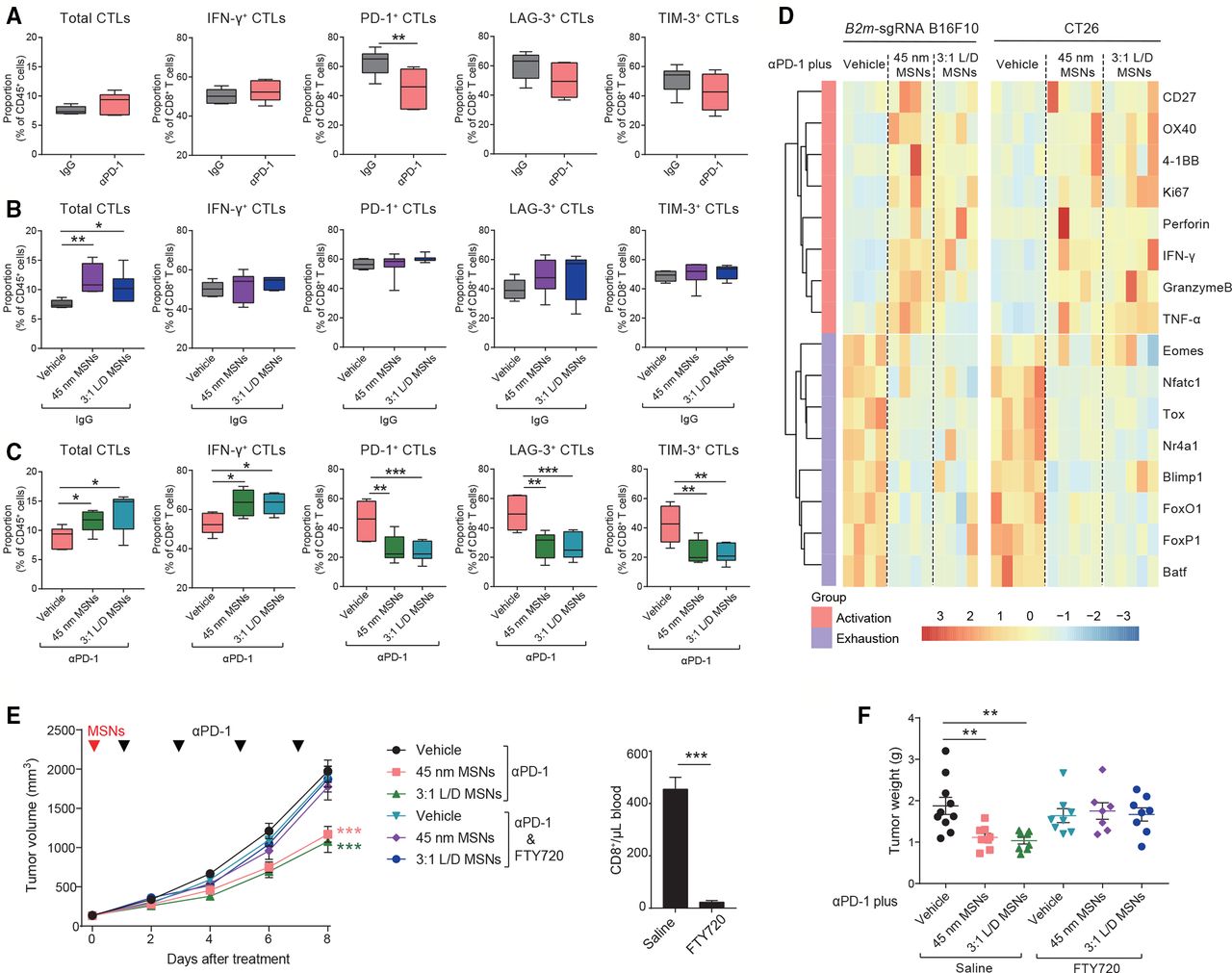
Considering that αPD-1 or MSNs alone did not show therapeutic efficacy in resistant tumors (figure 1 and online supplemental figure 1), we explored the potential synergistic effect of MSNs and αPD-1 on TME specified as tumorous CTLs. Early stage (day 2–3 after treatment) was focused to reveal the potential causative factors. We found that: (1) Consistent with treatment tolerance, neither the infiltration (total), cytotoxicity (IFN-γ+), nor exhaustion (LAG-3+, TIM-3+) of CTLs were influenced by αPD-1 alone (figure 3A). PD-1+ CTLs were reduced after αPD-1 treatment, which is in line with the effect of PD-1 antibody. (2) In contrast with αPD-1, although cytotoxicity and exhaustion were not affected, MSNs alone significantly increased the infiltrated CTLs in tumors (figure 3B), indicating that MSNs per se can inflame TME by promoting tumorous CTLs infiltration. (3) Furthermore, combined treatment resulted in a thorough CTLs activation with dramatically increased infiltration and cytotoxicity, while, reduced exhaustion (figure 3C). These results indicate that MSNs and αPD-1 synergistically prompted CTLs infiltration and activation in TME at the beginning of treatment.

MSNs drive CTLs infiltration to promote CTLs activation synergistically with αPD-1 at the beginning of treatment. (A–C) On day 2–3 after indicated treatments, the proportion of total CTLs and INF-γ, PD-1, LAG-3 and Tim-3 positive CTLs in B2m-sgRNA B16F10 tumor tissues was quantified by flow cytometry. (D) On day 3 after indicated treatments, the mRNA expression in CTLs isolated from B2m-sgRNA B16F10 or CT26 tumor tissues was shown. (E, F) Tumor growth (E) and tumor weights (F) of B2m-sgRNA B16F10 tumor-bearing mice treated with FTY720 to block T cell infiltration. Control mice received saline. Flow cytometry analysis quantified the numbers of CTLs collected from blood 3 days postinitial FTY720 administration. Error bars represent mean±SEM (n>6 per group in E). P value was calculated by unpaired Student’s t-test (*p<0.05, **p<0.01, ***p<0.001). CTLs, cytotoxic T-lymphocytes; INF-γ, interferon-γ; MSNs, mesoporous silica nanoparticles; PD-1, programmed cell death protein 1; TNFα, tumor necrosis factor-α.
To further illustrate the functional status of CTLs, we isolated CTLs from early-stage tumors (day 3 after treatment). On one hand, the levels of T cell activation markers (CD27, 4-1BB and OX40), proliferation marker (Ki67), and functional factors (Granzyme-B, Perforin, INF-γ, and TNF-α), were significantly increased by pretreatment with MSNs (figure 3D). On the other hand, transcriptional factors that establish and sustain T cell exhaustion (Tox, FoxO1, FoxP1, etc) were decreased under MSNs pretreatment (figure 3D). These results demonstrate that even at early stage of combine treatment, MSNs can inflame TME characterized with abundant high-activated and low-exhausted CTLs.
Since MSNs per se promoted CTLs infiltration at early stage, we further validated the role of CTLs infiltration in TME inflaming and antitumor responses. We utilized S1PR1 inhibitor, FTY720, to block T lymphocytes trafficking (figure 3E).19 Strikingly, the resensitizing effect of MSNs on resistant tumors to αPD-1 was completely abrogated in the context of FTY720 (figure 3E,F). Therefore, we conclude that MSNs, through promoting CTLs infiltration, inflamed TME and overcame anti-PD-1 resistance synergistically with αPD-1.
MSNs drive T cell-recruitment chemokines production in TAMs to promote CTLs infiltration
To explore how MSNs promoted tumorous CTLs infiltration, we analyzed chemokines trafficking immune cells in tumor tissues, and found that T cell-recruitment chemokines (Ccl5, Cxcl9 and Cxcl10) were significantly increased after treatment with spherical or rod-like MSNs plus αPD-1 (figure 4A). To depict the effector cell types in TME, chemokine-expressing cell populations were isolated from tumor tissues and the compositions were identified. As shown in figure 4B and online supplemental figure 6), macrophages (CD11b+F4/80+) dominantly engaged chemokines production, while other immune cells and non-immune cells only accounted for the minority. The influences of MSNs on chemokines in these cell types were further determined. At the beginning of treatment (day 2), in sorted macrophages (TAMs) rather non-immune cells, MSNs pretreatment significantly upregulated the expression of Ccl5, Cxcl9 and Cxcl10 even without αPD-1 therapy (figure 4C,D), not only confirming the infiltration-promoting effect of MSNs per se, but also suggesting the mediating role of TAMs in this process.
Supplemental material

MSNs promote the production of T cell-recruitment chemokines in macrophages. (A) The mRNA expression of CCL5, CXCL9, and CXCL10 in B2m-sgRNA B16F10 tumor tissues after indicated treatments was determined by qRT-PCR. (B) The major cell types that produced CCL5 and CXCL9 in B2m-sgRNA B16F10 tumor tissues were analyzed by flow cytometry. the right graph quantified the percentage of CCL5 and CXCL9 producing cell types. (C, D) The mRNA expression of CCL5, CXCL9 and CXCL10 in intratumoral macrophages (CD11b+F4/80+) (C) and non-immune cells (CD45−) (D) isolated from B2m-sgRNA B16F10 tumor tissues was determined by qRT-PCR. (E) Scheme of in vitro macrophages treatment and coincubation. RAW 264.7 cells were treated by 100 µg/mL MSNs for 12 hours. Cells were collected for gene expression assay, and supernatant was collected for T cell recruitment assay. (F) The mRNA expression of CCL5, CXCL9 and CXCL10 in RAW 264.7 cells after MSNs treatments was determined by qRT-PCR. (G) Quantification of migrated T cells recruited by the supernatant of macrophages after MSNs treatments. (H) Tumor growth of B2m-sgRNA B16F10 tumor-bearing mice treated with clodronate liposomes to deplete macrophages. Control mice received dimethyl sulfoxide. (I) The mRNA expression of CD8a, CCL5, CXCL9 and CXCL10 in tumor tissues after indicated treatments was determined by qRT-PCR. Error bars represent mean±SEM (n>7 per group in H). P value was calculated by unpaired Student’s t-test. (*P<0.05, **p<0.01, ***p<0.001). MSNs, mesoporous silica nanoparticles; PD-1, programmed cell death protein 1; FSC, forward scatter.
To confirm the mediation of macrophages for MSNs-induced T cell recruitment, we performed in vitro coincubation assays (figure 4E). After treatment with MSNs, macrophages were used to examine chemokine expressions, and the conditional medium were harvested for T cell recruitment assay. Following MSNs treatment, the expression of Ccl5, Cxcl9 and Cxcl10 in macrophages was significantly elevated (figure 4F); and macrophage medium-mediated CD8+ T cell recruitment was also markedly enhanced (figure 4G). We further determined the role of macrophages in MSNs-induced CTLs infiltration and antitumor effect in vivo. Clodronate liposomes can specifically deplete blood monocytes and tissue macrophages without affecting other immune cells.20 Macrophage depletion in mice by clodronate liposomes resulted in a complete abrogation of the resensitizing effect of MSNs (figure 4H). Importantly, tumorous CD8+ T cells infiltration (CD8a), and chemokines (Ccl5, Cxcl9, and Cxcl10) were no longer induced by MSNs in macrophage-depleted mice (figure 4I). The same phenomenon was observed in adoptive T cell therapy model (online supplemental figure 7), confirming the prerequisite of macrophages for MSNs-promoted CTLs infiltration and tumor sensitization.
Supplemental material
We further determined whether MSNs could drive macrophage polarization toward M1 in the TME. In B2m-sgRNA B16F10 and CT26 tumors, MSNs pretreatment promoted a shift of macrophages from M2 toward M1 type and increased the ratio of M1/M2 macrophages (online supplemental figure 8A,B). In H22 model, MSNs-treated tumor tissues showed a decline in M2 marker (arginase 1 (Arg-1)) and an increase in M1 marker (inducible nitric oxide synthase (iNOS)) (online supplemental figure 8C). These results indicate that MSNs regulate macrophages in terms of activity rather than quantity.
Supplemental material
These results suggest that MSNs drove the state transition of macrophages and macrophage-intrinsic chemokines expression promoted CTLs infiltration.
MSNs activate NF-κB signaling in tumors for production of T cell-recruitment chemokines
To investigate the mechanism of action that MSNs drove T cell-recruitment chemokines, we performed transcriptomics (RNA-seq) analysis using tumor tissues after MSNs and αPD-1 treatments. Across all samples in different groups, there was strong correlation among the expression of Ccl5, Cxcl9, and Cxcl10 (figure 5A), indicating that these chemokines showed the same pattern. Considering that same pattern genes generally share the regulatory module, we thus focused on genes with significantly positive association with Ccl5 (p<0.01) (figure 5B). Through cell-specific enrichment, macrophages were identified as the most highly responsive cell type (figure 5C), further supporting the above results that macrophages were the major source of T cell-recruitment chemokines. In transcriptional regulatory analysis, NF-κB complex accounted for the top two ranked transcriptional factors (figure 5D), indicating a leading regulation of NF-κB to Ccl5-correlated genes. Likewise, NF-κB signaling pathway and its related pathways (such as TLR signaling pathway, TNF signaling pathway) were significantly enriched (figure 5E). These results reveal NF-κB signaling as the most potential regulatory pathway for the expression of T cell-recruitment chemokines.

T cell-recruitment chemokines expression is associated with NF-κB signaling after MSNs treatment. (A) Correlation among the expression of T cell-recruitment chemokines across all B2m-sgRNA B16F10 tumor tissues. (B) Through transcriptomic analysis, all genes with significantly positive correlation with CCL5 were identified. (C) Cell-specific enrichment of CCL5-correlated genes. (D) Transcriptional regulatory enrichment of CCL5-correlated genes. (E) Top 10 signaling pathways by KEGG pathway enrichment of Ccl5-correlated genes. (F) In MSNs-treated tumors, the expression of genes in the top 10 Ccl5-related signaling pathways was presented by heatmap. (G) In tumor tissues, the activation changes of NF-κB, TNF and TLR signaling pathways in αPD-1 plus MSNs groups compared with αPD-1 alone were revealed by GSEA. (H) In tumor tissues after indicated treatments, the levels of proteins in TLR4, TNFR and NF-κB signaling were determined by Western blot. GSEA, Gene Set Enrichment Analysis; MSNs, mesoporous silica nanoparticles; PD-1, programmed cell death protein 1; TLR, Toll-like receptor; TNF, tumor necrosis factor.
To explore the influence of MSNs on these regulatory pathways, we compared the expression of genes in top 10 enriched pathways after different treatments. MSNs pretreatment, especially with αPD-1 therapy, markedly increased the overall expression of these genes (figure 5F), suggesting a potential activation induced by MSNs. From these top pathways, we further focused on NF-κB signaling and its upstream pathways, TLR pathway and TNF pathway, which also represent two typical receptors (TLRs and TNFRs) in macrophages. Compared with αPD-1 treatment alone, MSNs combined with αPD-1 significantly upregulated all these pathways (figure 5G). These results were confirmed in tumor tissues by western blot. As expected, the protein levels of TLR4 signaling (TLR4, MyD88, and TRAF6), TNFR1 signaling (TNFR1, TRADD, TRAF2, and RIP1), as well as NF-κB signaling (phospho-P65) were enhanced in MSNs plus αPD-1 treatment (figure 5H), suggesting that NF-κB and its upstream signaling pathways played key roles in the expression of T cell-recruitment chemokines after MSNs treatment.
MSNs activate TLR4/NF-κB in macrophages to overcome tumor resistance
We further explored how MSNs activated NF-κB pathway to overcome tumor resistance in vitro and in vivo. First, we examined the influence of MSNs in vitro and found that MSNs treatment markedly activated NF-κB signaling in Raw 264.7 cells (figure 6A). NF-κB signaling inhibition (JSH-23) in macrophages abrogated the promoting effect of MSNs on T cells recruitment. Following MSNs treatment, either macrophage-produced T cell-recruitment chemokines (figure 6B) or macrophage-recruited T cells (figure 6C) were dramatically compromised in NF-κB inhibited groups, suggesting that NF-κB signaling mediated MSNs-induced T cells recruitment. Second, we identified the responsible receptor for MSNs-induced NF-κB activation. MSNs enhanced the protein levels of TLR4 pathway in Raw 264.7 cells (figure 6A). Further, TLR4 blockade (Sparstolonin B) and TLR4 deficiency (TLR4-/-) in macrophages prevented MSNs-induced activation of NF-κB (figure 6D,E), as well as macrophage-produced T cells recruitment (figure 6F,G), suggesting that MSNs activated NF-κB in macrophages via TLR4. In the contrast, the protein levels of TNFR1 signaling were slightly affected by MSNs (online supplemental figure 9A). Anti-TNFR1 antibody (online supplemental figure 9B) and TNFR1 deficiency (TNFR1-/-) (online supplemental figure 9C–E) had little impact on NF-κB activation or T cells recruitment induced by MSNs. Finally, we determined whether MSNs overcame tumor resistance through TLR4/NF-κB pathway. In TLR4-/- mice, the resensitizing effect of MSNs to αPD-1 treatment was completely abolished (figure 6H,I). Moreover, tumorous CD8+ T cells infiltration (CD8a), cytotoxicity (IFN-γ), and chemokines (Ccl5, Cxcl9, and Cxcl10) were not influenced by MSNs any more (figure 6J), confirming the essential role of TLR4 in MSNs-induced CTLs infiltration, activation and tumor resensitization.
Supplemental material
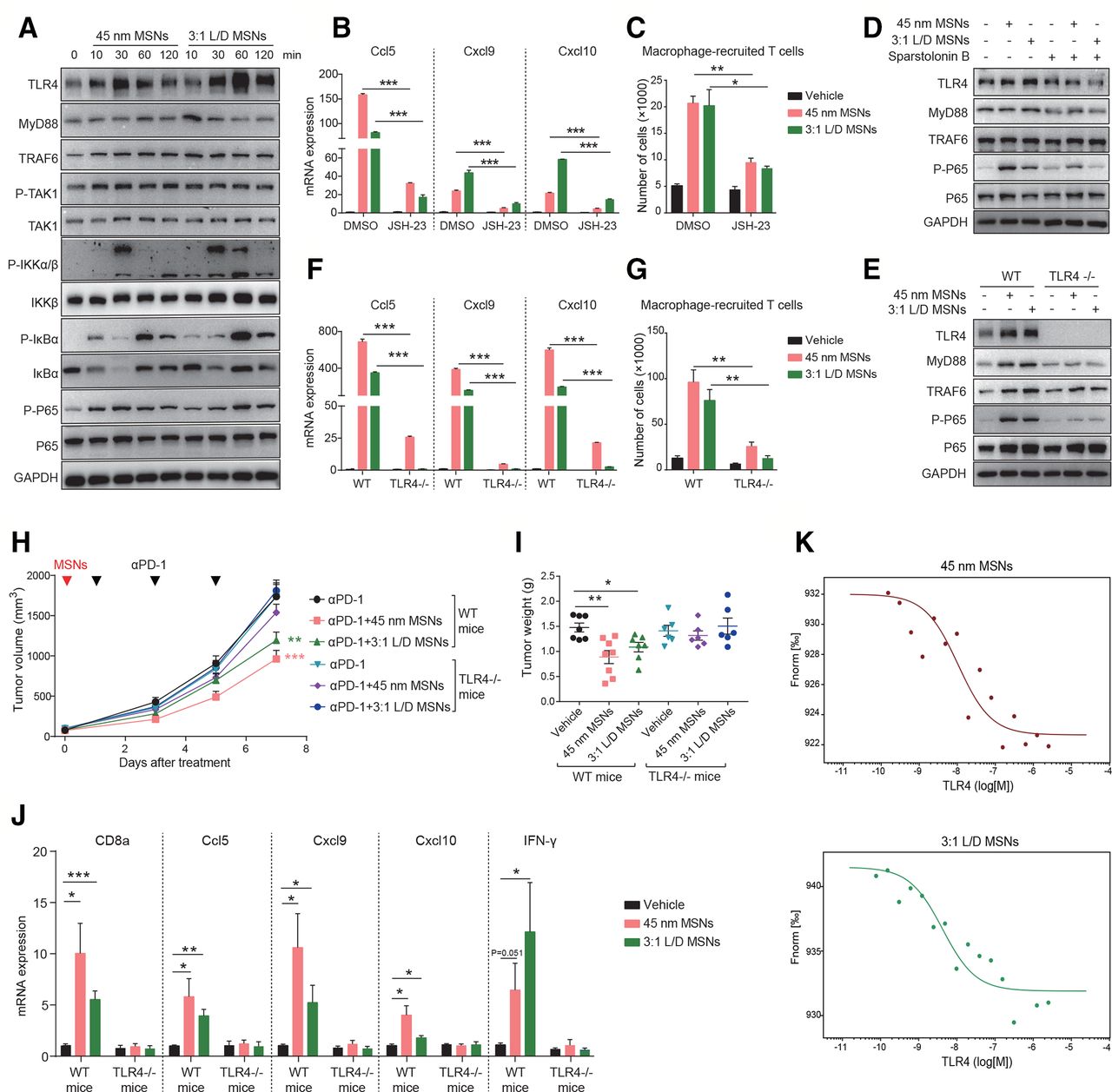
MSNs overcome tumor resistance through activating TLR4/NF-κB signaling in macrophages. (A) RAW 264.7 cells were treated with 100 µg/mL MSNs for different times and the protein levels were determined by Western blot. (B) RAW 264.7 cells were treated with 10 µM JSH-23 for 3 hours and then 100 µg/mL MSNs for another 12 hours. the mRNA expression of CCL5, CXCL9 and CXCL10 was determined by qRT-PCR. (C) RAW 264.7 cells were treated with 10 µM JSH-23 for 3 hours and then 100 µg/mL MSNs for another 12 hours. T cells recruited by the supernatant of macrophages were quantified through migration assay. (D) RAW 264.7 cells were treated with 100 µM TLR4 blockade sparstolonin B for 3 hours and then 100 µg/mL MSNs treatment for 1 hour. The protein levels were determined by Western blot. (E) Bone marrow-derived macrophages (BMDMs) from wide type (WT) and TLR4-deficient (TLR4-/-) mice were treated with 100 µg/mL MSNs for 1 hour. The protein levels were determined by Western blot. (F) WT and TLR4-/- BMDMs were treated 100 µg/mL MSNs treatment for 12 hours. The mRNA expression of CCL5, CXCL9 and CXCL10 was determined by qRT-PCR. (G) WT and TLR4-/- BMDMs were treated 100 µg/mL MSNs treatment for 12 hours. T cells recruited by the supernatant of macrophages were quantified through migration assay. (H, I) Tumor growth (H) and tumor weights (I) of B2m-sgRNA B16F10 tumors in WT and TLR4-/- mice after indicated treatments. (J) After indicated treatments, the mRNA expression of CD8a, CCL5, CXCL9, CXCL10 and IFN-γ in tumor tissues was determined by qRT-PCR. Error bars represent mean±SEM (n>5 per group in H). (K) The dose-response curves of MSNs with TLR4 in MST affinity analysis. The concentrations were 0.2 mg/mL for FITC-labeled MSNs and 0.1 nM–2.5 μM for TLR4 protein. P value was calculated by unpaired Student’s t-test. (*P<0.05, **p<0.01, ***p<0.001). MSNs, mesoporous silica nanoparticles; IFN-γ, interferon-γ; TLR4, Toll-like receptor 4.
To determine whether MSNs interact with TLR4 directly, we performed MST assay using purified TLR4 protein and FITC-labeled MSNs. As shown in figure 6K, we observed the variations of fluorescence signal along with the TLR4 concentration, suggesting a direct interaction between MSNs and TLR4. The dissociation constants (KD) of 45 nm MSNs and 3:1 L/D MSNs with TLR4 were calculated to be 10.5 and 4.2 nM, respectively, which representing the high binding affinity.21
Taken together, these results demonstrate that MSNs activate TLR4/NF-κB signaling in macrophages, promote T cell-recruitment chemokines production, drive CTLs tumorous infiltration, inflame the cold tumors and overcome the resistance to αPD-1 therapy (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic depiction of the synergistical effect of MSNs and PD-1 blockade on tumor immunotherapy. MSNs, mesoporous silica nanoparticles; PD-1, programmed cell death protein 1; TLR4, Toll-like receptor 4.
Discussion
Our results demonstrate that nanomaterials themselves can overcome resistance to anti-PD-1 therapy. This effect is correlated with particle diameter and aspect ratio parameters. Even only once injection, potent therapeutic effects are invoked as early as the second day after anti-PD-1 therapy, and immune memory persists until the end of treatment. By recruiting and activating CTLs, MSNs plus αPD-1 rapidly establish a T cell-inflamed TME and induce a long-lasting systemic antitumor immune response. Notably, as the representative nanomaterials, research on MSNs could shed light on the influences of physicochemical properties of materials on immunotherapy and improve the development and application of immunostimulant functional materials.
MSNs have been broadly explored in the biomedical field for two decades and represent remarkable biocompatibility, a critical consideration for clinical application.22 In vivo studies show that almost all MSNs (94.4%) injected into mice were excreted out through urine and feces within 4 days.23 The US Food and Drug Administration (FDA) approved Cornell dots (C-dots) as the first silica-based tumor diagnostic nanoparticles for stage I human clinical trials in 2011, indicating biosafety and massive potential for clinical viability of silica-based nanoparticulate systems. In this study, despite MSNs triggering significant T cell activation and enhancing the immunotherapeutic effect of PD-1 blockade, no discernible differences in mouse body weight were observed among treatment groups, implying efficient and safe actions. Both granular and rod-shaped MSNs were found to overcome tumor resistances to anti-PD-1 therapy, with potency shown to be stronger in smaller size and aspect ratio. This should be considered in future development of immune-activating materials. Notably, although MSNs can enhance CTLs infiltration in vivo, MSNs pretreatment combined with IgG did not elicit therapeutic efficacy in vivo, suggesting that in our experimental settings (only injection once before the day of αPD-1 treatment), MSNs alone were not enough to exert potent antitumor effects. However, MSNs and αPD-1 exerted a synergistic effect on T cell function in terms of exhaustion and cytotoxicity, which subsequently promoted efficacy and overcame resistance to anti-PD-1 therapy.
The molecular mechanisms of tumor resistance to immunotherapy remain poorly understood. Known mechanisms of resistance to anti-PD-1 therapy involve tumor cell-intrinsic and tumor cell-extrinsic factors.3 Tumor cell-intrinsic factors include absence of antigen presentation and insensibility to T cells, whereas tumor cell-extrinsic mechanisms include decreased infiltration of CTLs, increased levels of immunosuppressive cells, and enhanced inhibitory immune checkpoints (eg, PD-1, LAG-3 and TIM-3) on CTLs. Our data demonstrated that MSNs rapidly established a T cell-inflamed TME, characterized by abundant high-activated and low-exhausted CTLs, which were further showed to be essential in eliciting a response to anti-PD-1 therapy, and immunotherapeutic efficacy will be entirely negated if CTLs infiltration is blocked. Sufficient inflammation-inducing immune cells can overcome resistance to anti-PD-1 therapy, even in the absence of antigen presentation.24 Interestingly, in our study, MSNs increased the proportion of non-major histocompatibility complex (MHC)-I-restricted immune cells (such as CD4+ T cells and NK) in B2m-sgRNA B16F10 tumor (figure 2A). One explanation is that B2m-sgRNA B16F10 contained one-third of non-depleted cells due to multiple cell colonies, however, the potential roles of non-MHC-I-restricted immune cells cannot be completely excluded. Furthermore, our results showed CD8+ T cells are required for the resensitizing effect of MSNs. Therefore, whether the recognition-independent activities of CD8+ T cells are involved in the tumorous infiltration of other immune cells? To what extent do these immune cells contribute to the resistance reversal induced by MSNs? These questions are important and should be investigated in the future.
Chemokines play critical roles in T cell trafficking, which prompted us to investigate the chemokine-centric regulatory modules responsible for the recruitment of CTLs following MSNs treatment. Ccl5, Cxcl9 and Cxcl10 in cancer have been reported to correlate with CTLs infiltration.25 Our data indicated that macrophages accounted for the predominant source of Ccl5 and Cxcl9 in tumors. Most nanomaterials after injection are rapidly eliminated by the mononuclear phagocyte system, whereas macrophages comprise the major cell population.26 27 Previous studies have determined that MSNs are preferentially internalized by macrophages in vivo,28 29 suggesting that macrophages might be the main target cell of MSNs. Although the total proportion of macrophages in tumors did not always show increased trend following MSNs treatment, MSNs remarkably promoted the expression of Ccl5, Cxcl9 and Cxcl10 in macrophages both in vitro and in vivo, and subsequently enhanced CD8+ T cells recruitment by macrophages, suggesting that MSNs modulate macrophages in terms of activation rather than amount.
TLR4 and NF-κB signaling has been identified as the target pathway for the influence of MSNs on macrophages. NF-κB is a well-characterized transcription factor involving in the transcriptional regulation of T cell-recruitment chemokines (Ccl5, Cxcl9 and Cxcl10).30 31 There was a robust correlation between NF-κB signaling and T cell-recruitment chemokines in our data. Also, inhibition of NF-κB blocked MSNs-mediated T cell recruitment, suggesting that NF-κB signaling is responsible for CTLs tumorous infiltration under MSNs treatment. TLR4 was identified as a target of MSNs which further promoted NF-κB signaling. TLR4-based immunotherapy has been widely studied due to the crucial roles of TLR4 in both innate and adaptive immunity.32 TLR4 agonists, such as monophosphoryl lipid A, have been explored as potential adjuvants and obtained the FDA approval for clinical application in cancer therapy. In this study, MSNs upregulated TLR4, specifically triggered its downstream signaling in macrophages, and eventually overcame resistance to anti-PD-1 therapy, indicating that TLR4-based nanosystems could be considered as potential adjuvants for improving cancer immunotherapy. The size-relevant and morphology-relevant effects of MSNs on activating TLR4 in macrophages is worthy for further investigation in future studies.
Conclusion
In conclusion, this work demonstrates that MSNs trigger TLR4/NF-κB pathway in macrophages, drive the expression of T cell-recruitment chemokines, promote the tumorous infiltration of CTLs, and inflame immunologically cold tumors. By remodeling T cell-inflamed TME, MSNs plus PD-1 blockade overcome resistance to anti-PD-1 therapy in diameter-dependent and aspect ratio-dependent manners. This rational combination treatment efficiently inhibits tumor growth and recurrence with no obvious toxicity. This is the first report to demonstrate that biocompatible MSNs themselves (without additional functional transformation) can inflame cold tumors and overcome tumor resistance to anti-PD-1 therapy. Our findings provide rationale and preclinical evidence for the potential application of basic and enhanced functional MSNs in cancer immunotherapy.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Ethics approval
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute for Nutritional Sciences.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MS, PG, YY and LY contributed equally.
Contributors MS, PG, YY, ZJ and JL performed all in vivo and in vitro experiments. LY synthesised and characterized all nanomaterials used in the study. MS, QB and YC analyzed the data. MS and QB wrote the manuscript. YC, QB and HW supervized the study.
Funding This study was supported by grants from the National Key R&D Program of China (2018YFC2000700 and 2016YFA0203700), the National Natural Science Foundation of China (82030099, 81630086, 81973078, 51672303, and 51722211), Shanghai Public Health System Construction Three-Year Action Plan (GWV-10.1-XK15), Shanghai Municipal Human Resources and Social Security Bureau (2018060), Program of Shanghai Subject Chief Scientist (18XD1404300), and the International Cooperation Project of Guangzhou Development Zone (2017GH11), Innovative research team of high-level local universities in Shanghai.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.