
Abstract
Anthracycline chemotherapy maintains a prominent role in treating many forms of cancer. Cardiotoxic side effects limit their dosing and improved cancer outcomes expose the cancer survivor to increased cardiovascular morbidity and mortality. The basic mechanisms of cardiotoxicity may involve direct pathways for reactive oxygen species generation and topoisomerase 2 as well as other indirect pathways. Cardioprotective treatments are few and those that have been examined include renin angiotensin system blockade, beta blockers, or the iron chelator dexrazoxane. New treatments exploiting the ErbB or other novel pro-survival pathways, such as conditioning, are on the cardioprotection horizon. Even in the forthcoming era of targeted cancer therapies, the substantial proportion of today’s anthracycline-treated cancer patients may become tomorrow’s cardiac patient.
Similar content being viewed by others

Introduction
Cancer outcomes continue to improve due to earlier detection and newer targeted therapies, with anthracycline chemotherapy playing a major role in the modern era of cancer treatment. The serendipitous discovery of doxorubicin from Streptomyces peucetius and its precursor daunorubicin was a milestone in antibiotic overproduction techniques of the day [1]. Anthracyclines are listed among the World Health Organisation (WHO) model list of essential medicines [2]. Fifty years on from its discovery, anthracycline anti-tumour and cardiotoxic mechanisms alike continue to evoke considerable interest in basic science and clinical trials research.
Cancer now affects more than one in three people in their lifetime, and along with cardiovascular disease, they are the two leading causes of death in developed nations. Overall ten-year cancer survival stands at 50% across the twenty most common malignancies and approximately 80% or better in breast, lymphoma, melanoma and uterine cancers. These mortality trends reflect a broad improvement in survival rates in the developed economies [3]. In the United Kingdom survival trends have doubled in adults in the last 40 years and tripled in children since the 1960s [4]. Paradoxically, improved long term cancer survival has led to an increased awareness of the adverse cardiac effects of cancer treatment itself. These welcome improvements have thus shifted the care paradigm from cancer survival to cancer survivorship.
The Role of Anthracyclines – today’s Cancer Patients Are tomorrow’s Cardiac Patients
New biological and small molecule treatments have dramatically improved the outlook of many cancers over the last 15 years [5–7]. Even in this encouraging context, anthracycline chemotherapy regimens play a prominent role in many cancer treatments – e.g.32% of breast cancer patients [8], 57 to 70% of elderly lymphoma patients [9, 10], and 50 to 60% of childhood cancer survivors are treated with an anthracycline regimen [11]. Thus, with long term cancer survivorship there will be a substantial population of cancer patients who will continue to remain at risk of early cardiovascular morbidity and mortality due to their legacy anthracycline chemotherapy [12, 13].
Anthracyclines: Structure, Targets and Toxicity
The four most common anthracyclines are doxorubicin, daunorubicin, epirubicin and idarubicin (Fig. 1). Doxorubicin and daunorubicin were the first to be used in clinical practice. Epirubicin, a stereoisomer of doxorubicin, has an increased volume of distribution and longer half-life than doxorubicin (doxorubicin t½ = 1–3 h, epirubicin 31–35 h). Idarubicin, a derivative of daunorubicin, is more lipophilic and has a higher cellular uptake than daunorubicin.

The four anthracycline derivatives Doxorubicn (DOX), Daunarubicin (DNR), Epirubicin (EPI) and Idarubicin (IDA). The anthracyclines share a tetracyclic aglycone structure of four cyclohexane chains with a daunosamine sugar moiety at carbon C7 of ring a; adjacent quinone-hydroquinone groups in rings b and c; a methoxy substituent carbon C4 in ring D; a carbonyl group at C13; and a short side chain in C9. Doxorubicin and daunorubicin differ in their short chains. Doxorubicin has a primary alcohol, whereas daunorubicin has a methyl group. Epirubicin is derived from doxorubicin by axial-to-equitorial epimerisation of the hydroxyl group in the daunosamine moiety. Idarubicin is identical to daunorubicin except the 4-methoxy group in ring D is removed
Mechanisms of Action
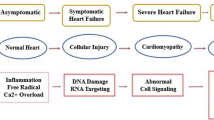
The exact mechanism of anthracycline-induced cardiotoxicity remains unclear, though it is likely to be multifactorial. Until recently, the most widely accepted hypothesis was that anthracyclines interfered with redox cycling, resulting in DNA damage due to reactive oxygen species (ROS) production [14]. More recently, topoisomerase 2 has been suggested to be the main mediator of cardiotoxicity, though other mechanisms contribute. (Fig. 2)

Doxorubicin staining shows sequestration in (L) cardiomyocytes and (R) malignant cervical cancer cells (courtesy Dr. I Piotrowska)
The Role of Reactive Oxidation Species
The quinone moiety of anthracyclines are susceptible to univalent reduction to a semiquinone radical by a number of cellular oxido-reductases. In myocardial cells, this is predominantly achieved via an enzymatic pathway involving NADH dehydrogenase (complex I) of the mitochondrial electron transport chain [15]. In the presence of molecular oxygen, the semiquinone auto-oxidises to generate the parent anthracycline and a superoxide anion [16]. This non-enzymatic pathway allows a self-perpetuating redox cycle to be established, resulting in the accumulation of superoxide anions. ROS levels may also be increased by free cellular iron and potentiating ferrous-ferric cycling of molecular iron [17]. The doxorubicin-iron complexes form toxic radical and reactive nitrogen species, resulting in increased nitrosative stress and mitochondrial dysfunction [18].
The Role of Topoisomerase 2β
DNA topoisomerases (Top) induce temporary single or double-stranded breaks to regulate the topological changes during DNA replication, transcription, recombination and chromatin remodelling [19]. In humans, Top2 is expressed as isoenzymes Top2α and Top2β [20]. Top2α is the most prevalent and is highly expressed in proliferating (malignant and non-malignant) cells. It is essential for chromosomal segregation and its expression varies during the cell cycle, peaking during G2/M phases [21]. Conversely, Top2β is more abundant in quiescent cells, such as adult mammalian cardiomyocytes, and its expression remains constant throughout the cell cycle.
Doxorubicin exerts its cytotoxic effect by intercalating DNA. Doxorubicin binds with DNA and topoisomerase 2 isoenzmes forming a ternary Top2-doxorubicin-DNA complex, which causes double-stranded DNA breaks. When bound to Top2α, the complex inhibits DNA replication; arrests the cell cycle in G1/G2; and induces apoptosis [22] as intended in proliferating malignant cells. Conversely, when bound to Top2β, mitochondrial dysfunction is triggered by the suppression of peroxisome proliferator-activated receptor (PPAR), which regulates oxidative metabolism [23], In adult mammalian cardiomyocytes, this leads to an activation of altered P53 tumour suppressor pathway, β-adrenergic signalling, impaired calcium handling, mitochondrial dysfunction and increased apoptosis. Without Top2β, doxorubicin cannot bind directly to DNA [22]. Animal studies with Top2β knockout (KO) mice have shown that the absence of Top2β protects against doxorubicin-induced cardiotoxicity [24, 25] partially due to reduced mitochondrial dysfunction [25].
ErbB2/ErbB4 and NRG-1 Signalling
Anthracyclines may also disrupt neuregulin-ErbB (NRG) receptor signalling to downstream cardiomyocyte pro-survival cascades involving the serine-threonine protein kinase B / AKT (whose origins stem from two human homologues of the retroviral oncogene v-akt [26]), mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K) and extracellular signal-related kinase (ERK1 / 2). NRG-1 controls cell fate decisions in neuronal, skeletal myocyte, glial cells and cardiomyocytes. The ErbB complexes themselves are important modulators of cell growth. Overexpression of the NRG-1 receptor subunit ErbB2 is an important driver in certain neoplasms dependent on ErbB3 and ErbB2 (i.e. HER2+ breast cancer). NRG-1 also binds ErbB4 and stimulates proliferation of differentiated mononucleated cardiomyocytes, leading to ErbB2/ErbB4 heterodimerization [27]. This leads to compensatory cellular hypertrophy, whereas deletion models demonstrate dilated cardiomyopathy and systolic dysfunction in the presence of pressure overload [28]. Anthracyclines appear to increase ErbB2 expression after chronic doxorubicin exposure, but decrease ErbB4 expression after acute doxorubicin dosing, potentially explaining the time course of anthracycline cardiomyocyte toxicity [18].
Other Targets
The Ras-related C3 botulinum toxin substrate 1 – guanosine triphosphate enzyme (Rac1 GTPase) subunit also contributes to anthracycline cardiotoxicity by fragmenting histone-associated DNA and activating the programmed cell death caspase-3 (cysteine-aspartic acid protease) enzyme via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation and ROS [29]. Other mechanisms include decreased c-Kit + cardiac progenitor cells following juvenile anthracycline exposure, leading to impaired compensatory mechanisms for stress and vascular injury repair, [30] and myofibril instability due to titin proteolysis and increased calcium sequestration, which may lead to diastolic dysfunction [28]. Together, these two mechanisms may partly explain the late cardiotoxicity seen in childhood cancer survivors manifest as early dilated cardiomyopathy followed by a period of compensated systolic function before a decompensated phenotype of restrictive physiology, decreased myocardial wall thickness and ejection fraction. [31].
Cardiotoxicity – in Search of A Consensus Definition
The cardiovascular side effects of anthracycline anti-tumour agents became apparent soon after their widespread use in the 1970s, but the more recent concept of ‘cardiotoxicity’ has no standard definition. The mechanisms, detection, prevention, and protection from anthracycline cardiotoxicity remain relevant today because of its long legacy of treated patients and because cumulative dose-related toxicity constrains treatment. We deconstruct ‘cardiotoxicity’ according to patient age, time course, and affected cardiac domain (clinical decompensation, structural change, biomarker rise, or arrhythmia).
Incidence
The incidence of cardiotoxicity varies according to how it is defined. Cardiotoxicity spans a continuum of frequency and severity, ranging in seriousness from overt clinical symptoms requiring urgent hospital admission, to an asymptomatic detectable structural change on cardiac imaging or new onset arrhythmia, to a measurable biomarker rise before symptomatic, structural or electrical change is detectable. These sub-types of cardiotoxicity occur as clinical decompensation in 2–4%, sub-clinical structural change in 9–11%, arrhythmia (e.g. AF) in >12%, and biomarker rise in 30–35% of cancer chemotherapy patients.
Cardiotoxicity, first observed in adult cancer patients as clinical congestive heart failure (CHF), characterised by pulmonary oedema, fluid overload, and effort intolerance, was initially reported in 1979 by Von Hoff et al. at 2.2% overall with a cumulative doxorubicin dose-dependent incidence of CHF of 3%, 7%, and 18% at 400, 550, and 700 mg/m2, respectively [32]. A further retrospective pooled analysis of three breast and lung cancer trials by Swain et al. documented higher rates of CHF – 4.7%, 26%, and 48% at 400, 550 and 700 mg/m2, respectively - providing the current basis to limit lifetime cumulative doxorubicin-equivalent exposure to ≤450 mg/m2 in patients without prior chest radiotherapy [33]. Further studies of biopsy-confirmed doxorubicin cardiomyopathy documented alarmingly poor prognosis – hazard ratio for fatal cardiotoxicity 3.46 and 50% mortality at two years – akin to restrictive and infiltrative cardiomyopathies [34].
Sub-clinical cardiotoxicity is commonly defined on cardiac imaging as clinically asymptomatic left ventricular systolic dysfunction (LVSD) with a fall in left ventricular (LV) ejection fraction (EF) by >10% points to a value of EF < 50% [35, 36]. When defined as radionuclide MUGA ejection fraction (EF) fall of more than 10% to an absolute value <50%, cardiotoxicity occurred with a cumulative incidences in 9%, 18%, 38%, and 65% of patients at 250, 350, 450 and 550 mg/m2, respectively, and Schwartz et al. classified 14.8% (220 of 1487) doxorubicin-treated cancer patients as high risk on the basis of abnormal falls in MUGA LVEF prior to the onset of clinical congestive cardiac failure, thereby establishing LVEF as predictive of clinical heart failure. A dose-escalation curve and reduction in %EF by MUGA scan were reported in breast cancer patients by Speyer et al., − 4% at 275–400 mg/m2, 15% at 400–500 mg/m2, 16% at 500–699 mg/m2, 18% at 800–899 mg/m2, and 21% at 900–999 mg/m2.
Smith et al. [11] reported significantly increased odds ratio for clinically symptomatic (OR 5.4 [95% CI 2.3–12.6]) and sub-clinical fall in LVEF (OR 6.25 [CI 2.58–15.13]) for anthracycline cardiotoxicity in their meta-analysis of 55 anthracycline cancer trials. Hequet et al. [35] documented a 27% incidence of 5-year sub-clinical cardiotoxicity in 39 of 141 adult lymphoma patients (mean doxorubicin dose 300 mg/m2, mean age 54 years), using echocardiographic criteria of fractional shortening (FS) < 25% (equivalent to EF < 50%). Predictors for sub-clinical cardiotoxicity were increasing age, male sex, doxorubicin dose and being overweight.
In the modern contemporary series by Cardinale et al., cardiotoxicity (defined as an echocardiographic LVEF decrease of more than 10% to EF < 50%) occurred in 9% of a mixed group of 2625 adult patients (aged 50 ± 13 years, 51% breast cancer, 28% Hodgkin lymphoma, 74% women) followed up for a median period of 5.2 years [37]. Of note, 98% of cases of asymptomatic LVEF decline were detected prospectively in the first 12 months of follow-up after chemotherapy, implicating early cardiotoxicity rather than a “late effects phenomenon” in adults as the dominant natural time course of anthracycline chemotherapy.
Relying solely on LVEF as a sub-clinical imaging marker of cardiotoxicity is problematic. The measurement itself is calculated by various echocardiographic techniques, each with its own limitations. Thavendiranathan et al. reported the minimum detectable change in EF by the same observer was 9%, 10%, and 4.8% by 2D bi-plane, tri-plane, and 3D methods; whereas the minimum detectable change in EF by different observers on different acquisitions (interobserver test-retest) was 13%, 16%, and 6% for 2D bi-plane, tri-plane and 3D methods [36]. Thus, the reproducibility and precision of echo-based LVEF measurement may be worse than the “asymptomatic 10% fall in EF” which is commonly used as the decision threshold to define cardiotoxicity (Table 1).
These limitations have led to development of more reliable measurements based on myocardial deformation that detect early structural change before a fall in EF. A relative change in 2D speckle-tracking echocardiographic global longitudinal strain (GLS) of 10 to 15% (95% CI = 8.3% - 14.6%) or an absolute GLS “greater” than −19% predicted cardiotoxicity defined as a fall in EF with sensitivity of 65–86% and specificity of 73–95%.
The Role of Cardiac Troponins
Cardiac troponin is central to the universal definition of acute coronary syndrome (ACS), and its release is predictive of prognosis in myocardial infarction and other forms of heart disease [39–46]. When measured as an early biomarker of cancer cardiotoxicity, troponin rise occurs consistently in 21% - 40% of patients after anthracycline chemotherapy, irrespective of assay type [47–49]. Though troponin-I and troponin-T levels are not directly comparable across the available assays, representative levels ranged from 11 to 120 ng/L in low-dose, and 160 to more than 1900 ng/L in high-dose anthracycline regimens (see Table 2) [56]. In the setting of cancer chemotherapy cardiotoxicity, the rise in troponin may quantify both cardiomyocyte apoptosis and myofibril degradation more than necrosis and predicts cardiotoxicity defined as fall in LVEF as well as major adverse cardiovascular events (MACE).
Baseline troponin levels may be elevated due to the burden of malignant disease itself. Auner et al. reported a baseline troponin-T levels >70 ng/L in 3.8% of 78 mixed acute myeloid and non-Hodgkin lymphoma patients [57]. Lipshultz et al. documented baseline increased troponin-T levels >10 ng/L in 10% of 119 paediatric acute lymphoblastic leukaemia, associated with a ten-fold increase in white-cell count (300 × 103/mm3 vs. 27 × 103/mm3) and significant blast crisis (89% vs. 57.5%) [53], reflecting the cardiotoxic effects of profound leucocytosis in the circulation, and in particular, the coronary circulation even before anthracycline exposure [58–60]. Cardiac troponin-T levels in the unprotected control cohort of the study were elevated in 50% of 76 patients, with multiple troponin-T elevations occurring in 37%.
Cancer itself may be arrhythmogenic, and its treatments may increase supraventricular tachyarrhythmias and atrial fibrillation [61, 62]. Baseline AF incidence in cancer was reported as 2.4% and new onset AF occurred in a further 1.8% in a large cohort of 24,125 cancer patients [63]. Early short term arrhythmia studies documented increased ventricular ectopy at 1 h post-infusion in 3%, rising to 24% during 1 to 24 hours of Holter monitoring.
Time Course of Cardiotoxicity
The time course of cardiotoxicity varies depending on patient age at time of exposure and the class effect of chemotherapy drugs, where childhood cancer survivors experience exponentially rising risk for cardiovascular events (a “late effect”), but older adults’ cardiovascular risk manifests earlier and is dependent on the number of conventional co-existing cardiac risk factors — hypertension in particular.
Kero et al. [64] reported increased hazard ratios (HR) 3.6 and 13.5 times that of age-matched sibling controls for cardiomyopathy in cancer survivors aged 0–19 years and 20–34 years, respectively, as well as HR 3.3 and 1.8 for ischaemic heart disease in the same age groups, respectively. Armstrong et al. have defined a long period of latency in childhood and young adult cancer survivors of 20 or more years in longitudinal cohort studies. Further evidence of individual cardiac risk and premature cardiac ‘ageing’ was documented in breast cancer survivors (treated with a mix of anthracyclines and radiotherapy). Peak oxygen consumption scores in breast cancer survivors were equivalent to scores in controls 20 years older – mean VO2 peak (mVO2) = 19.5 ml/kg/min in breast cancer survivors aged 50 years compared to mVO2 = 19.3 ml/kg/min in a cohort of healthy 70 year old controls [65]. Thus evidence of early cardiac aging in individual cancer survivors bears striking similarities to the epidemiological outcomes in adult survivors of childhood cancers.
Childhood Cancer Cardiotoxicity
Long-term outcomes in children treated for acute lymphoblastic leukaemia with or without dexrazoxane cardioprotection document a time course of sub-clinical left ventricular disease that is distinct to that of adults [66]. Cancer-treatment related cardiotoxicity is increased in younger (especially age < 7 years), female patients, chest radiotherapy, higher doses of anthracyclines, as well as higher body fat composition. The reasons for this are not completely understood. Anthracycline-mediated reduction in C-kit + cardiac progenitor cells resulting in impaired myocardial growth (and reduced LV mass) during increased somatic growth may amplify cardiotoxic vulnerability from the loss of myocytes during anthracycline exposure in childhood [67]. Autopsy studies in human hearts have documented preferential accumulation of doxorubicin in cardiac over skeletal and smooth muscle with gradual conversion to doxorubicinol [68]. Paediatric cancer treatments may be particularly cardiotoxic as neonatal murine heart mitochondria appear ‘primed’ for apoptosis via the B-cell lymphoma 2 / BCL-2 homology (BCL2-BH3) pathway compared to apoptosis-refractory adult mitochondria [69]. These putative mechanisms, together with diastolic dysfunction from increased calcium sequestration and titin proteolysis, may partially explain why paediatric cancer survivors demonstrate a decompensated phenotype after anthracycline exposure. Childhood cancer survivors demonstrate progressive sub-clinical left ventricular disease, as somatic growth outpaces paediatric cardiomyocyte growth, manifest as early dilated cardiomyopathy followed by a period of compensated systolic function and later restrictive physiology (normal to low cavity size, reduced ventricular wall thickness and fractional shortening), a distinctly different time course compared to adult cancer survivors.
Controversies in Cardiotoxicity: Type 1 Vs. Type 2 and the Early Vs. Late Paradox
The concept of reversible (‘type 1’) and irreversible (‘type 2’) cardiotoxicity was proposed with the introduction of HER2/ ErbB receptor blockers such as trastuzumab (Herceptin ®) [70]. Here the argument was that anthracycline cardiotoxicity was persistent, dose-related and irreversible (type 1), whereas trastuzumab cardiotoxicity was reversible, not dose-related and LV ‘function’ recoverable upon discontinuation of trastuzumab. This concept has not been without controversy because some cardioprotection studies suggest that anthracycline cardiotoxicity maybe reversible if detected within 3–6 months of a decline in ejection fraction [71, 72]. Furthermore, the typical regimen of trastuzumab was administered concomitantly with anthracyclines, though currently it is administered sequentially after anthracyclines, thereby confounding any lone effect of trastuzumab with anthracycline exposure.
Cardinale et al. prospectively followed 2625 cancer patients and documented a single linear time course for anthracycline cardiotoxicity defined as a fall in LVEF [37]. In this cohort, followed up for a median of 5.2 years, 98% of cardiotoxicity was documented in the first 12 months’ follow-up, without a bimodal “early” and subsequent “late effects” distribution, contrary to conventional wisdom – casting dispersions on the widely held belief of ‘early’ and ‘late’ cardiotoxicity in adult cancer survivors. Cardinale argued against the classic ‘late’ cardiotoxicity paradigm in adults, because prior studies detecting late phenomena were retrospective, and instead proposed that ‘late’ cardiotoxicity was ‘early’ cardiotoxicity that had escaped detection at an earlier stage.
Outcomes
The long term cardiovascular outcomes of cancer survivors vary according to several risk profiles. The predictors for cardiovascular outcomes after cancer survival include age at time of diagnosis, female gender in childhood cancer survivors, chest radiotherapy, time since cancer treatment [13] and anthracycline dosage [73]. Kero et al. reported increased HR for cardiomyopathy =4.6 (and strikingly high HR 13.5 for age 0–19 y) and ischaemic heart disease =1.8 compared to sibling controls for young adult cancer survivors aged <35 years.
The importance of cardiology follow-up for late cardiotoxicity due to chemotherapy is particularly important in childhood cancer survivors. The cumulative incidence of adverse cardiovascular outcomes by age 45 years in 10,724 childhood cancer survivors (acute lypmphoblastic leukaemia [ALL] 30%, Hodgkin Lymphoma [HL] 12.8%, Wilms tumour 9.6%, sarcoma 8.7%) are strikingly higher [73]. The incidence in childhood cancer survivors compared to sibling controls of coronary artery disease, heart failure, and valvular disease was 5.3% vs. 0.9%, 4.8% vs 0.3%, and 1.5% vs. 0.1%, respectively, signifying an increased relative risk (RR) between 6 and 16. Hypertension alone was associated with an increased RR of 5.6 of cardiac death. This substantial body of long term follow-up has produced an individualized childhood cancer survivor risk calculator [74].
Cardiotoxic morbidity and mortality occur at higher numerical epirubicin doses than for doxorubicin. Dose-related NYHA class II or worse congestive heart failure (CHF) incidence occurred in 1.9% at 800 mg/m2, 4% at 900 mg/m2, and 15% at >1000 mg/m2 in a series of 469 epirubicin-treated breast cancer patients [75]. CHF occurring at a median onset of 57 days with overall 38% mortality and median survival of 162 days and 125 days if patients had received radiotherapy.
Cardiac mortality is an increasingly important determinant of overall prognosis after cancer. Cardiovascular death was the leading cause of death surpassing recurrent breast cancer (15.9% vs. 15.1%) after median nine-year follow up in 63,566 female breast cancer patients [13]. Women were more likely to die of cardiovascular causes than from breast cancer as they aged, with cardiac death overtaking cancer death after nine years survivorship. In childhood cancer survivors, cardiac deaths accounted for 13% of deaths after 45 years’ survival, and the annualised cardiac death rate exceeded that of cancer recurrence (0.14% vs. 0.05%) after 30 years’ survivorship [76]. Thus with ever longer cancer survivorship, cardiovascular mortality supervenes cancer mortality.
Pregnancy Outcomes
Cancer is the second most common cause of death during the female reproductive years and it occurs in 1 in 1000 pregnancies with breast, cervical, lymphoma and leukaemia’s the most common malignancies [77]. Lymphoma occurs in 1 in 6000 pregnancies, and identified risks include increasing maternal age and HIV-related non-Hodgkin’s lymphoma in developed and developing countries, respectively [78]. Termination of pregnancy is recommended when malignancy is diagnosed during the first trimester due to the likelihood of aneuploidy and disrupted organogenesis, but until recently malignancy management in later pregnancy was based on small case report data. Pinnix et al. reported promising 5-year long term outcomes in lymphoma (HL and NHL) diagnosed during pregnancy. In a retrospective series of 39 pregnant women (median age 28 years), there were three terminations. In the remaining 36 pregnancies, 24 elected to have antenatal chemotherapy and 12 elected to defer treatment until after delivery. There were 4 miscarriages (two in first trimester, all four in the antenatal chemotherapy group), and the median gestational age of delivery was 37 weeks. There were no reported foetal anomalies. Progression free survival at 5 years was 74.7%, (overall survival =82.4%). This compared favourably with 5-year HL survival of 86% and NHL of 69.5% in women who were not pregnant [4].
The Search for Cardioprotective Therapies
A number of potential cardioprotective techniques and therapies have been explored ranging from modified anthracycline preparations, anti-oxidants, free radical scavengers, renin-angiotensin-system antagonists, cardioselective beta-blockers to statins. Whilst many show promise in animal studies, the clinical studies have had mixed results.
Modified analogues of doxorubicin such as epirubicin and liposomal doxorubicin are relatively less cardiotoxic than conventional doxorubicin. Epirubicin was associated with decreased odds ratio for clinical 0.39 (95%CI 0.20–0.78) and sub-clinical fall in LVEF (OR 0.30 (95% CI 0.16–0.57) [11]. In a meta-analysis, epirubicin had lower rates of clinical cardiotoxicity (OR 0.39, 0.2 to 0.78; p = 0.008; I2 = 0.5%) and subclinical cardiotoxicity (OR 0.30, 0.16 to 0.57; p < 0.001; I2 = 1.7%) than doxorubicin without compromising anti-tumour efficacy [11]. Liposomal doxorubicin also had lower rates of clinical cardiotoxicity (OR 0.18, 0.08 to 0.38; p < 0.001; I2 = 0%) and subclinical cardiotoxicity (OR 0.31, 0.20 to 0.48; p < 0.01; I2 = 48.5%) than doxorubicin, again, without compromising efficacy. Liposomal doxorubicin and epirubicin have similar levels of cardiotoxicity (RR 1.15, 0.47 to 2.84; p = 0.754) [11]. Continuous infusion doxorubicin was compared against bolus infusion in children with acute lymphoblastic leukaemia. There were similar levels of LV systolic dysfunction, cavity dilatation, reduced wall thickness and LV mass at 8 years, and thus no cardioprotection in children [79].
N-acetylcysteine has antioxidant properties and was therefore hypothesised to be of benefit due to the ROS hypothesis. However, clinical studies have failed to demonstrate a benefit from N-acetylcysteine in preventing or reversing doxorubicin-induced cardiomyopathy. In a study of doxorubicin-naïve patients with normal left ventricular function, patients given N-acetylcysteine 1 h before their first dose of doxorubicin had similar histological damage (tubular and mitochondrial area) on endomycardial biopsies taken at 4 and 24 h after doxorubicin administration to controls. In a small randomised controlled trial of 19 disease-free sarcoma patients with doxorubicin-induced cardiomyopathy, patients receiving N-acetylcysteine 5.5 g/m2 daily for 30 days (n = 11) showed no difference in left ventricular ejection fraction at rest or during exercise compared to controls [80]. The largest randomised controlled trial was performed by Myers et al. who randomised 54 patients with breast cancer, nodular lymphoma or metastatic soft tissue sarcoma to receive either N-acetylcysteine 5.5 g/m2 orally prior to doxorubicin or placebo. The rates of clinical heart failure were similar between the intervention and control groups (12.5% vs 10%, p = 0.77) [81].
Calcium antagonists have been studied in two small clinical trials. Milei et al. studied prenylamine in 26 adults with solid tumours undergoing doxorubicin chemotherapy. One patient in the control group (n = 13) and no patients in the intervention group (n = 13) developed cardiomyopathy [82]. Kraft et al. studied low-dose verapamil in adult patients with acute myeloid leukaemia [83]. One patient in the control group (n = 17) and no patients in the intervention group (n = 13) developed heart failure (no significant difference).
Amifostine, a broad-spectrum cytoprotective agent, showed promise in animal models, where it prevented doxorubicin-induced cardiotoxicity [84, 85]. However, this did not translate in a clinical study of 28 children with osteosarcoma treated with cisplatin and doxorubicin, where no cardiac benefit was observed. Moreover, amifostine was poorly tolerated, with 93% of patients suffering grade 3/4 vomiting compared to 7% in the control group.
Coenzyme Q10 was studied in a mixed cohort of 20 children with acute lymphoblastic lymphoma (ALL) and HL. Children in the control group had a reduction in interventricular septum wall thickening, measured by echocardiography, following anthracycline chemotherapy. The authors suggested a protective effect from coenzyme Q10, though this has not been replicated in larger studies [86]. Despite their promise for providing cardioprotection, free radical scavengers have failed to show protection in clinical studies. Probucol, a lipid-lowering agent and antioxidant, has shown to be protective against doxorubicin-induced cardiomyopathy without compromising antitumour efficacy in rats and mice.
Animal studies using dexrazoxane had mixed results in vivo. Deferiprone (Ferriprox) demonstrated anthracycline cardioprotection in a rat model [87, 88]. However, dexrazoxane is the only iron chelator that has been licenced for clinical use in (breast) cancer patients undergoing extended anthracycline dosing in excess of 300 mg/m2.
Dexrazoxane’s cardioprotective mechanism against anthracyclines was considered to be due to iron chelation, preventing anthracycline-iron binding and the subsequent ROS formation. However, cardioprotection seems to be exclusive to dexrazoxane and deferriprone rather than a class effect as other iron chelators, such as deferasirox, have not shown cardioprotection [89]. Dexrazoxane binds to topoisomerase-2, with cardioprotection conferred via top-2β, but the role of top-2α also formed the controversial hypothesis for potentially reduced anti-tumour efficacy and its role in malignant proliferation.
Dexrazoxane has largely been evaluated in women with advanced breast cancer and adults with sarcoma, and its current approval is for extended anthracycline dosing. A meta-analysis showed that dexrazoxane administration alongside doxorubicin or epirubicin reduced the rates of clinical cardiotoxicity (OR 0.21, 0.13 to 0.33; p < 0.000 l; I2 = 0%) and subclinical cardiotoxicity (RR 0.33, 0.20 to 0.55; p < 0.0001 l I2 = 0%) compared to doxorubicin or epirubicin alone [11]. Asselin et al. reported the cardioprotective effects of dexrazoxane vs doxorubicin-only in 537 children and adolescents with T-cell acute lymphoblastic leukaemia (T-ALL) or advanced lymphoblastic Non-Hodgkin lymphoma (NHL) [90]. At three years, LV function assessed by fraction shortening was significantly lower in the doxorubicin-only group than the dexrazoxane group (p = 0.05). Five-year event free survival and rate of secondary malignancies was not statistically different between the dexrazoxane group and doxorubicin-only group (76.7 ± 2.7% vs 76.0 ± 2.7%, p = 0.9, and 0.8 ± 0.5% vs 0.7 ± 0.5%, p = 0.17, respectively). Further meta-analysis of five randomised control trials suggested dexrazoxane was associated with a borderline increase in secondary malignant neoplasms that was not statistically significant (absolute incidence 2.7% [17/635] vs. 1.1% [7/610], p = 0.06).
In addition to their lipid-lowering effects, statins also have anti-inflammatory and anti-oxidant effects [91]. They have been postulated to increase anti-tumour efficacy but clinical studies in malignant melanoma and colorectal cancer have not observed this. In a small trial of patients undergoing anthracycline-containing chemotherapy regimes, patient were randomised to receive atorvastatin 40 mg (n = 20), commenced before their chemotherapy and continued for 6 months, or control (n = 20). The primary endpoint was reduction in LVEF to <50%. One patient (5%) in the statin arm and five patients (25%) in the control arm developed LVEF <50%, however, this did not reach statistical significance (p = 0.18). The mean change in LVEF, left ventricular end-diastolic diameter and end-systolic diameter (LVEDD and LVSD) from baseline to 6 months for the statin group and control group were 1.3 ± 3.8 vs −7.9 ± 8.0%, p < 0.001; −0.15 ± 4.0 vs 2.0 ± 3.3 mm, p = 0.021; and −1.35 ± 4.0 vs 2.1 ± 1.8 mm, p < 0.001, respectively [92]. In a retrospective, observational cohort study of 201 women receiving anthracyclines for newly diagnosed breast cancer, uninterrupted statin therapy had a reduced risk of heart failure compared to propensity-matched controls (hazard ratio 0.3, 95% CI: 0.1–0.9, p = 0.03). However, this finding may have been confounded by a greater ACE inhibitor and β-blocker use in the statin group compared to controls (38.8% vs 17.2%, p < 0.001 and 38.8% vs 14.9%, p < 0.001, respectively) [93].
Carvedilol is a non-selective β-antagonist with some α-antagonist effect. It also inhibits NADH dehydrogenase, found in complex I of the mitochondria, and has antioxidant activity. It has therefore received considerable interest in cardioprotection against anthracycline-induced cardiotoxicity and been studied in isolation and in combination with ACE inhibitors.
The OVERCOME trial (preventiOn of the left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive chemOtherapy for the treatment of Malignant hEmopathies) is the largest randomised controlled trial of combined ACE inhibitors and carvedilol [94]. Ninety adult patients with normal left ventricular ejection fraction (LVEF) and newly diagnosed haematological malignancy were randomised to either enalapril + carvedilol or control. The primary endpoints were changes in LVEF assessed by transthoracic echocardiography and cardiac magnetic resonance (CMR) at six months from baseline. The population comprised of 36 patients with acute leukaemia (acute myeloid leukaemia 30 and acute lymphoblastic leukaemia 6) and 54 were undergoing autologous stem cell transplant for Hodgkin disease (n = 9), non-Hodgkin lymphoma (n = 23), and multiple myeloma (n = 22). Doses of enalapril and carvedilol were similar to heart failure studies (8.2 ± 5.9 mg and 26.1 ± 18.2 mg, respectively). Left ventricular ejection fraction declined at six month in the control group on both echocardiography and CMR (−3.28% (95%CI: -5.49 to −1.07) and −3.04% (95%CI: -6.01 to −0.070, respectively), but was preserved in the intervention group (−0.17 (−2.24 to 1.90) on echocardiography and 0.36 (−2.41 to 3.13) on CMR). Interestingly, the pre-specified subgroup analysis showed that patients with acute leukaemia had a greater degree of LVEF decline, with mean − 6.4% (95%CI: -11.88 to −0.87) compared to −1.01% (95%CI: -4.46 to 2.45) in patients undergoing autologous stem cell transplant.
Prompt initiation of ACE inhibitors and β-blockers when LVEF declines is important for recovery of function [71, 95]. Cardinale et al. randomised patients with raised troponin I after high-dose anthracycline-containing chemotherapy to either enalapril (n = 56) or control (n = 58). Treatment was started one month after the completion of chemotherapy and continued for a year. The primary endpoint was a reduction in LVEF >10% to LVEF <50%. The maximum tolerated dose of enalapril was 16 ± 6 mg daily. Twenty five control subjects (43%) and no intervention subject had a reduction in LVEF to <50% (p < 0.001) [72]. Furthermore, their work documented that prompt initiation of treatment was an important determinant of LV functional recovery.
In their larger prospective cohort study of 2625 patients undergoin anthracycline chemotherapy, Cardinale et al. measured LVEF by serial echocardiography for a median follow-up of 5.2 years (quartile 1 to quartile 3, 2.6–8.0 years) [37]. Cardiotoxicity, defined as a reduction in LVEF of ≥10% to <50%, was seen in 226 (9%). The vast majority (98%) occurred within the first 12 months, with median time to development of 3.5 months (quartile 1 to quartile 3, 3–6 months) after completing chemotherapy. Prompt initiation of enalapril and carvedilol (n = 186) or enalapril alone (for those recruited before 1999, n = 40) at the time of detection of cardiotoxicity resulted in full recovery of LVEF, defined as return to baseline, in 25 (11%) and partial recovery, defined as an increase in LVEF ≥ 5%, in 160 (71%) patients [95]. This study also raised the intriguing observation that 98% of LV functional decline was detected in the first 12 months – challenging the paradigm of early vs. late cardiotoxicity observed in legacy retrospective studies.
Studies of carvedilol monotherapy are smaller. In adult females with non-metastatic breast cancer randomised to carvedilol (n = 30) or placebo (n = 40), commencing one week before starting doxorubicin and finishing one week after the final cycle, those taking carvedilol showed no change in their LV strain measurements from baseline to after chemotherapy, whereas patients taking placebo showed significant deterioration. Neither group showed significant deterioration in their LVEF [96]. Another small study randomised patients to carvedilol (n = 25) or placebo (n = 25) and demonstrated that the carvedilol group maintained LVEF, whereas the control group had a reduction in LVEF at 6 months compared to baseline (70.5 vs 69.37, p = 0.3, and 68.9 vs 52.3, p < 0.001, respectively).
Carvedilol has also shown a benefit in children aged 6–12 years with acute lymphoblastic leukaemia treated with doxorubicin [97]. Patients were randomised to carvedilol for five days before doxorubicin (n = 25) or doxorubicin alone (n = 25). Patients in the intervention arm had a higher fractional shortening (FS) one week after the last dose of doxorubicin compared to pre-chemotherapy (baseline 34.0 ± 4.5 vs 39.5 ± 6.3, p ≤ 0.05), whereas patients without carvedilol had a reduction in FS (40.0 ± 4.6 vs 33.5 ± 6.2, p ≤ 0.05). Diastolic parameters (E, A and E/A) were not different between the two groups. Similar findings were seen by Ewer between pre-doxorubicin and one-week after completion of chemotherapy in paediatric patients, aged 7 months to 15.5 years [98]. Nebivolol, a selective β1-antagonist with nitric oxide vasodilator properties, demonstratred increased contractility in animal studies, and cardioprotrection via preserved LVEF compared to placebo in patients with breast cancer receiving doxorubicin [99].
The recent PRADA trial (Prevention of cardiac dysfunction during adjuvant breast cancer therapy) compared the cardioproctive properties of candesartan and metoprolol in 120 patients with early breast cancer undergoing adjuvant FEC chemotherapy (fluorouracil, epirubin and cyclophosphamide) in a 2 × 2 factorial, randomised, placebo-controlled, double-blind study [100]. Patients were randomised to receive candesartan-metoprolol, candesartan-placebo, metoprolol-placebo or placebo-placebo. The primary endpoint was a change in cardiac magnetic resonance (CMR)-LVEF from baseline to the end of chemotherapy. Patients receving candesartan had no change in their LVEF (0.8, 95% CI: -0.04 to 1.9) after chemotherapy, whereas the non-candesartan group had LVEF decline of 2.6% (95% CI: 1.5 to 3.8, p = 0.026). No difference was seen between patients receiving metoprolol and metoprolol-naïve patients, 1.6% (95% CI: 0.4 to 2.8) and 1.8% (95% CI: 0.7 to 3.0), respectively. The PRADA results support cardioprotective properties on a statistically measurable scale, if not wholly clinically significant.
Remote ischaemic conditioning (RIC) is another potentially cardioprotective treatment that is currently under investigation in cancer patients [56]. This non-invasive non-pharmacological treatment is delivered via a blood pressure cuff as short bursts of ischaemia and reperfusion in a peripheral limb. RIC has been shown to protect the heart and other organs from subsequent severe ischaemic injury [101–107], and demonstrated lung protection in cancer surgery [103]. While the mechanism of RIC is not fully understood, it appears to involve a humoral and neural pathway that confers cardioprotection by activating innate pro-survival pathways that ultimately modulate common mechanisms in ischaemia reperfusion injury and anthracycline cardiotoxicity such as calcium overload, lipid peroxidation, ROS generation and mitochondrial function [108].
Conclusion
The cardiotoxic mechanisms of anthracyclines may involve the dual pathways of reactive oxidation species and topoisomerase 2-beta and a final common pathway of calcium overload, lipid peroxidation and mitochondrial dysfunction. There is no universally accepted definition of anthracycline cardiotoxicity, and thus its incidence varies according to how it is defined. Clinically symptomatic heart failure occurs in 2–4%, asymptomatic fall in LVEF in 9–11%, arrhythmia in 12% or more, and cardiac biomarker rise in 30–35% of treated patients. Predictors of cardiotoxicity include cumulative dose, cardiovascular risk factors and age of treatment.
It is essential that we continue to investigate ways of protecting the heart following cancer chemotherapy, At present the limited cardioprotective strategies available -- dexrazoxane, ACE-inhibitors, ARB, and beta-blockers -- are not in routine prophylactic use. New strategies are required to help combat the cardiotoxicity derived from anthracycline and other chemotherapies with novel agents such as NRG-1 that target ErbB receptor or non-pharmacological and non-invasive techniques, such as remote conditioning, which activate endogenous pro-survival pathways in the heart.
In the forthcoming era of personalised cancer medicine and targeted therapies, it may be tempting to consign the cardiac effects of anthracyclines to history. However the large population-at-risk, long latency in childhood cancer survivors, and prospectively increased cardiac risk in adult cancer survivors have implications for cancer survivorship as a cardio-oncology partnership. Thus, the increasing cancer survivor population represents a strong motivator to explore larger randomised controlled trials in cancer cardioprotection to prevent today’s cancer patient from becoming tomorrow’s cardiac patient.
References
Rimal H, Lee SW, Lee JH, Oh TJ. Understanding of real alternative redox partner of Streptomyces peucetius DoxA: prediction and validation using in silico and in vitro analyses. Arch Biochem Biophys. 2015;585:64–74.
Organization WH. WHO Model List of Essential Medicines. April 2015 ed2015.
Coleman MP, Forman D, Bryant H, Butler J, Rachet B, Maringe C, et al. Cancer survival in Australia, Canada, Denmark, Norway, Sweden, and the UK, 1995-2007 (the international cancer benchmarking partnership): an analysis of population-based cancer registry data. Lancet. 2011;377:127–38.
CancerResearchUK. Cancer mortality in the UK 2012. In: Cancer Statistics Report. London UK. 2014. www.cruk.org/cancerstats. Accessed 27th October 2014.
Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–52.
Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–98.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92.
Giordano SH, Lin YL, Kuo YF, Hortobagyi GN, Goodwin JS. Decline in the use of anthracyclines for breast cancer. J Clin Oncol. 2012;30:2232–9.
Nabhan C, Byrtek M, Rai A, Dawson K, Zhou X, Link BK, et al. Disease characteristics, treatment patterns, prognosis, outcomes and lymphoma-related mortality in elderly follicular lymphoma in the United States. Br J Haematol. 2015;170:85–95.
Chihara D, Westin JR, Oki Y, Ahmed MA, Do B, Fayad LE, et al. Management strategies and outcomes for very elderly patients with diffuse large B-cell lymphoma. Cancer. 2016;
Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, et al. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer. 2010;10:337.
Armstrong GT, Chen Y, Yasui Y, Leisenring W, Gibson TM, Mertens AC, et al. Reduction in late mortality among 5-year survivors of childhood cancer. N Engl J Med. 2016;374:833–42.
Patnaik JL, Byers T, DiGuiseppi C, Dabelea D, Denberg TD. Cardiovascular disease competes with breast cancer as the leading cause of death for older females diagnosed with breast cancer: a retrospective cohort study. Breast Cancer Res. 2011;13:R64.
Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–5.
Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. 1986;261:3060–7.
Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol. 2007;23:15–25.
Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014;124:617–30.
Hahn VS, Lenihan DJ, Ky B. Cancer therapy-induced cardiotoxicity: basic mechanisms and potential cardioprotective therapies. J Am Heart Assoc. 2014;3:e000665.
Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413.
Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–40.
Carpenter AJ, Porter AC. Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIalpha mutant human cell line. Mol Biol Cell. 2004;15:5700–11.
Tewey KMRT, Yang L, Halligan DB, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466–8.
Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–8.
Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839–46.
Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–42.
Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci U S A. 1987;84:5034–7.
Wadugu B, Kuhn B. The role of neuregulin/ErbB2/ErbB4 signaling in the heart with special focus on effects on cardiomyocyte proliferation. Am J Physiol Heart Circ Physiol. 2012;302:H2139–47.
Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8:459–65.
Ma J, Wang Y, Zheng D, Wei M, Xu H, Peng T. Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc Res. 2013;97:77–87.
Huang C, Zhang X, Ramil JM, Rikka S, Kim L, Lee Y, et al. Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularization resulting in greater susceptibility to stress-induced myocardial injury in adult mice. Circulation. 2010;121:675–83.
Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD, et al. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol. 2005;23:2629–36.
Von Hoff DD, Layard MW, Basa P, Davis Jr HL, Von Hoff AL, Rozencweig M, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710–7.
Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97:2869–79.
Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342:1077–84.
Hequet O, Le QH, Moullet I, Pauli E, Salles G, Espinouse D, et al. Subclinical late cardiomyopathy after doxorubicin therapy for lymphoma in adults. J Clin Oncol. 2004;22:1864–71.
Thavendiranathan P, Grant AD, Negishi T, Plana JC, Popovic ZB, Marwick TH. Reproducibility of echocardiographic techniques for sequential assessment of left ventricular ejection fraction and volumes: application to patients undergoing cancer chemotherapy. J Am Coll Cardiol. 2013;61:77–84.
Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. 2015;
Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, Asteggiano R, Galderisi M, Habib G, et al. Authors/task force members; ESC Committee for Practice Guidelines (CPG): ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: the task force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J. 2016;37:2768–2801.
Ahmed AN, Blonde K, Hackam D, Iansavichene A, Mrkobrada M. Prognostic significance of elevated troponin in non-cardiac hospitalized patients: a systematic review and meta-analysis. Ann Med. 2014;46:653–63.
Grinstein J, Bonaca MP, Jarolim P, Conrad MJ, Bohula-May E, Deenadayalu N, et al. Prognostic implications of low level cardiac troponin elevation using high-sensitivity cardiac troponin T. Clin Cardiol. 2015;38:230–5.
Grodin JL, Neale S, Wu Y, Hazen SL, Tang WH. Prognostic comparison of different sensitivity cardiac troponin assays in stable heart failure. Am J Med. 2014;
Hochholzer W, Valina CM, Stratz C, Amann M, Schlittenhardt D, Buttner HJ, et al. High-sensitivity cardiac troponin for risk prediction in patients with and without coronary heart disease. Int J Cardiol. 2014;176:444–9.
Kuster N, Monnier K, Baptista G, Dupuy AM, Badiou S, Bargnoux AS, et al. Estimation of age- and comorbidities-adjusted percentiles of high-sensitivity cardiac troponin T levels in the elderly. Clin Chem Lab Med. 2014;
Qian G, Wu C, Zhang Y, Chen YD, Dong W, Ren YH. Prognostic value of high-sensitivity cardiac troponin T in patients with endomyocardial-biopsy proven cardiac amyloidosis. J Geriatr Cardiol. 2014;11:136–40.
Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD. Third universal definition of myocardial infarction. Circulation. 2012;126:2020–35.
Yiu KH, Lau KK, Zhao CT, Chan YH, Chen Y, Zhen Z, et al. Predictive value of high-sensitivity troponin-I for future adverse cardiovascular outcome in stable patients with type 2 diabetes mellitus. Cardiovasc Diabetol. 2014;13:63.
Auner HW, Tinchon C, Linkesch W, Tiran A, Quehenberger F, Link H, et al. Prolonged monitoring of troponin T for the detection of anthracycline cardiotoxicity in adults with hematological malignancies. Ann Haematol. 2003;82:218–22.
Cardinale D, Sandri MT, Colombo A, Colombo N, Boeri M, Lamantia G, et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circ. 2004;109:2749–54.
Kilickap S, Barista I, Akgul E, Aytemir K, Aksoyek S, Aksoy S, et al. cTnT can be a useful marker for early detection of anthracycline cardiotoxicity. Ann Oncol. 2005;16:798–804.
Cardinale D, Sandri MT, Martinoni A, Tricca A, Civelli M, Lamantia G, et al. Left ventricular dysfunction predicted by early troponin I release after high-dose chemotherapy. J Am Coll Cardiol. 2000;36:517–22.
Cardinale D, Sandri MT, Martinoni A, Borghini E, Civelli M, Lamantia G, et al. Myocardial injury revealed by plasma troponin I in breast cancer with high-dose chemotherapy. Ann Oncol. 2002;13:710–5.
Sandri MT, Cardinale D, Zorzino L, Passerini R, Lentati P, Martinoni A, et al. Minor increases in plasma troponin I predict decreased left ventricular ejection fraction after high-dose chemotherapy. Clin Chem. 2003;49:248–52.
Lipshultz SE, Rifai N, Dalton VM, Levy DE, Silverman LB, Lipsitz SR, et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med. 2004;351:145–53.
Haney S, Cresti N, Verrill M, Plummer C. Cardiac troponin release following standard dose anthracycline-based adjuvant chemotherapy (abstr). Eur Heart J. 2013;34(Suppl 1):1074.
Katsurada K, Ichida M, Sakuragi M, Takehara M, Hozumi Y, Kario K. High-sensitivity troponin T as a marker to predict cardiotoxicity in breast cancer patients with adjuvant trastuzumab therapy. Springerplus. 2014;3:620.
Chung R, Maulik A, Hamarneh A, Hochhauser D, Hausenloy DJ, Walker JM, et al. Effect of remote ischaemic conditioning in oncology patients undergoing chemotherapy: rationale and design of the ERIC-ONC study--a single-center, blinded. Randomized Controlled Trial Clin Cardiol. 2016;39:72–82.
Auner HW, Tinchon C, Linkesch W, Halwachs-Baumann G, Sill H. Correspondence re: O. J. Arola et al., acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer res., 60: 1789-1792, 2000. Cancer Res. 2001;61:2335–6.
Colovic N, Bogdanovic A, Virijevic M, Vidovic A, Tomin D. Acute myocardial infarction during induction chemotherapy for acute MLL t(4;11) leukemia with lineage switch and extreme leukocytosis. Srp Arh Celok Lek. 2015;143:734–8.
Landolfi R, Di Gennaro L, Barbui T, De Stefano V, Finazzi G, Marfisi R, et al. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood. 2007;109:2446–52.
Schmid-Schonbein GW. The damaging potential of leukocyte activation in the microcirculation. Angiology. 1993;44:45–56.
Hidalgo JD, Krone R, Rich MW, Blum K, Adkins D, Fan MY, et al. Supraventricular tachyarrhythmias after hematopoietic stem cell transplantation: incidence, risk factors and outcomes. Bone Marrow Transplant. 2004;34:615–9.
Kilickap S, Barista I, Akgul E, Aytemir K, Aksoy S, Tekuzman G. Early and late arrhythmogenic effects of doxorubicin. South Med J. 2007;100:262–5.
Hu YF, Liu CJ, Chang PM, Tsao HM, Lin YJ, Chang SL, et al. Incident thromboembolism and heart failure associated with new-onset atrial fibrillation in cancer patients. Int J Cardiol. 2013;165:355–7.
Kero AE, Jarvela LS, Arola M, Malila N, Madanat-Harjuoja LM, Matomaki J, et al. Cardiovascular morbidity in long-term survivors of early-onset cancer: a population-based study. Int J Cancer. 2014;134:664–73.
Jones LW, Courneya KS, Mackey JR, Muss HB, Pituskin EN, Scott JM, et al. Cardiopulmonary function and age-related decline across the breast cancer survivorship continuum. J Clin Oncol. 2012;30:2530–7.
Lipshultz SE, Scully RE, Lipsitz SR, Sallan SE, Silverman LB, Miller TL, et al. Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol. 2010;11:950–61.
Lipshultz SE, Lipsitz SR, Mone SM, Goorin AM, Sallan SE, Sanders SP, et al. Female sex and drug dose as risk factors for late cardiotoxic effects of doxorubicin therapy for childhood cancer. N Engl J Med. 1995;332:1738–43.
Stewart DJ, Grewaal D, Green RM, Mikhael N, Goel R, Montpetit VA, et al. Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer Res. 1993;13:1945–52.
Sarosiek KA, Letai A. Directly targeting the mitochondrial pathway of apoptosis for cancer therapy using BH3 mimetics - recent successes, current challenges and future promise. FEBS J. 2016;283:3523–33.
Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat Rev Cardiol. 2010;7:564–75.
Cardinale D, Colombo A, Lamantia G, Colombo N, Civelli M, De Giacomi G, et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. J Am Coll Cardiol. 2010;55:213–20.
Cardinale D, Colombo A, Sandri MT, Lamantia G, Colombo N, Civelli M, et al. Prevention of high-dose chemotherapy-induced cardiotoxicity in high-risk patients by angiotensin-converting enzyme inhibition. Circulation. 2006;114:2474–81.
Armstrong GT, Oeffinger KC, Chen Y, Kawashima T, Yasui Y, Leisenring W, et al. Modifiable risk factors and major cardiac events among adult survivors of childhood cancer. J Clin Oncol. 2013;31:3673–80.
StJude. Children's Hospital CCSS CHF Risk Calculator. 2015. https://ccss.stjude.org/tools-and-documents/calculators-and-other-tools/ccss-chf-risk-calculator.html. Accessed 13/06/16 2016.
Ryberg M, Nielsen D, Skovsgaard T, Hansen J, Jensen BV, Dombernowsky P. Epirubicin cardiotoxicity: an analysis of 469 patients with metastatic breast cancer. J Clin Oncol. 1998;16:3502–8.
Mertens AC, Liu Q, Neglia JP, Wasilewski K, Leisenring W, Armstrong GT, et al. Cause-specific late mortality among 5-year survivors of childhood cancer: the childhood cancer survivor study. J Natl Cancer Inst. 2008;100:1368–79.
Pavlidis NA. Coexistence of pregnancy and malignancy. Oncologist. 2002;7:279–87.
Pereg D, Koren G, Lishner M. The treatment of Hodgkin’s and non-Hodgkin’s lymphoma in pregnancy. Haematologica. 2007;92:1230–7.
Lipshultz SE, Miller TL, Lipsitz SR, Neuberg DS, Dahlberg SE, Colan SD, et al. Continuous versus bolus infusion of doxorubicin in children with ALL: long-term cardiac outcomes. Pediatrics. 2012;130:1003–11.
Dresdale AR, Barr LH, Bonow RO, Mathisen DJ, Myers CE, Schwartz DE, et al. Prospective randomized study of the role of N-acetyl cysteine in reversing doxorubicin-induced cardiomyopathy. Am J Clin Oncol. 1982;5:657–63.
Myers C, Bonow R, Palmeri S, Jenkins J, Corden B, Locker G, et al. A randomized controlled trial assessing the prevention of doxorubicin cardiomyopathy by N-acetylcysteine. Semin Oncol. 1983;10:53–5.
Milei J, Marantz A, Ale J, Vazquez A, Buceta JE. Prevention of adriamycin-induced cardiotoxicity by prenylamine: a pilot double blind study. Cancer Drug Deliv. 1987;4:129–36.
Kraft J, Grille W, Appelt M, Hossfeld DK, Eichelbaum M, Koslowski B, et al. Effects of verapamil on anthracycline-induced cardiomyopathy: preliminary results of a prospective multicenter trial. Haematol Blood Transfus. 1990;33:566–70.
Nazeyrollas P, Frances C, Prevost A, Costa B, Lorenzato M, Kantelip JP, et al. Efficiency of amifostine as a protection against doxorubicin toxicity in rats during a 12-day treatment. Anticancer Res. 2003;23:405–9.
Dragojevic-Simic VM, Dobric SL, Bokonjic DR, Vucinic ZM, Sinovec SM, Jacevic VM, et al. Amifostine protection against doxorubicin cardiotoxicity in rats. Anti-Cancer Drugs. 2004;15:169–78.
Iarussi D, Auricchio U, Agretto A, Murano A, Giuliano M, Casale F, et al. Protective effect of coenzyme Q10 on anthracyclines cardiotoxicity: control study in children with acute lymphoblastic leukemia and non-Hodgkin lymphoma. Mol Asp Med. 1994;15:s207–12.
Ammar el SM, Said SA, Suddek GM, El-Damarawy SL. Amelioration of doxorubicin-induced cardiotoxicity by deferiprone in rats. Can J Physiol Pharmacol. 2011;89:269–76.
Popelova O, Sterba M, Simunek T, Mazurova Y, Guncova I, Hroch M, et al. Deferiprone does not protect against chronic anthracycline cardiotoxicity in vivo. J Pharmacol Exp Ther. 2008;326:259–69.
Hasinoff BB, Patel D, Wu X. The oral iron chelator ICL670A (deferasirox) does not protect myocytes against doxorubicin. Free Radic Biol Med. 2003;35:1469–79.
Asselin BL, Devidas M, Chen L, Franco VI, Pullen J, Borowitz MJ, et al. Cardioprotection and safety of dexrazoxane in patients treated for newly diagnosed T-cell acute lymphoblastic leukemia or advanced-stage lymphoblastic non-Hodgkin lymphoma: a report of the Children's oncology group randomized trial pediatric oncology group 9404. J Clin Oncol. 2016;34:854–62.
Schupp N, Schmid U, Heidland A, Stopper H. Rosuvastatin protects against oxidative stress and DNA damage in vitro via upregulation of glutathione synthesis. Atherosclerosis. 2008;199:278–87.
Acar Z, Kale A, Turgut M, Demircan S, Durna K, Demir S, et al. Efficiency of atorvastatin in the protection of anthracycline-induced cardiomyopathy. J Am Coll Cardiol. 2011;58:988–9.
Seicean S, Seicean A, Plana JC, Budd GT, Marwick TH. Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy: an observational clinical cohort study. J Am Coll Cardiol. 2012;60:2384–90.
Bosch X, Rovira M, Sitges M, Domenech A, Ortiz-Perez JT, de Caralt TM, et al. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: the OVERCOME trial (preventiOn of left ventricular dysfunction with enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of malignant hEmopathies). J Am Coll Cardiol. 2013;61:2355–62.
Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. 2015;131:1981–8.
Tashakori Beheshti A, Mostafavi Toroghi H, Hosseini G, Zarifian A, Homaei Shandiz F, Fazlinezhad A. Carvedilol administration can prevent doxorubicin-induced cardiotoxicity: a double-blind randomized trial. Cardiology. 2016;134:47–53.
El-Shitany NA, Tolba OA, El-Shanshory MR, El-Hawary EE. Protective effect of carvedilol on adriamycin-induced left ventricular dysfunction in children with acute lymphoblastic leukemia. J Card Fail. 2012;18:607–13.
Ewer MS, Ali MK, Gibbs HR, Swafford J, Graff KL, Cangir A, et al. Cardiac diastolic function in pediatric patients receiving doxorubicin. Acta Oncol. 1994;33:645–9.
Kaya MG, Ozkan M, Gunebakmaz O, Akkaya H, Kaya EG, Akpek M, et al. Protective effects of nebivolol against anthracycline-induced cardiomyopathy: a randomized control study. Int J Cardiol. 2013;167:2306–10.
Gulati G, Heck SL, Ree AH, Hoffmann P, Schulz-Menger J, Fagerland MW, et al. Prevention of cardiac dysfunction during adjuvant breast cancer therapy (PRADA): a 2 × 2 factorial, randomized, placebo-controlled, double-blind clinical trial of candesartan and metoprolol. Eur Heart J. 2016;37:1671–80.
Bautin AE, Galagudza MM, Datsenko SV, Tashkhanov DM, Marichev AO, Bakanov A, et al. Effects of remote ischemic preconditioning on perioperative period in elective aortic valve replacement. Anesteziol Reanimatol. 2014:11–7.
Davies WR, Brown AJ, Watson W, McCormick LM, West NE, Dutka DP, et al. Remote ischemic preconditioning improves outcome at 6 years after elective percutaneous coronary intervention: the CRISP stent trial long-term follow-up. Circ Cardiovasc Interv. 2013;6:246–51.
Li C, Xu M, Wu Y, Li YS, Huang WQ, Liu KX. Limb remote ischemic preconditioning attenuates lung injury after pulmonary resection under propofol-remifentanil anesthesia: a randomized controlled study. Anesthesiology. 2014;121:249–59.
Pryzklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation. 1993;87:893–9.
Sloth AD, Schmidt MR, Munk K, Kharbanda RK, Redington AN, Schmidt M, et al. CONDI investigators. Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur Heart J. 2014;35:168–75.
White SK, Frohlich GM, Sado DM, Maestrini V, Fontana M, Treibel TA, et al. Remote ischemic conditioning reduces myocardial infarct size and edema in patients with ST-segment elevation myocardial infarction. JACC Cardiovasc Interv. 2014;14:01073–5.
Zuo B, Wang F, Song Z, Xu M, Wang G. Using remote ischemic conditioning to reduce acute kidney injury in patients undergoing percutaneous coronary intervention: a meta-analysis. Curr Med Res Opin. 2015:1–20.
Pickard JM, Davidson SM, Hausenloy DJ, Yellon DM. Co-dependence of the neural and humoral pathways in the mechanism of remote ischemic conditioning. Basic Res Cardiol. 2016;111:50.
Acknowledgements
This work was supported by funding from the Biomedical Research Centre (grant number BRC233/CM/SD/101320). This manuscript was undertaken at University College London Hospitals / University College London (UCLH/UCL) who received a proportion of funding from the Department of Health’s National Institute for Health Research Biomedical Research Centres funding scheme.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors (JVM, RC, AM, IP, JMW, DMY) declare that they have no conflicts of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This article does not contain any studies conducted by the authors and thus there are no direct funding disclosures. Two authors (RC, IP) were supported by funding from the Biomedical Research Centre (grant number BRC233/CM/SD/101320). This manuscript was undertaken at University College London Hospitals / University College London (UCLH/UCL) who received a proportion of funding from the Department of Health’s National Institute for Health Research Biomedical Research Centres funding scheme.
Informed Consent
Not applicable to this article-- as this article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
McGowan, J.V., Chung, R., Maulik, A. et al. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc Drugs Ther 31, 63–75 (2017). https://doi.org/10.1007/s10557-016-6711-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-016-6711-0