
Abstract
Chemotherapy and radiation therapy (RT) are standard therapeutic modalities for patients with cancer, including breast cancer. Historic studies examining tissue and cellular responses to RT have predominantly focused on damage caused to proliferating malignant cells leading to their death. However, there is increasing evidence that RT also leads to significant alterations in the tumor microenvironment, particularly with respect to effects on immune cells infiltrating tumors. This review focuses on tumor-associated immune cell responses following RT and discusses how immune responses may be modified to enhance durability and efficacy of RT.
Similar content being viewed by others

Introduction
A role for leukocytes in solid tumor development has long been suspected [1]; however, only recently have immune-competent spontaneous models of human cancer development enabled mechanistic evaluation of leukocytes to determine if their presence in solid tumors is coincidental or functional. With the advent of mouse models of multi-stage neoplastic progression, it is now clear that lymphoid and myeloid cells can either restrain or propel cancer development, depending on their maturation state, as well as the local microenvironment regulating their bioeffector phenotype [2–4]. A role for immune cells as mediators of therapeutic response in cancers has only recently been explored [5, 6].
Chemotherapy (CTX) and radiation therapy (RT) remain as part of the standard therapeutic armament for patients with cancer, including breast cancer (BC) [7]. Both CTX and RT impact growing cancers through their ability to induce cell death by disrupting various parameters of cell biology necessary for survival. However, recent data has emerged demonstrating that the type of cell death induced by cytotoxic therapy is significant with regards to the type of immune response elicited within a tissue [8]. These studies have revealed that effectiveness of CTX and RT may in part depend on whether cell death (induced by cytotoxic therapy) is “sensed” by leukocytes [5, 9, 10]. Leukocytes detect cell death through immune-based receptors for molecules released by dying cells (often termed “danger signals”), such as toll-like receptor (TLR)-4 and its ligands including the high-mobility group box protein (HMGB) 1 [11]. Detection of danger signals in tissues by leukocytes activates an immune response involving cells of the innate (myeloid and natural killer cells) and adaptive (T and B cell) lineages. This review will focus on immunologic consequences of RT and discusses emerging data indicating that therapeutic reprogramming of immune responses in tumors may regulate efficacy and durability of RT.
Immune Cells and Cancer Development
Cancer research has primarily focused on the role of activating and/or inactivating mutations in genes regulating aspects of cell proliferation or cell death. Much of that research has been geared towards understanding how these activating and/or inactivating mutations support the multi-step model of tumorigenesis where progressive accumulation of genetic changes in somatic cells act as drivers of cancer development [12]. This neoplastic cell-intrinsic perspective of multi-stage tumorigenesis overlooks progressive alterations in the tumor microenvironment that also regulate cancer development [13]. Solid tumors contain neoplastic and non-neoplastic stromal cells embedded in a dynamic extracellular matrix (ECM) microenvironment. Cellular components of tumor stroma include hematogenous and lymphatic vascular cells, infiltrating and resident leukocytes, various populations of fibroblasts and mesenchymal support cells unique to each tissue microenvironment. Clinical and experimental studies have established that chronic infiltration of neoplastic tissue by leukocytes, i.e., chronic inflammation, promotes development and/or progression of solid tumors. However, the organ-specific cellular and molecular programs that favor pro-tumor, as opposed to anti-tumor immunity by leukocytes remain incompletely understood.
Retrospective clinical studies have revealed an increased presence of extra follicular B cells, T regulatory (Treg) cells, high ratios of CD4/CD8 or TH2/TH1 T lymphocytes in primary tumors or in draining lymph nodes that correlate with tumor grade, stage and overall survival (OS) [14–22]. Lymphocyte density in pretreatment biopsies has also been found to represent an independent predictor of complete pathologic response following anthracycline and taxane-based chemotherapy [23]. On the other hand, high densities of macrophages in BC stroma, and some other solid tumors, correlates with increased vascular density and worse clinical outcome [24–29]. Infiltration of macrophages inside tumor nests however, particularly when CD8+ cytotoxic lymphocytes (CTL) are also present, correlates with increased overall survival (OS) [30]. These differences might be explained in part by the realization that macrophages exert either pro- or anti-tumor bioactivities depending on the types of cytokines to which they are exposed. Macrophages exposed to TH1 cytokines including interferon (IFN)γ, tumor necrosis factor (TNF)α, and granulocyte monocyte-colony stimulating factor (GM-CSF) exhibit enhanced cytotoxic activity, production of pro-inflammatory cytokines and antigen presentation [31, 32]. On the other hand, macrophages exposed to TH2 cytokines such as interleukin (IL)-4 and -13, immune complexes or immunosuppressive cytokines [31] instead block CTL activity and promote angiogenesis and tissue remodeling [33, 34]. Thus, the presence of leukocytes can exert either a positive or negative force depending on the functional properties they possess.
Radiation Therapy and Activation of Stress Response Pathways
Historically, RT was thought to induce an immunosuppressive microenvironment largely based on experimental studies with whole body irradiation revealing lower levels of circulating lymphocytes resulting from increased radiation sensitivity of bone marrow as compared to other tissues [35]. Fas, a cell surface proapototic protein and member of the tumor necrosis factor receptor (TNF-R) family, contains an intracellular "death domain" that is activated following ligand binding that subsequently leads to apoptosis. Fas is expressed on many cell types including lymphocytes and is upregulated in response to cell damage. Activation of Fas-mediated cell death is a mechanism by which immune cells eliminate damaged cells, including those damaged by RT [36]. Thus, while whole body radiation is “immunosuppressive” due to triggering widespread apoptosis of immune cells via Fas, focal radiation such as that used for treatment of many types of solid tumors instead has limited immunosuppressive side effects, and may actually promote changes in the local tumor microenvironment that paradoxically enhance infiltration and activation of multiple immune cell types (Fig. 1) that may either foster, and/or suppress tumor development [2].

Leukocyte infiltration following RT. Representative tissue sections of mammary carcinomas stained with hematoxylin and eosin (A) following isolation from a murine mammary carcinoma 96 h after receiving 5 Gy of localized gamma irradiation. CD45+ staining (red) of adjacent tissue sections (B) demonstrates extent of leukocyte infiltration following RT. The percentage change in CD45+ cell infiltration was assessed by flow cytometry of whole tumor cell suspensions revealing a significant increase in CD45+ cells following RT.
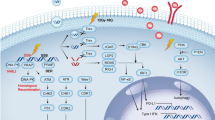
At the most simplistic level, a main mechanism by which ionizing radiation mediates a biologic effect is via generation of free radicals that lead to genotoxic (DNA) damage, and subsequent activation of stress-response pathways through activation of the DNA damage pathway ataxia telangiectasia mutated (ATM). Activation of the ATM protein pathway following RT involves activation of p53 and nuclear factor (NF)-κB transcription factors [37, 38]. NF-κB can also be activated independently of DNA damage through radiation-induced activation of TNFR-associated factors (TRAFs) [39, 40]. NF-κB directly regulates expression of molecules that promote a “pro-inflammatory” immune response, including TNF-α [41], interleukin (IL)-1 [42], chemokines such as CCL5 [43]; adhesion molecules including intracellular adhesion molecule (ICAM)-1 [44, 45], E-selectin [46] and vascular cell adhesion molecule (VCAM)-1 [47], as well as major histocompatability complex (MHC) molecules (Fig. 2) [48], and expression of several anti-apoptotic genes including Bax and Bcl-2 [49–52]. Signaling cascades induced by radiation through ATM/NF-κB, in addition to the direct cell death resulting from radiation damage, stimulates influx and activation of leukocytes leading to a productive immune response.

Schematic of the immune response to RT. Tumor cells respond to ionizing RT by upregulating cytokines (TNFα, IL-1α/β and IL-6), adhesion molecules (ICAM-1, VCAM-1, E-selectin) and MHC Class I. Death of tumor cells also generates release of inflammatory molecules HMGB1 and ATP. This response recruits macrophages and DCs to tumors where they then receive activation signals resulting in their migration to draining lymph nodes where APCs (macrophages and dendritic cells) present tumor-derived antigens and stimulate T cell responses. Tumor-specific T cells then re-infiltrate tumors and induce death of damaged malignant cells.
Immune Responses to Radiation
RT and Cytokine Expression
Cytokines are peptide-type regulatory proteins, such as the interleukins and lymphokines, released by immune cells leading to generation of an immune response. Some cytokines act to inhibit immune responses, e.g. IL-10 and transforming growth factor (TGF)-β, or instead stimulate immune responses, e.g., TNF-α or IL-1 [53]. TNF-α and IL-1 are pro-inflammatory cytokines that also mediate leukocyte recruitment in tumors [53, 54]. In the 1980’s, Hallahan and colleagues reported that TNF-α mRNA and protein levels were increased in human sarcoma cells following RT, an effect that sensitized tumor cells to radiation-induced cell death [55]. Macrophage-derived IL-1α and IL-1β have also been found increased in response to RT in vivo following sublethal total body irradiation [56–58], as also have IL- 6 [59] and TGF-β [60]. Consequences resulting from the release of these cytokines are recruitment and activation of leukocytes from peripheral blood and extravasation into tissue (tumor) parenchyma as is illustrated in Fig. 1.
Adhesion Molecules Regulated by RT
Adhesion molecules are proteins located on the cell surface that mediate interaction with other cells or extracellular matrix. Cell adhesion molecules such as ICAM-1, E-selectin and VCAM-1 are upregulated on endothelial cells during inflammation and are critical for leukocyte trafficking across endothelial barriers [61]. Vascular endothelial cells within tumor vessels respond to RT by upregulation of ICAM-1 and E-selectin and thereby facilitate leukocyte arrest and adhesion prior to transmigration [62]. Blockade of CD11b, the ligand for ICAM-1, in a transplantable murine squamous carcinoma model significantly reduced tumor-infiltration by CD11b+ myeloid cells following RT resulting in diminished tumor growth [63]. Similarly, examination of tumor tissue removed from head and neck cancer patients following RT revealed marked increase in endothelial ICAM-1 expression, in concert with increased β2 integrin-positive myeloid cell infiltration [64]. Other adhesion molecules are also regulated by RT including VCAM-1 in melanoma in an interferon (IFN)γ-dependent manner [65].
Chemokines and RT
Chemokines are a family of small chemotactic cytokines that regulate directional migration of cells expressing a cognate chemokine receptor. While some chemokines are important for homeostatic circulation of leukocytes, others are induced following tissue damage. Two important chemokines regulated by RT are CXCL16 and SDF-1. Using a murine model of mammary carcinogenesis, Matsumura and colleagues reported that CXCL16, which is upregulated in tumors following RT, induced recruitment and activation of T cells expressing CXCR6, the ligand for CXCL16. Mice deficient for CXCR6 exhibited decreased CD8+ T cell recruitment in tumors and decreased RT responsiveness [66]. Murine melanoma, fibrosarcoma and colon carcinoma cell lines in vitro upregulate CXCL16 in response to RT indicating that CXCL16 expression may be a common response across many tumor types [67]. Thus, radiation-induced CXCL16 is an important mechanism by which RT promotes CD8+ T cell infiltration leading to tumor suppression.
Stromal cell-derived factor (SDF)-1α is also upregulated following RT in bone marrow-derived cells [68] and cell lines derived from brain tumors [69]. Using an in vivo model, Kozin and colleagues observed that lung and breast xenograft tumors responded with increased CD11b+F4/80+ macrophage infiltration following RT that was dependent on expression of SDF-1α. Inhibition of the SDF-1α pathway with a small molecule inhibitor blocking the interaction of SDF-1α and CXCR4 prevented infiltration of macrophages and significantly delayed tumor regrowth following RT [70]. Studies such as these indicate that RT upregulates expression of some chemokines (CXCL16 and SDF-1α) that can in turn regulate presence of either tumor suppressive lymphocytes (CD8+ T cells), or tumor-promoting cells such as macrophages.
RT and Antigen Presentation
Once leukocytes have migrated into sites of tissue damage in response to cytokines and chemokines, functional antigen-presenting cells (APC) are required for a productive anti-tumor T cell response to ensue. APCs capture antigens, and following processing, present them on their cell surface via MHC. T cells recognize antigens bound to MHC and respond by proliferating and generating anti-tumor T cells responses. Lugade and colleagues, utilizing a transplantable murine model of melanoma, reported increased expression of MHC class I on tumor cells following RT [71], a response also observed on GL261 glioma tumor cells [72], indicating that RT enhances tumor cell recognition by T cells through upregulation of MHC class I on the surface of tumor cells, as well as on the surface of APCs [73, 74]. Increased presence of radiation-specific peptides has also been identified as a mechanism whereby tumor-specific T cell responses are elicited by RT [75], a mechanism that also contributes to enhanced anti-tumor immunity.
RT-Induced Immunogenic Cell Death
Radiation of tumor cells generally produces two responses: proliferative arrest (which in the case of senescence is indefinite) or cell death. Tumor cell death can occur by several mechanisms including apoptosis, necrosis, autophagy or mitotic catastrophe. Apoptosis is a stereotyped pattern of morphological changes involving chromatin condensation (pyknosis), nuclear fragmentation (karyorhexis), shrinkage of cytoplasm, blebbing of plasma membranes, and final disintegration of cells into membrane-surrounded apoptotic bodies [76, 77]. While often observed in vitro, apoptosis is rarely seen in vivo since dying cells are efficiently recognized, engulfed and eradicated by neighboring cells before they enter the late stages of the apoptotic process [78]. Necrosis is characterized by cell swelling followed by rupture of plasma membranes and subsequent spillage of cellular contents into intercellular spaces. Autophagy is marked by sequestration of large parts of the cytoplasm in autophagic vacuoles typically before cells undergo apoptosis. Finally, mitotic catastrophe is described by prolonged mitotic arrest with associated micro- and/or multinucleation prior to undergoing death. Radiation-mediated cell death is generally thought to occur primarily through either apoptosis or mitotic catastrophe.
The notion that immunogenic-mediated cell death is also an important aspect of RT response has been demonstrated by several groups. Apetoh and colleagues immunized mice with tumor cells previously exposed to either chemotherapy or RT, and then re-challenged them with the tumor cells and monitored for tumor growth [11]. Immunization with tumor cells treated with either chemotherapy or RT prevented regrowth of tumors in ~30% of mice as compared to mice immunized with untreated tumor cells. When cells were harvested from draining lymph nodes in immunized mice, and rechallenged ex vivo, only lymph node cells from mice immunized with tumor cells treated with RT produced IFN-γ in response to re-challenge. Protective immunization in this scenario was dependent on the presence of TLR-4 on dendritic cells (DCs) and its ligand HMGB1, both released by tumor cells following RT [11]. Two other factors, calreticulin and ATP, also significantly contribute to immunogenic cell death, in a manner similar to HMGB1, where cell death triggers rapid translocation of calreticulin to the surface of cells thereby promoting antigen presentation by dying cells and DCs [79, 80]. Mice previously vaccinated with irradiated tumor cells engineered to express an siRNA against calreticulin exhibit a greatly reduced immune response to challenge as compared to irradiated cells alone [81].
Cytotoxic therapies (chemotherapy and RT) induce rapid release of ATP from cells. ATP acts on the P2X(7) purinergic receptor expressed by DCs, leading to activation of the NOD-like receptor family, pyrin domain containing-3 protein (NLRP3)-dependent caspase-1 activation complex (also known as the inflammasome). Inflammasome activation leads to release of pro-inflammatory cytokines such as IL-1β, which are important for priming T cells. When components of this pathway (NLRP3, caspase-1 or IL-1R) are absent, reduced T cell responses towards cells killed by chemotherapy or RT are observed [82], thus indicating that release of ATP from dying cells is a critical aspect of immunogenic cell death and anti-tumor immunity.
Further support for the importance of immunogenic cell death mediated by HMGB1 and TLR-4 bearing DCs has been provided by retrospective evaluation of a cohort of breast cancer patients treated with adjuvant anthracyclines following resection. Women harboring an Asp299Gly TLR-4 polymorphism exhibited reduced response to HMGB1, and a significantly higher rate of metastatic disease [11]. Interestingly, similar to HMGB1, breast cancer patients treated with anthracycline who harbored a loss-of-function allele in P2RX7 (Glu496Ala) exhibited significantly worse metastasis-free survival as compared to patients with wildtype alleles [82].
RT and Activation of Innate Immune Programs
Cells of the innate and adaptive lineages work in concert to provide rapid and effective responses to a wide variety of pathogens. While cells of the innate lineage provide an immediate and pre-programmed response, response by cells of the adaptive lineage are delayed but instead are antigen-specific and lead to prolonged memory [83]. Innate leukocytes, including DCs, macrophages, natural killer (NK) cells and mast cells, are referred to as “first responders” to inflammatory mediators, largely based on the fact that they are often prestationed in tissues.
Naïve DCs continually sample antigens and migrate to draining lymph nodes for antigen presentation to T cells following their activation by “inflammatory mediators”. RT induces opposing responses in tumors with regards to DCs: directly-irradiated DCs are less effective APCs, however, the tumor microenvironment generated by RT enhances APC capabilities of DCs. In vitro examination of human DCs has revealed that RT induced a “tolerogenic” phenotype by decreasing the amount of IL-12 produced by mature DCs, leading to decreased naïve CD8+ T cell priming [84]. However, when DCs are adoptively transferred into tumors in combination with RT and chemotherapy in vivo, complete regression of tumors is enhanced [85]. Thus, while intratumoral DCs present at the time of chemotherapy or RT yield reduced immune responsiveness, the environment created by the RT fosters enhanced DC activation and enhanced anti-tumor immunity.
Tumor-infiltrating macrophages, derived from circulating monocytes, make up a substantial component of the leukocyte infiltrate in solid tumors [86]. Macrophages exhibit either anti- or pro- tumoral bioactivities dependent on the cytokines, chemokines and soluble mediators they are exposed to [87, 88]. Given this duality, it is not surprising that the effect of radiation on macrophages is complex with evidence that radiation can support either their anti- or pro-tumor properties. Using human macrophage-derived cell lines, Lambert and colleagues observed that RT enhanced macrophage cytolytic activity [89]. Other groups have reported that low dose whole-body RT increased expression of TLR4/MD2 and CD14 expression on murine peritoneal macrophages, leading to increased secretion of anti-tumor cytokines including IL-12 and IL-18, thus indicating that RT increases anti-tumor potential of macrophages [90]. Despite the evidence that RT can stimulate cytolytic activity and anti-tumor cytokine production in macrophages, there also exists extensive literature indicating that macrophages also promote resistance to RT. In orthotopically-transplanted sarcoma and carcinomas, presence of macrophages was inversely correlated with tumor regression following RT [91]. In melanoma, local RT of implanted tumors increased the number of APCs in draining lymph nodes and increased the number of CD11b+ cells in tumors [71]. CD11b+ myeloid cells (a portion of which are macrophages) contribute growth factors such as vascular endothelial growth factor (VEGF) and matrix metalloproteinase-9 (MMP-9) that supports angiogenic programs in growing tumors [92]. Preventing influx of CD11b+ myeloid cells following RT results in enhanced RT effects [63, 93] likely due to their increased expression of T cell suppressive molecules iNOS and arginase I [94, 95]. Thus, while radiation can stimulate macrophage cytolytic activity and anti-tumor cytokine production, this may be insufficient to inhibit tumor growth if there is simultaneous recruitment or activation of macrophages harboring dominant pro-tumor properties.
NK cells are lymphoid cells that, unlike B and T cells, do not possess specific antigen receptors, and thus are considered innate lineage cells. NK cells play an important role in tumors by targeting malignant cells by direct cytolysis and secretion of potent immune mediators including several cytokines and chemokines [96]. Examination of tumor cells exposed to ionizing radiation in vitro indicates that RT induces expression of NKG2D ligands, an activating receptor for NK cells [97, 98]. Other pro-inflammatory stress molecules released by dying cells include heat shock protein 70 (Hsp70), a stress response protein with a role in binding defective proteins and presenting them on the surface of cells [99]. When exposed to RT, pancreatic and colon carcinoma cell release Hsp70, thereby targeting them for lysis by NK cells [100]. That NKG2D ligands and Hsp 70 render cells more susceptible to NK-cell mediated cytolysis indicates that RT-stimulated NK activity may be an important component of RT-induced immune responsiveness.
Mast cells are pre-stationed in many tissues where they act as important sentinel cells capable of mounting rapid responses to tissue damage. Mast cells also accumulate in tissues undergoing angiogenesis, wound healing and tissue repair. During these processes, they secrete angiogenic factors, such as vascular endothelial growth factor (VEGF), and other inflammatory mediators such as histamine, heparin cytokines, chemokines, proteases and lipid mediators [101]. Heissig and colleagues reported that low-dose irradiation fostered mast cell-dependent vascular regeneration in a limb ischemia model where RT promoted VEGF production by mast cells in a matrix metalloproteinase-9 (MMP-9)-dependent manner [102]. RT, through MMP-9 up-regulated by VEGF in stromal and endothelial cells, induced release of Kit-ligand (KitL) and promoted migration of mast cells from bone marrow to the ischemic site [102] similar to RT effects in the thoracic cavity where mast cell density increased in bronchoalveolar lavage fluid [103]. Influx of mast cells following RT is blocked by treatment with imatinib [104], a small molecule tyrosine kinase inhibitor with activity against Kit, platelet derived growth factor receptor (PDGFR) and Abelson Murine Leukemia Viral Oncogene Homolog (Abl) [105]. Imatinib treatment inhibited proliferation and induced apoptosis of mast cells [106] and increased efficacy of RT in several murine tumor models [107]. Given that low-dose RT fosters mast cell-dependent vascular regeneration during limb ischemia model, it seems reasonable to conclude that increased recruitment and activation of mast cells following RT and subsequent alterations in ischemic microenvironments and activation of angiogenic programs may paradoxically foster tumor growth.
RT and Adaptive Immunity
In experimental rodent models of cancer development, e.g. brain, sarcoma, lung and breast, RT alone or in combination with DC or immunostimulatory therapies enhanced generation of anti-tumor responses mediated by cytotoxic T cells [66, 72, 108]. RT alone can also stimulate anti-tumor T cell-based immunity when given at high-doses by increasing the number of activated CD8+ T cells [109–111]. In 4 T1 mammary tumors, recruitment of cytotoxic T cells is dependent on CXCR6, a receptor for CXCL16. RT in combination with anti-CTLA-4 mAB increases recruitment of CXCR6+ CD8+ T cells [66]. Given that patients undergoing RT for prostate cancer exhibited detectable anti-tumor CD4+ and CD8+ T cells responses following RT that were undetectable prior to therapy [112], it stands to reason that in addition to genotoxic damage, induction of anti-tumor immunity via T cell activation represents an important mechanisms mediating the efficacy of RT-based therapy.
Radiation and Immunotherapy
Preclinical Models
Since one consequence of RT in tissues is induction of various immune -mediated programs, several groups have explored the potential of augmenting RT responsiveness with immunotherapeutics (immunostimulatory cytokines, DC-based therapy and antibodies targeting T cell costimulatory pathways) engineered to bolster anti-tumor immunity.
Immunostimulatory cytokines including IL-2, IL-12 and TNF-α [54] have been used in combination with RT to stimulate anti-tumor T cell responses. Addition of these pro-inflammatory cytokines enhances RT efficacy by bolstering cytotoxic T cell responses [113–117]. Interestingly, IL-3, a cytokine that activates monocytes and mast cells [118], delays tumor growth in response to RT [119]. The enhanced tumor inhibition observed by combining cytokines with RT provides evidence that effectiveness of RT depends on immune-mediated mechanisms that can be targeted effectively to enhance overall RT response. Based on this postulate, several groups have attempted to increase the presence of antigen-presenting DCs in tumors. Intratumoral injection of CpG oligodeoxynucleotides that activate TLR9 on macrophages and DCs resulted in increased RT response and resistance to a second challenge with the same tumor, thus indicating development of a durable immune response [120].
Antigen-presentation on the surface of DCs to T cells requires both MHC and costimulatory molecules, B7 molecules and OX40 [121, 122]. Strategies to enhance costimulatory molecules in combination with RT have been employed in a transgenic model of colon carcinogenesis. Carcinoma cells were engineered to express a human antigen (CEA) – when RT was given in combination with a viral vaccine expressing CEA and T cell co-stimulatory molecules, complete tumor regression was observed accompanied by anti-tumor CD4+ and CD8+ T cell infiltration [123]. Inhibition of tumor growth and enhanced overall survival was also observed in a murine sarcoma model when RT was given in combination with an agonistic antibody for OX40, a costimulatory molecule found on activated T cells that stimulates T cell proliferation and differentiation [122]. Inhibition of CTLA-4 costimulation also enhanced effectiveness of RT in 4 T1 mammary carcinomas carcinoma resulting in diminished metastasis and increased survival [124, 125]; however, RT dose and timing were critical with regards to anti-CTLA-4 therapy [126]. Despite these numerous successful preclinical trials demonstrating efficacy of immune-modulation therapy in combination with RT, only a few clinical studies have been initiated to date.
Clinical Studies
Clinical evaluation of RT in combination with immunotherapeutic strategies are currently being conducted for prostate, melanoma and liver carcinomas. RT has been evaluated in conjunction with drugs that inhibit androgen production resulting in enhanced autoantibody responses in 15–30% of prostate cancer patients [127] and correlating with previous studies indicating that anti-androgen therapy also increases T cell activity due to thymic regrowth [128, 129]. Vaccination of prostate cancer patients with recombinant viral-based vaccines expressing prostate-specific antigen (PSA), in combination with the costimulatory molecule B7-1 and standard RT to the prostate (70 Gy of RT in 1.8 to 2.0 Gy fractions), resulted in a three-fold increase in PSA-specific T cells and evidence of generating T cells against other prostate-specific antigens in 76% of patients [130]. Immunogenicity of irradiated tumor cells in patients with melanoma was examined in which autologous irradiated melanoma cells engineered to express GM-CSF, a white blood cell growth factor, were injected into patients resulting in a significant anti-tumor immune response leading to tumor regression in 50% of patients [131]. The synergy between RT and DCs has been further evaluated in a small study of patients with hepatoma where DCs were injected intratumorally following a single-dose of RT leading to development of tumor-specific immune responses in 30% of patients [132]. Though these clinical studies involve small numbers of non-randomized patients, they present compelling findings indicating that the durability of RT may be enhanced by combinatorial therapy with selective immune-based therapeutics.
Conclusions
Though many cancer patients will receive RT, it is clear that clinician-scientists are only just beginning to understand the full spectrum of biologic responses resultant from RT. RT clearly influences multiple immune-based programs in tissues, some of which lead to durable tumor regression, whereas others propel tumor development. It seems reasonable to conclude that identifying pathways mediating activation of myeloid-based protumor immunity induced by RT, will encourage development of novel therapeutics that suppress those activities to effectively bolster RT responses. Moreover, blockade of these protumor immune-based pathways may also present the opportunity to then combine RT with anti-tumor immunotherapeutics to yield effective and durable suppression of tumors, resulting in improved outcomes for patients with cancer.
References
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45.
de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37.
Bui JD, Schreiber RD. Cancer immunosurveillance, immunoediting and inflammation: independent or interdependent processes? Curr Opin Immunol. 2007;19(2):203–8.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44.
Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8(1):59–73.
Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10(7):718–26.
Harris J, Part G. Breast cancer. Overview. In: Gunderson L, Tepper J, editors. Clinical radiation oncology. 2nd ed. Philadelphia: Elsevier Churchill Livingstone; 2007.
Locher C, Conforti R, Aymeric L, et al. Desirable cell death during anticancer chemotherapy. Ann NY Acad Sci. 2010;1209(1):99–108.
Tesniere A, Apetoh L, Ghiringhelli F, et al. Immunogenic cancer cell death: a key-lock paradigm. Curr Opin Immunol. 2008;20(5):504–11.
Haynes NM, van der Most RG, Lake RA, Smyth MJ. Immunogenic anti-cancer chemotherapy as an emerging concept. Curr Opin Immunol. 2008;20(5):545–57.
Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–9.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
Laconi E, Doratiotto S, Vineis P. The microenvironments of multistage carcinogenesis. Semin Cancer Biol. 2008;18(5):322–9.
Coronella JA, Telleman P, Kingsbury GA, Truong TD, Hays S, Junghans RP. Evidence for an antigen-driven humoral immune response in medullary ductal breast cancer. Cancer Res. 2001;61(21):7889–99.
Punt CJ, Barbuto JA, Zhang H, Grimes WJ, Hatch KD, Hersh EM. Anti-tumor antibody produced by human tumor-infiltrating and peripheral blood B lymphocytes. Cancer Immunol Immunother. 1994;38(4):225–32.
Coronella-Wood JA, Hersh EM. Naturally occurring B-cell responses to breast cancer. Cancer Immunol Immunother. 2003;52(12):715–38.
Shimokawara I, Imamura M, Yamanaka N, Ishii Y, Kikuchi K. Identification of lymphocyte subpopulations in human breast cancer tissue and its significance: an immunoperoxidase study with anti-human T- and B-cell sera. Cancer. 1982;49(7):1456–64.
Fernandez Madrid F. Autoantibodies in breast cancer sera: candidate biomarkers and reporters of tumorigenesis. Cancer Lett. 2005;230(2):187–98.
Lee YT, Sheikh KM, Quismorio Jr FP, Friou GJ. Circulating anti-tumor and autoantibodies in breast carcinoma: relationship to stage and prognosis. Breast Cancer Res Treat. 1985;6(1):57–65.
Chin Y, Janseens J, Vandepitte J, Vandenbrande J, Opdebeek L, Raus J. Phenotypic analysis of tumor-infiltrating lymphocytes from human breast cancer. Anticancer Res. 1992;12(5):1463–6.
Kohrt HE, Nouri N, Nowels K, Johnson D, Holmes S, Lee PP. Profile of immune cells in axillary lymph nodes predicts disease-free survival in breast cancer. PLoS Med. 2005;2(9):e284.
Bates GJ, Fox SB, Han C, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. 2006;24(34):5373–80.
Denkert C, Loibl S, Noske A, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010;28(1):105–13.
Tsutsui S, Yasuda K, Suzuki K, Tahara K, Higashi H, Era S. Macrophage infiltration and its prognostic implications in breast cancer: the relationship with VEGF expression and microvessel density. Oncol Rep. 2005;14(2):425–31.
Steidl C, Lee T, Shah SP, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362(10):875–85.
Bolat F, Kayaselcuk F, Nursal TZ, Yagmurdur MC, Bal N, Demirhan B. Microvessel density, VEGF expression, and tumor-associated macrophages in breast tumors: correlations with prognostic parameters. J Exp Clin Cancer Res. 2006;25(3):365–72.
Chen JJ, Lin YC, Yao PL, et al. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol. 2005;23(5):953–64.
Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010;21(1):41–8.
Campbell MJ, Tonlaar NY, Garwood ER, et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res Treat. Sep 15 2010.
Kawai O, Ishii G, Kubota K, et al. Predominant infiltration of macrophages and CD8(+) T Cells in cancer nests is a significant predictor of survival in stage IV nonsmall cell lung cancer. Cancer. 2008;113(6):1387–95.
Mantovani A, Sica A, Locati M. New vistas on macrophage differentiation and activation. Eur J Immunol. 2007;37(1):14–6.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69.
Mantovani A, Allavena P, Sica A. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: role in tumour progression. Eur J Cancer. 2004;40(11):1660–7.
Ruffell B, Denardo DG, Affara NI, Coussens LM. Lymphocytes in cancer development: Polarization towards pro-tumor immunity. Cytokine Growth Factor Rev. 2010;21:3–10.
Appelbaum FR. The influence of total dose, fractionation, dose rate, and distribution of total body irradiation on bone marrow transplantation. Semin Oncol. 1993;20(4 Suppl 4):3–10. quiz 11.
Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30(2):180–92.
Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9(10):759–69.
Wu ZH, Miyamoto S. Many faces of NF-kappaB signaling induced by genotoxic stress. J Mol Med. 2007;85(11):1187–202.
Rashi-Elkeles S, Elkon R, Weizman N, et al. Parallel induction of ATM-dependent pro- and antiapoptotic signals in response to ionizing radiation in murine lymphoid tissue. Oncogene. 2006;25(10):1584–92.
Li N, Karin M. Ionizing radiation and short wavelength UV activate NF-kappaB through two distinct mechanisms. Proc Natl Acad Sci USA. 1998;95(22):13012–7.
Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med. 1990;171(1):35–47.
Mori N, Prager D. Transactivation of the interleukin-1alpha promoter by human T-cell leukemia virus type I and type II Tax proteins. Blood. 1996;87(8):3410–7.
Wickremasinghe MI, Thomas LH, O’Kane CM, Uddin J, Friedland JS. Transcriptional mechanisms regulating alveolar epithelial cell-specific CCL5 secretion in pulmonary tuberculosis. J Biol Chem. 2004;279(26):27199–210.
Caldenhoven E, Coffer P, Yuan J, et al. Stimulation of the human intercellular adhesion molecule-1 promoter by interleukin-6 and interferon-gamma involves binding of distinct factors to a palindromic response element. J Biol Chem. 1994;269(33):21146–54.
van de Stolpe A, Caldenhoven E, Stade BG, et al. 12-O-tetradecanoylphorbol-13-acetate- and tumor necrosis factor alpha-mediated induction of intercellular adhesion molecule-1 is inhibited by dexamethasone. Functional analysis of the human intercellular adhesion molecular-1 promoter. J Biol Chem. 1994;269(8):6185–92.
Schindler U, Baichwal VR. Three NF-kappa B binding sites in the human E-selectin gene required for maximal tumor necrosis factor alpha-induced expression. Mol Cell Biol. 1994;14(9):5820–31.
Iademarco MF, McQuillan JJ, Rosen GD, Dean DC. Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1). J Biol Chem. 1992;267(23):16323–9.
Johnson DR, Pober JS. HLA class I heavy-chain gene promoter elements mediating synergy between tumor necrosis factor and interferons. Mol Cell Biol. 1994;14(2):1322–32.
Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651.
Brach MA, Hass R, Sherman ML, Gunji H, Weichselbaum R, Kufe D. Ionizing radiation induces expression and binding activity of the nuclear factor kappa B. J Clin Invest. 1991;88(2):691–5.
Criswell T, Leskov K, Miyamoto S, Luo G, Boothman DA. Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene. 2003;22(37):5813–27.
Lee SJ, Dimtchev A, Lavin MF, Dritschilo A, Jung M. A novel ionizing radiation-induced signaling pathway that activates the transcription factor NF-kappaB. Oncogene. 1998;17(14):1821–6.
Germano G, Allavena P, Mantovani A. Cytokines as a key component of cancer-related inflammation. Cytokine. 2008;43(3):374–9.
Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9(5):361–71.
Hallahan DE, Spriggs DR, Beckett MA, Kufe DW, Weichselbaum RR. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc Natl Acad Sci USA. 1989;86(24):10104–7.
Baker WH, Limanni A, Chang CM, Jackson WE, Seemann R, Patchen ML. Comparison of interleukin-1 alpha gene expression and protein levels in the murine spleen after lethal and sublethal total-body irradiation. Radiat Res. 1995;143(3):320–6.
O’Brien-Ladner A, Nelson ME, Kimler BF, Wesselius LJ. Release of interleukin-1 by human alveolar macrophages after in vitro irradiation. Radiat Res. 1993;136(1):37–41.
Hong JH, Chiang CS, Tsao CY, Lin PY, McBride WH, Wu CJ. Rapid induction of cytokine gene expression in the lung after single and fractionated doses of radiation. Int J Radiat Biol. 1999;75(11):1421–7.
Okunieff P, Chen Y, Maguire DJ, Huser AK. Molecular markers of radiation-related normal tissue toxicity. Cancer Metastasis Rev. 2008;27(3):363–74.
Calveley VL, Khan MA, Yeung IW, Vandyk J, Hill RP. Partial volume rat lung irradiation: temporal fluctuations of in-field and out-of-field DNA damage and inflammatory cytokines following irradiation. Int J Radiat Biol. 2005;81(12):887–99.
Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7(10):803–15.
Hallahan D, Kuchibhotla J, Wyble C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996;56(22):5150–5.
Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci USA. 2010;107(18):8363–8.
Handschel J, Prott FJ, Sunderkotter C, Metze D, Meyer U, Joos U. Irradiation induces increase of adhesion molecules and accumulation of beta2-integrin-expressing cells in humans. Int J Radiat Oncol Biol Phys. 1999;45(2):475–81.
Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180(5):3132–9.
Matsumura S, Wang B, Kawashima N, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181(5):3099–107.
Matsumura S, Demaria S. Up-regulation of the pro-inflammatory chemokine CXCL16 is a common response of tumor cells to ionizing radiation. Radiat Res. 2010;173(4):418–25.
Zong ZW, Cheng TM, Su YP, et al. Recruitment of transplanted dermal multipotent stem cells to sites of injury in rats with combined radiation and wound injury by interaction of SDF-1 and CXCR4. Radiat Res. 2008;170(4):444–50.
Tabatabai G, Frank B, Mohle R, Weller M, Wick W. Irradiation and hypoxia promote homing of haematopoietic progenitor cells towards gliomas by TGF-beta-dependent HIF-1alpha-mediated induction of CXCL12. Brain. 2006;129(Pt 9):2426–35.
Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70(14):5679–85.
Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174(12):7516–23.
Newcomb EW, Demaria S, Lukyanov Y, et al. The combination of ionizing radiation and peripheral vaccination produces long-term survival of mice bearing established invasive GL261 gliomas. Clin Cancer Res. 2006;12(15):4730–7.
Liao YP, Wang CC, Butterfield LH, et al. Ionizing radiation affects human MART-1 melanoma antigen processing and presentation by dendritic cells. J Immunol. 2004;173(4):2462–9.
Cao ZA, Daniel D, Hanahan D. Sub-lethal radiation enhances anti-tumor immunotherapy in a transgenic mouse model of pancreatic cancer. BMC Cancer. 2002;2:11.
Reits EA, Hodge JW, Herberts CA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–71.
Galluzzi L, Maiuri MC, Vitale I, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14(7):1237–43.
Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11.
Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2(12):965–75.
Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61.
Perez CA, Fu A, Onishko H, Hallahan DE, Geng L. Radiation induces an antitumour immune response to mouse melanoma. Int J Radiat Biol. 2009;85(12):1126–36.
Obeid M, Panaretakis T, Joza N, et al. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14(10):1848–50.
Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–8.
Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327(5963):291–5.
Merrick A, Errington F, Milward K, et al. Immunosuppressive effects of radiation on human dendritic cells: reduced IL-12 production on activation and impairment of naive T-cell priming. Br J Cancer. 2005;92(8):1450–8.
Moyer JS, Li J, Wei S, Teitz-Tennenbaum S, Chang AE. Intratumoral dendritic cells and chemoradiation for the treatment of murine squamous cell carcinoma. J Immunother. 2008;31(9):885–95.
Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–7.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
Lambert LE, Paulnock DM. Modulation of macrophage function by gamma-irradiation. Acquisition of the primed cell intermediate stage of the macrophage tumoricidal activation pathway. J Immunol. 1987;139(8):2834–41.
Shan YX, Jin SZ, Liu XD, Liu Y, Liu SZ. Ionizing radiation stimulates secretion of pro-inflammatory cytokines: dose-response relationship, mechanisms and implications. Radiat Environ Biophys. 2007;46(1):21–9.
Milas L, Wike J, Hunter N, Volpe J, Basic I. Macrophage content of murine sarcomas and carcinomas: associations with tumor growth parameters and tumor radiocurability. Cancer Res. 1987;47(4):1069–75.
Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13(3):193–205.
Meng Y, Beckett MA, Liang H, et al. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010;70(4):1534–43.
Tsai CS, Chen FH, Wang CC, et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys. 2007;68(2):499–507.
Doedens AL, Stockmann C, Rubinstein MP, et al. Macrophage expression of HIF-1α suppresses T cell function and promotes tumor progression. Cancer Res. 2010;70:7465–75.
Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. 2010;142(6):847–56.
Kim JY, Son YO, Park SW, et al. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp Mol Med. 2006;38(5):474–84.
Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436(7054):1186–90.
Sherman M, Multhoff G. Heat shock proteins in cancer. Ann NY Acad Sci. 2007;1113:192–201.
Gehrmann M, Marienhagen J, Eichholtz-Wirth H, et al. Dual function of membrane-bound heat shock protein 70 (Hsp70), Bag-4, and Hsp40: protection against radiation-induced effects and target structure for natural killer cells. Cell Death Differ. 2005;12(1):38–51.
Porta C, Larghi P, Rimoldi M, et al. Cellular and molecular pathways linking inflammation and cancer. Immunobiology. 2009;214(9–10):761–77.
Heissig B, Rafii S, Akiyama H, et al. Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J Exp Med. 2005;202(6):739–50.
Majori M, Poletti V, Curti A, Corradi M, Falcone F, Pesci A. Bronchoalveolar lavage in bronchiolitis obliterans organizing pneumonia primed by radiation therapy to the breast. J Allergy Clin Immunol. 2000;105(2 Pt 1):239–44.
Thomas DM, Fox J, Haston CK. Imatinib therapy reduces radiation-induced pulmonary mast cell influx and delays lung disease in the mouse. Int J Radiat Biol. 2010;86(6):436–44.
Sherbenou DW, Druker BJ. Applying the discovery of the Philadelphia chromosome. J Clin Invest. 2007;117(8):2067–74.
Gleixner KV, Rebuzzi L, Mayerhofer M, et al. Synergistic antiproliferative effects of KIT tyrosine kinase inhibitors on neoplastic canine mast cells. Exp Hematol. 2007;35(10):1510–21.
Oertel S, Krempien R, Lindel K, et al. Human glioblastoma and carcinoma xenograft tumors treated by combined radiation and imatinib (Gleevec). Strahlenther Onkol. 2006;182(7):400–7.
Demaria S, Ng B, Devitt ML, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58(3):862–70.
Gough MJ, Crittenden MR, Sarff M, et al. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. J Immunother. 2010;33(8):798–809.
Lee Y, Auh SL, Wang Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114(3):589–95.
Takeshima T, Chamoto K, Wakita D, et al. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: its potentiation by combination with Th1 cell therapy. Cancer Res. 2010;70(7):2697–706.
Tabi Z, Spary LK, Coleman S, Clayton A, Mason MD, Staffurth J. Resistance of CD45RA- T cells to apoptosis and functional impairment, and activation of tumor-antigen specific T cells during radiation therapy of prostate cancer. J Immunol. 2010;185(2):1330–9.
Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9(7):480–90.
Lee J, Moran JP, Fenton BM, et al. Alteration of tumour response to radiation by interleukin-2 gene transfer. Br J Cancer. 2000;82(4):937–44.
Yamini B, Yu X, Pytel P, et al. Adenovirally delivered tumor necrosis factor-alpha improves the antiglioma efficacy of concomitant radiation and temozolomide therapy. Clin Cancer Res. 2007;13(20):6217–23.
Lohr F, Hu K, Haroon Z, et al. Combination treatment of murine tumors by adenovirus-mediated local B7/IL12 immunotherapy and radiotherapy. Mol Ther. 2000;2(3):195–203.
Seetharam S, Staba MJ, Schumm LP, et al. Enhanced eradication of local and distant tumors by genetically produced interleukin-12 and radiation. Int J Oncol. 1999;15(4):769–73.
Aldinucci D, Olivo K, Lorenzon D, et al. The role of interleukin-3 in classical Hodgkin's disease. Leuk Lymphoma. 2005;46(3):303–11.
Oh YT, Chen DW, Dougherty GJ, McBride WH. Adenoviral interleukin-3 gene-radiation therapy for prostate cancer in mouse model. Int J Radiat Oncol Biol Phys. 2004;59(2):579–83.
Mason KA, Ariga H, Neal R, et al. Targeting toll-like receptor 9 with CpG oligodeoxynucleotides enhances tumor response to fractionated radiotherapy. Clin Cancer Res. 2005;11(1):361–9.
Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev. 2009;229(1):126–44.
Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol. 2009;29(3):187–201.
Chakraborty M, Abrams SI, Coleman CN, Camphausen K, Schlom J, Hodge JW. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004;64(12):4328–37.
Demaria S, Kawashima N, Yang AM, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11(2 Pt 1):728–34.
Pilones KA, Kawashima N, Yang AM, Babb JS, Formenti SC, Demaria S. Invariant natural killer T cells regulate breast cancer response to radiation and CTLA-4 blockade. Clin Cancer Res. 2009;15(2):597–606.
Dewan MZ, Galloway AE, Kawashima N, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15(17):5379–88.
Nesslinger NJ, Sahota RA, Stone B, et al. Standard treatments induce antigen-specific immune responses in prostate cancer. Clin Cancer Res. 2007;13(5):1493–502.
Roden AC, Moser MT, Tri SD, et al. Augmentation of T cell levels and responses induced by androgen deprivation. J Immunol. 2004;173(10):6098–108.
Johnke RM, Edwards JM, Kovacs CJ, et al. Response of T lymphocyte populations in prostate cancer patients undergoing radiotherapy: influence of neoajuvant total androgen suppression. Anticancer Res. 2005;25(4):3159–66.
Gulley JL, Arlen PM, Bastian A, et al. Combining a recombinant cancer vaccine with standard definitive radiotherapy in patients with localized prostate cancer. Clin Cancer Res. 2005;11(9):3353–62.
Soiffer R, Lynch T, Mihm M, et al. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc Natl Acad Sci USA. 1998;95(22):13141–6.
Chi KH, Liu SJ, Li CP, et al. Combination of conformal radiotherapy and intratumoral injection of adoptive dendritic cell immunotherapy in refractory hepatoma. J Immunother. 2005;28(2):129–35.
Acknowledgements
The authors acknowledge support from the American Board of Radiology to SLS, and grants from the NIH/NCI and Department of Defense (W81XWH-06-1-0416, PR080717) to LMC.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Shiao, S.L., Coussens, L.M. The Tumor-Immune Microenvironment and Response to Radiation Therapy. J Mammary Gland Biol Neoplasia 15, 411–421 (2010). https://doi.org/10.1007/s10911-010-9194-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-010-9194-9