
Abstract
Factor H (FH) binds apoptotic cells to limit the inflammatory potential of complement. Here we report that FH is actively internalized by apoptotic cells to enhance cathepsin L-mediated cleavage of endogenously expressed C3, which results in increased surface opsonization with iC3b. In addition, internalized FH forms complexes with nucleosomes, facilitates their phagocytosis by monocytes and induces an anti-inflammatory biased cytokine profile. A similar cytokine response was noted for apoptotic cells coated with FH, confirming that FH diminishes the immunogenic and inflammatory potential of autoantigens. These findings were supported by in vivo observations from CFH−/− MRL-lpr mice, which exhibited higher levels of circulating nucleosomes and necrotic cells than their CFH+/+ littermates. This unconventional function of FH broadens the established view of apoptotic cell clearance and appears particularly important considering the strong associations with genetic FH alterations and diseases such as systemic lupus erythematosus and age-related macular degeneration.
Similar content being viewed by others


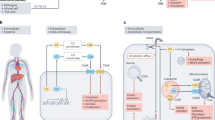
Main
Factor H (FH), one of the most abundant plasma proteins, is the major soluble inhibitor of the alternative complement pathway. Apoptotic cells bind FH while triggering complement activation by binding of C1 complex, which ensures efficient opsonization and removal of apoptotic debris but prevents excessive complement activation and inflammation.1, 2 Dysregulation of complement contributes significantly to the pathology of many diseases.3 Recently, novel roles and intracellular location for complement have been identified suggesting that its functions exceed our current understanding.4
After previously identifying the ligands for FH on the apoptotic cell surface as dsDNA, histones and annexin A2,5 we sought to explore the functional consequences of the FH-apoptotic cell interaction in the context of the chronic autoimmune disorder systemic lupus erythematosus (SLE) and age-related macular degeneration (AMD). Aberrant apoptosis and impaired clearance of apoptotic cells are of central importance in the pathogenesis of SLE and lead to formation of autoantibodies.6, 7, 8 Anti-chromatin autoantibodies are a hallmark of SLE and anti-annexin A2 autoantibodies are also frequently observed in SLE patients.9 Interestingly, the corresponding autoantigens are exactly those that we identified as ligands for FH on the apoptotic cell surface, suggesting a possibility of disturbance of FH function in SLE. Glomerulonephritis is one typical manifestation of human SLE10 and mutations in FH and another complement inhibitor CD46 are associated with earlier onset of nephritis in SLE patients.11 FH-deficient (CFH−/−) mice spontaneously develop membranoproliferative glomerulonephritis, which is dependent on C3 activation.12 When crossed with the MRL-lpr mouse strain, a model of lupus, the originated CFH−/− MRL-lpr mice exhibit accelerated lupus nephritis and die at younger age than their CFH+/+ MRL-lpr littermates.13, 14
AMD is the leading cause of visual impairment in the elderly, and the FH polymorphism Y402H is the major genetic risk factor for AMD development.15, 16 The presence of drusen (extracellular deposits of debris) between the retinal pigmented epithelium (RPE) and the choroid of the macula is characteristic of AMD. Local inflammation including complement has a key role in drusen biogenesis17 and there is increased apoptosis of RPE, photoreceptor and inner nuclear layer cells in AMD.18
By investigating the significance of FH-apoptotic cell interaction in these two settings, we found that FH is specifically internalized in an active manner by early apoptotic cells and that it increases the cleavage and deposition of endogenous C3, facilitating opsonization. Further, FH binds to nucleosomes in and released from apoptotic cells and alters the cytokine profile of phagocytes after nucleosome uptake, in an anti-inflammatory direction.
Results
FH is actively internalized by various cell types
FH labeled with pH-sensitive dye pHrodo was time- and temperature-dependently internalized by Jurkat T cells (Figures 1a and b). To exclude passive diffusion, internalization of other apoptotic cell-binding proteins such as C4b-binding protein, C1q and protein S was tested. None of these became internalized at physiological concentrations as shown for protein S, which is only half the size of FH (Figures 1a and b). This strongly suggests that FH is specifically internalized by apoptotic Jurkat T cells.

FH gets actively internalized by various cell types. (a and b) Protein internalization by apoptotic Jurkat T cells incubated for 2, 10 and 30 min with 125 μg/ml pHrodo-labeled FH and 25 μg/ml protein S at 37 °C (a) and 4 °C (b) assessed by flow cytometry. (c) FH and protein S internalization by apoptotic RPE cells as in a. (d) FH internalization by early (annexin A5 positive) and late apoptotic (annexin A5 and Via-Probe positive) cell populations of Jurkat T cells. (a–d) Representative histograms of n=3 independent experiments. (e) Localization of 30 μg/ml AlexaFluor488-labeled FH (green) in apoptotic Jurkat T cells after 5 and 30 min incubation at 37 °C as well as 30 min incubation after pre-treatment with 30 mM NaN3 assessed by confocal microscopy. Nuclei counterstained with propidium iodine (red). Representative images of n=3 independent experiments. Scale bars, 50 and 10 μm for zoomed images
To assess whether FH internalization is a general phenomenon, RPE cells were rendered apoptotic and incubated with pHrodo-labeled FH and protein S. The specific internalization of FH but not protein S was confirmed (Figure 1c).
Next, we investigated which apoptotic cell populations internalize FH. We previously demonstrated that late apoptotic cells bind FH with the strongest intensity whereas the binding to live cells is negligible.1, 5 As anticipated of an active process, early (annexin A5 positive) but not late (annexin A5 and Via-Probe positive) apoptotic cells internalized FH (Figure 1d).
To further visualize FH internalization, apoptotic Jurkat T cells were incubated with AlexaFluor488-labeled FH (green) while the nuclei were counterstained with propidium iodine (red). After 5 min incubation, FH was bound to the cell surface of almost all apoptotic cells (Figure 1e). Of note, apoptosis induction always resulted in a mixed population consisting of ~45% early and 45% late apoptotic/secondary necrotic as well as 10% live cells. After 30 min incubation, approximately half of the apoptotic cells had internalized FH while no interaction of FH with live cells was observed. The presence of FH throughout the depth of the cell was confirmed with z-stacks (data not shown). Confirming the flow cytometric observations, FH internalization did not occur or was significantly delayed when the apoptotic cells were incubated with FH at 4°C (data not shown) or in the presence of 30 mM NaN3 (Figure 1e). These results reveal that apoptotic cells specifically internalize FH in an active, energy-dependent manner.
Endogenous C3 is cleaved and deposited on the surface upon apoptosis
C3 endogenously expressed by many different cell types19 is cleaved upon complement activation into the anaphylatoxin C3a and the opsonin C3b. In serum, surface-deposited C3b will be further cleaved into iC3b by the serine protease factor I. We confirmed using qPCR that both Jurkat T cells and RPE cells expressed endogenous C3 (Figures 2a and b). To further study C3 expression and cellular distribution at the protein level, flow cytometry was utilized. The expression of intracellular C3, determined with an anti-C3d Ab that binds to all C3 forms and cleavage fragments, did not change significantly upon apoptosis induction (Figure 3c). However, the cell surface C3/C3d deposition increased dramatically on cells rendered apoptotic for at least 4 h (Figure 2d). Neo-epitope-specific antibody revealed that C3 was converted to iC3b. The intracellular iC3b level was already significantly increased after 2 h in apoptotic cells and increased further until 7.5 h after induction (Figure 2e). The cell surface deposition was likewise significantly increased already after 2 h and increased further until 7.5 h induction (Figure 2f). These results clearly demonstrate that C3 is endogenously present in Jurkat T cells and that it is cleaved into the opsonin iC3b, which becomes exposed on the surface upon apoptosis induction.

Endogenous C3 is expressed by live and apoptotic Jurkat T and RPE cells, and FH increases its apoptosis-induced cleavage and deposition. (a and b) Level of C3 mRNA expression in live, 4 and 6 h apoptotic Jurkat T cells (a) and RPE cells (b) relative to GAPDH and HPRT1 housekeeping gene expressions. Values are means±S.D. from n=3 independent repetitions. (c–f) Intracellular and cell surface expression of C3 and iC3b in live and apoptotic (induced for 2–7.5 h in serum-free medium) Jurkat T cells assessed by flow cytometry. Data are means±S.D. from n=4 independent experiments, P-values refer to live cells. (g–j) Cell surface expression of iC3b on 6 h apoptotic Jurkat T and RPE cells that were pre-incubated for 30 min with 150 μg/ml FH determined by flow cytometry. Results are displayed as representative histograms (g and h) of n=3 (Jurkat T cells) and n=4 (RPE cells) independent experiments and means±S.D. (i and j). *P<0.05, **P<0.01, ***P<0.001. gMFI, geometrical mean fluorescent intensity; HPRT1, hypoxanthine phosphoribosyltransferase 1; BG, background

FH is stable within apoptotic cells and acts as cofactor for CTSL in the cleavage of C3. (a) FH presence in lysates of 4 h apoptotic Jurkat T cells incubated with 125I-labeled FH for indicated time points. 125I-FH incubated in buffer for 20 h was applied as positive control. Displayed is one representative gel of n=3 independent experiments. (b) Influence of FH on CTSL-mediated cleavage of C3. 125I-labeled C3 was incubated with 23 μg/ml of the activated CTSL in the presence of increasing concentrations of FH. CTSL-specific inhibitor was added to an additional sample with highest FH concentration. Densitometric analysis is presented as means±S.D. of n=3 independent experiments, P-value refers to sample without addition of FH. One representative gel is displayed. (c) Binding of FH to CTSL. Increasing concentrations of FH were incubated with immobilized CTSL, osteoadherin (positive control) and prothrombin (negative control). Data are means±S.D. of n=3 independent experiments, P-values for CTSL refer to negative control. **P<0.01, ***P<0.001. ctr, control; CTSL-inh, cathepsin L-inhibitor; Neg, negative; Pos, positive
FH increases cleavage and deposition of endogenous C3
To investigate if endogenous C3 is affected by internalized FH, Jurkat T cells were first pre-incubated with FH and then rendered apoptotic still in the presence of FH. Strikingly, significantly more iC3b became exposed on the cell surface in the presence of FH (Figures 2g and i). A similar impact of FH on iC3b exposure was confirmed for apoptotic RPE cells (Figures 2h and j).
FH is stable within apoptotic cells and acts as cofactor for cathepsin L in the cleavage of C3
To estimate the time frame in which FH might function within apoptotic cells, its stability was determined by incubating 125I-labeled FH with apoptotic Jurkat T cells and no degradation was observed (Figure 3a). This indicates that FH is stable within the apoptotic cells over a period of at least 44 h.
The lysosomal cysteine protease cathepsin L (CTSL) cleaves C3 into C3a and C3b upon T-cell activation.19 Upon apoptosis induction, cathepsins are released into the cytosol and remain functional as the cytosolic pH decreases.20 To investigate a potential effect of FH on CTSL-mediated cleavage of C3, a fixed concentration of activated CTSL was incubated with 125I-labeled C3 in the presence of increasing concentrations of FH. CTSL did not cleave C3 in the absence of FH at the chosen concentration (Figure 3b). However, the cleavage of C3 into C3b was increased in a concentration-dependent manner by addition of FH. Upon addition of a CTSL-specific inhibitor, no cleavage was observed even in the presence of the highest FH concentration, proving the specificity of the reaction. C3b was not cleaved further into iC3b even after overnight incubation (data not shown).
The interaction between FH and CTSL was further studied using a direct binding assay. FH bound weakly, but specifically and in a dose-dependent manner to immobilized CTSL (Figure 3c) as well as positive control5 but not negative control (prothrombin).
Internalized FH is bound to nucleosomes
To study whether FH also binds to nucleosomes intracellularly, cells were pre-incubated with buffer, FH or α1-antitrypsin (negative control) and then rendered apoptotic for 24 h still in the presence of the proteins. Cell supernatants, lysed cytosolic fractions as well as purified mononucleosomes, mononucleosomes incubated with FH and purified FH or histone H2 were separated on an agarose gel. Ethidium bromide was used to visualize the DNA content (red signal). The gel was then blotted to a membrane and proteins were detected using specific antibodies (green signal) against FH (Figure 4a) or histone H2B (Figure 4b). The generated images were overlaid to detect co-migration. Most FH present in the cell supernatant exhibited the same mobility as the purified FH (Figure 4a). However, FH in the cytosolic fraction exhibited a different mobility, indicating that it is present inside the cells and bound to other components (arrowhead #1). FH incubated with mononucleosomes also migrated differently in the gel than purified FH alone (arrowhead #2). Purified histone H2A/B did not enter the gel, but histones in complex, as present in the cytosolic fraction, ran further into the gel (Figure 4b, arrowhead #3). Interestingly, a DNA band at the same height as the FH present in the cytosolic fraction appeared on the gel indicating that a higher molecular weight DNA fragment was interacting with FH (arrowhead #4).

FH binds to nucleosomes and induces in vitro nucleosome release in apoptotic Jurkat T and RPE cells. (a–b) Detection of FH–nucleosome complexes by modified western blot. Jurkat T cells were pre-incubated with 170 μg/ml proteins for 1 h and then rendered apoptotic. Supernatants and lysates were collected after 24 h. DNA content was stained with ethidium bromide and visualized in red. Immunoblotting for FH (a) and histone H2B (b) is visualized in green. DNA and protein images were overlaid and colocalization is displayed in yellow. One representative overlay of n=3 independent experiments is shown. (c and d) Detection of FH–nucleosome complexes by ELISA. The samples were prepared as in (a) but with 75 μg/ml proteins and applied to sandwich ELISA. Data are means±S.D. of n=3 independent experiments. (e and f) Nucleosome release analyzed by Cell Death Detection ELISA. Samples were prepared as in (c), but additionally for RPE cells. Supernatants and lysates were collected after 48 h. Data are means±S.D. of n=4 (Jurkat T cells) and n=3 (RPE cells) independent experiments. (g) Phagocytosis of nucleosomes. Purified pHrodo-labeled nucleosomes (3.3 μg) were pre-incubated with buffer, 10% NHS, FH-depleted serum, 150 μg/ml FH or α1-antitrypsin for 1 h and incubated with peripheral human monocytes for 1 h and analyzed by flow cytometry. Depicted are means±S.D. of n=4 independent experiments, P-values refer to buffer control. *P<0.05, **P<0.01, ***P<0.001. α1-AT, α1-antitrypsin; Nuc, nucleosomes; NHS, normal human serum; SN, supernatant
To confirm the interaction of FH with nucleosomes, the same samples were analyzed using a sandwich ELISA capturing with an anti-histone Ab and detecting with an anti-FH Ab. Complexes of FH and histones were only detected in the supernatant (Figure 4c) and cytosolic fractions (Figure 4d) where FH was added before apoptosis induction.
FH induces nucleosome release in vitro
To investigate whether the presence of FH during the progression of apoptosis modifies nucleosome release, the nucleosome content in the cell supernatants was determined by ELISA. Unexpectedly, FH significantly increased the nucleosome release to the cell supernatant in both cell lines (Figures 4e and f). α1-antitrysin was used as negative control.
FH facilitates phagocytosis of nucleosomes
To test if FH affects phagocytosis of released nucleosomes by purified human peripheral blood monocytes we used flow cytometry. Nucleosomes pre-incubated with normal human serum were significantly better phagocytosed than buffer pre-incubated nucleosomes (Figure 4g). Pre-incubation with purified FH resulted in a similar phagocytosis rate as human serum. The phagocytosis rate of nucleosomes pre-incubated with FH-depleted serum or α1-antitrypsin did not differ from nucleosomes pre-incubated with buffer. Together, these results demonstrate that FH is crucial for serum-mediated phagocytosis of nucleosomes.
FH regulates in vivo nucleosome and apoptotic cell clearance in MRL-lpr mice
To test whether CFH−/− MRL-lpr mice exhibit altered nucleosome levels in serum compared to CFH+/+ MRL-lpr mice, we used a nucleosome ELISA. The nucleosome level in CFH+/+ MRL-lpr mice was not different from C57BL/6 mice (Figure 5a). However, CFH−/− MRL-lpr mice exhibited significantly increased nucleosome levels in their blood compared with CFH+/+ MRL-lpr mice at an age of 12 weeks. This difference was even more pronounced in the 17–19-week old mice (Figure 5b).

FH regulates in vivo nucleosome release in MRL-lpr mice. (a) Blood nucleosome level in C57BL/6 (n=5), CFH+/+ MRL-lpr (n=6) and CFH−/− MRL-lpr (n=6) mice at 12 weeks age determined by Cell Death Detection ELISA. (b) Blood nucleosome level in 17–19-week old CFH+/+ MRL-lpr (n=6) and CFH−/− MRL-lpr (n=6) mice determined as in (a). (c–e) Flow cytometric assessment of live (c), apoptotic (d) and secondary necrotic (e) T lymphocytes (CD3+) in CFH+/+ MRL-lpr (n=4), CFH−/− MRL-lpr (n=4) and congenic MRL/+ control mice (n=4). (a–e) Values are depicted as means±S.D. *P<0.05, **P<0.01, ***P<0.001
Further, the amount of live T lymphocytes (CD3+) was lower (Figure 5c) and the amount of apoptotic T lymphocytes significantly higher (Figure 5d) in CFH+/+ MRL-lpr mice than in congenic MRL/+ mice. CFH−/− MRL-lpr mice exhibited significantly less apoptotic (Figure 5d) but significantly more late apoptotic/secondary necrotic T lymphocytes (Figure 5e) than their CFH+/+ littermates. A similar pattern was observed for total leukocytes (data not shown). Absence of FH therefore leads to an increase in both late apoptotic/secondary necrotic cells and nucleosomes in autoimmune MRL-lpr mice, indicating impaired clearance of apoptotic debris.
FH bound to nucleosomes and apoptotic cells changes the cytokine profile released by phagocytes after uptake
To further investigate the role of FH on monocyte responses after phagocytosis of nucleosomes, purified mononucleosomes were pre-incubated with medium, FH or α1-antitrypsin (negative control) and added to peripheral human monocytes for 20 h. IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-17, G-CSF, GM-CSF, IFN-γ, MCP-1 and TNF-α were released by monocytes after phagocytosis of nucleosomes, whereas monocytes kept in medium alone did not release these mediators. Pre-incubation of the nucleosomes with FH strongly enhanced the release of the anti-inflammatory cytokine IL-10 in comparison with nucleosomes incubated in buffer or with α1-antitrypsin (Figure 6a). A similar pattern was observed for IL-2 and IL-4, albeit these were present in lower concentrations. The release of the chemoattractant IL-8 was not affected by FH. Of the pro-inflammatory cytokines, IL-1β, IL-6 and TNF-α, only the latter was increased in the presence of FH, whereas the others were unaltered (Figure 6b). IFN-γ, GM-CSF and IL-17 were significantly upregulated, whereas the chemokine MCP-1 was downregulated in the presence of FH (Figure 6c). Accordingly, IL-10 was strongly upregulated also when monocytes were incubated with apoptotic cells pre-incubated with FH, but not pre-incubated with buffer and negative control (Figure 6d).

FH bound to nucleosomes and apoptotic cells modifies the cytokine profile released by monocytes after phagocytosis. (a–c) Cytokine release profile of monocytes after uptake of mononucleosomes. Nucleosomes were pre-incubated with cell medium, 150 μg/ml FH or α1-antitrypsin and incubated with monocytes. Supernatants were collected after 20 h and cytokine levels were determined with Bio-Plex Pro human cytokine 17-plex assay. Data are means±S.D. from n=3 independent experiments determined in one multiplex assay. (d) IL-10 release of monocytes after uptake of apoptotic Jurkat T and RPE cells. Cells were pre-incubated as in (a–c) and incubated with monocytes (ratio 3:1). Cytokine profiles were determined as in (a–c). Data are means and separate values of n=2 independent experiments measured in one multiplex assay. *P<0.05, **P<0.01, ***P<0.001. α1-AT, α1-antitrypsin
FH bound to nucleosomes induces tyrosine phosphorylation of Siglec-3 and Siglec-9 in monocytes
To assess the mechanism by which FH is involved in altering the monocyte responses after phagocytosis of nucleosomes we used a tyrosine phosphorylation array. Jurkat T-cell-derived nucleosomes pre-incubated with FH or medium alone were incubated with peripheral human monocytes for 20 h and cell lysates from three donors were pooled and assessed for tyrosine phosphorylation of 59 phospho-immunoreceptors with proteome profiler arrays (Figure 7a). Siglec-3 and Siglec-9 were strongly phosphorylated in the presence of FH (Figure 7b).

FH bound to nucleosomes induces tyrosine phosphorylation of Siglec-3 and Siglec-9 in monocytes and directly binds to Siglec-9. (a and b) Tyrosine phosphorylation of monocyte immunoreceptors after uptake of Jurkat T-cell-derived nucleosomes. Nucleosomes were pre-incubated with medium or 150 μg/ml FH and incubated with monocytes. Cell lysates were collected after 20 h, pooled from three donors and tyrosine phosphorylation was determined with human phospho-immunoreceptor proteome profiler arrays. (a) One representative array set of n=2 independent repetitions. (b) Fold change of FH over control of receptors with higher phosphorylation intensity than PBS background in at least one of the two repetitions. Data are means and separate values of n=2 independent experiments. (c) Immunoprecipitation for Siglec-9 with following WB for FH. Purified FH (25 ng) was applied as positive control. One representative blot is shown. Fcγ RIIA, Fcγ receptor IIA; FcRH, Fc receptor homolog; ILT2, inhibitory receptors Ig-like transcript 2; monos, monocytes; Nuc, nucleosomes
FH binds directly to Siglec-9
To investigate the interaction of FH and Siglec-9, peripheral human monocytes were incubated with medium, FH alone and FH bound to nucleosomes. Immunoprecipitation for Siglec-9 and following WB for FH showed clearly that FH can directly interact with Siglec-9 and that the interaction is not dependent on the presence of nucleosomes (Figure 7c).
Discussion
Complement is essential to promote clearance of apoptotic cells21 and inefficient clearance can break tolerance and initiate autoimmunity.22 FH bound to apoptotic cells prevents excessive complement activation but allows opsonization in order to ensure efficient removal of dying cells. In the current study, we show that FH is actively internalized by apoptotic cells, whereby it regulates cleavage and deposition of endogenous C3 at the cell surface in the form of the potent opsonin iC3b, thereby facilitating phagocytosis. In addition, FH bound to nucleosomes facilitates their phagocytosis by human monocytes, and alters the cytokine profile produced by monocytes after phagocytosis toward those considered anti-inflammatory.
FH was not only internalized by Jurkat and RPE cells but also by additional leukocyte cell lines (Namalwa B cells and monocytic THP-1 cells). To our knowledge, active internalization of a specific serum protein by a dying cell is an unexplored phenomenon. The early apoptotic cell membrane remains intact hence excluding the passive transfer of FH into the cytosol. This was also confirmed by the lack of internalization of other tested apoptotic cell-binding proteins, including protein S, which is smaller than FH. Sodium azide blocks all ATP-dependent processes within a cell and strongly diminished FH internalization, confirming an active energy-dependent process.
C3, a major ligand for FH, can be produced by a variety of different cells.4 We sought interactions of internalized FH with other intracellular complement proteins, and after observing that C3 mRNA is still expressed in apoptotic cells, we revealed a concentration-dependent increase in C3 cleavage and iC3b deposition on the cell surface in both Jurkat T and RPE cells when FH was present during apoptosis induction. C3 is stored in the endosomes of T cells and may be cleaved into biologically active C3a and C3b upon T-cell activation by the lysosomal cysteine protease CTSL.19 CTSL and other lysosomal proteases are released into the cytosol upon apoptosis induction.23 The fact that FH does not become degraded within the apoptotic cells implies that it remains functional after internalization. Indeed, we found that FH acts as cofactor for CTSL-mediated cleavage of C3 into C3b, which may be due to direct binding of FH to CTSL. However, even in the presence of FH, it appears that CTSL cannot process C3b to iC3b. The binding of FH to C3b is known to impart a conformational alteration in C3b, which results in a 15-fold higher affinity of factor I for C3b.24 QPCR analysis revealed that both apoptotic Jurkat T and RPE cells express detectable amounts of factor I mRNA (data not shown). Thus the additional cleavage step into iC3b might be performed by factor I or by other proteases, during apoptosis induction.
Nucleosomes are major autoantigens and patients suffering from SLE have increased levels of nucleosomes in their blood, which are positively correlated with disease activity, particularly renal involvement.25 During apoptosis, nucleosomes are released to the cytosol, where we now show they become coated with internalized FH. Unexpectedly, FH increased the release of nucleosomes to the cell supernatant in apoptotic cells. This process was previously suggested to prevent autoimmunity due to faster clearance of released nucleosomes.26 Further, FH directly facilitates nucleosome phagocytosis. The in vivo findings that CFH−/− MLR-lpr mice had higher levels of serum nucleosomes in comparison with CFH+/+ MLR-lpr mice are consistent with our findings. However, it is important to note that the CFH−/− MLR-lpr mice exhibit, due to spontaneous consumption prevented normally by FH, significantly lower plasma C3 levels than their CFH+/+ littermates. Thus it is hard to judge whether the levels of circulating nucleosomes and necrotic cells are higher in CFH−/− MLR-lpr mice due to a direct effect of FH or diminished opsonization with iC3b or both.
Importantly, we found that FH not only facilitates iC3b deposition on apoptotic cells but also alters cytokine responses from monocytes, incubated with nucleosomes and apoptotic cells, in an anti-inflammatory direction. Furthermore, the FH-nucleosome induced release of GM-CSF would result in an increased production of granulocytes and monocytes and ensure the presence of professional phagocytes at the site of nucleosome release. Increased levels of IFN-γ could also stimulate the differentiation of monocytes into macrophages. The mechanism by which FH alters monocyte response appears to be in part mediated by enhanced stimulation of Siglec-3 and Siglec-9. These are inhibitory receptors of the sialic acid-binding Ig-like lectin family, which attenuate immune responses and dampen inflammation.27 FH binds to sialic acid28 and seems to facilitate the interaction of cis sialic acids and siglec receptors on the monocytes, which enhances their inhibitory response.
There is a significant association between the CFH genotype, and incidences of AMD and chronic renal disease29, 30 and these two seemingly disparate diseases may have a clinical and biological relationship. This fits very well with our hypothesis that FH internalization has an important role in facilitating opsonization, particularly in tissues such as kidneys and eyes. Both diseases are associated with an increased amount of local apoptotic cells and complement-mediated chronic inflammation. Since many autoantigens found in SLE are previously identified FH ligands, it is possible that the blockade of FH ligands by antibodies results in decreased FH internalization, leading to diminished opsonization with iC3b and release of nucleosomes without FH. This would explain the inefficient removal of apoptotic cells and would also lead to complement-mediated inflammation. The same outcome could occur when FH expression is diminished or its function altered due to mutations or polymorphisms in the gene. Interestingly, we showed previously that binding of FH to annexin A2 and histones was affected by the AMD-associated polymorphism in FH.5 This new role of FH is endorsed by a recent study showing that the expression of human complement FH prevents AMD-like retina damage and kidney abnormalities in aged CFH−/− mice.31
In conclusion, we have found a new intriguing function of the complement inhibitor FH in apoptotic cell clearance and handling of potential autoantigens (nucleosomes). FH becomes internalized by apoptotic cells and induces intracellular cleavage of C3, which becomes exposed on the cell surface in form of the potent opsonin iC3b, providing an additional removal signal to phagocytes. In addition, FH binds to nucleosomes, facilitates their removal and ensures release of an anti-inflammatory cytokine profile by phagocytes after uptake of nucleosomes and apoptotic cells. This new function is of significant importance where complement is dysfunctional and clearance of apoptotic cells inefficient.
Materials and Methods
Healthy donors, cells and induction of cell death
Blood samples were obtained with ethical approval (Dnr. 2013/846). Human peripheral blood monocytes were isolated by density gradient centrifugation using LymphoPrep (Axis-Shield, Oslo, Norway) following positive selection using CD14 microbeads (Milteny Biotech, Bergisch Gladbach, Germany). Jurkat T-cell clone E61 and retinal pigment epithelia cell line ARPE-19 (ATCC) were grown in RPMI 1640 and DME/F12 media (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% heat-inactivated FCS (Gibco; Life Technologies Europe BV, Stockholm, Sweden). Jurkat T-cells were rendered apoptotic using 1 μM staurosporine (Sigma-Aldrich, St. Louis, MO, USA) and RPE cells using 5 mM H2O2 (Merck, Millipore, Darmstadt, Germany) for 4 h in corresponding medium without FCS.
Mice
CFH−/− C57BL/6 mice12 (generously provided by Dr. Marina Botto, Imperial College, UK) were backcrossed (ten crosses) onto the MRL/MRL/+-Tnfrsf6lpr /2J mice (stock number 006825) (Jackson Laboratories, Bar Harbor, ME, USA). CFH+/− MRL-lpr mice were intercrossed to generate CFH−/−, CFH+/− and CFH+/+ MRL-lpr mice used in these studies, which maintained the Tnfrsf6lpr/lpr genotype in all animals.14 Littermate CFH−/−, CFH+/− and CFH+/+ MRL-lpr mice were studied at 12 and 17–19 weeks of age. These studies were approved by the Animal Care and Use Committee at University of Buffalo.
Proteins, antibodies, nucleosomes, inhibitors and sera
Details are included in Supplementary Materials and Methods.
Internalization studies
Apoptotic cells were incubated with pHrodo-labeled proteins at 4 °C and 37 °C for indicated time points. Reaction was stopped by transferring cells on ice and washing with NaN3-containing buffer. Internalization was measured in a CyFlow Space (Sysmex-Partec, Norderstedt, Germany) flow cytometer and analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
Confocal microscopy
Apoptosis was directly induced on SuperFrostPlus object slides (Menzel, Braunschweig, Germany) and indicated samples were pre-incubated with 30 mM NaN3 for 10 min. All samples were incubated with 30 μg/ml AlexaFluor488-labeled FH for 30 min at 37 °C, fixed and nuclei were counterstained with propidium iodine. Cells were mounted with DakoCytomation fluorescent mounting medium (Dako, Stockholm, Sweden) and images were obtained with a LSM 510 Meta confocal microscope using a × 63 oil objective and Zen 2009 software (Zeiss, Oberkochen, Germany).
Quantitative RT-PCR
TaqMan gene expression assays (Applied Biosystems; Life Technologies Europe BV, Stockholm, Sweden) were performed according to the manufacturer’s instruction: C3 (Hs_00163811_m1), GAPDH (Hs_00984230_m1) and HPRT1 (Hs99999909_m1). C3 gene expression level was determined using the ΔCt method after normalization with the geometric mean of the two housekeeping genes.32
Flow cytometry
Intracellular and cell surface staining and measurement of apoptosis and necrosis were carried out with standard techniques (Supplementary Experimental Procedures).
FH stability test
FH was labeled with 125I according to the chloramine T technique.33 Apoptotic Jurkat T cells were incubated for indicated time points with 125I-labeled FH. Cells were lysed, proteins separated on 10% SDS-PAGE under reducing conditions and radioactive signal analyzed using Typhoon FLA 9500 Phosphorimager (GE Healthcare, Life Sciences, Logan, UT, USA).
Degradation assay
C3 was labeled with 125I, CTSL was activated in 45 mM citric acid, 154 mM dibasic sodium phosphate, 1.56 mM l-cysteine, pH 6.0 (resembling cytosolic pH of apoptotic cells) for 30 min at 37 °C. Twenty-three μg/ml activated CTSL were incubated with increasing concentrations of FH, 150 μg/ml C3met, 40 nM CTSL inhibitor (optionally) and 125I-C3 for 3 h at 37 °C. The reaction was stopped by addition of reducing Laemmli buffer. Cleavage fragments were separated on 4–15% Criterion Gradient Gel (Bio-Rad, Hercules, CA, USA) and analyzed as above.
Modified western blot
Jurkat T cells and nucleosomes were pre-incubated with FH, α1-antitrypsin or medium for 1 h at 37 °C. Apoptosis was induced with 0.75 μM staurosporine for 24 h. The supernatant was collected after centrifugation for 10 min at 200 × g. The cytoplasmic fraction was collected after 30 min lysis of the cell pellet. Samples were loaded on agarose gel. DNA was stained with ethidium bromide and photographed with ChemiDoc MP system (Bio-Rad). Samples were blotted onto a nitrocellulose membrane (Pall) using a vacuum blotter (Boekel/Appligene, Feasterville, PA, USA). FH was detected using goat anti-human FH Ab and histones using rabbit anti-human histone H2B Ab following HRP-labeled secondary Abs. Membranes were photographed and the images were overlaid with the DNA images.
Binding assay and ELISA
Maxisorp microtiter plates (Nunc; Life Technologies Europe BV, Stockholm, Sweden) were coated with CTSL, osteoadherin, prothrombin and rabbit anti-human histone H2B Ab over night at 4 °C. After blocking, increasing concentrations of FH or samples prepared as for the modified WB were incubated for 1 h at room temperature. FH binding/FH-histone complexes were detected with goat anti-human FH Ab as above.
Nucleosome release was determined by Cell Death Detection ELISAPLUS assay (Roche, Basel, Switzerland) according to the manufacturer’s instruction.
Phagocytosis assay
pHrodo-labeled mononucleosomes were first pre-incubated with FH, α1-antitrypsin or medium for 1 h at 37 °C and then added to monocytes for an additional hour. Monocytes were stained with mouse anti-human CD14-APC and phagocytosis was determined as gMFI of pHrodo-signal in CD14+ monocytes.
Cytokine profiling
Apoptotic cells and mononucleosomes (10 μg) were pre-incubated with FH, α1-antitrypsin or medium for 45 min at 37 °C and added to monocytes (3 × 105/sample). Supernatants were collected after 20 h and supplemented with 0.5% BSA. Cytokine levels were determined in a Bio-Plex 200 system using the Bio-Plex Pro human cytokine 17-plex assay according to the manufacturer’s instructions.
Tyrosine phosphorylation of immunoreceptors
Jurkat T-cell-derived nucleosomes (100 μg) were pre-incubated with FH or medium for 45 min at 37 °C and added to monocytes (3 × 106/sample). Cells were lysed after 20 h and lysates of three donors were pooled for each array. Tyrosine phosphorylation of 59 immunoreceptors was determined by human phospho-immunoreceptor proteome profiler array (R&D Systems) according to the manufacturer’s instructions.
Immunoprecipitation
Monocytes were incubated for 20 h with medium, 150 μg/ml FH alone and 150 μg/ml FH that was pre-incubated for 45 min with Jurkat T-cell-derived nucleosomes. Cells were lysed and Siglec-9 was pulled-down with Protein G Sepharose (GE Healthcare) coated with anti-Siglec-9 Ab. Samples and purified FH were separated on 10% SDS-PAGE under reducing conditions and blotted onto PVDV membrane (Bio-Rad) using Turbo Trans Blot (Bio-Rad). FH was detected by WB as described above.
Statistical analysis
Analyses were performed on GraphPad Prism. Data are presented as mean±S.D. and analyzed with two-tailed t-test, one-way or two-way analysis of variance followed by Dunnett’s, Tukey’s or Bonferroni’s multiple comparison post test, as appropriate. All experiments including monocytes were considered to be repeated measures, since different donors were used for repetitions. P-values <0.05 denoted statistical significance and are displayed as: *P<0.05, **P<0.01, ***P<0.001.
Abbreviations
- α1-AT:
-
alpha-1 antitrypsin
- AF:
-
AlexaFluor
- AMD:
-
age-related macular degeneration
- BG:
-
background
- Cyt:
-
cytosol
- CTSL:
-
cathepsin L
- FH:
-
Factor H
- gMFI:
-
geometrical mean fluorescent intensity
- HPRT1:
-
hypoxanthine phosphoribosyltransferase 1
- NHS:
-
normal human serum
- Nuc:
-
nucleosomes
- RPE:
-
retinal pigment epithelial
- SLE:
-
systemic lupus erythematosus
- SN:
-
supernatant
References
Trouw LA, Bengtsson AA, Gelderman KA, Dahlback B, Sturfelt G, Blom AM . C4b-binding protein and factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack. J Biol Chem 2007; 282: 28540–28548.
Botto M, Walport MJ . C1q, autoimmunity and apoptosis. Immunobiology 2002; 205: 395–406.
Sjoberg AP, Trouw LA, Blom AM . Complement activation and inhibition: a delicate balance. Trends Immunol 2009; 30: 83–90.
Kolev M, Friec GL, Kemper C . Complement - tapping into new sites and effector systems. Nat Rev Immunol 2014; 14: 811–820.
Leffler J, Herbert AP, Norstrom E, Schmidt CQ, Barlow PN, Blom AM et al. Annexin-II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J Biol Chem 2010; 285: 3766–3776.
Mevorach D, Mascarenhas JO, Gershov D, Elkon KB . Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med 1998; 188: 2313–2320.
Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR . Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum 1998; 41: 1241–1250.
Radic M, Herrmann M, van der Vlag J, Rekvig OP . Regulatory and pathogenetic mechanisms of autoantibodies in SLE. Autoimmunity 2011; 44: 349–356.
Ao W, Zheng H, Chen XW, Shen Y, Yang CD . Anti-annexin II antibody is associated with thrombosis and/or pregnancy morbidity in antiphospholipid syndrome and systemic lupus erythematosus with thrombosis. Rheumatol Int 2011; 31: 865–869.
Lech M, Anders HJ . The pathogenesis of lupus nephritis. J Am Soc Nephrol 2013; 24: 1357–1366.
Jonsen A, Nilsson SC, Ahlqvist E, Svenungsson E, Gunnarsson I, Eriksson KG et al. Mutations in genes encoding complement inhibitors CD46 and CFH affect the age at nephritis onset in patients with systemic lupus erythematosus. Arthritis Res Ther 2011; 13: R206.
Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ et al. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet 2002; 31: 424–428.
Bao L, Haas M, Boackle SA, Kraus DM, Cunningham PN, Park P et al. Transgenic expression of a soluble complement inhibitor protects against renal disease and promotes survival in MRL/lpr mice. J Immunol 2002; 168: 3601–3607.
Bao L, Haas M, Quigg RJ . Complement factor H deficiency accelerates development of lupus nephritis. J Am Soc Nephrol 2011; 22: 285–295.
Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA 2005; 102: 7227–7232.
Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005; 308: 385–389.
Anderson DH, Mullins RF, Hageman GS, Johnson LV . A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol 2002; 134: 411–431.
Dunaief JL, Dentchev T, Ying GS, Milam AH . The role of apoptosis in age-related macular degeneration. Arch Ophthalmol 2002; 120: 1435–1442.
Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013; 39: 1143–1157.
Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC . Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol 2000; 2: 318–325.
Holers VM . Complement and its receptors: new insights into human disease. Annu Rev Immunol 2014; 32: 433–459.
Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M . The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol 2010; 6: 280–289.
Repnik U, Cesen MH, Turk B . The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol 2013; 5: a008755.
DiScipio RG . Ultrastructures and interactions of complement factors H and I. J Immunol 1992; 149: 2592–2599.
Williams RC Jr, Malone CC, Meyers C, Decker P, Muller S . Detection of nucleosome particles in serum and plasma from patients with systemic lupus erythematosus using monoclonal antibody 4H7. J Rheumatol 2001; 28: 81–94.
Stephan F, Marsman G, Bakker LM, Bulder I, Stavenuiter F, Aarden LA et al. Cooperation of factor VII-activating protease and serum DNase I in the release of nucleosomes from necrotic cells. Arthritis Rheumatol 2014; 66: 686–693.
Pillai S, Netravali IA, Cariappa A, Mattoo H . Siglecs and immune regulation. Annu Rev Immunol 2012; 30: 357–392.
Meri S, Pangburn MK . Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci USA 1990; 87: 3982–3986.
Xing C, Sivakumaran TA, Wang JJ, Rochtchina E, Joshi T, Smith W et al. Complement factor H polymorphisms, renal phenotypes and age-related macular degeneration: the Blue Mountains Eye Study. Genes Immun 2008; 9: 231–239.
Klein R, Knudtson MD, Lee KE, Klein BE . Serum cystatin C level, kidney disease markers, and incidence of age-related macular degeneration: the Beaver Dam Eye Study. Arch Ophthalmol 2009; 127: 193–199.
Ding JD, Kelly U, Landowski M, Toomey CB, Groelle M, Miller C et al. Expression of human complement factor h prevents age-related macular degeneration-like retina damage and kidney abnormalities in aged cfh knockout mice. Am J Pathol 2015; 185: 29–42.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3: RESEARCH0034.
Greenwood FC, Hunter WM, Glover JS . The preparation of I-131-labelled human growth hormone of high specific radioactivity. Biochem J 1963; 89: 114–123.
Acknowledgements
We thank Dr. Ben King, Lund University, for language revision of the manuscript and Dr. Christiane Desel, University of Oxford, for discussion of our cytokine data. Jennifer Mytych and Karolina I. Smoląg are students of Biotechnology at University of Rzeszow, Poland. This study was supported by the Swedish Research Council (K2012-66X-14928-09-5), Cancerfonden, Foundations of Österlund, Crafoord, Greta and Johan Kock, Lars Hiertas Minne, Tore Nilsson, King Gustav V's 80th Anniversary, Knut and Alice Wallenberg, Inga-Britt and Arne Lundberg and grants for clinical research (ALF and from the Skåne University Hospital).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by L Zitvogel
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Martin, M., Leffler, J., Smoląg, K. et al. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ 23, 903–911 (2016). https://doi.org/10.1038/cdd.2015.164
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.164
This article is cited by
-
Complosome — the intracellular complement system
Nature Reviews Nephrology (2023)
-
Potential clinical utility of liquid biopsies in ovarian cancer
Molecular Cancer (2022)
-
Smoking induces sex-specific changes in the small airway proteome
Respiratory Research (2021)
-
The HSP GRP94 interacts with macrophage intracellular complement C3 and impacts M2 profile during ER stress
Cell Death & Disease (2021)
-
Tipping the balance: intricate roles of the complement system in disease and therapy
Seminars in Immunopathology (2021)
