
Abstract
Regulatory T cells hold promise as targets for therapeutic intervention in autoimmunity, but approaches capable of expanding antigen-specific regulatory T cells in vivo are currently not available. Here we show that systemic delivery of nanoparticles coated with autoimmune-disease-relevant peptides bound to major histocompatibility complex class II (pMHCII) molecules triggers the generation and expansion of antigen-specific regulatory CD4+ T cell type 1 (TR1)-like cells in different mouse models, including mice humanized with lymphocytes from patients, leading to resolution of established autoimmune phenomena. Ten pMHCII-based nanomedicines show similar biological effects, regardless of genetic background, prevalence of the cognate T-cell population or MHC restriction. These nanomedicines promote the differentiation of disease-primed autoreactive T cells into TR1-like cells, which in turn suppress autoantigen-loaded antigen-presenting cells and drive the differentiation of cognate B cells into disease-suppressing regulatory B cells, without compromising systemic immunity. pMHCII-based nanomedicines thus represent a new class of drugs, potentially useful for treating a broad spectrum of autoimmune conditions in a disease-specific manner.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Santamaria, P. The long and winding road to understanding and conquering type 1 diabetes. Immunity 32, 437–445 (2010)
Babbe, H. et al. Clonal expansions of CD8+ T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 192, 393–404 (2000)
Firestein, G. S. Evolving concepts of rheumatoid arthritis. Nature 423, 356–361 (2003)
Sakaguchi, S. et al. Foxp3+CD25+CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 212, 8–27 (2006)
Zhou, X. et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nature Immunol. 10, 1000–1007 (2009)
Komatsu, N. et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nature Med. 20, 62–68 (2014)
Bailey-Bucktrout, S. L. et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 39, 949–962 (2013)
Gagliani, N. et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nature Med. 19, 739–746 (2013)
McLarnon, A. IBD: regulatory T-cell therapy is a safe and well-tolerated potential approach for treating refractory Crohn's disease. Nature Rev. Gastroenterol. Hepatol. 9, 559 (2012)
Desreumaux, P. et al. Safety and Efficacy of Antigen-Specific Regulatory T-Cell Therapy for Patients With Refractory Crohn's Disease. Gastroenterology 143, 1207–1217 (2012)
Roncarolo, M. G., Gregori, S., Lucarelli, B., Ciceri, F. & Bacchetta, R. Clinical tolerance in allogeneic hematopoietic stem cell transplantation. Immunol. Rev. 241, 145–163 (2011)
Tsai, S. et al. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity 32, 568–580 (2010)
Stratmann, T. et al. Susceptible MHC alleles, not background genes, select an autoimmune T cell reactivity. J. Clin. Invest. 112, 902–914 (2003)
Kamanaka, M. et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 25, 941–952 (2006)
Mukherjee, R., Wagar, D., Stephens, T., Le-Chan, E. & Singh, B. Identification of CD4+ T cell-specific epitopes of islet-specific glucose-6-phosphatase catalytic subunit-related protein: a novel β cell autoantigen in type 1 diabetes. J. Immunol. 174, 5306–5315 (2005)
Burton, B. R. et al. Sequential transcriptional changes dictate safe and effective antigen-specific immunotherapy. Nature Commun. 5, 4741–4747 (2014)
Getts, D. R. et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nature Biotechnol. 30, 1217–1224 (2012)
Pot, C. et al. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol. 183, 797–801 (2009)
Spensieri, F. et al. Human circulating influenza-CD4+ICOS1+IL-21+ T cells expand after vaccination, exert helper function, and predict antibody responses. Proc. Natl Acad. Sci. USA 110, 14330–14335 (2013)
Hale, J. S. et al. Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity 38, 805–817 (2013)
Sato, K. et al. Marked induction of c-Maf protein during Th17 cell differentiation and its implication in memory Th cell development. J. Biol. Chem. 286, 14963–14971 (2011)
Saraiva, M. et al. Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity 31, 209–219 (2009)
Verdaguer, J. et al. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J. Exp. Med. 186, 1663–1676 (1997)
Wang, J. et al. In situ recognition of autoantigen as an essential gatekeeper in autoimmune CD8+ T cell inflammation. Proc. Natl Acad. Sci. USA 107, 9317–9322 (2010)
Yang, J. et al. Islet-specific glucose-6-phosphatase catalytic subunit-related protein-reactive CD4+ T cells in human subjects. J. Immunol. 176, 2781–2789 (2006)
Holst, J. et al. Generation of T-cell receptor retrogenic mice. Nature Protocols 1, 406–417 (2006)
Reijonen, H. et al. Detection of GAD65-specific T cells by major histocompatibility complex class II tetramers in type 1 diabetic patients and at-risk subjects. Diabetes 51, 1375–1382 (2002)
Yang, J. et al. CD4+ T cells from type 1 diabetic and healthy subjects exhibit different thresholds of activation to a naturally processed proinsulin epitope. J. Autoimmun. 31, 30–41 (2008)
Moore, A., Grimm, J., Han, B. & Santamaria, P. Tracking the recruitment of diabetogenic CD8+ T cells to the pancreas in real time. Diabetes 53, 1459–1466 (2004)
Giuliani, F. et al. Additive effect of the combination of glatiramer acetate and minocycline in a model of MS. J. Neuroimmunol. 158, 213–221 (2005)
Leavenworth, J. W., Tang, X., Kim, H. J., Wang, X. & Cantor, H. Amelioration of arthritis through mobilization of peptide-specific CD8+ regulatory T cells. J. Clin. Invest. 123, 1382–1389 (2013)
Acknowledgements
We thank S. Thiessen, J. DeLongchamp, J. Erickson, J. Luces, R. Barasi and K. Umeshappa for technical contributions; L. Kennedy, L. Robertson and Y. Liu for flow cytometry; F. Jirik for help with histological analyses of arthritic mice; J. Elliott and K. Suzuki for Meso Scale measurements; M. Fritzler for Luminex; and P. Colarusso for assistance with microscopy. This work was funded by the Canadian Institutes of Health Research (CIHR), the Diabetes Research Foundation, the Juvenile Diabetes Research Foundation (JDRF), the Canadian Diabetes Association (CDA), the Multiple Sclerosis Society of Canada (MSSC), the Brawn Family Foundation, National Research Council of Canada–Industrial Research Assistance Program (NRC-IRAP), Instituto de Investigaciones Sanitarias Carlos III (ISCIII) Integrated Projects of Excellence and FEDER, the Ministerio de Economia y Competitividad of Spain (MINECO), the European Association for the study of diabetes (EASD), the Sardà Farriol Research Programme, and the European Community’s Seventh Framework Programme. X.C.C. was supported by studentships from the AXA Research Fund and the endMS network. P.A. was supported by the endMS network. J.B. was suported by a Rio Hortega fellowship and by a grant from the Spanish Society for Diabetes. S.T. was supported by a studentship from the Alberta Heritage Foundation of Medical Research (AHFMR). J.W. was funded by a fellowship from the CDA. P.Se. is an investigator of the Ramon y Cajal reintegration program and is supported by a JDRF Career Development Award. P.Sa. is a Scientist of the Alberta Innovates-Health Solutions and a scholar of ISCIII. The JMDRC is supported by the Diabetes Association (Foothills) and the CDA.
Author information
Authors and Affiliations
Contributions
X.C.-C. executed most of the experiments in Figs 1, 3a–d, h–j, 4a–f, Supplementary Table 1 and Extended Data Figs 1b–n, 2, 3, 5a–f, n–t 6, with contributions from J.Y., P.A., S.T. and J.W., and contributed to writing the manuscript with P.Sa. J.Y. executed the experiments in Figs 3e–g, 4g–i and Extended Data Figs 1a, c–f, 2h and 5u, v. P.A. executed all of the experiments described in Figs. 2, 3b, and Extended Data Figs 4 and 5g–m. J.B. recruited T1D patients and healthy controls and performed the experiments leading to Fig. 5, Extended Data Fig. 7 and Supplementary Table 2 under the supervision of P.Se. S.S., Y.Y. and A.M. produced nanoparticles and pMHC–NP conjugates for the study. C.F. produced 2.5mi/IAg7 class II monomers for mechanistic experiments. S.A. and M.K. contributed to the execution of the EAE experiments, and analysed histological sections for histopathological features of CNS inflammation and demyelination under the supervision of V.W.Y. E.J. provided human T1D-relevant pMHC class II monomers and tetramers. N.G., C.I. and T.S. produced the pMHC class II monomers used for the studies on the reversal of T1D. P.Sa. designed the study, supervised and coordinated its execution and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
P.Sa. is the scientific founder of Parvus Therapeutics Inc. and has a financial interest in the company.
Extended data figures and tables
Extended Data Figure 1 Sustained expansion of cognate TR1-like CD4+ T cells by pMHCII–NP therapy restores normal glucose homeostasis in diabetic NOD mice by suppressing antigen presentation and the activation of non-cognate autoreactive T cells in the PLNs and the progression of insulitis.
a, Top left, expansion of cognate CD4+ T cells by 2.5mi/IAg7–NPs in anti-CD25 mAb-treated NOD Foxp3-eGFP mice. Data correspond to 8-week-old mice treated three times a week with 500 μg of a depleting anti-CD25 mAb i.p. or control anti-HPRN mAbs, followed by 10 doses of 2.5mi/IAg7–NPs starting at 10 weeks of age (two doses per week; n = 4 mice each). Bottom, the tetramer+CD4+ T cells from anti-CD25 mAb-treated mice express TR1 markers. Right, percentage of circulating FOXP3+eGFP+CD4+ (top) and CD25+CD4+ cells (bottom). b, Tetramer+CD4+ T cells sorted from 2.5mi/IAg7–NP-treated mice proliferate and produce IL-10 and, to a lesser extent IFNγ in response to stimulation with 2.5mi peptide-pulsed DCs (n = 3 mice). c, Representative cell surface CD49b and LAG-3 profiles on tetramer+CD4+ T cells from BDC2.5 NOD Foxp3-eGFP mice compared with tetramer−CD4+ T cells from transgenic or wild-type NOD mice (n = 4). d, Upregulation of CD49b and LAG-3 on anti-CD3/anti-CD28 mAb-activated BDC2.5 CD4+ T cells from BDC2.5 NOD Foxp3-eGFP mice in response to 2.5mi/IAg7–NP (25 μg pMHC per ml) versus 2.5mi peptide (10 μg ml−1) or 2.5mi/IAg7 monomers (25 μg pMHC per ml). e, Upregulation of eGFP (IL-10) in anti-CD3/anti-CD28 mAb-activated BDC2.5 CD4+ T cells from BDC2.5 NOD Il10GFP mice in response to 2.5mi/IAg7–NP as a function of CD49b and LAG-3 expression. f, Expression of eGFP (IL-10) in the CD4+ T cells of 2.5mi/IAg7–NP-treated NOD Il10GFP mice (2 doses per week for 5 weeks) as a function of CD49b and LAG-3 expression (left, representative profiles; right, eGFP MFI values) (n = 8). g, Proliferation of CFSE-labelled 8.3-TCR-transgenic CD8+ T cells (IGRP206–214/NRP–V7-specific) in response to 2.5mi/NRP–V7–peptide-pulsed or unpulsed DCs in the presence of tetramer− or tetramer+ CD4+ T cells from 2.5mi/IAg7-NP-treated mice and in the presence or absence of cytokine-blocking mAbs, rat IgG (negative control) or 1-methyl-l-tryptophan (1-MT; an IDO inhibitor). Data correspond to average of proliferated cells in 3–7 experiments per condition. h, Changes in blood glucose levels of spontaneously hyperglycaemic (>11 mM) female NOD mice treated with 2.5mi/IAg7–NP, IGRP4–22/IAg7–NP, IGRP128–145/IAg7–NP or HEL14-22/IAg7–NP (n = 6–9 per group), IGRP4–22 peptide or IGRP4–22 peptide–NPs (n = 9, 4–5 each). Mice received two doses per week until irreversibly hyperglycaemic or normoglycaemic for 4 consecutive weeks, at which point treatment was withdrawn. i, Incidence and timing of disease relapse in hyperglycaemic female NOD mice rendered stably normoglycaemic by treatment with 2.5mi/IAg7–NP, IGRP4-22/IAg7–NP or IGRP128–145/IAg7–NPs upon treatment withdrawal (after 4 consecutive weeks of normoglycaemia). Data correspond to responder mice in Fig. 1g. j, Post-prandial serum insulin levels in pMHC–NP-treated mice that reverted to normoglycaemia until 50 weeks of age (n = 6) versus newly diabetic (n = 12) and non-diabetic age-matched untreated controls (n = 10). k, Intra-peritoneal glucose tolerance tests (IPGTT) of the mice in h. l, Areas under the curve (AUC) in the IPGTTs shown in k. m, IPGTT serum insulin levels corresponding to the mice in k. n, Proliferation of CFSE-labelled IGRP206–214-reactive 8.3-CD8+ T cells in the PLNs compared with MLNs of 2.5mi/IAg7–NP-treated mice that reverted to normoglycaemia until 50 weeks of age, non-diabetic age-matched untreated controls and newly diabetic mice. Left panels show representative FACS profiles. Right panel compares percentages of proliferated cells in the PLNs after subtraction of the background proliferation values in non-draining MLNs (n = 6–8 mice per group). P values were calculated by Mann–Whitney U-test, log-rank (Mantel–Cox) test or two-way ANOVA. Data are averages ± s.e.m.
Extended Data Figure 2 Nanoparticles coated with different T1D-relevant pMHCII complexes expand cognate TR1-like CD4+ T cells in vivo to similar extent, regardless of epitope dominance or role of the target T-cell specificity in the disease process.
a, Percentage of tetramer+CD4+ T cells in the PLN, MLN and bone marrow (BM) of 2.5mi/IAg7–NP-treated mice that reverted to normoglycaemia until 50 weeks of age (n = 5–6 mice per lymphoid organ) or relapsed (n = 1–2) compared with newly diabetic (n = 5–6) and non-diabetic age-matched untreated controls (n = 4–6). b, Percentage of tetramer+CD4+ T cells in the splenic CD4+ T cells of 2.5mi/IAg7–NP-treated mice that reverted to normoglycaemia until 50 weeks of age or of age-matched non-diabetic untreated mice, stained with two T1D-relevant but non-cognate pMHCII tetramers (n = 3–4 per group). c, Percentage of tetramer+CD4+ T cells in blood, spleen, PLN, MLN and bone marrow of IGRP4–22/IAg7–NP-treated mice that reverted to normoglycaemia until 50 weeks of age (n = 5–6 mice per lymphoid organ) compared with newly diabetic (n = 5–8) and non-diabetic age-matched untreated controls (n = 4–6). d, Percentage of tetramer+CD4+ T cells in blood, spleen, PLN, MLN and bone marrow of IGRP128–145/IAg7–NP-treated mice that reverted to normoglycaemia until 50 weeks of age (n = 5–7 mice per lymphoid organ) compared with newly diabetic (n = 4–7) and non-diabetic age-matched untreated controls (n = 5–7). e, Representative IGRP4–22/IAg7, IGRP128–145/IAg7 and GPI282–292/IAg7 tetramer staining profiles for splenic CD4+ T cells from IGRP4–22/IAg7–NP- and IGRP128–145/IAg7–NP-treated compared with untreated NOD mice. f, Percentages of blood CD4+ T cells of IGRP4–22/IAg7–NP- or IGRP128–145/IAg7–NP-cured, HEL14–22/IAg7–NP-treated and age-matched non-diabetic untreated mice stained with non-cognate pMHCII tetramers (n = 3–7 per group). g, The tetramer+CD4+ T cells of mice treated with IGRP128–145/IAg7–NP (top) and IGRP4-22/IAg7–NP (bottom) proliferate and produce IL-10 specifically in response to stimulation with IGRP4–22 or IGRP128–145-peptide-pulsed DCs, respectively (n = 3 mice each). cpm, counts per minute. h, Percentages of IGRP4–22/IAg7 tetramer+CD4+ T cells in blood, spleen, PLN, MLN and bone marrow of NOD mice at the onset of hyperglycaemia or upon treatment with IGRP4–22/IAg7–NPs, or IGRP4–22 peptide or IGRP4–22 peptide-coated nanoparticles (n = 5–9 mice per organ). P values were calculated by Mann–Whitney U-test. Data are averages ± s.e.m.
Extended Data Figure 3 EAE-relevant pMHCII–NPs expand cognate IL-10-secreting TR1-like CD4+ T cells and ameliorate established clinical and pathological signs of EAE.
a, b, Changes in the average weights of C57BL/6 mice immunized with pMOG35–55 and treated with pMOG38–49/IAb–NPs or uncoated nanoparticles starting on days 14 (a) or 21 (b) after immunization. c, Percentage of pMOG38–49/IAb tetramer+CD4+ T cells in peripheral lymph nodes, bone marrow and central nervous system (CNS) of mice from a and b. d, The tetramer+CD4+ T cells of pMOG38–49/IAb–NP-treated mice proliferate and produce IL-10 and, to a lesser extent, IFNγ in response to stimulation with pMOG38–49 peptide-pulsed DCs. e, Left and middle, representative luxol fast blue (LFB)/H&E cerebellum staining images from untreated and treated mice from b showing presence of inflammatory foci and areas of demyelination (red arrows). Right, average number of inflammatory foci per section. Data corresponds to 4 untreated and 5 treated mice. f, Representative LFB/H&E-stained spinal cord sections from mice in b. Data were compared with Mann–Whitney U-test. Data are averages ± s.e.m.
Extended Data Figure 4 EAE- or CIA-relevant pMHCII–NPs expand cognate TR1-like CD4+ T cells and ameliorate clinical and pathological signs of EAE or CIA in HLA-DR4-IE-transgenic C57BL/6 IAbnull or C57BL/10.M mice.
a, Changes in the average EAE scores of HLA-DR4-IE-transgenic C57BL/6 IAbnull mice immunized with hPLP175–192 or hMOG97–108 and treated with hPLP175–192 /DR4-IE or hMOG97–108/DR4-IE–NPs or uncoated nanoparticles starting on the day when mice reached a score of 1.5 (to synchronize the groups for disease activity) (n = 3–4 per group). b, Percentage of tetramer+CD4+ T cells in spleen, blood, cervical and inguinal LNs and CNS of mice from a. Data correspond to 4 pMHC–NP-treated and 6 control-NP-treated mice. c, Changes in the average weights of HLA-DR4–IE-transgenic C57BL/6 IAbnull mice from a, immunized with hPLP175–192 or hMOG97–108 and treated with hPLP175–192/DR4-IE–NPs, hMOG97–108/DR4-IE–NPs or uncoated nanoparticles when the mice reached a score of 1.5. d, LFB/H&E staining of the cerebellum of HLA-DR4-IE-transgenic C57BL/6 IAbnull mice from a showing reductions in inflammation and demyelination in mice treated with hPLP175–192 /DR4-IE or hMOG97–108/DR4-IE–NPs compared with controls. e, Percentage of tetramer+CD4+ T cells in lymph nodes and bone marrow of the mice in Fig. 2a (C57BL/10.M HLA-DR4-IE mice immunized with bovine collagen) at the end of follow-up (10 doses, 5 weeks). f, Changes in the average weights of HLA-DR4-IE-transgenic C57BL/6 IAbnull mice immunized with hPLP175–192 from Fig. 2f. g, Representative LFB/H&E staining of the cerebellum of HLA-DR4-IE-transgenic C57BL/6 IAbnull mice immunized with hPLP175–192 and treated with hPLP175–192/DR4-IE–NPs, hMOG97–108/DR4-IE–NPs, hMOG97–108 peptide i.v. or s.c. (8 μg per dose), hMOG97–108/DR4-IE monomer (25 μg per dose), hMOG97–108 peptide–NPs (using the molar equivalent of peptide delivered via pMHC–NPs; 0.68 μg per dose), or hMOG97–108 peptide–MPs (15 μg peptide per dose) compared with mice left untreated or treated with uncoated NPs or MPs (at the same NP/MP number). h, Changes in the average EAE scores and body weights of HLA-DR4-IE-transgenic C57BL/6 IAbnull mice immunized with hPLP175–192 in response to treatment with hMOG97–108 peptide i.v. or s.c. (8 μg per dose16), hMOG97–108/DR4-IE monomer (25 μg per dose), hMOG97–108 peptide–NPs (0.68 μg peptide per dose), hMOG97–108 peptide–MPs (15 μg peptide per dose17), or a single dose of hMOG97–108 peptide–MPs (15 μg peptide17) compared with mice left untreated or treated with uncoated NPs or MPs (at the same NP/MP number) (n = 4–5 per group). The cohort of mice treated with one dose had to be terminated after 2.5 weeks, owing to rapid progression of disease. i, Percentages of tetramer+CD4+ T cells in spleen, blood, cervical and inguinal LNs and bone marrow of mice from h (n = 3–9 per group). Data were compared with Mann–Whitney U-test or two-way ANOVA. Data are averages ± s.e.m.
Extended Data Figure 5 Disease reversal by pMHC–NPs is driven by the TR1 cytokines IL-21, IL-10 and TGF-β and involves several downstream cellular targets.
a, Changes in blood glucose levels in diabetic NOD mice (>11 mM) treated with IGRP4–22/IAg7–NPs and blocking anti-IL-10, anti-IFNγ or anti-TGF-β mAbs or anti-HRPN rat-IgG (n = 4–6 per group). b, c, Percentages of tetramer+CD4+ T cells in the spleens (b), and proliferation of CFSE-labelled 8.3-CD8+ T cells in the PLNs verus MLN of the mice from Fig. 3a at the end of follow up (c). d, Changes in blood glucose in hyperglycaemic NOD, NOD Il10−/− and NOD Ifng−/− mice (n = 3−6 per group) in response to 2.5mi/IAg7–NPs. e, f, Percentages of tetramer+CD4+ T cells in the spleens (e), and proliferation of CFSE-labelled 8.3-CD8+ T cells in the PLNs versus MLN of the mice from d at the end of follow up (f). g, EAE scores of mice treated with pMHC–NPs and rat-IgG or blocking mAbs (n = 4 per group). h, LFB/H&E staining of the cerebellum of HLA-DR4-IE-transgenic C57BL/6 IAbnull mice from g, highlighting differences in inflammation and demyelination in mice treated with hPLP175–192/DR4-IE–NPs and rat-IgG versus blocking anti-IL-10, anti-TGF-β or anti-IL-21R mAbs. i, Changes in the average body weights of HLA-DR4-IE-transgenic C57BL/6 IAbnull mice from g. j, Percentage of tetramer+CD4+ T cells in spleen, blood and inguinal LNs of mice from g (n = 4 per group). k, l, Changes in the average EAE scores (k) and body weights (l) of C57BL/6 Il27r−/− mice immunized with pMOG35–55 and treated with pMOG38–49/IAb–NPs or uncoated nanoparticles starting on the day when mice reached a score of 1.5 (to synchronize the groups for disease activity) (n = 7 and 4, respectively). m, Representative IBA1 and LFB/H&E stainings of the cerebellum and the corresponding relative rank scores of mice from k (n = 3 and 4, respectively). n, Percentage of tetramer+CD4+ T cells in spleen, blood, inguinal LNs and bone marrow of mice from k (left), and representative CD49b and LAG-3 staining profiles of tetramer+ versus tetramer− cells (right). o, Percentage of B220+ cells in the PLNs or MLNs of 2.5mi/IAg7–NP- or HEL14-22/IAg7–NP-treated mice (n = 4 per group). p, Correlation between the percentages of PLN and splenic B220+ cells and 2.5mi/IAg7 tetramer+CD4+ T cells in additional cohorts of mice treated with 2.5mi/IAg7–NPs, over a range of total pMHC dose (0.75–25 μg of total pMHC) (n = 24–28). q, Left, in vitro proliferation of CFSE-labelled BDC2.5 CD4+ T cells against 2.5mi or GPI282–292 peptide-pulsed B cells purified from the PLNs or MLNs of untreated NOD mice or mice treated with 2.5mi/IAg7–NPs (n = 5–6 per group). Right, representative CFSE dilution profiles. Briefly, profiles show the extent of CFSE dilution in CFSE-labelled BDC2.5 CD4+ T cells cultured in the presence of 2.5mi or GPI282–292 peptide-pulsed B cells purified from the PLNs or MLNs of untreated or 2.5mi/IAg7–NP-treated NOD mice. r, PLN-derived B cells (105) from 2.5mi/IAg7–NP-treated mice secrete IL-10 ex vivo in response to LPS (1 μg ml−1). Data correspond to 6 pMHC-treated and 5 untreated NOD mice. s, t, Changes in the percentages of 2.5mi (PKH26-labelled) compared with GPI282–292 peptide-pulsed (CFSE-labelled) B cells (s) or DCs (t) 7 days after transfer (at 1:1 ratio) into untreated or 2.5mi/IAg7–NP-treated NOD mice. Histograms show averaged ratios for each cell type and condition (n = 3–4 mice per cell type and condition). u, Percentages of CD5+CD1dhieGFP+B220+ cells in mice treated as in Fig. 3b plus blocking Abs (n = 4 each). v, LPS-stimulated PLN B cells from NOD mice treated with 10 doses of 2.5mi/IAg7–NPs suppress the proliferation of CFSE-labelled BDC2.5 CD4+ T cells by 2.5mi peptide-pulsed DCs in vitro, as compared to LPS-stimulated PLN B cells from untreated controls. x, Percentage of CD19+CD3− cells in blood before and after 3 doses of 250 μg of anti-CD20 mAb (n = 4). y, 2.5mi/IAg7–NP-induced upregulation of IL-21 and IL-10 mRNA in memory eGFP− BDC2.5 CD4+ T cells from BDC2.5-TCR-transgenic NOD Foxp3-eGFP donors in NOD Thy1a hosts (n = 5). P values were calculated by Pearson correlation, Mann–Whitney U-test or two-way ANOVA. Data are averages ± s.e.m.
Extended Data Figure 6 Effects of cytokine blockade or genetic deficiency on the cytokine profile of cognate CD4+ T cells expanded by 2.5mi/IAg7–NPs.
n = 3 mice each. Data are averages ± s.e.m.
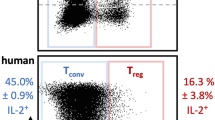
Extended Data Figure 7 Human T1D-relevant pMHCII–NPs, but not free peptide or peptide-coated nanoparticles or microparticles, expand cognate TR1-like CD4+ T cells in human PBMC-engrafted NSG hosts.
a, FACS profiles (cognate versus control tetramer staining in hCD4+ T cells) of samples from mice identified as responders in Supplementary Table 2. Numerical data on tetramer+ T cells are presented on Supplementary Table 2. b, Representative FACS profiles (cognate versus control tetramer staining in splenic hCD4+ T cells) of human healthy control PBMC-engrafted NSG hosts treated with IGRP13-25/DR3–NPs (left), or human T1D PBMC-engrafted NSG hosts treated with IGRP13–25 peptide, IGRP13–25 peptide-coated nanoparticles, IGRP13–25 peptide-coated microparticles, or left untreated (right). See Fig. 5 legend for details.
Extended Data Figure 8 Schematic of the proposed mode of operation of pMHCII-based nanomedicines.
pMHCII-coated NPs (pMHC–NP, lacking costimulatory molecules) promote the differentiation of disease-primed (antigen-experienced) IFNγ-producing CD4+ TH1-cells into memory TR1-like CD4+ T cells followed by systemic expansion. This differentiation process (but not the subsequent expansion) requires both IFNγ and IL-10, whereas IL-27 is dispensable. The pMHC–NP-expanded (mono-specific) autoreactive TR1-like CD4+ T cells then suppress other autoreactive T-cell responses by secreting IL-21, IL-10 and TGF-β, which act on local APCs (B cells, CD11c+ and CD11b+ cells) that have captured the cognate autoantigen and thus present cognate pMHCII complexes to the expanded TR1-like cells. This interaction inhibits the proinflammatory function of the targeted APCs and blocks their ability to present other pMHC class I and class II complexes to non-pMHC–NP-cognate autoreactive T-cell specificities (note that the local APCs uptake both cognate and non-cognate autoantigens shed into the milieu simultaneously). Suppression of antigen-presentation requires IL-10 and TGF-β but not IFNγ or IL-21. Furthermore, cognate interactions between the pMHC–NP-expanded TR1 CD4+ T cells and autoreactive B cells specific for the cognate autoantigen (able to display the cognate pMHCII complex on the surface) promotes their differentiation into Breg cells in an IL-21-dependent manner, which contribute to promote local immunosuppression, likely by secreting IL-10. Suppression of antigen presentation selectively targets APCs displaying the cognate pMHC, but as local APCs that capture the cognate autoantigen also capture other autoantigens simultaneously, the autoregulatory CD4+ T cells expanded by pMHC–NPs blunt the presentation of other autoantigenic pMHC complexes to a broad range of autoreactive T cells. This suppression is disease-specific and self-limiting.
Supplementary information
Supplementary Tables 1 and 2
This file comprises: Table 1 - Transcriptional profile of pMHC-NP-expanded CD4+ T-cells. (a) qRT-PCR for a panel of 384 immunological markers in 2.5mi/IAg7 tetramer+ versus tetramer– CD4+ T-cells sorted from NOD mice treated with 10 doses of 2.5mi/IAg7-NPs from 10-15 weeks of age (n=3 and 4 samples, respectively). The cells were stimulated in vitro with anti-CD3/anti-CD28 mAb-coated Dynabeads before RNA collection. Panel summarizes the most significant differences. (b) qRT-PCR for 8 TR1-relevant markers, including markers that were not represented in the primer set used in a. Data correspond to four additional 2.5mi/IAg7 tetramer+ and seven tetramer– CD4+ T-cell samples; Table 2 - Human T1D donors and outcome of pMHC-NP, peptide, peptide-NP and peptide-MP therapy in PBMC-engrafted NSG hosts. See text for details. (PDF 729 kb)
Rights and permissions
About this article

Cite this article
Clemente-Casares, X., Blanco, J., Ambalavanan, P. et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 530, 434–440 (2016). https://doi.org/10.1038/nature16962
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature16962
This article is cited by
-
Tolerance-inducing therapies in coeliac disease — mechanisms, progress and future directions
Nature Reviews Gastroenterology & Hepatology (2024)
-
Comparison of Different Stabilizing Agents and Antigen Attachment Methods on Iron Oxide Nanoparticles for Vaccination
Biomedical Materials & Devices (2024)
-
An engineered Fc fusion protein that targets antigen-specific T cells and autoantibodies mitigates autoimmune disease
Journal of Neuroinflammation (2023)
-
Single cell transcriptomics shows that malaria promotes unique regulatory responses across multiple immune cell subsets
Nature Communications (2023)
-
An injectable subcutaneous colon-specific immune niche for the treatment of ulcerative colitis
Nature Biomedical Engineering (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
