
Abstract
Immunological memory is a cardinal feature of adaptive immunity and an important goal of vaccination strategies. Here we highlight advances in the understanding of the diverse T lymphocyte subsets that provide acute and long-term protection from infection. These include new insights into the transcription factors, and the upstream 'pioneering' factors that regulate their accessibility to key sites of gene regulation, as well as metabolic regulators that contribute to the differentiation of effector and memory subsets; ontogeny and defining characteristics of tissue-resident memory lymphocytes; and origins of the remarkable heterogeneity exhibited by activated T cells. Collectively, these findings underscore progress in delineating the underlying pathways that control diversification in T cell responses but also reveal gaps in the knowledge, as well as the challenges that arise in the application of this knowledge to rationally elicit desired T cell responses through vaccination and immunotherapy.
Similar content being viewed by others

Main
Advances in the understanding of T lymphocyte memory have revealed the extraordinary diversification potential of adaptive immunity. Classic textbook definitions of immunological memory highlight the key properties of long-term remembrance of previous exposure to antigen as more rapid and robust responses upon re-exposure to antigen, due to the enhanced frequency of pathogen-specific cells and acquired functional properties. More specialized definitions of memory T cells often also include specific characteristics, such as antigen-independent persistence and self-renewal, which highlights an important conceptual difference between 'immunological memory' and a 'memory cell'. For many years it has been clear that memory T cells are not a single cell type but instead exhibit considerable heterogeneity from phenotypic, functional, anatomic and developmental perspectives. In particular, the developmental origins of memory T cells and the developmental relationships between different subsets of T cells have been among the more controversial concepts in the field. The answers to the questions of which signals and pathways give rise to distinct types of memory T cells are of central importance for the optimization of vaccine design and immunotherapies for cancer and other diseases. The goal of this Review is to summarize and contextualize findings describing the diversity of effector and memory T cells and the origins of this diversity. We will focus on the CD8+ T cell response but will also discuss various topics in the context of what is known about CD4+ T cells when relevant.
Heterogeneity of effector and memory lymphocyte subsets
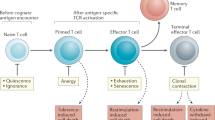
In response to pathogen infection, naive T lymphocytes undergo activation and proliferation, giving rise to progeny with effector and memory fates that are able to mediate immediate and long-term protection. In this Review we use the terms 'effector' and 'memory' to refer to antigen-experienced lymphocytes that are present before microbe clearance and long after microbe clearance, respectively. Such a broad, temporal definition acknowledges data showing that cells with memory potential arise during the acute phase of an immune response1,2 and that certain protective functions generally attributed to 'effector' cells, such as the secretion of inflammatory cytokines and cytolytic activity, are shared with certain subsets of memory T lymphocytes3.
Heterogeneity among memory lymphocytes in their surface-receptor expression, effector function, location and trafficking properties has long been recognized3,4, with the description of at least four distinct subsets of memory T lymphocytes: central memory T cells (TCM cells), effector memory T cells (TEM cells), tissue-resident memory T cells (TRM cells) and stem memory T cells ( Box 1 ). The effector and memory lymphocyte subsets are generally considered to be cellular 'fates,' while cells that are engaged in the process of differentiating toward one of these subsets are considered to be in transient 'states'. The term 'fate' suggests a lack of plasticity that is implicit in the term 'state.' However, it should be appreciated that there is evidence for interconversion between memory subsets5, and it remains unknown whether cells seemingly destined for death may retain the ability to change this outcome. Indeed, external influences, including the presence of inflammation, signaling via the T cell antigen receptor (TCR) and cytokines have been shown to be strong determinants of T lymphocyte differentiation6.
It is well appreciated that in addition to the heterogeneity of memory T cells, heterogeneity of cell fate among the progeny of activated T lymphocytes also exists in the effector T cell population. For CD4+ T lymphocytes, diversity among fate 'choices' includes various effector subsets, such as the helper T cell subsets TH1, TH2, TH17 and TH9, as well as follicular helper T cells (TFH cells) and induced regulatory T cells (Treg cells)7, each of which serves a specialized function and is adapted to counter a specific class of pathogen8 or to counterbalance excessive activation of the immune system. While CD8+ T lymphocytes may exhibit less functional diversity (i.e., type 2 or interleukin 17 (IL-17)-producing ('type 17') CD8+ T cells are not typically observed), heterogeneity in terms of other functional attributes and phenotypic markers has been increasingly recognized and may distinguish the efficacy of CD8+ T cell populations in distinct settings. Effector CD8+ T cells can exhibit heterogeneity in their ability to produce individual cytokines, such as interferon-γ (IFN-γ), tumor-necrosis factor (TNF) or interleukin 2 (IL-2), or β-chemokines or to co-produce multiple cytokines and chemokines, which correlates with improved protection against infection9 and enhanced anti-tumor immunity10. Additional heterogeneity may exist for CD8+ T cells with respect to newly synthesized perforin versus pre-formed perforin and/or coexpression of perforin and various granzymes11. Notably, functional profiling of CD8+ effector and memory populations is further complicated, as these may also depend on pathogen type as well. For example, the number and frequency of antigen-specific effector and memory CD8+ cell subsets that express the lectin-like receptor KLRG1 and the cytokine receptor IL-7R are vastly different in the response to different pathogens, such as Listeria monocytogenes or vesicular stomatitis virus, and this can be attributed to the unique inflammatory environment generated by each infection12. This pathogen-specific heterogeneity can have profound consequences; in a study using a prime-boost strategy to induce CD8+ T cell–mediated protection against lethal infection with influenza A virus, vaccinia virus expressing influenza virus nucleoprotein induced a protective CXCR3hi CD8+ memory population in the lungs, while L. monocytogenes expressing influenza virus nucleoprotein did not13. Thus, understanding the key influences that regulate the heterogeneity of effector and memory cells will greatly inform vaccine strategies.
Along with showing functional heterogeneity, effector CD8+ T lymphocytes exhibit nearly endless diversity in their expression of surface molecules that can serve as phenotypic markers of antigen-experienced cells14. A current challenge for the field is to determine which of these phenotypic markers, individually or in combination, represent indicators of cell 'fate' versus those that simply reflect cell 'state'. In other words, can the determinants of true lineage-defining events be distinguished from transiently expressed phenotypic markers that may simply reflect an individual T cell's exposure to environmental signals? In the effector CD8+ T cell population, for example, it is possible to identify, to some degree, individual lymphocytes that will be, or already have been, programmed to survive and differentiate into memory CD8+ T cells versus those that are terminal and are programmed to undergo apoptosis? CD8+ T lymphocytes with high expression of IL-7R and low expression of KLRG1 (IL-7RhiKLRG1lo) during acute infection have a tenfold greater likelihood of survival after infection is cleared than do their KLRG1hiIL-7Rlo counterparts, which leads to the conclusion that the IL-7Rhi effector population includes an intermediate stage of memory differentiation, while the KLRG1hi population represents terminally differentiated lymphocytes destined for death1. It should be appreciated, however, that these markers, while useful, do not fully capture the degree of heterogeneity of effector cells, as additional phenotypic and functional heterogeneity can be observed in both IL-7Rhi subsets and KLRG1hi subsets15. KLRG1hi cells are readily detectable after the resolution of infection16, and the receptors KLRG1 or IL-7R themselves are neither necessary nor sufficient for terminal differentiation or the development of memory lymphocytes17,18, respectively. Moreover, the IL-7Rhi subset seems to retain plasticity, while the KLRG1hi subset does not, because the former 'memory precursor' subset can also give rise to TRM cells or exhausted CD8+ T cells, while the KLRG1hi subset cannot19,20.
Phenotypic heterogeneity in the effector T cell population at the peak of the adaptive immune response raises the possibility that other molecules expressed even earlier might be used to predict or might influence the eventual fate of naive lymphocytes activated by infection. Indeed, CD8+ T lymphocytes with high expression of the transcriptional regulator Id3 on day 5 of pathogen infection exhibit a transcriptional program similar to that of long-lived memory cells21. Moreover, the cytokine receptor IL-2Rα can be an early determinant of T lymphocyte fate specification. As early as the second cell division, differences in the abundance of IL-2Rα expression in CD4+ T lymphocytes responding to infection influences the TH1 cell–versus–TFH cell fate 'choice' by controlling the relative expression of the transcription factors Blimp-1 and Bcl-6 (refs. 22,23). Similarly, differences in IL-2Rα expression on day 3 of pathogen infection24,25, and as early as the first cell division26, may influence the eventual fate of recently activated CD8+ T lymphocytes, in part through the differences in the induction of Blimp-1 (refs. 27,28). IL-2Rαhi CD8+ T cells are more likely to undergo apoptosis and IL-2Rαlo CD8+ T cells exhibit an increased likelihood to survive after clearance of infection, although it should be noted that IL-2 signals are dispensible for optimal memory development29. Together, these observations suggest that the signals that direct activated CD8+ T lymphocytes toward or away from the long-lived memory fate may not be absolute but instead may operate along a gradient, such that small amounts of a particular signal may promote or be permissive for one fate, while large amounts of the same signal could foster a different fate. Such a concentration gradient–based model of CD8+ T lymphocyte differentiation is consistent with other developmental models of fate determination30,31.
Lessons about T cell subset heterogeneity from single-cell studies
Various transcriptional profiling studies have driven many of the fundamental discoveries discussed in the preceding sections12,32,33. By comparing the gene expression of CD8+ T lymphocytes during the course of well-characterized infections, these studies have identified genes, groups of genes and pathways central to the specification of terminal differentiation versus long-lived memory. Notably, these studies have shown that resting, long-lived memory CD8+ or CD4+ populations share greater than 95% similarity in gene expression with their naive counterparts and with each other34. Furthermore, most genes whose expression is higher in memory cells than in naive cells are also expressed by effector CD8+ T cells, which leaves only a relatively short list of genes that are uniquely induced in memory cells12,32,33. Two meta-analyses using data from the Immunological Genome Project or from 386 publicly available gene-expression profiles comparing naive, effector and memory CD8+ T cells have focused on identifying transcriptional regulators of memory signature genes32,35. These studies have picked out many of the factors discussed above as being key to the generation of effector and memory cell populations yet have also identified a small number of transcriptional regulators not yet linked to memory cell formation; these studies provide the basis for future work32,35. However, so far, no lineage-defining genes have been established for long-lived memory T cells, which perhaps reinforces the idea that a complex transcriptional network rather than a 'master regulator' promotes the memory state or memory fate, or perhaps indicates the limits of current experimental strategies. The collective experimental data showing substantial heterogeneity among the progeny of activated T lymphocytes, moreover, may mask less dramatic but nonetheless key changes in gene expression or changes in expression in individual cells, which places some constraints on the interpretation of data generated by population-level profiling.
For initial investigation into the origins of such diversity, the following approaches have been used to probe the potential fate of cellular progeny derived from a single lymphocyte: adoptive transfer of a single T lymphocyte into a congenic recipient36,37; a limiting-dilution strategy in which polyclonal T lymphocytes are adoptively transferred into a congenic recipient to achieve the transfer of a single antigen-specific T cell38,39; and a 'DNA-barcoding' technology that enables the generation of naive T cells carrying unique genetic tags that allow them to be analyzed and distinguished40,41. Initial studies have demonstrated that a single, naive CD8+ T lymphocyte is able to give rise to both effector lymphocytes and memory lymphocytes36,40, providing evidence for a 'one naive cell, multiple fates' model of T lymphocyte diversification. Subsequent studies have generated additional insights, revealing that individual T lymphocytes exhibit highly disparate tendencies to yield effector progeny heterogeneous in their proliferative capacity, cytokine production and expression of phenotypic markers, including KLRG1 and IL-7R37,38,39,41. This heterogeneity is evident among polyclonal T lymphocytes bearing different TCRs as well as among T cells bearing identical TCRs38,39. Furthermore, adoptive transfer of a single TCM cell also gives rise to this heterogeneity42. Moreover, the degree of heterogeneity appears to be somewhat tissue specific, with effector lymphocytes in the intestinal lamina propria exhibiting less phenotypic diversity than splenic T cells following oral infection38. Finally, heterogeneity in the TH1 cell–versus–TFH cell fate 'choice' has been observed for CD4+ T lymphocytes responding to infection, which was influenced by TCR affinity and dwell times of complexes of peptide and major histocompatibility complex39.
Together, the findings reported above from multiple groups suggest that a single, activated T lymphocyte exhibits a remarkable capacity to yield heterogeneous progeny, but that individual lymphocytes do so with a high degree of variability. The precise mechanisms that control this variability remain unknown, but several non–mutually exclusive possibilities probably contribute to this. First, TCR avidity, peptide–major histocompatibility complex dwell times and subsequent strength of TCR signal can influence the type of progeny derived from a single T lymphocyte39; other factors that influence strength of signal, such as costimulation, probably also contribute to this (Fig. 1a). Second, differences in the microenvironments encountered by the progeny of an individual activated lymphocyte, owing to the type of infection, antigen abundance, cytokine milieu, site of infection and even region in the same spleen or lymph node, can affect eventual fate 'choices'6. Third, differences in gene expression in otherwise identical naive T lymphocytes at the time of their activation, due to stochastic fluctuations43 or disparities in tonic TCR signaling44, might also contribute to this. Finally, the observation that single T cells have distinct tendencies to yield heterogeneous progeny does not argue for or against the possibility of asymmetric T lymphocyte division, whereby an activated T cell unequally partitions fate determinants during mitosis, resulting in daughter cells that exhibit a predisposition toward disparate fates from their inception45,46. Indeed, the type of division (asymmetric or symmetric) that a recently activated T lymphocyte undergoes during an immune response, controlled by TCR affinity47 and expression of adhesion molecules45, may represent yet another mechanism that can shape the heterogeneity of cell fates that arise.

(a) Many disparate factors can serve to influence cell-fate specification in the first 24–72 hours following T cell activation; these include TCR signal strength, costimulation, inflammatory cytokines, tissue microenvironment, metabolic regulators and intermediates, 'pioneering' and transcription factors and the mode of cellular division (symmetric or asymmetric). It should be emphasized that these inputs are not mutually exclusive and act simultaneously in concert, yielding heterogeneous progeny that continue to integrate cumulative signals. APC, antigen-presenting cell. (b) Categories (top) and prominent examples of each category (below) of factors that can promote 'effector-like' or 'memory-like' differentiation (left margin). FAO, fatty acid oxidation; SRC, spare respiratory capacity; OxPhos, oxidative phosphorylation.
Another key question raised by the single-cell adoptive-transfer studies discussed above, particularly since heterogeneous progeny were studied 7–10 days after initiation of the immune response, is the timing with which daughter cells of a single T lymphocyte begin to adopt distinct differentiation states. While genomic profiling studies have examined lymphocytes before the peak of the adaptive immune response32, these data are based on analyses of bulk cellular populations and thus do not distinguish the gene-expression patterns of individual T cells. Advances that couple microfluidics with quantitative RT-PCR, however, enable the use of single-cell transcriptional profiling approaches in studying diverse biological processes, including embryonic development48 and induced pluripotent stem-cell differentiation49, and have revealed heterogeneity observed only at the single-cell level. A study of the differentiation of CD8+ T lymphocytes using a similar single-cell analysis approach has revealed transcriptional heterogeneity as early as the first cellular division in vivo and has discerned gene-expression signatures predictive of the eventual fate of the daughter cells26. Early induction of expression of the genes encoding the chemokine receptor CXCR3 and the chemokine CCL5 was predictive of the long-lived memory fate, whereas early expression of the genes encoding IL-2Rα, the lectin Gal-1 and the transcriptional repressor Zeb2 and downregulation of expression of the genes encoding the transcription factor TCF-7 and L-selectin were predictive of the terminally differentiated effector fate. The presence of distinct transcriptional patterns in lymphocytes that have undergone their first division suggests an early diversification of T lymphocyte fates. Collectively, these studies make a compelling argument for the integration of single-cell adoptive-transfer and single-cell analysis approaches in future studies of T lymphocyte fate specification.
Transcriptional and epigenetic regulation of differentiation
An area that has received considerable attention in the past few years is the identification of transcription factors that regulate the differentiation of effector and memory T cells. Indeed, a growing list of transcription factors has been linked to various aspects of effector and/or memory T cell biology (Table 1). Each of these transcription factors has potentially important effects on gene expression in effector and/or memory T cells, imparting specialized functional activity. An important conceptual theme that has emerged from these studies, moreover, is the idea that pairs of transcription factors operate in opposing ways to facilitate the terminal-effector–versus–memory CD8+ T lymphocyte fates (Fig. 1b). For example, large amounts of the transcription factor T-bet foster the differentiation of effector CD8+ T cells toward the terminally differentiated KLRG1hi fate15,50. Notably, the amount of T-bet expressed can be positively regulated by inflammatory signals such as IL-12 (ref. 15), which provides a direct link between the amount of inflammation present and further expansion of the pool of terminal-effector cells51,52. This link between inflammation and T-bet provides a parsimonious way to connect the potential severity of infection to the need for more effector cells to fight the pathogen. Genetic deletion of T-bet essentially abrogates formation of the KLRG1hi subset of effector CD8+ T cells with little effect on the IL-7Rhi subset15. The transcription factor Eomes is highly homologous to T-bet, especially in the DNA-binding domain, but seems to foster the development of memory CD8+ T cells rather than KLRG1hi effector CD8+ T cells53,54. Genetic deletion of Eomes has a relatively modest effect on the effector pool but results in memory attrition55 and failure to generate self-renewing TCM cells56. Thus, this related pair of transcription factors operates to 'preferentially' foster the differentiation of terminal effector cells versus TCM cells. Notably, the amount of T-bet and Eomes is key, and a gradient effect clearly exists in which increasing amounts of T-bet foster increasing differentiation into terminal effector cells15. This counterbalance also applies to the transcription factor pair Id2 and Id3 (refs. 21,57) and the pair Blimp-1 and Bcl-6 (refs. 27,28,58,59) and perhaps other pairs.
While the paradigm outlined above has been delineated mainly from studies of CD8+ T cells, these same transcription factors also seem to influence CD4+ T cell memory. Indeed, the pair Blimp-1 and Bcl-6 has been reported to influence effector versus memory subsets of CD4+ T cells27,28,58. The association of Bcl-6 with CD4+ T cell memory also has important potential implications, given the key role of this transcription factor in the biology of TFH cells60,61,62. Indeed, one key distinction between the biology of CD8+ T cells and that of CD4+ T cells is that the transcription factors associated with different functional lineages of CD4+ T cells, often called 'master regulators', are known, as follows: T-bet for TH1 cells, GATA-3 for TH2 cells, RORγt for TH17 cells, and Bcl-6 for TFH cells8. It remains unclear whether there are two layers of transcriptional control in CD4+ T cells that specify effector fate versus memory fate as well as functional specialization. In other words, is there a Blimp-1+GATA-3+ effector TH2 cell population and a Bcl-6+GATA-3+ memory TH2 cell population? Or do T-bet, GATA-3 and RORγt also impart information about effector and memory fate for CD4+ T cells? While studies have defined phenotypic and functional subsets of effector CD4+ T cells that correlate with distinct effector and memory fates, understanding of the transcriptional control of CD4+ T cell memory generally lags behind understanding of such control of CD4+ T cell functional specification.
The classic notion that a single transcription factor or pair of transcription factors controls gene expression is clearly an oversimplification. It is well appreciated that key transcription factors such as those described above often operate with numerous locally bound cofactors and in partnership with more distally bound transcriptional regulators63,64. A published study has systematically mapped combinatorial interactions among known transcription factors and has correlated this with tissue-specific expression; these data have led to the estimation that tissue-type specification would be accomplished by networks of approximately 15 transcription factors, many of which are broadly expressed 'facilitators' of gene expression65. This favors the conclusion that cell specification is accomplished not by specific expression of single master regulators but by cell type–restricted interactions among a network of transcription factors expressed uniquely in combination. In other words, a particular transcription factor may be expressed in multiple tissues, but only the coexpression and colocalization of a specific set of transcription factors in a given tissue enables their interaction and the specification of a unique fate. This requires a broader look at the transcription factors discussed above and a more complete description of the combination of factors that regulate gene expression in CD4+ or CD8+ effector cells for full understanding of how memory lineages are specified. To begin to define some of this complexity in the control of gene expression and differentiation, several network-based approaches have been applied to the elucidation of T cell differentiation32,66,67,68,69,70. These studies have provided considerable insight in the types of regulatory programs used by T cells during effector and memory differentiation and have also revealed central 'hub' genes and pathways that are probably pivotal in fate determination and/or characteristic properties of different types of T cells.
It is tempting to associate individual transcription factors that control a key function of a cell type with the developmental control of a subset of cells with that function. For example, T-bet has long been thought to be the lineage-defining transcription factor for TH1 cells, with similar inferences made about GATA-3 and TH2 cells8. Work from several groups using network-based and epigenetic approaches has prompted a reevaluation of this concept, which has highlighted the importance of the relative accessibility of key regulatory elements such as enhancers in establishing cell lineage characteristics. A study examining the epigenetic 'landscape' of TH1 and TH2 cells has identified key lineage-specific open enhancer regions by defining chromatin accessibility and binding of the histone acetyltransferase p300 (ref. 71) and has observed that the enhancer 'landscape' of TH1 cells or TH2 cells is not substantially perturbed by deletion of T-bet or GATA-3, respectively, although transcription of mRNA encoding the lineage-defining cytokines is reduced in cells deficient in T-bet and GATA-3. These data are consistent with an emerging theme also observed for macrophages and other cell types72 that it is the epigenetic enhancer 'landscape', rather than individual 'effector transcription factors', that more accurately defines cell fate. The data indicate that while transcription factors such as T-bet and GATA-3 control expression of some of the key genes associated with TH1 and TH2 cells, the fundamental identity of these lineages can be established by other transcriptional events, which suggests an upstream 'pioneering factor' that regulates accessibility of the entire transcriptional network to key sites of gene regulation (Fig. 2).

(a) 'Pioneer' transcription factors enter closed chromatin at enhancers and/or promoters of key lineage-specific genes. They displace histones and/or recruit histone-modifying enzymes (such as histone acetyltransferases (HAT)) and other transcription factors. 'Pioneer' factors can be either individual transcription factors or complexes. Epigenetically open chromatin at enhancers and promoters then can be acted on by 'effector' transcription factors that actively turn gene transcription on and off (in cooperation with other core or generic transcriptional control proteins). Ac, acetylation. (b) In the absence of 'pioneer' factor function, an epigenetic 'landscape' of accessibility of lineage-specific genes is not established. Effector transcription factors, even if expressed, cannot access lineage-specific genes to regulate expression.
Indeed, studies of CD4+ T cells66,73,74 and CD8+ T cells75 indicate that the transcription factors BATF and IRF4 might serve the 'pioneering' role noted above, although true biochemical evidence of 'pioneering' activity to displace nucleosomes and initiate chromatin opening does not yet exist. Loss of either BATF75,76,77 or IRF4 (refs. 76,78,79,80,81) greatly perturbs the early phases of the CD8+ T cell immune response, which results in collapse of the effector phase after initial proliferation. Interestingly, this is accompanied by disrupted regulation of a large percentage of genes associated with CD8+ T cell activation, including genes encoding key transcription factors and molecules that control metabolism, as well as molecules associated with effector function75. BATF and IRF4 have largely overlapping gene targets, and the binding of each to genes appears to be dependent on the other in the context of CD4+ TH2 cells and TH17 cells, dendritic cells and B cells74,82,83, which suggests they function as a complex. In various lymphocyte lineages, BATF and IRF4 are fundamental in establishing the proper expression of many genes induced by transcription factors, such as T-bet, that drive the effector programs. Although these initial studies provide an important step forward, additional work to establish the 'pioneering' function of BATF-IRF4 in regulating chromatin accessibility during the CD8+ T cell response is needed to elucidate how the activation of a naive T cell establishes a chromatin 'landscape' that reveals appropriate regulatory regions that allow stimulus-driven transcription factors to promote lineage-specific gene expression. As discussed above, there is robust evidence that cells with strong memory potential are established very early in the immune response; how this may be influenced by early modifications to gene accessibility is a key question.
An additional challenge in linking the role of individual transcription factors to the specification of memory and effector lineages is the fact that in comparisons of the expression of many of these transcription factors, the differences between the terminal KLRG1hi population and the IL-7Rhi population that contains the memory cell precursors are rather small. For example, for T-bet expression, the difference between these two populations at the peak of the immune response is approximately twofold15, which illustrates again that cell-fate specification probably involves more complexity and/or network interactions than can be orchestrated by a single transcription factor. Thus, an important component of understanding how heterogeneity arises in the effector population and contributes to memory formation will probably include a description of the enhancers and regulatory regions of different CD8+ T cell subsets, as well as their accessibility at different stages of the immune response, and identification of the many factors that bind those regions. Although there are excellent studies that have examined epigenetic changes at individual genes in effector T cells versus memory T cells84,85,86 and a handful of landmark surveys of the overall epigenetic 'landscape' of effector and memory cells87, understanding of epigenetics in T cell memory remains incomplete. Thus, a major challenge for the field will be to determine how to integrate this information with data on the function of transcription factors such as those described above, and microRNAs and long noncoding RNAs, to generate comprehensive understanding of the events that determine heterogeneous lymphocyte fates.
Memory in situ
Much of the understanding of the gene-expression programs that govern the formation and function of memory populations has been gleaned from studies of recirculating populations of TEM cells and TCM cells, in which TEM cells were assumed to represent memory populations that provide frontline defenses in tissues should reinfection occur. However, subsequent studies have highlighted the fact that in most tissues, a prominent population of long-lived CD8+ T cells that is distinct from the TEM cell populations and does not recirculate is seeded after infection88,89,90,91,92. These tissue-resident memory cells are poised at sites of potential reinfection, such as the intestinal, genital and respiratory mucosa and skin, and coordinate the initial response to pathogens, providing a substantial boost to immunity in the tissue by directly recognizing antigen, recruiting circulating memory cells and diminishing pathogen load in the earliest phase of infection88,89,90. Indeed, published work indicates that these TRM cells may even supplant, or at least augment, innate immunity in the speed with which they recognize and respond to new infections. The activation of TRM cells by antigen and subsequent production of IFN-γ can induce local expression of the integrin ligand VCAM-1 by vascular epithelium, which promotes the trafficking of leukocytes to the inflamed tissue, including the recruitment of B cells and recirculating memory T cells, which rapidly escalates the local response90,93. Further, the production of TNF and IL-2 by TRM cells enhances the maturation of dendritic cells and activation of natural killer cells, respectively93, which demonstrates the broad effect of this small memory population. The activation of TRM cells and IFN-γ production can also induce a gene-expression program indicative of an 'anti-pathogen' response by local tissue cells94, and such triggering of TRM cells can provide local protection from subsequent viral infection independently of antigen recognition93. While it is difficult to generalize, as TRM cells have been studied in a range of tissues and infections that probably have idiosyncratic differences, a model for the differentiation of CD8+ TRM cells is emerging nonetheless. To summarize, in a primary immune response, TRM cells arise early when peripheral tissues are seeded from the pool of recently activated T cells that gain access to the tissues due to altered expression of chemokine receptors, selectins and/or integrins20,94,95. Some of these T cells then take up permanent residence in these nonlymphoid tissues and further differentiate into TRM cells in situ20,88,95,96,97. Interestingly, in one experimental system it was the KLRG1lo subset of effector CD8+ T cells that gave rise to TRM cells98, which suggests that these cells may have the developmental plasticity to give rise to distinct types of memory CD8+ T cells. While TRM cells may accumulate most substantially near the site of original infection, enhanced protection against vaccinia virus, lymphocytic choriomeningitis virus and herpes simplex virus at distant sites indicate that broad dissemination of these cells in the tissues provides enhanced protection systemically88,89,90. Much less is known about tissue-resident populations of CD4+ T cells, which are not as abundant and show localization different from that of CD8+ TRM cells95, although studies suggest that CD4+ TRM cells provide enhanced protection from lethal infection with herpes virus and may be maintained by interactions with tissue macrophages99.
It is thought that signals induced by such cytokines as TGF-β, IL-15, IL-33 and TNF in T cells entering the tissues can induce acquisition of the TRM cell phenotype, with high expression of the activation marker CD69, high expression of the integrin CD103 (αEβ7) in some tissues, and low expression of the cytokine receptor CD122 (IL-2Rβ), the activation marker CD62L, the transcription factor KLF2, the chemokine receptor CCR7, the surface marker Ly6C and the receptor for sphingosine 1-phosphate (S1P1)), as well as functional characteristics (elevated granzyme B and cytokine production). TGF-β has a key role in establishing and sustaining CD69+CD103+ TRM cells in a range of tissues by helping to induce the expression of CD103 and integrins that mediate retention96,98,100. Deficiency in the gene encoding CD69, CD103, the receptor for TGF-β or IL-15 substantially impairs the generation of a tissue-resident memory population and/or the ability to sustain such a population. Interestingly, TGF-β produced by Treg cells supports the accumulation of CD103+ TRM cells in the central nervous system during infection with West Nile virus, which suggests an important interaction between Treg cells and tissue-resident memory cells. In addition to promoting the expression of key molecules, TGF-β, along with IL-33 and TNF, causes the downregulation of the gene encoding KLF2 and thus of its target S1P1 (ref. 101). Forced expression of S1P1 by CD8+ T cells prevents the establishment of tissue-resident cells. Thus, entry into the tissues can trigger a switch whereby cells downregulate the receptor that would promote their return to circulation and induce the TRM cell phenotype.
There is still much to be learned about how the TRM cell population is established and sustained and how these mechanisms may relate to the pathways that control the differentiation of recirculating memory populations. The general assumption that memory T cells in the tissues are terminally differentiated and derive from TEM cells has not been borne out by studies of TRM cells; during the effector phase, TRM cells differentiate from KLRG1lo cells once in the skin98, and TRM cells harvested from gut epithelium can give rise to both recirculating cell populations and TRM cell populations following rechallenge102, which suggests plasticity in commitment to the memory subsets and reinforcement of TRM cell characteristics by tissue-derived signals. Consistent with the proposal that tissue-specific signals drive the specification of TRM cells, microarray analysis has revealed that the gene-expression signatures of TRM cells derived from brain, gut and skin are highly related, sharing a common core signature distinct from those of other resident populations and recirculating populations derived from the same tissues or spleen (TEM cells and TCM cells)98,103. Interestingly, expression of the genes encoding Eomes and TCF7, both associated with long-lived memory T cell populations in the spleen and lymph nodes, is substantially lower in TRM cells than in circulating memory populations. However, TRM cells do not have high expression of the genes encoding Blimp-1, SIP1 or T-bet, as might be expected of terminally differentiated cells. Thus, TRM cells do represent a unique, long-lived antigen-experienced population that shows evidence of a distinct gene-expression program. Understanding which specific microenvironmental signals influence the specification and longevity of this population is a key priority, as these cells are exposed to unique metabolic cues, nutrient and O2 levels and inflammatory signals compared with those to which recirculating memory T cell populations are exposed.
Metabolic state and memory T lymphocyte differentiation
The integration of signals mediated by TCRs, costimulatory molecules and cytokines initiates and guides the population expansion and differentiation of naive T cells into effector and memory populations104,105, both of which have substantial functional and phenotypic heterogeneity. One of the more remarkable aspects of T cell responses following infection or vaccination is that quiescent naive T cells transition to a state of extraordinarily rapid proliferation and increase in biomass. After a lag of 24–36 hours following their initial activation, CD8+ T cells can divide at least every 4–8 hours for the next 5–7 days, which leads to an increase in cell number of 100- to 10,000-fold. The single-cell studies described above36,40 indicate that because only a fraction of the naive pool gives rise to the numerical majority of the resulting effector pool, these division rates may in fact be underestimates. The extensive proliferation and production of effector molecules must be accompanied by substantial alterations in metabolism, including major changes in how energy is generated and the production of biomolecules that support cell growth. The metabolic pathways and changes that accompany activation, as well as the role of the mTOR, Akt and PI(3)K kinase pathways that affect these changes, have been reviewed in detail104,106,107,108. Here we will focus on how these changes may influence memory-cell differentiation. Naive T cells are presumed to require basal levels of energy to support their survival, which are thought to be derived from pyruvate oxidation and mitochondria-dependent fatty acid β-oxidation, in which the catabolism of fatty acids into acetyl-CoA can be used to fuel oxidative phosphorylation and drive ATP production109,110. 'Oxidative phosphorylation' refers to the mitochondrial generation of ATP by the oxidation of metabolic intermediates (NADH and FADH2 generated from acetyl-CoA in the tricarboxylic acid (TCA) cycle), which establishes a proton gradient in the mitochondrial matrix and drives ATP synthesis. Within the first 24 hours following T cell activation, a rapid increase in glycolysis and glutamine oxidation driven by c-Myc-dependent metabolic reprogramming occurs, at least in part due to upregulation of numerous genes encoding molecules necessary for these processes109,110. At later time points, activity of the transcriptional regulator HIF ('hypoxia inducible factor') downstream of mTOR may have a role in promoting the glycolytic program105,111. In the effector phase, aerobic glycolysis becomes the more prominent metabolic pathway, although effector cells also continue to derive energy from oxidative phosphorylation. As the immune response wanes, antigen-specific cells either die or return to a quiescent state and adopt a metabolic profile more similar to that of naive cells, whereby oxidative phosphorylation is the dominant pathway that meets the attenuated energy requirements of the memory T cell population108,110,112. Several lines of experimentation have shown that sustained glycolytic activity can restrain the formation of memory cells, while inhibition of glycolysis can alter T cell effector functions and may promote the development of memory cells113. These data have inspired a new appreciation for the central importance of metabolic programming in regulating differentiation and the generation of the immune response (Fig. 3). There is accumulating evidence that changes in metabolic status and activity do not merely affect the bioenergetic and biosynthetic needs of T cells but may also influence gene expression, thereby integrating the outcomes of cellular growth with function and cell fate.

mTOR is activated by environmental cues and signaling via receptors (TCRs, costimulatory receptors and cytokine receptors) in T cells via kinase-dependent pathways, including PI(3)K-Akt and PDK1. mTOR regulates cellular growth, survival and metabolism via multiple mechanisms, including the induction of glycolysis through the stabilization of HIF-1α, as well as lipid and protein biosynthesis. mTOR promotes translation initiation and protein synthesis via phosphorylation of ribosomal protein S6 kinase and eIF4E-binding proteins. The AMPK complex is activated when cellular energy levels decrease (ATP:AMP) and suppresses cell growth by blocking biosynthetic pathways and inhibiting mTOR. AMPK can induce fatty acid oxidation and suppress glycolysis. mTOR can induce activity of HIF, a transcription factor that coordinates the cellular response to low oxygen tension, including induction of the expression of many molecules required for glycolysis. HIF is a heterodimeric complex of either HIF-1α or HIF-2α with HIF-1β, which is constitutively expressed. The stability of HIFα protein is post-transcriptionally regulated by oxygen availability via iron-dependent PHDs, which tag HIF for recognition by ubiquitination dependent on the von Hippel-Lindau tumor suppressor VHL. The stabilization of HIF mRNA and protein in cells of the immune system can be induced by hypoxia as well as by signaling via TCRs, cytokine receptors and Toll-like receptors, whereby it can promote (both directly and indirectly) effector functions, including microbicidal activity by macrophages, cytokine production by TH17 cells, and expression of effector molecules such as IFN-γ and granzyme B. As an example of a metabolic enzyme that can also function as an RNA-binding protein and regulate mRNA translation, GAPDH has been shown to regulate the translation of mRNA encoding effector molecules such as IFN-γ. GAPDH is engaged as a metabolic enzyme during glycolysis; however, when not engaged in glycolysis and when the cell generates ATP via oxidative phosphorylation, GAPDH can bind the 3′ untranslated region of cytokine-encoding mRNA and diminish translation. Illustrative of how broadly molecules generated by different metabolic pathways may affect gene expression is the activity of products of the TCA cycle. For example, accumulation of α-ketoglutarate, succinate and fumarate produced as part of the TCA cycle can lead to product-mediated inhibition of PHDs and HIF activation and also affect epigenetic modifications of histones and DNA, including decreased methylation. Acetyl-CoA can provide donor acetyl groups that facilitate histone acetylation.
Two key observations brought the study of metabolism to the forefront in the discussion of memory T cell differentiation114,115. Inhibition of mTOR by treatment with rapamycin and activation of AMPK by treatment with metformin, an inhibitor of mTOR that promotes fatty acid catabolism, promotes the formation of memory cells and 'rescues' the impaired survival of T cells deficient in the adaptor TRAF6 (refs. 114,115). These observations suggested the exciting possibility that the differentiation of lymphocytes into memory cells can be pharmacologically manipulated and sparked a series of studies supporting the idea that a key aspect of the differentiation of CD8+ T cells into memory cells is the switch from glycolytic metabolism to fatty acid oxidation. Memory CD8+ T cells show greater oxygen consumption, a surrogate of oxidative phosphorylation, than that of naive or effector cells, but even more notable, they have a greater maximal capacity for oxygen consumption or mitochondrial spare respiratory capacity, an indication of their ability to generate energy under stress116. This enhanced spare respiratory capacity of memory CD8+ T cells has been shown to be due to mitochondrial fatty acid oxidation and has been hypothesized to support the survival of memory CD8+ T cells and their ability to respond rapidly upon reinfection116,117. IL-15, which is known to support the survival and homeostasis of memory CD4+ or CD8+ T cells, promotes mitochondrial biogenesis. Similarly, overexpression of the carnitine palmitoyl transferase CPT1, a rate-limiting component of mitochondrial fatty acid oxidation, also favors the accumulation of memory CD8+ T cells116. Additional evidence that the generation of memory T cells is dependent on AMPK-induced oxidative phosphorylation has been provided by the study of AMPK-deficient CD8+ T cells, which mount a normal primary response but, after a second exposure to antigen, are defective in their ability to undergo population expansion compared with that of wild-type cells118. Such studies draw a strong correlation between memory differentiation and the reliance of CD8+ T cells on mitochondrial oxidative phosphorylation to supply cellular energy and indicate that this is a driving and perhaps required aspect of memory T cell formation.
Complementary studies have shown that manipulation of the effector CD8+ T cell metabolic profile also affects the differentiation of effector and memory subsets during the CD8+ T cell response. While constitutive activation of glycolysis by forced expression of the glycolytic enzyme PGAM1 limits long-term survival following infection, inhibition of glycolysis with 2-deoxyglucose during activation promotes memory-like gene expression and the accumulation of CD8+ T cells that can respond to infection or protect against tumors119. Effector CD8+ T cells with elevated HIF activity, due to genetic deletion of its negative regulator VHL, also show dramatically enhanced glycolytic activity and effector function, accompanied by a sustained effector state even in chronic infection, during which wild-type cells undergo progressive exhaustion120. Inhibition of glycolysis with 2-deoxyglucose or mTOR activity can impair the production of effector molecules (IFN-γ, granzyme B and perforin) and suppress many of the HIF-mediated changes in expression111,120,121,122. Bcl-6, a memory T cell–associated transcription factor, has been shown to repress glycolytic molecule–encoding genes known to be targets of HIF in CD4+ or CD8+ effector cells34, which provides a transcriptional link to the metabolic changes observed during memory formation. Thus, in multiple contexts, promoting mitochondrial fatty acid oxidation or suppressing glycolysis favors the emergence of antigen-experienced CD8+ T cells with prolonged survival and antigen recall; conversely, driving glycolysis supports a more effector-like fate.
Such observations have led to the hypothesis that the generation of memory CD8+ T cells requires metabolic reprogramming to mitochondrial fatty acid oxidation and that this further supports the rapid recall response of memory cells116. However, achieving a mechanistic understanding of how metabolism may direct differentiation promises to be a complex task. As metabolic activity is integrally linked to proliferation, survival and biomolecule synthesis, manipulating aspects of glycolysis or fatty acid oxidation probably affects these fundamental cellular processes known to influence memory formation and may further affect different effector and memory T cell populations to varying degrees. However, multiple lines of evidence have shown that the metabolic state can directly affect gene expression and influence signaling pathways that control T cell fate108,110. For example, the availability of AMP or ATP in the cell can alter the phosphorylation of a range of molecules and promote the activity of AMPK, which in turn can inhibit mTOR and diminish cap-dependent mRNA translation due to its effect on the translation-initiation inhibitor 4E-BP1, the guanine nucleotide–exchange factor eIF2B and S6 kinase. Further, metabolites generated via the TCA cycle can act in a range of contexts that may modify gene expression, including altering the activity of prolyl-hydroxylases (PHDs) (and thus HIF activity), DNA hydroxylases (and thus DNA methylation) and histone demethylases (and thus histone methylation)108. While many potential mechanisms may be involved, an example of such regulation involves 2-oxoglutarate, which is an intermediate of the TCA cycle and a cofactor for histone demethylases that contain a JmjC binding domain, TET 5-methylcytosine hydroxylases and PHD/Eg1N prolyl-4-hydroxylases. Levels of 2-oxoglutarate can thus affect the epigenetic regulation of gene expression and PHD-HIF activity. Notably, mutations have been discovered in the gene encoding isocytrate dehydrogenase that lead to production of the 'oncometabolite' 2-hydroxyglutarate in place of 2-oxoglutarate; this leads to changes in the activity of JmjC, TET and PHDs, which suggests a direct link between altered metabolic intermediates in this pathway, histone demethylation and cell differentiation or transformation123,124. Furthermore, acetyl-CoA, which is derived from the TCA cycle and is important for lipid biosynthesis, can also provide donor acetyl groups and influence the acetylation of proteins mediated by histone acetyltransferases, which links growth factor–induced metabolism to the regulation of gene expression to support cell growth125. The observation that glycolysis promotes IFN-γ production by CD4+ T cells in part by diverting the glycolytic enzyme GAPDH from its binding to the 3′ untranslated region of IFN-γ mRNA further shows the complexity that arises from the repurposing of the enzymes and intermediates of metabolic processes to post-transcriptional regulatory functions126.
At present, the metabolic regulation of CD4+ T cell lineages is understood mainly in the context of effector helper T cell lineages105. The TH1, TH2 and TH17 cell lineages show upregulation of expression of the glucose transporter GLUT1 and glycolysis upon activation to a greater degree than do Treg cells, which display enhanced lipid oxidation127,128. Conditions that favor mitochondrial respiration, such as the addition of fatty acids, rapamycin or metformin, apparently favor the generation of Treg cells and, similarly, inhibition of glycolysis blocks the generation of effector helper T cells but not that of Treg cell populations. Notably, for differentiating TH17 cells, the induction of HIF-1α-driven glycolysis and expression of RORγt by mTOR is essential for the proper induction of this population128,129. Deficiency in the gene encoding mTOR also impairs the generation of TH1, TH2 and TH17 cell lineages and can divert cells to the Treg cell lineage, which is consistent with the promotion of glycolytic flux and effector function by mTOR130. However, there is minimal information that addresses how the metabolic programming of effector CD4+ T cell lineages may converge with the differentiation of memory CD4+ T cells; clarity on this topic will require a better definition of the precursors of memory CD4+ T cells and the ability to monitor these subsets in vivo.
For a field that has viewed cell fate 'decisions' mainly through the lens of gene expression, it is evident that a new perspective must evolve that considers metabolic state and metabolite levels, as well as the epigenetic 'landscape', which may influence T cell differentiation and memory formation via diverse mechanisms. As the field moves to integrate the existing understanding of cell fate determination as controlled by signaling pathways mediated by antigen receptors, cytokine receptors and costimulatory molecules with the metabolic pathways that underpin all aspects of cell viability, it will probably be crucial to study T cells in situ and ex vivo as much as possible, which will allow more accurate determination of what does happen rather than what can happen. Furthermore, parsing the degree of proliferation or survival observed in a condition that manipulates metabolic activity from the decreasing potential model of memory formation can be problematic; enhanced TCR or inflammatory signals promote greater proliferation and differentiation, which require a greater supply of energy and materials. Thus, correlating more glycolytic activity with effector cells and, conversely, more oxidative phosphorylation with memory cells makes sense. The pressing question of the moment is as follows: to what degree do these drive the process of memory formation?
Concluding thoughts
As a productive response to infection or malignancy is forged, T cells traffic from sites of priming in secondary lymphoid organs through a broad range of tissue microenvironments where they mediate the response to infection, experiencing a range of oxygen tensions, nutrients, inflammatory mediators, antigen load and survival factors. For example, activated effector CD8+ T cells home to infected tissues, where they are exposed to antigen presented by infected cells, hypoxia and nutrient deprivation as the result of tissue damage, as well as cytokines such as TNF and IFN-γ produced by neighboring cells of the immune system. A T cell programmed for functional activity in the draining lymph node must integrate these environmental cues into the gene-expression networks that regulate effector capacity and differentiation. Thus, hypoxia and TCR-mediated signals may result in an elevated abundance of HIF, which supports glycolysis and effector function, while exposure to TGF-β, IL-33 and TNF as cells enter the tissue may suppress KLF2, which results in inhibition of inhibiting S1P1 and egress, and thus support the formation of TRM cells. Furthermore, the variable induction of signals that results from gradients of exposure due to the timing of entrance into the tissues and position relative to that of other inflammatory cells and infection must be overlaid with the cumulative set of signals received during priming. Analyzing the complex inputs that influence the bias toward terminal differentiation versus longer-lived T cell fates on a population level may be misleading, yet doing so with single-cell resolution currently remains a challenge. The ability to 'read out' transcriptional activity, the activation of signaling pathways, gene expression and chromatin modifications with systems that report specific signaling pathways and transcriptional activity by individual cells and deep sequencing from small numbers of cells is increasingly possible. Applying such techniques in conjunction with approaches for evaluating metabolic activity and function in vivo will be necessary to elucidate the events that determine lymphocyte fate and reveal the origins of the tremendous heterogeneity in phenotype and function observed in effector and memory T cell populations. Identifying the key memory subsets that contribute to providing optimal protection for specific pathogens acquired through distinct routes and sites of infection will be a crucial challenge.
It is an exciting time for immunology, with the introduction of some of the most powerful drugs for the treatment of cancer based on immunomodulation. There has also been considerable promise in the areas of vaccination, treatments for autoimmunity and understanding, and in some cases, gene therapy–based reversal of primary immunodeficiencies. Advances in '-omics' approaches that allow the analysis of smaller numbers of cells and even single cells means it is now possible to directly investigate in humans many of the questions posed above. Indeed, it is possible to take advantage of numerous immunologically based interventions in humans to answer specific and mechanistic questions about T cell differentiation, memory and transcriptional programs or networks. Moreover, combination therapies designed to complement one treatment approach (i.e., checkpoint blockade in cancer) with a second, often mechanistically distinct therapeutic approach may offer novel opportunities for investigating how epigenetic or metabolic pathways intersect with immunoregulatory pathways to enhance rational immunomodulation in humans. Ultimately, a major goal should be to be able to capitalize on the molecular understanding of effector and memory T cell differentiation to specifically 'tune' vaccines and immunotherapies to elicit distinct memory T cell states and fates.
References
Kaech, S.M. et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4, 1191–1198 (2003). References 1 and 15 show that populations of effector CD8+ T cells expressing KLRG1 or IL-7R show enrichment for terminal effector cells or cells that will become memory lymphocytes, respectively.
Huster, K.M. et al. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc. Natl. Acad. Sci. USA 101, 5610–5615 (2004).
Sallusto, F., Lenig, D., Forster, R., Lipp, M. & Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999).
Hamann, D. et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 186, 1407–1418 (1997). References 3 and 4 provide evidence for phenotypic and functional heterogeneity in memory T lymphocytes.
Wherry, E.J. et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4, 225–234 (2003).
Kaech, S.M. & Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12, 749–761 (2012).
Josefowicz, S.Z., Lu, L.F. & Rudensky, A.Y. Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 30, 531–564 (2012).
Zhu, J., Yamane, H. & Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 28, 445–489 (2010).
Seder, R.A., Darrah, P.A. & Roederer, M. T-cell quality in memory and protection: implications for vaccine design. Nat. Rev. Immunol. 8, 247–258 (2008).
Yuan, J. et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc. Natl. Acad. Sci. USA 105, 20410–20415 (2008).
Makedonas, G. et al. Perforin and IL-2 upregulation define qualitative differences among highly functional virus-specific human CD8 T cells. PLoS Pathog. 6, e1000798 (2010).
Obar, J.J. et al. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J. Immunol. 187, 4967–4978 (2011).
Slutter, B., Pewe, L.L., Kaech, S.M. & Harty, J.T. Lung airway-surveilling CXCR3hi memory CD8+ T cells are critical for protection against influenza A virus. Immunity 39, 939–948 (2013).
Newell, E.W., Sigal, N., Bendall, S.C., Nolan, G.P. & Davis, M.M. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity 36, 142–152 (2012).
Joshi, N.S. et al. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295 (2007).
Olson, J.A., McDonald-Hyman, C., Jameson, S.C. & Hamilton, S.E. Effector-like CD8+ T cells in the memory population mediate potent protective immunity. Immunity 38, 1250–1260 (2013).
Grundemann, C. et al. The NK receptor KLRG1 is dispensable for virus-induced NK and CD8+ T-cell differentiation and function in vivo. Eur. J. Immunol. 40, 1303–1314 (2010).
Goldrath, A.W. et al. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J. Exp. Med. 195, 1515–1522 (2002).
Angelosanto, J.M., Blackburn, S.D., Crawford, A. & Wherry, E.J. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol. 86, 8161–8170 (2012).
Carbone, F.R., Mackay, L.K., Heath, W.R. & Gebhardt, T. Distinct resident and recirculating memory T cell subsets in non-lymphoid tissues. Curr. Opin. Immunol. 25, 329–333 (2013).
Yang, C.Y. et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat. Immunol. 12, 1221–1229 (2011).
Choi, Y.S. et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34, 932–946 (2011).
Pepper, M., Pagan, A.J., Igyarto, B.Z., Taylor, J.J. & Jenkins, M.K. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity 35, 583–595 (2011).
Kalia, V. et al. Prolonged interleukin-2Rα expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 32, 91–103 (2010).
Pipkin, M.E. et al. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90 (2010).
Arsenio, J. et al. Early specification of CD8+ T lymphocyte fates during adaptive immunity revealed by single-cell gene-expression analyses. Nat. Immunol. 15, 365–372 (2014).
Kallies, A., Xin, A., Belz, G.T. & Nutt, S.L. Blimp-1 transcription factor is required for the differentiation of effector CD8+ T cells and memory responses. Immunity 31, 283–295 (2009).
Rutishauser, R.L. et al. Transcriptional repressor Blimp-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31, 296–308 (2009).
Williams, M.A., Tyznik, A.J. & Bevan, M.J. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature 441, 890–893 (2006).
Driever, W. & Nusslein-Volhard, C. The bicoid protein determines position in the Drosophila embryo in a concentration-dependent manner. Cell 54, 95–104 (1988).
Driever, W. & Nusslein-Volhard, C. A gradient of bicoid protein in Drosophila embryos. Cell 54, 83–93 (1988).
Best, J.A. et al. Transcriptional insights into the CD8+ T cell response to infection and memory T cell formation. Nat. Immunol. 14, 404–412 (2013).
Wherry, E.J. et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007).
Oestreich, K.J. et al. Bcl-6 directly represses the gene program of the glycolysis pathway. Nat. Immunol. 15, 957–964 (2014).
Hu, G. & Chen, J. A genome-wide regulatory network identifies key transcription factors for memory CD8+ T-cell development. Nat. Commun. 4, 1–14 (2013).
Stemberger, C. et al. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity 27, 985–997 (2007). References 36 and 40 show that a single activated CD8+ T cell can give rise to both effector T cells and memory T cells.
Buchholz, V.R. et al. Disparate individual fates compose robust CD8+ T cell immunity. Science 340, 630–635 (2013).
Plumlee, C.R., Sheridan, B.S., Cicek, B.B. & Lefrancois, L. Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity 39, 347–356 (2013).
Tubo, N.J. et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 153, 785–796 (2013).
Gerlach, C. et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J. Exp. Med. 207, 1235–1246 (2010).
Gerlach, C. et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science 340, 635–639 (2013).
Graef, P. et al. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8+ central memory T cells. Immunity 41, 116–126 (2014).
Levine, J.H., Lin, Y. & Elowitz, M.B. Functional roles of pulsing in genetic circuits. Science 342, 1193–1200 (2013).
Palmer, M.J., Mahajan, V.S., Chen, J., Irvine, D.J. & Lauffenburger, D.A. Signaling thresholds govern heterogeneity in IL-7-receptor-mediated responses of naive CD8+ T cells. Immunol. Cell Biol. 89, 581–594 (2011).
Chang, J.T. et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science 315, 1687–1691 (2007). This paper shows that an activated CD8+ T cell can undergo asymmetric division to give rise to the effector and memory fates.
Chang, J.T. et al. Asymmetric proteasome segregation as a mechanism for unequal partitioning of the transcription factor T-bet during T lymphocyte division. Immunity 34, 492–504 (2011).
King, C.G. et al. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity 37, 709–720 (2012).
Guo, G. et al. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev. Cell 18, 675–685 (2010).
Buganim, Y. et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150, 1209–1222 (2012).
Intlekofer, A.M. et al. Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J. Exp. Med. 204, 2015–2021 (2007).
Badovinac, V.P., Porter, B.B. & Harty, J.T. CD8+ T cell contraction is controlled by early inflammation. Nat. Immunol. 5, 809–817 (2004).
Badovinac, V.P., Messingham, K.A., Jabbari, A., Haring, J.S. & Harty, J.T. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat. Med. 11, 748–756 (2005).
Pearce, E.L. et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302, 1041–1043 (2003).
Intlekofer, A.M. et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 6, 1236–1244 (2005).
Banerjee, A. et al. Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol. 185, 4988–4992 (2010).
Paley, M.A. et al. Technical Advance: Fluorescent reporter reveals insights into eomesodermin biology in cytotoxic lymphocytes. J. Leukoc. Biol. 93, 307–315 (2013).
Cannarile, M.A. et al. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nat. Immunol. 7, 1317–1325 (2006).
Shin, H. et al. A role for the transcriptional repressor Blimp-1 in CD8+ T cell exhaustion during chronic viral infection. Immunity 31, 309–320 (2009).
Cui, W., Liu, Y., Weinstein, J.S., Craft, J. & Kaech, S.M. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity 35, 792–805 (2011).
Johnston, R.J. et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010 (2009).
Nurieva, R.I. et al. Bcl6 mediates the development of T follicular helper cells. Science 325, 1001–1005 (2009).
Yu, D. et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31, 457–468 (2009).
Tata, J.R. Signalling through nuclear receptors. Nat. Rev. Mol. Cell Biol. 3, 702–710 (2002).
Lam, M.T. et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498, 511–515 (2013).
Ravasi, T. et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 140, 744–752 (2010). This study indicates that cell fate specification may be achieved by tissue-restricted interactions among multiple transcription factors rather than by a single master regulator.
Ciofani, M. et al. A validated regulatory network for Th17 cell specification. Cell 151, 289–303 (2012).
Doering, T.A. et al. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37, 1130–1144 (2012).
Mingueneau, M. et al. The transcriptional landscape of αβ T cell differentiation. Nat. Immunol. 14, 619–632 (2013).
Schmidl, C. et al. The enhancer and promoter landscape of human regulatory and conventional T-cell subpopulations. Blood 123, e68–e78 (2014).
Shih, H.Y. et al. Transcriptional and epigenetic networks of helper T and innate lymphoid cells. Immunol. Rev. 26, 23–49 (2014).
Vahedi, G. et al. STATs shape the active enhancer landscape of T cell populations. Cell 151, 981–993 (2012). This paper suggests that it is the epigenetic enhancer landscape, rather than individual transcription factors, that specifies cell fate.
Ostuni, R. et al. Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171 (2013).
Li, P. et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546 (2012).
Glasmacher, E. et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 338, 975–980 (2012).
Kurachi, M. et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 15, 373–383 (2014).
Grusdat, M. et al. IRF4 and BATF are critical for CD8+ T-cell function following infection with LCMV. Cell Death Differ. 21, 1050–1060 (2014).
Kuroda, S. et al. Basic leucine zipper transcription factor, ATF-like (BATF) regulates epigenetically and energetically effector CD8 T-cell differentiation via Sirt1 expression. Proc. Natl. Acad. Sci. USA 108, 14885–14889 (2011).
Man, K. et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat. Immunol. 14, 1155–1165 (2013).
Nayar, R. et al. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J. Immunol. 192, 5881–5893 (2014).
Yao, S. et al. Interferon regulatory factor 4 sustains CD8+ T cell expansion and effector differentiation. Immunity 39, 833–845 (2013).
Raczkowski, F. et al. The transcription factor Interferon Regulatory Factor 4 is required for the generation of protective effector CD8+ T cells. Proc. Natl. Acad. Sci. USA 110, 15019–15024 (2013).
Tussiwand, R. et al. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 490, 502–507 (2012).
Murphy, T.L., Tussiwand, R. & Murphy, K.M. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat. Rev. Immunol. 13, 499–509 (2013).
Wei, G. et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 (2009).
Youngblood, B. et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity 35, 400–412 (2011).
Zediak, V.P., Johnnidis, J.B., Wherry, E.J. & Berger, S.L. Cutting edge: persistently open chromatin at effector gene loci in resting memory CD8+ T cells independent of transcriptional status. J. Immunol. 186, 2705–2709 (2011).
Wei, G. et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity 35, 299–311 (2011).
Gebhardt, T. et al. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 10, 524–530 (2009).
Jiang, X. et al. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 483, 227–231 (2012).
Schenkel, J.M., Fraser, K.A., Vezys, V. & Masopust, D. Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 14, 509–513 (2013). References 88, 89, 90, and 97 show that a noncirculating, resident population of CD8+ memory T cells is established in tissues following multiple infections, which provides enhanced protection from reinfection.
Turner, D.L. et al. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 7, 501–510 (2014).
Sathaliyawala, T. et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 38, 187–197 (2013).
Schenkel, J.M. et al. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346, 98–101 (2014).
Ariotti, S. et al. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 346, 101–105 (2014).
Shin, H. & Iwasaki, A. Tissue-resident memory T cells. Immunol. Rev. 255, 165–181 (2013).
Casey, K.A. et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 188, 4866–4875 (2012).
Masopust, D. et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 207, 553–564 (2010).
Mackay, L.K. et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 14, 1294–1301 (2013).
Iijima, N. & Iwasaki, A. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346, 93–98 (2014).
Zhang, N. & Bevan, M.J. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696 (2013).
Skon, C.N. et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 14, 1285–1293 (2013).
Masopust, D., Vezys, V., Wherry, E.J., Barber, D.L. & Ahmed, R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J. Immunol. 176, 2079–2083 (2006).
Wakim, L.M. et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 189, 3462–3471 (2012).
Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 12, 325–338 (2012).
Pollizzi, K.N. & Powell, J.D. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 14, 435–446 (2014).
Ghesquiere, B., Wong, B.W., Kuchnio, A. & Carmeliet, P. Metabolism of stromal and immune cells in health and disease. Nature 511, 167–176 (2014).
Powell, J.D. & Delgoffe, G.M. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 33, 301–311 (2010).
Pearce, E.L., Poffenberger, M.C., Chang, C.H. & Jones, R.G. Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454 (2013).
Wang, R. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Wang, R. & Green, D.R. Metabolic checkpoints in activated T cells. Nat. Immunol. 13, 907–915 (2012).
Finlay, D.K. et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 209, 2441–2453 (2012).
MacIver, N.J., Michalek, R.D. & Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283 (2013).
Gubser, P.M. et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 14, 1064–1072 (2013).
Pearce, E.L. et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107 (2009).
Araki, K. et al. mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112 (2009). References 114 and 115 show that regulation of mTOR and AMPK activity alters differentiation and suggest that a switch from energy derived from glycolysis to energy derived from mitochondria-supported oxidative phosphorylation accompanies and may promote the formation of CD8+ memory T cells.
van der Windt, G.J. et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78 (2012).
O'Sullivan, D. et al. Memory CD8 T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88 (2014).
Rolf, J. et al. AMPKa1: a glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 43, 889–896 (2013).
Sukumar, M. et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 123, 4479–4488 (2013).
Doedens, A.L. et al. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 14, 1173–1182 (2013).
Cham, C.M., Driessens, G., O'Keefe, J.P. & Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 38, 2438–2450 (2008).
Cham, C.M. & Gajewski, T.F. Glucose availability regulates IFN-γ production and p70S6 kinase activation in CD8+ effector T cells. J. Immunol. 174, 4670–4677 (2005).
Losman, J.A. & Kaelin, W.G. Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 27, 836–852 (2013).
Lu, C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012).
Wellen, K.E. et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009).
Chang, C.H. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013).
Michalek, R.D. et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011).
Shi, L.Z. et al. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 (2011).
Dang, E.V. et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell 146, 772–784 (2011). References 128 and 129 illustrate a role for HIF activity in regulating the subset differentiation of CD4+ helper T cells and suggest that HIF-regulated gene expression and glycolytic activity supports the generation of T H 17 cells.
Delgoffe, G.M. et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 12, 295–303 (2011).
Masson, F. et al. Id2-mediated inhibition of E2A represses memory CD8+ T cell differentiation. J. Immunol. 190, 4585–4594 (2013).
Mollo, S.B., Ingram, J.T., Kress, R.L., Zajac, A.J. & Harrington, L.E. Virus-specific CD4 and CD8 T cell responses in the absence of Th1-associated transcription factors. J. Leukoc. Biol. 95, 705–713 (2013).
Zhou, X. et al. Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity 33, 229–240 (2010).
Hess Michelini, R., Doedens, A.L., Goldrath, A.W. & Hedrick, S.M. Differentiation of CD8 memory T cells depends on Foxo1. J. Exp. Med. 210, 1189–1200 (2013).
Kim, M.V., Ouyang, W., Liao, W., Zhang, M.Q. & Li, M.O. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity 39, 286–297 (2013).
Sullivan, J.A., Kim, E.H., Plisch, E.H., Peng, S.L. & Suresh, M. FOXO3 regulates CD8 T cell memory by T cell-intrinsic mechanisms. PLoS Pathog. 8, e1002533 (2012).
Zhou, X. & Xue, H.H. Cutting edge: generation of memory precursors and functional memory CD8+ T cells depends on T cell factor-1 and lymphoid enhancer-binding factor-1. J. Immunol. 189, 2722–2726 (2012).
Fearon, D.T., Manders, P. & Wagner, S.D. Arrested differentiation, the self-renewing memory lymphocyte, and vaccination. Science 293, 248–250 (2001).
Turtle, C.J., Swanson, H.M., Fujii, N., Estey, E.H. & Riddell, S.R. A distinct subset of self-renewing human memory CD8+ T cells survives cytotoxic chemotherapy. Immunity 31, 834–844 (2009).
Gattinoni, L. et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 15, 808–813 (2009).
Zhang, Y., Joe, G., Hexner, E., Zhu, J. & Emerson, S.G. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat. Med. 11, 1299–1305 (2005).
Acknowledgements
We thank members of our laboratories for discussions, and L. Shaw for assistance with figures. Supported by the US National Institutes of Health (OD008469, DK093507 and AI095277 to J.T.C.; AI072117 and AI067545 to A.W.G.; and AI083022, AI082630, AI095608, AI05343, AI112521 and HHSN266200500030C to E.J.W.), the Leukemia and Lymphoma Society (A.W.G.) and the Howard Hughes Medical Institute (J.T.C.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Chang, J., Wherry, E. & Goldrath, A. Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15, 1104–1115 (2014). https://doi.org/10.1038/ni.3031
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.3031
This article is cited by
-
Precise surface functionalization of PLGA particles for human T cell modulation
Nature Protocols (2023)
-
Seasonal plasticity in immunocompetent cytokines (IL-2, IL-6, and TNF-α), myeloid progenitor cell (CFU-GM) proliferation, and LPS-induced oxido-inflammatory aberrations in a tropical rodent Funambulus pennanti: role of melatonin
Cell Stress and Chaperones (2023)
-
Regulation of activated T cell survival in rheumatic autoimmune diseases
Nature Reviews Rheumatology (2022)
-
‘Stem-like’ precursors are the fount to sustain persistent CD8+ T cell responses
Nature Immunology (2022)
-
The transcription factor Fli1 restricts the formation of memory precursor NK cells during viral infection
Nature Immunology (2022)
