
Key Points
-
Toll-like receptors (TLRs) and their signalling pathways have therapeutic potential for the treatment of inflammation, cancer, infection, allergy and autoimmunity.
-
The elucidation of the structure of several TLRs may aid medicinal chemistry in the rational design of small-molecule agonists and antagonists.
-
Agonists for TLR4, TLR7, TLR8 and TLR9 are being tested as adjuvants for infectious diseases and in certain cancers. There is also potential for some of these agents in the de-sensitization of airways in allergic asthma and allergy.
-
TLR antagonists are being investigated for the treatment of inflammatory diseases. Examples here include the inhibition of TLR7 and TLR9 in systemic lupus erythematosus, inhibition of TLR4 in sepsis and asthma or allergy, and inhibition of TLR2 in ischaemia and reperfusion injury.
-
Emerging findings on the underlying mechanism of TLR action might also lend themselves to therapeutic manipulation. Examples here include modulation of microRNAs, the exploitation of viral proteins to prevent nuclear factor-κB signalling and the development of compounds that interfere with the ubiquitination of signalling molecules.
Abstract
There is a growing interest in the targeting of Toll-like receptors (TLRs) for the prevention and treatment of cancer, rheumatoid arthritis, inflammatory bowel disease and systemic lupus erythematosus (SLE). Several new compounds are now undergoing preclinical and clinical evaluation, with a particular focus on TLR7 and TLR9 activators as adjuvants in infection and cancer, and inhibitors of TLR2, TLR4, TLR7 and TLR9 for the treatment of sepsis and inflammatory diseases. Here, we focus on TLRs that hold the most promise for drug discovery research, highlighting agents that are in the discovery phase and in clinical trials, and on the emerging new aspects of TLR-mediated signalling — such as control by ubiquitination and regulation by microRNAs — that might offer further possibilities of therapeutic manipulation.
Similar content being viewed by others

Main
Infection, inflammation, autoimmune diseases and cancer all involve complex signalling cascades that contain many possible drug targets. Despite this complexity it has proven possible to target a single protein — for example the pro-inflammatory cytokine tumour necrosis factor (TNF)1 — and achieve a dramatic therapeutic effect. Our understanding of the molecular components that regulate innate immunity and inflammation and lead to the induction of cytokines, such as TNF, has seen remarkable progress since the discovery of the Toll-like receptors (TLRs) almost 13 years ago. These receptors are prototypical pattern recognition receptors (PRRs), which recognize conserved microbial signature molecules known as either pathogen-associated molecular patterns (PAMPs) or microbe-associated molecular patterns (MAMPs), because they recognize any microbe regardless of its degree of pathogenicity2. Their ability to initiate and propagate inflammation makes them attractive therapeutic targets. However, as with many targets for anti-inflammatory agents, the approach is somewhat empirical. The initial discoveries around the biology of TLRs allowed companies to start developing modulators and there is preclinical evidence of their therapeutic potential. We are now at the stage of evaluating such molecules in human diseases; given the extensive literature on TLRs in disease, we can be optimistic that targeting them will prove useful in the clinic.
Key features of TLRs
TLRs recognize conserved structures of microbes and endogenous (host-derived) molecules. TLRs that recognize bacterial and fungal components are localized on the cell surface, whereas TLRs that recognize viral or microbial nucleic acids are localized to intracellular membranes such as endosomes or phagosomes. Thus, different TLRs are amenable to targeting by different types of agents. Cell surface TLRs can be targeted by small molecules (for example, eritoran that inhibits TLR4) and antibodies (for example, OPN-305, which targets TLR2), whereas the intracellular nucleic-acid sensing TLRs require targeting with modified oligonucleotides. Box 1 describes the ligand specificity and expression of TLRs in different cell types. NOD-like receptors (NLRs) are a related family of proteins that sense microbial products or the products of damaged cells, such as ATP and uric acid. NLRs also have potential as targets for new anti-inflammatory agents and are compared with TLRs in Box 2.
Progress has been made in the elucidation of the structures of particular TLRs with their ligands. The interface between lipopolysaccharide (LPS), MD-2 and TLR4 contains hydrophobic interactions, hydrogen bonds and charge interactions, particularly with the phosphates on LPS3,4 (Figs. 1, 2). Similarly, the molecular interactions between the acyl chains in the triacylated lipopeptide that binds the TLR1–TLR2 dimer have been described, as have the interfaces between the TLR3 ectodomain and the sugar phosphate backbone of the double stranded RNA (dsRNA) ligand. These molecular details should be useful in efforts to rationally design TLR antagonists5,6,7.

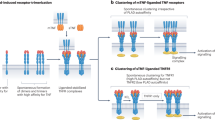
a | Toll-like receptor (TLR)1 and TLR2 form a heterodimer in response to triacylated lipopeptides like Pam3CSK4. Two thioester-linked lipid chains of Pam3CSK4 bind to a pocket in TLR2 and a single amide-linked lipid chain is inserted into a small channel in TLR1, forming a bridge between the two TLR molecules. The adaptor proteins MAL and MYD88 are then recruited through Toll-interleukin-1 resistance (TIR) domain–TIR interactions leading to activation of interleukin-1 receptor-associated kinase 4 (IRAK4) followed by IRAK1, and ultimately NF-κB which leads to the induction of pro-inflammatory cytokines. b | Lipopolysaccharide (LPS) binding induces the formation of a symmetric M-shaped TLR4–MD-2–LPS multimer composed of two copies of the complex. Five of the six lipid chains of lipid-A in the LPS molecule are buried within the hydrophobic pocket of MD-2 and the sixth chain, which remains exposed, interacts with the phenylalanines of TLR4. The activation of TLR4 leads to the recruitment of MAL and MYD88 and the activation of the serine/threonine kinase IRAK4, which engages with the NF-κB and mitogen-activated protein kinase cascades, leading to the induction of pro-inflammatory cytokines. TLR4 can also recruit TRIF (TIR domain-containing adaptor-inducing interferon-β) and TRAM (TRIF-related adaptor molecule) from endosomes, which leads to the activation of the transcription factor IRF3 and the induction of genes such as those encoding Type I interferons (IFNs)8. c | Double stranded RNA (dsRNA) from replicating viruses binds to the TLR3 dimers found in cell endosomes and bring the two horseshoe-shaped molecules together forming a homodimer. The TLR3 ectodomain interacts with the sugar phosphate backbone of the RNA not with the bases. The protein–protein interactions occur at the leucine-rich repeat (LRR)-carboxy-terminal domain of TLR3 molecules. TLR3 also recruits TRIF, which activates a similar pathway as TLR4.

Some of the interactions between ligands, Toll-like receptors (TLRs) and adaptor molecules may be amenable to targeting with small molecules. TLR4 residues are shown in green and blue, MD-2 residues are shown in grey and lipopolysaccharide (LPS) residues are shown in orange. R2 (LPS) is the sixth acyl chain in the lipid-A portion of LPS. It is the only chain that is exposed and interacts with TLR4. Data from Ref. 3.
The key signalling output from the receptor–ligand interactions is the tightening of the interface between the Toll–interleukin-1 resistance (TIR) domains on each TLR, which is responsible for signalling. This presumably leads to a conformational change or creates a new surface that allows the recruitment of adaptor proteins (which also contain TIR domains) and triggers signalling. There are differences in adaptor usage between different TLRs, which may permit specific modulation with therapeutic agents (Fig. 1). For example for TLR2 and TLR4 signalling, MAL and MYD88 lead to the activation of the serine/threonine kinase interleukin-1 receptor-associated kinase 4 (IRAK4), which engages with the nuclear factor-κB (NF-κB) and mitogen-activated protein (MAP) kinase cascades and leads to the induction of pro-inflammatory cytokines. TLR4 also recruits TRAM and TRIF in endosomes, which leads to the activation of the transcription factor IRF3 and the induction of genes such as those encoding Type I interferons8. TLR3 also recruits TRIF, which activates a similar pathway.
Implication of TLRs in disease
TLRs fulfil many of the criteria that are required to be considered potential therapeutic targets. These include overexpression in disease, knockout mice being resistant to disease in disease models, ligands exacerbating inflammation in disease models and genetic differences in TLRs (or their signalling proteins) correlating with risk of disease. However, whether it is overexpression of a given TLR or enhanced TLR-mediated signalling that is important for disease pathogenesis has yet to be demonstrated. Although there are few examples of effective interventions that target TLRs in human disease, one example of success is imiquimod, which has been approved for use in humans for several years for the treatment of genital warts, actinic keratosis and non-melanoma skin cancers9,10. Although purported to act as a TLR7 and TLR8 agonist, it has never actually been shown to act directly on these receptors11,12.
Perhaps the strongest evidence that TLRs represent good therapeutic targets comes from human genetics studies13. Many polymorphisms in genes that encode TLRs and their signalling molecules have been associated with human disease progression and susceptibility (Table 1). Genetic polymorphisms provide a compelling link to disease, but it is still unclear how useful these genetic differences will be for predicting the relative importance of a given TLR as a drug target for inflammation or infection. A greater understanding of such polymorphisms in innate immune signalling pathways and associated pathologies will aid targeting novel therapies to the relevant patient populations.
Although the roles of TLRs in human disease are still not fully understood, there are significant in vitro and animal model data to support roles for particular TLRs in disease initiation and progression. Mice that are deficient in particular TLRs have been studied in models of human inflammatory diseases, such as arthritis, multiple sclerosis and inflammatory bowel disease. Are TLRs better validated than other targets for new anti-inflammatory agents? At this stage we would have to conclude that this is not the case as similar criteria are met by other targets such as adhesion molecules and other receptors. However, because TLR activation occurs early in the cascade of events that give rise to inflammation, there might be an advantage in blocking them as they might be close to the initiating events that give rise to chronic inflammation. Because TLRs also detect products released from inflamed tissue, there may be a 'vicious cycle' of TLR activation that generates inflammatory products that feed back on TLRs, leading to further activation. Breaking such a positive feedback loop may be a highly effective strategy to limit inflammation.
Given the central role that molecules in the TLR pathways have in the innate immune response, there has been a focus on identifying antagonists and agonists to enhance the immune response, particularly in the context of vaccine adjuvancy in infectious diseases and therapeutic vaccine potential in cancer. The LPS derivative monophosphoryl lipid-A (MPL), the outer membrane protein from the haemophilia influenza B vaccine and Freund's complete adjuvant all act through TLRs14. Tables 2, 3, 4 summarize the TLR drug pipeline, from preclinical candidates through to those in the clinic and approved for use. Figure 3 illustrates the structures of some small molecular weight TLR drugs.

Imiquimod, purported to be a Toll-like receptor 7 (TLR7) agonist, is launched for papillomavirus infection. CRX-527 is a TLR4 agonist, but its development has been suspended. OM-174 is a TLR2 and TLR4 agonist that is in Phase I trails for cancer. TAK-242 is a TLR4 antagonist, but its development has been discontinued. Eritoran is a TLR4 antagonist that is in Phase III trials for sepsis. Ibudilast (AV4II) is a TLR4 antagonist that is in Phase II trials for opioid dependence and has been launched (Banyu/Kyorin) as an ophthalmic formulation for the treatment of ocular inflammation.
The downside of activating TLRs is not clear at present. Overactivation of pathways could give rise to unwanted effects, including autoimmunity and tissue fibrosis, although to date no ill-effects have been reported in animal models or in clinical trials15,16.
TLR agonists for cancer
TLR7, TLR8 and TLR9. A combination of empirical approaches to develop immunostimulatory molecules, with no knowledge of the actual targets, and the identification of TLRs as targets for some of these molecules, has led to the design of specific and potent immunostimulatory molecules. In the 19th century William Coley observed the antitumour effect of repeated injections of toxins from dead Gram-positive bacterium Streptococcus pyogenes and Gram-negative bacterium Serratia marcescens (known as Coley's toxin)17,18. It was later confirmed that bacterial DNA and LPS are the bacterial products involved in the antitumour activity and that the activation of the TLR9 and TLR4 signalling pathways leads to cytokine production, the stimulation of adaptive immunity and activation of natural killer and cytotoxic T cells19. The possibility of using TLR ligands as therapies to stimulate an immune response and fight disease is currently under investigation.
One of the first and most successful drugs targeting TLRs is imiquimod. It acts through a TLR7–MYD88-dependent pathway to cause secretion of the pro-inflammatory cytokine IFNα, which has antiviral and antitumour action. The production of IL-6 and TNF-α is also induced by imiquimod20.
Single stranded RNA-based TLR7 and TLR8 agonists are being developed for the treatment of cancer and infectious diseases. Early studies showed that the activation of TLR7 or TLR8 can reverse the suppressive function of regulatory T cells. When combined with the ability of TLR7 and TLR8 to activate dendritic cells, this effect results in a strong antitumour response21. The ability of the agonists to activate TLR7 and/or TLR8 depends on modifications made to the ligand's RNA sequence. In compounds in which the natural guanosine nucleotide was substituted with 7-deazaguanosine both TLR7 and TLR8 were activated22. However, another compound in which two RNA molecules were attached by their 3′ end only activated TLR8. Only the 7-deazaguanosine-containing compound activated both TLR7 and TLR8 in monkeys; this might help in the design of future structure–activity relationship studies. When used in combination with chemotherapeutic agents, the agonists had antitumour effects in a mouse non-small cell lung cancer model and the dual TLR7/TLR8 agonist showed antitumour activity in an IFNγ-dependent manner. In mice, non-human primates and in human cells the RNA-based TLR7 and TLR8 agonists induce T-helper 1 cell (Th1) responses, which could be useful for targeting tumours. A small-molecule-based TLR7 agonist, 852A, is also being developed for different types of cancer. The compound stimulates dendritic cells to produce multiple cytokines including IFNα in vitro and in vivo23,24. It is currently being evaluated in a Phase II clinical trial for treatment of inoperable melanoma25.
CpG-based oligonucleotides that target TLR9 signalling are also being evaluated as potential anticancer drugs. They mimic unmethylated bacterial DNA, the natural ligand for TLR9. Immune modulatory oligonucleotides (IMOs) that stimulate TLR9 signalling are being developed. IMO-2055 has anticancer activity in a mouse model when used as a monotherapy and this activity was amplified when it was used in combination with chemotherapeutic agents (NCT00729053)26,27. Its mechanism of action remains unclear, but the activation of the TLR9 signalling pathway by compounds such as IMOs enhances the antitumour T-cell response28. In a Phase I trial that evaluated IMO-2055 for safety and immunological activity alone and in combination with the chemotherapy agents gemcitabine or irinotecan, the IMO was much more efficacious when used in combination. This compound is currently being analysed in two Phase Ib trials, the first trial in patients with non-small cell lung carcinoma in combination with bevacizumab and erlotinib (NCT00633529) and the second in patients with colorectal cancer in a combination with cetuximab and irinotecan (NCT00719199)28.
MGN-1703 and MGN-1706 are double stem-loop immunomodulating adjuvants that consist of non-coding DNA and are being developed as anticancer TLR9 agonists. Individually these compounds have anticancer effects in preclinical models29 and positive results were obtained in a Phase I clinical trial of MGN-1703 in prostate and colorectal cancer (see Further information). Immunostimulatory sequences (ISS) are comprised of short DNA sequences that enhance the activity of TLR9 and enhance the production of T-helper 1 cells (Th1), which lead to the production of memory T cells and a Th1 response, and are also under investigation. ISS can be linked to antigens or used alone to suppress T-helper 2 cell (Th2) response. The lead compound ISS1018 induces the production of immunoglobulin and IFNα in vitro by B cells, and of IFNβ, IL-12 and TNF-α by plasmacytoid dendritic cells17. The ISS1018 oligonucleotide sequence is being used in clinical trials alone and combined with antigens to combat non-Hodgkin's lymphoma and other cancers as well as allergy (NCT00251394, NCT00403052)30. Agatolimod is another CpG-based oligonucleotide therapy that is being developed to treat renal cancer, allergy, asthma and hepatitis B infection31. It is currently in a Phase II clinical trial for treatment of melanoma, cutaneous T-cell, Non-Hodgkin, follicle centre and marginal zone B-cell lymphomas (NCT00091208). However, evaluation was discontinued in non-small cell lung carcinoma when it did not show increased efficacy over standard chemotherapy32.
TLR3. The TLR3-ectodomain binds as a dimer to dsRNA from replicating viruses that is 40–50 nucleotides in length, but only at an acidic pH indicating its endosomal location6. Its crystal structure revealed that it interacts with the sugar phosphate backbone of the RNA, rather than with the bases. Surprisingly, the conformation of TLR3 does not change when it binds dsRNA; the dsRNA brings the two horseshoe-shaped molecules together, which facilitates a protein–protein interaction at the carboxy-terminal domain. Synthetic dsRNA stimulates an IFN-mediated anticancer immune response and induces apoptosis in human breast cancer cells in a TLR3-dependent manner33.
The dsRNA mimic IPH-3102 activates NF-κB signalling and type I IFN responses in vitro and destroys melanoma and breast cancer cells. It is being developed for the treatment of breast cancer and as a vaccine adjuvant. Inn February 2009 IPH-3102 had successfully shown proof-of-concept in animal models34.
TLR2 and TLR4. Lipid-A is the biologically active part of the LPS molecule produced by bacteria such as Escherichia coli. The binding of LPS to TLR4 induces the formation of a symmetric M-shaped TLR4–MD-2–LPS multimer that is composed of two copies of the complex. Five of the six lipid chains of lipid-A are buried within the hydrophobic pocket of MD-2 and the sixth chain, which remains exposed, interacts with the phenylalanines of TLR4. The two phosphates in lipid-A also interact with each TLR4 chain3. Subtle differences in the structure of lipid-A affect its bioactivity, with different lipid-A structures able to act as TLR agonists or antagonists21. It is well known that the controlled activation of either TLR2 or TLR4 induces the production of TNF-α, as well as increased production of inducible nitric oxide synthase (iNOS) which induces apoptosis of chemotherapy-resistant tumour cells. A purified, water soluble diphosphorylated and triacetylated lipid-A derived from E. coli known as OM-174 acts as a TLR4 agonist. It reduces tumour growth, increases IFNγ production and prolongs the survival of mice35,36,37.
To trigger an immune response, dying tumour cells need to emit signals that cause dendritic cells to mature and elicit phagocytosis. A recent study showed that the TLR4 signalling pathway can mediate such a signal38. Individuals with breast cancer who carried a TLR4 loss-of-function allele relapsed much quicker following chemotherapy and radiotherapy compared with those carrying two wild-type copies, demonstrating the clinical relevance of this pathway. As the immune response produced in response to dying tumour cells is essential for a successful cancer therapy, TLR4 agonists may be exploited to enhance the immunogenicity of current chemotherapeutic regimens. It is possible that activation of TLR4 by an endogenous ligand — which could be produced by the tumour itself or by tissue responses to tumour products — also may contribute to this effect.
TLR2 binds a wider array of ligands than any other member of the TLR family. The heterodimerization of TLR2 with either TLR1 or TLR6 is essential for the recognition of bacterial lipopeptides and lipoproteins, such as the synthetic triacylated Pam3CSK4, lipoteichoic acid (LTA) found on Staphylococcus aureus and MALP-2, the ligand from Mycoplasma spp. Triacylation of lipopeptides is required for TLR1 and TLR2 signalling, and diacylation of lipopeptides is required for TLR2 and TLR6 signalling. Pam3CSK4 simultaneously binds to TLR1 and TLR2 resulting in an M-shaped heterodimer similar to TLR4. The crystal structure revealed that two thioester-linked lipid chains of Pam3CSK4 bind to a pocket in TLR2 and a single amide-linked lipid chain is inserted into a small channel in TLR1, thus forming a bridge between the two TLR molecules7.
The TLR2 agonist SMP-105 that consists of cell-wall skeleton components, such as mycolic acids, and peptidoglycans from Mycobacterium bovis (strain BCG/Tokyo), is approved for the treatment bladder cancer, although there is little information on its mechanism of action in this indication39. The compound has shown strong adjuvant and antitumour activities. In mice, SMP-105 activates NF-κB in a TLR2-dependent and TLR4-independent manner. Upon administration of the compound, TLR2 knockout mice showed impairment of TNF-α and IL-6 production as well as reduced tumour growth40.
TLR5. A TLR5 agonist made from flagellin from Salmonella enterica, CBLB502, that activates NF-κB and inhibits the pro-apoptotic p53 pathway (while not interfering with the antitumour effect of radiation), is being developed for the treatment of cancer. Studies in mice have shown that there is added efficacy in stopping tumour growth when flagellin is combined with CpG oligonucleotides41,42.
PolyTLR agonists. Cadi-05 is being developed as a polyTLR agonist and polyantigenic vaccine consisting of autoclaved mycobacteria that activate a number of TLRs. It is being evaluated for treatment of tuberculosis and advanced melanoma, prostate and bladder cancers. In a Phase I clinical trial, five out of nine patients who received Cadi-05 in combination with radiotherapy became asymptomatic and showed no disease recurrence 2 years later. This treatment also reduced the size of tumours in mice and inhibited growth in cell lines43. However, a Phase I/II trial examining safety in patients with Stage III or Stage IV melanoma was terminated when no additional efficacy was shown (NCT00675727).
Although there is empirical evidence for a given TLR as a good target for cancer therapy, a compelling rationale is often lacking in human clinical trials. Many cancer patients are immunosuppressed due to traditional antitumour therapies, and it is difficult to produce a strong positive innate immune response. Nevertheless, there is optimism that the combination of innate immune stimulatory compounds and anticancer agents will ultimately lead to a successful anticancer therapy.
TLR agonists for viral and bacterial infections
TLR7, TLR8 and TLR9. Because TLRs are crucial in the recognition of viral and bacterial pathogens such as hepatitis and influenza, vaccines and therapies that activate these receptors and lead to a pro-inflammatory response and subsequent destruction of the invading pathogen are sought. The current treatment of respiratory syncytial virus, hepatitis C infection and other viral infections is administration of IFNα in combination with the antiviral drug ribavarin. However, this treatment has adverse side-effects including neutropaenia (low white-blood cell counts), thrombocytopaenia (low platelet counts) and anaemia (low red-blood cell counts)44.
Resiquimod (R-848) is currently being evaluated for treating hepatitis C and other viral infections45,46. It signals through the TLR7 and TLR8 pathways, inducing IFNα, IL-12 and TNF-α. Resiquimod also activates natural killer cells, indirectly inducing IFNγ, and therefore may promote the development of antigen-specific cell-mediated responses47. Phase III trials of the drug for treatment of herpes simplex virus were suspended due to lack of efficacy and although a Phase II study for the treatment of hepatitis C infection demonstrated a decreased viral load, the trial showed adverse effects similar to IFNα treatment46. A small-molecule TLR7 agonist, ANA773, is under investigation for the potential treatment of hepatitis C infection and cancer. In animal studies, every other day dosing led to good immune stimulation, and recent results from a Phase I clinical trial revealed that high-dose treatment led to the induction of an immune response and a decrease in viral load48.
IMO-2125 is a TLR agonist that targets TLR9, inducing high and sustained levels of IFNα and other cytokines, as well as activating T-cells and natural killer cells in non-human primates26. It is being evaluated in a Phase I clinical trial in patients with hepatitis C who do not respond well to the current standard of care (NCT00728936). The possibility of using IMO-2125 in combination with ribavarin is also being explored in treatment-naive patients (NCT00990938). SD-101 and HEPLISAV (hepatitis B surface antigen) are compounds designed to stimulate TLR9 for the potential treatment of hepatitis C and hepatitis B infections49. SD-101 is being tested for the treatment of hepatitis C and a Phase Ib trial is ongoing (NCT00823862). In addition, a hepatitis B virus ISS which also targets TLR9 (comprised of CpG-based ISS1018 and HEPLISAV) has demonstrated the potential to provide more rapid and increased protection against hepatitis B infection compared with standard IFNα plus ribavarin therapy in nine different clinical trials49,50.
The potential for added efficacy through the combination of CpG oligonucleotides such as CpG-7909 or CpG-10109 (AV7909) with the only Food and Drug Administration (FDA)- approved vaccine to prevent anthrax infection (Biothrax; Emergent BioSolutions), is being explored31. Again the approach here seems to be empirical. In a Phase I/II clinical trial that evaluated the safety and immunogenicity of the AV7909 vaccine, the addition of CpG-7909 to Biothrax increased peak antiprotective antigen (anti-PA) titres 6-fold and reduced the time to peak titre compared with Biothrax alone. Additionally, only two doses of AV7909 with Biothrax were required to elicit the same serum anti-PA IgG levels achieved by three doses of Biothrax alone51.
TLR4. As mentioned previously, MPL has been approved in Europe as a vaccine adjuvant and is a component of the hepatitis B vaccine and the human papillomavirus virus vaccine52. It is less toxic than LPS and specifically activates the TRAM/TRIF pathway in TLR4 signalling, which leads to induction of IFNβ and regulation of CD80/86 (Refs 53, 54, 55). This effect on CD80/86 seems to be a key aspect of adjuvancy.
TLR3. Rintatolimod (also know as poly I:C) is a TLR3 agonist being developed for the treatment of chronic fatigue syndrome (CFS), hepatitis B and hepatitis C infection, HIV, influenza, severe acute respiratory syndrome and cancer56. It is a dsRNA molecule, which was first synthesized in the 1970s and is purported to regulate levels of cellular RNAse L, an enzyme that is required to fight off invading viruses and is known to be elevated in patients with CFS57.
TLR5. Vaccine adjuvants that act as TLR5 agonists are also under investigation to treat viral infections. VAX-102 is a protein therapeutic that links flagellin from Salmonella typhimurium (STF2) with the ectodomain of matrix protein 2 (M2e), which is a highly conserved region in different virus strains including influenza A58. This vaccine adjuvant could potentially protect against all strains of seasonal and pandemic influenza. VAX-102 has demonstrated positive effects against lethal influenza challenge in mice and a recent Phase I trial in healthy volunteers demonstrated safety in humans (NCT00603811). Further clinical studies are planned and the effects of VAX-102 against West Nile virus, Japanese encephalitis and Listeria spp. infection are also being examined in animal models.
There is great potential in using the adjuvancy effect of TLR agonists to treat viral and bacterial infections. From the data generated so far, it seems that the combination of standard therapies and adjuvant therapies that stimulate TLR signalling will be the most successful in the treatment of infections.
Agonists for allergy, asthma and autoimmunity
TLR7, TLR8 and TLR9. The current anti-inflammatory treatment for allergies and asthma relies largely on steroids and antibodies. The primary feature of an allergic response is usually a strong Th2 response which can be counterbalanced by the induction of TLR signalling; this would induce a cytokine response and activate antigen presenting cells, leading to a Th1 response59. Agonists that activate TLRs, particularly TLR7, TLR8 and TLR9 would be expected to induce a strong Th1 response and have been a focus for novel therapeutics in these disease areas60.
Intranasal, inhaled administration of the TLR9 agonist QAX-935 (IMO-2134) to monkeys prevented an increase in airway resistance and inflammation, and induced the expression of several IFN-dependent genes that are indicative of an effective Th1 response34,61. Further investigation into the efficacy of this compound should resume in 2010. The CpG DNA-based TLR9 agonists AVE0675 and SAR-21609 are also being examined for the treatment of asthma, and asthma and viral respiratory tract infection (either alone or in combination with specific allergen immunotherapies), respectively. AVE0675 inhalation monotherapy had been approved for a Phase I clinical trial for safety in patients with allergy (see Further information and Refs 43, 62 for reviews). DNA-based immunomodulatory sequences (DIMs) — synthetic oligonucleotides that activate TLR9 — are strong inducers of IFNβ. Thus, activation of TLR9 could be beneficial in patients with multiple sclerosis who are on IFNβ therapy and have experienced adverse side effects or tolerance to the therapy. In a rat herpes simplex encephalitis model, DIMS9054 showed positive effects on survival. DIMS0150 (Kappaproct; InDex Pharmaceuticals) has orphan drug designation in Europe and is currently approved for clinical use as a single dose administration in patients with steroid resistant or dependent ulcerative colitis. It has completed its third Phase II clinical trial63.
Other TLRs are being explored for their potential role in allergy and asthma. TLR8 agonists act as potent stimulators of Th1 polarizing cytokines such as IFNγ and IL-12, and are being developed for the potential treatment of allergic rhinitis. In preclinical studies, VTX-1463 reduced allergic responses through the suppression of a Th2-mediated allergic response. When administered intranasally, it resulted in a decrease in the number of eosinophils and less congestion. Clinical evaluation of this compound is expected in early 2010. VTX-2337 is another compound under development for its potential in the treatment of cancer and allergy and is currently in a Phase I trial in patients with late stage cancer (NCT00688415). The TLR7 agonist AZD8848 (DSP-3025) is being assessed for its ability to elicit long-term disease remission in allergic disorders such as bronchial asthma and allergic rhinitis. It has anti-inflammatory potential and is currently being tested in a Phase I clinical trial (NCT00770003) with recruitment underway for a Phase II trial (NCT00999466).
TLR4. Pollinex Quattro is a novel vaccine which contains ragweed pollen extract adsorbed onto L-tyrosine, combined with MPL to stimulate the TLR4 signalling pathway64. The vaccine was well tolerated and reduced allergy symptoms in rats. The drug is currently in a Phase III clinical trial, in which a reduction in seasonal allergies was observed65.
TLR activation has been a high priority for therapeutic intervention in a broad spectrum of human disease and vaccination regimes. This is supported by the clinical evaluation of oral and inhaled low molecular weight compounds, lipid analogues, protein- and nucleotide-based agonists towards TLR2, TLR3, TLR4, TLR5, TLR7, TLR8 and TLR9. The results of these ongoing clinical trials are eagerly awaited.
TLR antagonists
In addition to the development of agonists, there has also been significant research on the potential for TLR antagonists as novel therapeutics. Such compounds could be used to treat diseases such as sepsis, SLE and rheumatoid arthritis, in which the immune system is inappropriately overactive. The inhibition of the pro-inflammatory cytokine TNF by biopharmaceuticals has proven to be remarkably effective in the treatment of diseases such as rheumatoid arthritis1. If rheumatoid arthritis is aggressively treated early with a combination of therapies, patients show remarkable improvements, and if the combination therapy continues there is significant inhibition of joint destruction66,67,68. Despite this success, there is still a clear unmet need for therapies for chronic inflammatory diseases.
TLR7, TLR8 and TLR9. Current treatments of SLE and rheumatoid arthritis include the use of antimalarial drugs such as hydroxychloroquine (HCQ), which is now known to act as a TLR9 antagonist and as a lesser TLR7 and TLR8 antagonist. Derivatives and small-molecule analogues of chloroquine and quinacrine suppress activation of an immune response69,70. The quinazoline derivative CPG-52364 is an anti-inflammatory TLR antagonist which specifically inhibits TLR7, TLR8 and TLR9 and inhibits disease progression of SLE and other autoimmune diseases in animal models43,71. It showed a marked increase in potency compared with the use of HCQ alone, but when used as a combination therapy it had added efficacy by preventing the formation of antiDNA antibodies in SLE-prone mice. CPG-52364 is currently being evaluated in a Phase I clinical trial (NCT00547014)43
Immunoregulatory sequences (IRS) are short DNA sequences, that can act as bifunctional inhibitors of TLR7 and TLR9. The most promising compound of this type is IRS-954 (DV-1079), which prevented SLE progression in SLE-prone mice and reduced serum levels of nucleic-acid specific antibodies72. Administration of the drug to HIV-stimulated peripheral blood monocyte cells also led to a decrease in IFNα production, suggesting a potential therapeutic opportunity for treating HIV infection73. IMO-3100, another dual antagonist which blocks TLR7 and TLR9 is being tested for the treatment of inflammatory and autoimmune disease, such as SLE, rheumatoid arthritis, multiple sclerosis, psoriasis and colitis. A recent study that evaluated the effect of IMO-3100 on gene expression in human peripheral blood monocytes found that the drug inhibited TLR ligand-induced increases in the expression of TNF-α, IFNα and IL-17 (Ref. 74).
TLR4. The effort to inhibit lipid-A binding to TLR4 has received a boost with the recent reporting of the crystal structure of the TLR4–MD-2–LPS complex3. One of the most advanced TLR4 antagonists is the synthetic lipodisaccharide, eritoran (E5564) which reduces the effects of LPS inhalation and sepsis in animal models75,76. The structure of TLR4–MD-2 with eritoran suggests that the basis for antagonism is due to its inability to cause the 5å shift at position F126 in MD-2, which is seen with LPS3. Such molecular details should be very useful in the effort to design additional antagonists. Eritoran decreases the production of pro-inflammatory cytokines in LPS-stimulated cultured human monocytes77 and in a Phase II clinical trial eritoran reduced the mortality rate due to sepsis by 6.4% compared with the placebo group78. This result led to the initiation of a Phase III trial (NCT00334828). Eritoran needs to be administered as either a continuous infusion or by repeated intravenous injections to keep the levels of active drug high enough to elicit a therapeutic effect79.
Variations of lipid-A, notably CRX-527, which has a short second acyl chain, have reduced agonistic activity80,81. Lipid-IVa is an intermediate in the synthesis of lipid-A, however unlike lipid-A it inhibits TLR signalling in humans by competing with LPS for binding to MD-2 (Ref. 82). The basis for decreased potency of CRX-527 or the antagonistic effect of lipid-IVa is not known, but could be determined by solving their crystal structures and modelling the interaction of each bound to the TLR4–MD-2 complex and comparing them with LPS bound to the complex. TAK-242 also targets TLR4-dependent signalling, although the precise target is not known. Development of this compound was discontinued during a Phase III clinical trial for sepsis83 because the drug's profile did not meet the criteria required to support continued development, not because of drug safety issues (NCT00633477). It is not known whether this was related to the target or the class of drug.
The TLR4 antagonist ibudilast (AV411) is being studied in advanced clinical trials for the treatment of chronic pain and addiction withdrawal84. Ibudilast suppresses pro-inflammatory cytokines such as TNF-α and IL-6, and may induce the anti-inflammatory cytokine IL-10. The mechanism underlying the induction of IL-10 is unclear but it is speculated that ibudilast suppresses the activation of glial cells and thus, neuroinflammation85.
TLR4 antagonistic antibodies are being developed to block immune signalling in diseases associated with an excessive immune response. The most advanced is NI-0101, which interferes with the dimerization of TLR4 by binding to an epitope on TLR4. 1A6 is an antibody that recognizes TLR4–MD-2 and when it was administered to a murine dextran sulfate sodium (DSS) colitis model at the same time as DSS, it delayed the development of colitis and reduced the inflammatory response. However, when the antibody was administered during the recovery stage of the disease it impaired mucosal healing86. This suggests that blocking TLR4 may offer improvement in the uncontrolled inflammation of colitis by reducing antigen-presenting cell infiltrate and pro-inflammatory cytokine production in a time-dependent manner. Finally, the monoclonal antibody 15C1 that neutralizes LPS-induced signalling in the presence or absence of MD-2 by a novel Fc receptor-mediated mechanism has been reported87.
TLR2. OPN-305 is a TLR2-specific monoclonal antibody that inhibits TLR2-mediated pro-inflammatory cytokine production and is being tested for the potential treatment of inflammatory diseases. Recent data suggest a potential role for TLR2 in SLE, diabetes and Alzheimer's disease88,89. Positive results have been seen with the antibody in a murine model of myocardial ischaemia and reperfusion injury and other inflammatory and autoimmune disease models90. In 2009 the compound was granted orphan status for the prevention of the ischaemia and reperfusion injury associated with organ transplantation. The first human trials are expected to begin in 2010. In addition, OPN-401, a viral protein-derived peptide that inhibits TLR4-dependent signalling is also in preclinical development (see Further information). Both TLR2 and TLR4 have been implicated in ischaemia and reperfusion injury in different tissues, notably in the heart and brain91,92,93.
A novel technique that combines immunoprecipitation with systematic evolution of ligands by exponential enrichment (SELEX) to identify and characterize immune regulating oligonucleotides was used to facilitate the screening of high-affinity DNA or RNA molecules that bind to TLRs (Ref. 94). Molecules that bind to TLR2 were identified in this study and the most efficient was AP177. This molecule efficiently antagonized TLR2 signalling, inhibited NF-κB activity and suppressed the secretion of IL-6 and IL-8 cytokine from cells challenged with the TLR2-specific ligand SA-LTA or the TLR1 and TLR2 ligand PAM3CSK4, but less so when cells were stimulated with FSL-1 (a TLR2 and TLR6 ligand). AP177 seems to directly bind to TLR2 and competitively block ligand binding to TLR2 as assessed by its inability to block LPS signalling through TLR4. The technique developed in this study could potentially be used to identify other molecules that affect TLR signalling and alter the subsequent immune response.
The future of targeting TLRs
Well characterized approaches — ligand mimetics to activate TLRs and antibodies or small molecules to inhibit them — are being used to identify novel therapeutics that target TLRs. The structure of several TLRs may now aid medicinal chemistry in the rational design of small-molecule agonists and antagonists. However there is also likely to be significant potential in non-traditional approaches to drug discovery, such as the use of microRNAs (miRNAs). A key role for miRNAs in the regulation of genes involved in immune defence is being uncovered95. They are unique molecules because their expression is cell- and/or tissue-specific. Some miRNA are negative regulators of TLR signalling, acting as a brake on the pathway, whereas others act as positive regulators. They act by binding to messenger RNA (mRNA) of a target gene, promoting degradation of the mRNA or inhibiting translation of the protein. Because miRNAs are generally only about 22 nucleotides in length they offer potential as drug therapies. Their deregulation has been identified in cancers and viral infections. Immuno-miRs are miRNAs that act on specific immune regulatory genes. Figure 4 details two examples of immuno-miRs and the genes they target. miR-146 targets two proteins involved in TLR signalling, TRAF6 and IRAK1, and is upregulated in response to LPS, negatively regulating the mRNA levels of both TRAF6 and IRAK1 (Ref. 96). miR-155 is upregulated in response to LPS and targets the Src homology-2 domain-containing inositol 5-phosphatase 1 (SHIP-1), which is a negative regulator of NF-κB signalling97,98. We have recently shown that LPS increases miR-21 levels in cells targeting the IL-10 inhibitor PDCD4, leading to an increase in IL-10 production152. Because miRNAs fine-tune gene expression, the effects of therapeutically administering a single miRNA may impact on the expression of many target genes. This may manifest itself clinically as a safety issue or an efficacy benefit; however, these potential off-target effects have not deterred researchers from exploring miRNA therapies. Several companies are developing approaches to target miRNAs, notably with so–called antagomirs and locked nucleic acid miRNA inhibitors95. These approaches, if used to target TLR-regulated miRNA could have therapeutic benefit.

Manipulating microRNA expression using antagomirs or locked nucleic acid molecules in cells will have an effect on the expression of target gene messanger RNA levels. miR-146 targets tumour necrosis factor receptor (TNFR)-associated factor 6 (TRAF6) and interleukin (IL)-1 receptor-associated kinase 1 (IRAK1), which are important molecules in the Toll-like receptor (TLR) signalling pathway96. The elimination of miR-146 levels could lead to an adjuvant effect in cancer or infection. The elimination of miR-155 whose target is SH2-containing inositol 5-phosphatase 1 (SHIP-1) could also have an adjuvant effect by releasing the break on the phosphoinositide 3 kinase (PI3K) pathway97. The elimination of miR-21 would inhibit IL-10 production and have an immunostimulatory effect152. Mimicking these miRNAs would have an anti-inflammatory effect. LPS, lipopolysaccharide; NF-kB, nuclear factor-kB; PIP3, phosphatidylinositol-3,4,5-triphosphate; TRAM, TRIF-related adaptor molecule.
Another possible novel approach has come from work highlighting the manipulation of TLR signalling by viral nucleic acids and proteins99,100. Vaccinia virus produces several proteins that target TLR signalling. The protein A52R sequesters IRAK2 and TRAF6 from the pathway and prevents NF-κB activation100. A peptide termed P13 has been synthesized from the A52R protein that blocks cytokine secretion from cells in vitro101. The peptide also effectively inhibited both in vitro and in vivo LPS-induced inflammatory responses and enhanced survival of mice compared with control treatment. The use of peptides that take advantage of viral evasion of the immune system is being evaluated by Opsona Therapeutics.
Kinases have been a fruitful area for small-molecule drug discovery and the IRAKs are potential innate immunity targets awaiting therapeutic evaluation. The best characterized member of the family is IRAK4 (Ref. 102). Mice that express a catalytically inactive mutant of IRAK4 are completely resistant to septic shock and show impaired IL-1 activity103. Dendritic cells from these mice don't produce IFNα in response to TLR7, TLR8 or TLR9 agonists or following infection by influenza virus. Furthermore, these mice are resistant to joint inflammation and destruction, and also bone destruction, in a model of arthritis, supporting the targeting of IRAK4 for chronic inflammation104. IRAK2 has also recently been shown to be involved in sustained NF-κB activation. Importantly, its kinase activity is required, indicating that inhibition of IRAK2 could be anti-inflammatory105.
Phosphorylation coupled to ubiquitination are key covalent modifications in innate immune signalling that lead to activation of NF-κB5. Recent research has shown that modulating these modifications in cell signalling cascades can be beneficial in the treatment of cancer, neurodegenerative disease, inflammation and viral infections. Proteins targeted for degradation are tagged with a chain of ubiquitin molecules by ligation to lysine residues. K48-linked polyubiquitin is thought to be the primary form that is recognized by the proteasome for degradation, whereas K63-linked ubiquitination is regulatory106. Ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s) and ubiquitin ligases (E3s) all have a role in the process of ubiquitination in the cell. In the TLR signalling pathway, TRAF6 has E3 ubiquitin-ligase activity and preclinical in vitro studies have demonstrated that inhibitors of TRAF6 block auto-ubiquitination as well as cell proliferation107. In addition, Pellino3 promotes K63-linked ubiquitination of IRAK1, allowing the recruitment of NF-κB essential modifier (NEMO) and the IκB kinase (IKK) complex. The interaction between IRAK1 and TRAF6 then allows the recruitment of TAK1, which is found in close proximity to IKK2, leading to the activation of the IKK complex5. Agents that target Pellino3 would interfere with this process. Figure 5 illustrates molecules involved in TLR signalling that when inhibited could have an effect on the inflammatory response. There has been significant preclinical activity seeking inhibitors of the ubiquitination pathway, but this has yet to produce a strong clinical candidate. Nevertheless, the mechanism remains attractive for the treatment of inflammation.

Inhibition of the kinase activity of IL-1 receptor-associated kinase 1 (IRAK1), IRAK2 or IRAK4 will block downstream signalling pathways. Ubiquitination has been shown to have an important regulatory role in Toll-like receptor (TLR) signalling. Inhibiting Pellinos will block ubiquitination of IRAK1, which will in turn limit the recruitment of nuclear factor-kB (NF-kB) essential modulator (NEMO). Inhibition of TNFR-associated factor 6 (TRAF6) auto-ubiquitination should block transforming growth factor-β-activated protein kinase 1 (TAK1) recruitment. Finally, inhibiting the cellular inhibitors of apoptosis (cIAPs) will block K48-linked ubiquitination of TRAF3, inhibiting the pathway131. IKK, inhibitor of kB kinase; LPS, lipopolysaccharide; SMAC, second mitochondria-derived activator of caspase; TRAM, TRIF-related adaptor molecule; TAB, TAK1-binding protein; Ub, ubiquitin.
Inhibitors of apoptosis (IAPs) are another group of molecules with great therapeutic potential108. IAPs are upregulated in many cancers and exert their effect by inhibiting caspases. They are regulated by an endogenous inhibitor: second mitochondria-derived activator of caspase (SMAC) and the identification of SMAC mimetics represents a novel therapeutic approach109,110. Figure 5 illustrates the potential effect of SMAC mimetics on TLR signalling. SMAC binds to IAPs and disrupts their ability to inhibit caspases. cIAP1 and cIAP2 also associate with TRAF1, TRAF2 and TRAF6 to participate in NF-κB activation by TNF-α, CD40 and IL-1 receptor/TLR signalling molecules. Importantly, cIAP1 seems to mediate K48-linked ubiquitination of TRAF3, which has been shown to be an inhibitor of TLR signalling111. Inhibition of cIAPs would therefore be predicted to block TRAF3 degradation and inhibit signalling by TNF-α, CD40 and IL-1 receptor/TLR molecules. Using crystal structures and molecular docking it has been possible to design small-molecule SMAC mimetics to cIAP which have shown promising effects in vitro108. Such mimetics kill malignant cells by promoting TNF-mediated apoptosis and might have potent anti-inflammatory effects.
Details of the TLR signalling pathways continue to be uncovered. While we await the outcome of on-going clinical evaluation of novel TLR agonists and antagonists it is clear that there are a number of new opportunities to target innate immune signalling. The emergence of novel regulatory molecules such as miRNA and protein targeting by ubiquitination also present exciting new opportunities for drug discovery and development. Both preclinical and clinical data support our belief that the ability to specifically target key processes in innate immunity might prevent uncontrolled infection and limit inflammation in multiple diseases.
References
O'Neill, L. Targeting signal transduction as a strategy to treat inflammatory diseases. Nature Rev. Drug Discov. 5, 549–563 (2006).
Koropatnick, T. A. et al. Microbial factor-mediated development in a host–bacterial mutualism. Science 306, 1186–1188 (2004).
Park, B. S. et al. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 458, 1191–1195 (2009). References 3, 6 and 7 are the first studies to illustrate the crystal structures for TLR4, TLR3 and the TLR1–TLR2 heterodimer with their specific ligands.
Dziarski, R. & Gupta, D. Role of MD-2 in TLR2- and TLR4-mediated recognition of Gram-negative and Gram-positive bacteria and activation of chemokine genes. J. Endotoxin Res. 6, 401–405 (2000).
Carpenter, S. & O'Neill, L. A. Recent insights into the structure of Toll-like receptors and post-translational modifications of their associated signalling proteins. Biochem. J. 422, 1–10 (2009).
Liu, L. et al. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 320, 379–381 (2008).
Jin, M. S. et al. Crystal structure of the TLR1–TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130, 1071–1082 (2007).
Kawai, T. & Akira, S. TLR signaling. Cell Death Differ. 13, 816–825 (2006).
Somani, N. & Rivers, J. K. Imiquimod 5% cream for the treatment of actinic keratoses. Skin Therapy Lett. 10, 1–6 (2005).
Stockfleth, E., Christophers, E., Benninghoff, B. & Sterry, W. Low incidence of new actinic keratoses after topical 5% imiquimod cream treatment: a long-term follow-up study. Arch. Dermatol. 140, 1542 (2004).
Larange, A., Antonios, D., Pallardy, M. & Kerdine-Romer, S. TLR7 and TLR8 agonists trigger different signaling pathways for human dendritic cell maturation. J. Leukoc. Biol. 85, 673–683 (2009).
Lysa, B. et al. Gene expression in actinic keratoses: pharmacological modulation by imiquimod. Br. J. Dermatol. 151, 1150–1159 (2004).
Schwartz, D. & Cook, D. Polymorphisms of the Toll-like receptors and human disease. Clin. Infect. Dis. 41 (Suppl. 7), 403–407 (2005).
Ishii, K. J. & Akira, S. Toll or toll-free adjuvant path toward the optimal vaccine development. J. Clin. Immunol. 27, 363–371 (2007).
Ehlers, M. & Ravetch, J. V. Opposing effects of Toll-like receptor stimulation induce autoimmunity or tolerance. Trends Immunol. 28, 74–79 (2007).
Meneghin, A. & Hogaboam, C. M. Infectious disease, the innate immune response, and fibrosis. J. Clin. Invest. 117, 530–538 (2007).
Rakoff-Nahoum, S. & Medzhitov, R. Toll-like receptors and cancer. Nature Rev. Cancer 9, 57–63 (2009).
Cho, Y. J., Ahn, B. Y., Lee, N. G., Lee, D. H. & Kim, D. S. A combination of E. coli DNA fragments and modified lipopolysaccharides as a cancer immunotherapy. Vaccine 24, 5862–5871 (2006).
Wei, M. Q., Mengesha, A., Good, D. & Anne, J. Bacterial targeted tumour therapy — dawn of a new era. Cancer Lett. 259, 16–27 (2008).
Hemmi, H. et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nature Immunol. 3, 196–200 (2002).
O'Neill, L. A., Bryant, C. E. & Doyle, S. L. Therapeutic targeting of toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol. Rev. 61, 177–197 (2009).
Lan, T. et al. Stabilized immune modulatory RNA compounds as agonists of Toll-like receptors 7 and 8. Proc. Natl Acad. Sci. USA 104, 13750–13755 (2007).
Dudek, A. Z. et al. First in human phase I trial of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced cancer. Clin. Cancer Res. 13, 7119–7125 (2007).
Dummer, R. et al. An exploratory study of systemic administration of the toll-like receptor-7 agonist 852A in patients with refractory metastatic melanoma. Clin. Cancer Res. 14, 856–864 (2008).
Harrison, L. I., Astry, C., Kumar, S. & Yunis, C. Pharmacokinetics of 852A, an imidazoquinoline Toll-like receptor 7-specific agonist, following intravenous, subcutaneous, and oral administrations in humans. J. Clin. Pharmacol. 47, 962–969 (2007).
Agrawal, S. & Kandimalla, E. R. Synthetic agonists of Toll-like receptors 7, 8 and 9. Biochem. Soc. Trans. 35, 1461–1467 (2007).
Goodchild, A. et al. Primary leukocyte screens for innate immune agonists. J. Biomol. Screen 14, 723–730 (2009).
Krieg, A. M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 27, 161–167 (2008).
Kochling, J. et al. Anti-tumor effect of DNA-based vaccination and dSLIM immunomodulatory molecules in mice with Ph+ acute lymphoblastic leukaemia. Vaccine 26, 4669–4675 (2008).
Vollmer, J. et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur. J. Immunol. 34, 251–262 (2004).
Dorn, A. & Kippenberger, S. Clinical application of CpG-, non-CpG-, and antisense oligodeoxynucleotides as immunomodulators. Curr. Opin. Mol. Ther. 10, 10–20 (2008).
Schmidt, C. Clinical setbacks for toll-like receptor 9 agonists in cancer. Nature Biotech. 25, 825–826 (2007).
Salaun, B., Coste, I., Rissoan, M. C., Lebecque, S. J. & Renno, T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 176, 4894–4901 (2006).
Panter G., K. A., Jerala R. Therapeutic applications of nucleic acids as ligands for Toll-like receptors. Curr. Opin. Mol. Ther. 11, 133–145 (2009).
D'Agostini, C. et al. Antitumour effect of OM-174 and cyclophosphamide on murine B16 melanoma in different experimental conditions. Int. Immunopharmacol. 5, 1205–1212 (2005).
Garay, R. P. et al. Cancer relapse under chemotherapy: why TLR2/4 receptor agonists can help. Eur. J. Pharmacol. 563, 1–17 (2007).
De Ridder, M. et al. The radiosensitizing effect of immunoadjuvant OM-174 requires cooperation between immune and tumor cells through interferon-γ and inducible nitric oxide synthase. Int. J. Radiat. Oncol. Biol. Phys. 66, 1473–1480 (2006).
Apetoh, L. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nature Med. 13, 1050–1059 (2007).
Simons, M. P., O'Donnell, M. A. & Griffith, T. S. Role of neutrophils in BCG immunotherapy for bladder cancer. Urol. Oncol. 26, 341–345 (2008).
Murata, M. Activation of Toll-like receptor 2 by a novel preparation of cell wall skeleton from Mycobacterium bovis BCG Tokyo (SMP-105) sufficiently enhances immune responses against tumors. Cancer Sci. 99, 1435–1440 (2008).
Burdelya, L. G. et al. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science 320, 226–230 (2008).
Sfondrini, L. et al. Antitumor activity of the TLR-5 ligand flagellin in mouse models of cancer. J. Immunol. 176, 6624–6630 (2006).
Parkinson, T. The future of toll-like receptor therapeutics. Curr. Opin. Mol. Ther. 10, 21–31 (2008).
Schalm, S. W. et al. Ribavirin enhances the efficacy but not the adverse effects of interferon in chronic hepatitis C. Meta-analysis of individual patient data from European centers. J. Hepatol. 26, 961–966 (1997).
Mark, K. E. et al. Topical resiquimod 0.01% gel decreases herpes simplex virus type 2 genital shedding: a randomized, controlled trial. J. Infect. Dis. 195, 1324–1331 (2007).
Pockros, P. J. et al. Oral resiquimod in chronic HCV infection: safety and efficacy in 2 placebo-controlled, double-blind phase IIa studies. J. Hepatol. 47, 174–182 (2007).
Caron, G. et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-γ production by memory CD4+ T cells. J. Immunol. 175, 1551–1557 (2005).
Kronenberger, B. & Zeuzem, S. Current and future treatment options for HCV. Ann. Hepatol. 8, 103–112 (2009).
Barry, M. & Cooper, C. Review of hepatitis B surface antigen-1018 ISS adjuvant-containing vaccine safety and efficacy. Expert Opin. Biol. Ther. 7, 1731–1737 (2007).
Harandi, A. M., Davies, G. & Olesen, O. F. Vaccine adjuvants: scientific challenges and strategic initiatives. Expert Rev. Vaccines 8, 293–298 (2009).
Gu, M., Hine, P. M., James Jackson, W., Giri, L. & Nabors, G. S. Increased potency of BioThrax anthrax vaccine with the addition of the C-class CpG oligonucleotide adjuvant CPG 10109. Vaccine 25, 526–534 (2007).
Krieg, A. M. Toll-free vaccines? Nature Biotech. 25, 303–305 (2007).
Mata-Haro, V. et al. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316, 1628–1632 (2007). This study showed that the vaccine adjuvant MPLA has a bias towards TRIF-dependent signalling, which has important implications for the development of future therapies.
Gavin, A. L. et al. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science 314, 1936–1938 (2006).
van Duin, D., Medzhitov, R. & Shaw, A. C. Triggering TLR signaling in vaccination. Trends Immunol. 27, 49–55 (2006).
Jasani, B., Navabi, H. & Adams, M. Ampligen: a potential toll-like 3 receptor adjuvant for immunotherapy of cancer. Vaccine 27, 3401–3404 (2009).
Navabi, H. et al. A clinical grade poly I:C-analogue (Ampligen) promotes optimal DC maturation and Th1-type T cell responses of healthy donors and cancer patients in vitro. Vaccine 27, 107–115 (2009). A poly I:C analogue which stimulates dendritic cell maturation and IL-12 and induces much lower levels of IL-10 compared with poly I:C, has shown great promise as an adjuvant for cancer therapies.
Huleatt, J. W. et al. Potent immunogenicity and efficacy of a universal influenza vaccine candidate comprising a recombinant fusion protein linking influenza M2e to the TLR5 ligand flagellin. Vaccine 26, 201–214 (2008). This study showed that the use of a TLR-specific ligand coupled to an influenza protein can offer widespread protection against strains of influenza A.
Kanzler, H., Barrat, F. J., Hessel, E. M. & Coffman, R. L. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nature Med. 13, 552–559 (2007).
Gangloff, S. C. & Guenounou, M. Toll-like receptors and immune response in allergic disease. Clin. Rev. Allergy Immunol. 26, 115–125 (2004).
Kline, J. N. & Krieg, A. M. Toll-like receptor 9 activation with CpG oligodeoxynucleotides for asthma therapy. Drug News Perspect. 21, 434–439 (2008).
Heijink, I. H. & Van Oosterhout, A. J. Strategies for targeting T-cells in allergic diseases and asthma. Pharmacol. Ther. 112, 489–500 (2006).
Pastorelli, L., Pizarro, T. T., Cominelli, F. & Vecchi, M. Emerging drugs for the treatment of ulcerative colitis. Expert Opin. Emerg. Drugs 14, 505–521 (2009).
Baldrick, P., Richardson, D., Woroniecki, S. R. & Lees, B. Pollinex Quattro Ragweed: safety evaluation of a new allergy vaccine adjuvanted with monophosphoryl lipid A (MPL) for the treatment of ragweed pollen allergy. J. Appl. Toxicol. 27, 399–409 (2007).
DuBuske L. M., C. M.a.H. T. Significant reduction in combined symptom and medication score compared with placebo following MPL-adjuvanted uSCIT in patients with seasonal grass pollen allergy. J. of Allergy Clin. Immunol. 123, S216–S216 (2009).
Feldmann, M. Translating molecular insights in autoimmunity into effective therapy. Annu. Rev. Immunol. 27, 1–27 (2009).
Klareskog, L. et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet 363, 675–681 (2004).
van der Heijde, D. et al. Inhibition of radiographic progression with combination etanercept and methotrexate in patients with moderately active rheumatoid arthritis previously treated with monotherapy. Ann. Rheum. Dis. 68, 1113–1118 (2009). This study demonstrates that the use of a combination therapy consisting of the antiTNF drug etanercept and methotrexate is much more efficacious in treating patients with moderate rheumatoid arthritis than either therapy alone.
Macfarlane, D. E. & Manzel, L. Antagonism of immunostimulatory CpG-oligodeoxynucleotides by quinacrine, chloroquine, and structurally related compounds. J. Immunol. 160, 1122–1131 (1998).
Sun, S., Rao, N. L., Venable, J., Thurmond, R. & Karlsson, L. TLR7/9 antagonists as therapeutics for immune-mediated inflammatory disorders. Inflamm. Allergy Drug Targets 6, 223–235 (2007).
Lipford G. et al. Selective Toll-like receptor 7/8/9 antagonists for the oral treatment of autoimmune diseases. American College of Rheumatology 2007 Annual Scientific Meeting (2007). ACR website [online], (2007).
Barrat, F. J., Meeker, T., Chan, J. H., Guiducci, C. & Coffman, R. L. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur. J. Immunol. 37, 3582–3586 (2007). This study showed that in plasmacytoid dendritic cells isolated from patients with SLE, the TLR7 and TLR9 inhibitor IRS954 inhibited the production of IFNα in response to DNA and RNA from viruses. The compound prevented disease progression in SLE-prone mice.
Pawar, R. D. et al. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J. Am. Soc. Nephrol. 18, 1721–1731 (2007).
Jiang, W., Bhagat, L., Yu, D., Kandimalla, E. R. & Agrawal, S. IMO-3100, an antagonist of Toll-like receptors 7 and 9, modulates gene expression and regulatory networks induced by ligands. J. Immunol. 182, 48.25 (2009).
Mullarkey, M. et al. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J. Pharmacol. Exp. Ther. 304, 1093–1102 (2003).
Savov, J. D. et al. Toll-like receptor 4 antagonist (E5564) prevents the chronic airway response to inhaled lipopolysaccharide. Am. J. Physiol. Lung Cell. Mol. Physiol. 289, L329–L337 (2005).
Czeslick, E., Struppert, A., Simm, A. & Sablotzki, A. E5564 (Eritoran) inhibits lipopolysaccharide-induced cytokine production in human blood monocytes. Inflamm. Res. 55, 511–515 (2006).
Bennett-Guerrero, E. et al. A phase II, double-blind, placebo-controlled, ascending-dose study of Eritoran (E5564), a lipid A antagonist, in patients undergoing cardiac surgery with cardiopulmonary bypass. Anesth. Analg. 104, 378–383 (2007).
Wasan, K. M. et al. Influence of plasma cholesterol and triglyceride concentrations and eritoran (E5564) micelle size on its plasma pharmacokinetics and ex vivo activity following single intravenous bolus dose into healthy female rabbits. Pharm. Res. 25, 176–182 (2008).
Cluff, C. W. et al. Synthetic toll-like receptor 4 agonists stimulate innate resistance to infectious challenge. Infect. Immun. 73, 3044–3052 (2005).
Amlie-Lefond, C. et al. Innate immunity for biodefense: a strategy whose time has come. J. Allergy Clin. Immunol. 116, 1334–1342 (2005).
Ulevitch, R. J. Therapeutics targeting the innate immune system. Nature Rev. Immunol. 4, 512–520 (2004).
Ii, M. et al. A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-Chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol. Pharmacol. 69, 1288–1295 (2006).
Ledeboer, A. et al. The glial modulatory drug AV411 attenuates mechanical allodynia in rat models of neuropathic pain. Neuron Glia Biol. 2, 279–291 (2006).
Ledeboer, A., Hutchinson, M. R., Watkins, L. R. & Johnson, K. W. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opin. Investig. Drugs 16, 935–950 (2007).
Ungaro, R. et al. A novel toll-like receptor 4 (TLR4) antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. Am. J. Physiol. Gastrointest Liver Physiol. 296, G1167–G1179 (2009).
Dunn-Siegrist, I. et al. Pivotal involvement of Fcγ receptor IIA in the neutralization of lipopolysaccharide signaling via a potent novel anti-TLR4 monoclonal antibody 15C1. J. Biol. Chem. 282, 34817–34827 (2007).
Chen, K. et al. Toll-like receptors in inflammation, infection and cancer. Int. Immunopharmacol. 7, 1271–1285 (2007).
Urbonaviciute, V. et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J. Exp. Med. 205, 3007–3018 (2008).
Arslan, F., de Kleijn, D. P., Timmers, L., Doevendans, P. A. & Pasterkamp, G. Bridging innate immunity and myocardial ischemia/reperfusion injury: the search for therapeutic targets. Curr. Pharm. Des 14, 1205–1216 (2008).
Chong, A. J. et al. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J. Thorac. Cardiovasc. Surg. 128, 170–179 (2004).
Hua, F. et al. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res. 1262, 100–108 (2009).
Tang, S. C. et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl Acad. Sci. USA 104, 13798–13803 (2007).
Chang, Y. C., Kao, W. C., Wang, W. Y., Yang, R. B. & Peck, K. Identification and characterization of oligonucleotides that inhibit Toll-like receptor 2-associated immune responses. FASEB J. 23, 3078–3088 (2009). This paper shows the development of a novel technique for identifying antagonistic TLR2 aptamers. Functional peptides were identified using a NF-κB reporter assay. One of the identified molecules reduced NF-κB by 80%.
Sheedy, F. J. & O'Neill, L. A. Adding fuel to fire: microRNAs as a new class of mediators of inflammation. Ann. Rheum. Dis. 67 (Suppl. 3), 50–55 (2008).
Taganov, K. D., Boldin, M. P., Chang, K. J. & Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl Acad. Sci. USA 103, 12481–12486 (2006). This is the first study to show a miRNA can have a direct role on NF-κB signalling through the targeting of IRAK1 and TRAF6.
O'Connell, R. M., Chaudhuri, A. A., Rao, D. S. & Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl Acad. Sci. USA 106, 7113–7118 (2009).
Tili, E. et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-α stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 179, 5082–5089 (2007).
Harte, M. T. et al. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 197, 343–351 (2003).
McCoy, S. L., Kurtz, S. E., Macarthur, C. J., Trune, D. R. & Hefeneider, S. H. Identification of a peptide derived from vaccinia virus A52R protein that inhibits cytokine secretion in response to TLR-dependent signaling and reduces in vivo bacterial-induced inflammation. J. Immunol. 174, 3006–3014 (2005).
Tsung, A. et al. A novel inhibitory peptide of Toll-like receptor signaling limits lipopolysaccharide-induced production of inflammatory mediators and enhances survival in mice. Shock 27, 364–369 (2007).
Buckley, G. M. et al. IRAK-4 inhibitors. Part III: a series of imidazo[1,2-a]pyridines. Bioorg Med. Chem. Lett. 18, 3656–3660 (2008).
Cohen, P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 21, 317–324 (2009).
Koziczak-Holbro, M. et al. The critical role of kinase activity of interleukin-1 receptor-associated kinase 4 in animal models of joint inflammation. Arthritis Rheum. 60, 1661–1671 (2009).
Kawagoe, T. et al. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nature Immunol. 9, 684–691 (2008).
Nalepa, G., Rolfe, M. & Harper, J. W. Drug discovery in the ubiquitin-proteasome system. Nature Rev. Drug Discov. 5, 596–613 (2006).
Guedat, P. & Colland, F. Patented small molecule inhibitors in the ubiquitin proteasome system. BMC Biochem. 8 (Suppl. 1), S14 (2007).
Cossu, F. et al. Structural basis for bivalent smac-mimetics recognition in the IAP protein family. J. Mol. Biol. 392, 630–644 (2009).
Cossu, F. et al. Designing Smac-mimetics as antagonists of XIAP, cIAP1, and cIAP2. Biochem. Biophys. Res. Commun. 378, 162–167 (2009).
Vince, J. E. et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693 (2007). One of the first studies to show that IAP antagonists or SMAC mimetics could induce apoptosis of cancer cells by stimulating NF-κB and TNFα.
Vallabhapurapu, S. et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nature Immunol. 9, 1364–1370 (2008). This paper describes an alternative NF-κB activation pathway that involves an inhibitor of TLR signalling, TRAF2.
Latz, E. et al. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2–CD14 complex in a process that is distinct from the initiation of signal transduction. J. Biol. Chem. 277, 47834–47843 (2002).
Hughes, A. L. & Piontkivska, H. Functional diversification of the toll-like receptor gene family. Immunogenetics 60, 249–256 (2008).
Underhill, D. et al. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401, 811–815 (1999).
Medzhitov, R. & Janeway, C. J. The Toll receptor family and microbial recognition. Trends Microbiol. 8, 452–456 (2000).
Muzio, M. et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J. Immunol. 164, 5998–6004 (2000).
Zarember, K. A. & Godowski, P. J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 168, 554–561 (2002).
Strandskog, G., Ellingsen, T. & Jorgensen, J. B. Characterization of three distinct CpG oligonucleotide classes which differ in ability to induce IFN α/β activity and cell proliferation in Atlantic salmon (Salmo salar L.) leukocytes. Dev. Comp. Immunol. 31, 39–51 (2007).
Geddes, K., Magalhaes, J. G. & Girardin, S. E. Unleashing the therapeutic potential of NOD-like receptors. Nature Rev. Drug Discov. 8, 465–479 (2009).
Proell, M., Riedl, S. J., Fritz, J. H., Rojas, A. M. & Schwarzenbacher, R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS ONE 3, e2119 (2008).
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J. & Alnemri, E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Koizumi, K. et al. Single nucleotide polymorphisms in the gene encoding the major histocompatibility complex class II transactivator (CIITA) in systemic lupus erythematosus. Ann. Rheum. Dis. 64, 947–950 (2005).
Macaluso, F. et al. Polymorphisms in NACHT-LRR (NLR) genes in atopic dermatitis. Exp. Dermatol. 16, 692–698 (2007).
Magitta, N. F. et al. A coding polymorphism in NALP1 confers risk for autoimmune Addison's disease and type 1 diabetes. Genes Immun. 10, 120–124 (2009).
McGovern, D. P. et al. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum. Mol. Genet. 14, 1245–1250 (2005).
Skinningsrud, B. et al. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J. Clin. Endocrinol. Metab. 93, 3310–3317 (2008).
Weidinger, S. et al. Association of NOD1 polymorphisms with atopic eczema and related phenotypes. J. Allergy Clin. Immunol. 116, 177–184 (2005).
Noguchi, E., Homma, Y., Kang, X., Netea, M. G. & Ma, X. A Crohn's disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nature Immunol. 10, 471–479 (2009).
Ogura, Y. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411, 603–606 (2001).
Maksimovic, L. et al. New CIAS1 mutation and anakinra efficacy in overlapping of Muckle-Wells and familial cold autoinflammatory syndromes. Rheumatology 47, 309–310 (2008).
Zapata, J. M. et al. Lymphocyte-specific TRAF3 transgenic mice have enhanced humoral responses and develop plasmacytosis, autoimmunity, inflammation, and cancer. Blood 113, 4595–4603 (2009).
Bochud, P. Y. et al. Toll-like receptor 2 (TLR2) polymorphisms are associated with reversal reaction in leprosy. J. Infect. Dis. 197, 253–261 (2008).
Thuong, N. T. et al. A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun. 8, 422–428 (2007).
Lorenz, E., Mira, J., Cornish, K., Arbour, N. & Schwartz, D. A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect. Immun. 68, 6398–6401 (2000).
Agnese, D. et al. Human toll-like receptor 4 mutations but not CD14 polymorphisms are associated with an increased risk of Gram-negative infections. J. Infect. Dis. 186, 1522–1525 (2002).
Kiechl, S. et al. Toll-like receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 347, 185–192 (2002).
Sheedy, F. J., Marinou, I., O'Neill, L. A. & Wilson, A. G. The Mal/TIRAP S180L and TLR4 G299D polymorphisms are not associated with susceptibility to, or severity of, rheumatoid arthritis. Ann. Rheum. Dis. 67, 1328–1331 (2008).
Gewirtz, A. T. et al. Dominant-negative TLR5 polymorphism reduces adaptive immune response to flagellin and negatively associates with Crohn's disease. Am. J. Physiol. Gastrointest Liver Physiol. 290, G1157–G1163 (2006).
Castiblanco, J. et al. TIRAP (MAL) S180L polymorphism is a common protective factor against developing tuberculosis and systemic lupus erythematosus. Infect. Genet. Evol. 8, 541–544 (2008).
Khor, C. C. et al. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nature Genet. 39, 523–528 (2007).
Nejentsev, S. et al. Analysis of association of the TIRAP (MAL) S180L variant and tuberculosis in three populations. Nature Genet. 40, 261–262; author reply 262–263 (2008).
Ramasawmy, R. et al. Heterozygosity for the S180L variant of MAL/TIRAP, a gene expressing an adaptor protein in the toll-like receptor pathway, is associated with lower risk of developing chronic chagas cardiomyopathy. J. Infect. Dis. 199, 1838–1845 (2009).
Ferwerda, B. et al. Functional and genetic evidence that the Mal/TIRAP allele variant 180L has been selected by providing protection against septic shock. Proc. Natl Acad. Sci. USA 106, 10272–10277 (2009).
Demirci, F. Y. et al. Association of a common interferon regulatory factor 5 (IRF5) variant with increased risk of systemic lupus erythematosus (SLE). Ann. Hum. Genet. 71, 308–311 (2007).
Ferreiro-Neira, I. et al. Opposed independent effects and epistasis in the complex association of IRF5 to SLE. Genes Immun. 8, 429–438 (2007).
Tewfik, M. A. et al. Polymorphisms in interleukin-1 receptor-associated kinase 4 are associated with total serum IgE. Allergy 64, 746–753 (2009).
Day, N. et al. Interleukin receptor-associated kinase (IRAK-4) deficiency associated with bacterial infections and failure to sustain antibody responses. J. Pediatr. 144, 524–526 (2004).
Picard, C. et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 299, 2076–2079 (2003).
Medvedev, A. E. et al. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J. Exp. Med. 198, 521–531 (2003).
Picard, C. et al. Inherited human IRAK-4 deficiency: an update. Immunol. Res. 38, 347–352 (2007).
Dhiman, N. et al. Associations between SNPs in toll-like receptors and related intracellular signaling molecules and immune responses to measles vaccine: preliminary results. Vaccine 26, 1731–1736 (2008).
Sheedy, F. J. et al. Negative regulation of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nature Immunol. 11, 141–147 (2010).
Acknowledgements
E.H. would like to thank the Health Research Board and L.A.J.O. thanks Science Foundation Ireland for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
L.A.J.O is a cofounder of Opsona Therapeutics, which uses immune system research to develop new drugs and vaccines.
Related links
Related links
DATABASES
OMIM
ClinicalTrials.gov
FURTHER INFORMATION
Mologen AG's clinical development programe MGN-1703
Glossary
- Tumour necrosis factor.
-
(TNF). A pro-inflammatory cytokine involved in immune response. It induces apoptosis and inflammation.
- MD-2
-
A cell surface molecule that is associated with TLR4. It is required for immune signalling in response to lipopolysaccharide.
- Toll–interleukin-1 resistance (TIR) domain
-
A conserved intracellular domain of Toll and IL-1 families of receptors, which is also present in the adaptor molecules MYD88, MAL, TRIF and TRAM.
- Interleukin-1 receptor-associated kinase 4
-
(IRAK4). A kinase that interacts with MYD88 and TRAF6 and is crucial for TLR signalling.
- Nuclear factor-κB
-
(NF-κB). A transcription factor that is translocated to the nucleus following the stimuation of cell surface receptors by microbe-associated molecular patterns.
- Type I interferons
-
A class of cytokines, produced by plasmacytoid dendritic cells, that includes interferon α and β.
- Monophosphoryl lipid-A
-
(MPL). A non-toxic derivative of lipid-A from Gram-negative lipopolysaccharide.
- Coley's toxin
-
A mixture of lipopolysaccharides and bacterial DNA products named after William Coley, who in 1893 first used dead bacteria to improve the survival of inoperable patients with cancer.
- Regulatory T cells
-
A class of T cells that suppresses the immune response and prevents the development of autoimmune disorders.
- Structure–activity relationship
-
A medicinal chemistry approach that involves the correlation of structural features with the activity of compounds in a given assay to improve potency and/or efficacy.
- T-helper 1 cell (Th1) response
-
An immune response involved in cell-mediated immunity that is produced when dendritic cells interact with pre-T-helper cells to produce IFNγ, IL-1, IL-2 and TNF-β, and leads to the activation of macrophages.
- CpG-based oligonucleotides
-
Oligonucleotides with cytosine–phosphate–guanosine-containing motifs that are found mainly in bacterial and viral DNA and that induce an immune response through activation of TLR9. There are three classes of CpG-oligonucleotides, CpG-A, CpG-B and CpG-C.
- Immune modulatory oligonucleotides
-
(IMOs). DNA or RNA based oligonucleotides of varying length that are used to target TLR7, TLR8 or TLR9.
- T-helper 2 cell (Th2) response
-
Part of the humoral immune response that helps B-cells produce IL-4, IL-5, IL-6, IL-9, IL-10 and IL-13, which evoke a strong antibody response and are required for the production of IgE. This leads to eosinophil and mast-cell accumulation and an allergic response.
- Lipid-A
-
The biologically active part of LPS, its hydrophobic nature anchors LPS to the membrane.
- Flagellin
-
A protein constituent of a bacterium's flagellum that specifically activates TLR5.
- Ribavarin
-
An antiviral pro-drug that when metabolized resembles RNA purine molecules. It impedes DNA and RNA metabolism and interferes with viral replication. It is active against many viruses including influenza and hepatitis B.
- CD80/86
-
A cell surface protein found on activated B-cells and monocytes that provides the necessary stimulation to prime T cells. When this complex binds to CTLA-4 expressed on the surface of T helper cells, the T-cell response is reduced.
- Ectodomain of matrix protein 2
-
(M2e). A highly conserved region of the influenza A virus that consists of 23 amino acids of the M2 protein (the third integral membrane protein of the virus).
- Lipid-IVa
-
A precursor in the biosynthetic pathway for lipopolysaccharide that acts as a TLR4 antagonist.
- Systematic evolution of ligands by exponential enrichment
-
(SELEX). Also known as in vitro evolution. This process is used to produce either RNA or DNA oligonucleotides that specifically bind to a ligand. The process starts with a large library of oligonucleotides that are then exposed to the ligand. The process is repeated and the oligonucleotide sequence amplified until an aptamer that has a high affinity for the ligand is produced.
- microRNAs
-
(miRNAs). Small 20–23 nucleotide non-coding RNA molecules that fine-tune gene expression by targeting specific messanger RNA.
- Locked nucleic acid miRNA inhibitor
-
Nucleic acid analogues in which the ribose ring is 'locked' by a methylene bridge, resulting in increased thermal stability and discrimination with respect to target nucleic acids.
- Ubiquitination
-
A post-translational modification involving 3 enzymes: E1 the ubiquitin-activating enzyme, E2 the ubiquitin-conjugating enzyme and E3 the ubiquitin protein ligase. Tagging a molecule with K48-linked ubiquitins targets it for degradation by the proteasome
- Pellino3
-
A protein with E3 ubiquitin ligase activity that activates the p38 MAP kinase pathway to negatively regulate TLR signalling.
- NF-κB essential modifier
-
(NEMO). The regulatory subunit of the IκB kinase complex that phosphorylates inhibitors of NF-κB, leading to their degradation and the translocation of NF-κB into the nucleus.
- IκB kinase (IKK) complex
-
A complex that contains two catalytic subunits, IKKα and IKKβ, and the regulatory subunit NEMO that acts as a master regulator of NF-κB activation.
- Inhibitors of apoptosis
-
(IAPs). Endogenous inhibitors of programmed cell death or apoptosis, which bind caspases and inhibit their activation.
- Second mitochondria-derived activator of caspase
-
(SMAC). A protein that promotes cytochrome-c-dependent cell activation by eliminating IAP, which leads to cell apoptosis.
Rights and permissions
About this article
Cite this article
Hennessy, E., Parker, A. & O'Neill, L. Targeting Toll-like receptors: emerging therapeutics?. Nat Rev Drug Discov 9, 293–307 (2010). https://doi.org/10.1038/nrd3203
Issue Date:
DOI: https://doi.org/10.1038/nrd3203
This article is cited by
-
Bacteria-based multiplex system eradicates recurrent infections with drug-resistant bacteria via photothermal killing and protective immunity elicitation
Biomaterials Research (2023)
-
Vaccine adjuvants: mechanisms and platforms
Signal Transduction and Targeted Therapy (2023)
-
Hederacoside C ameliorates colitis via restoring impaired intestinal barrier through moderating S100A9/MAPK and neutrophil recruitment inactivation
Acta Pharmacologica Sinica (2023)
-
A review of immune modulators and immunotherapy in infectious diseases
Molecular and Cellular Biochemistry (2023)
-
A Novel Multiepitope Vaccine Against Bladder Cancer Based on CTL and HTL Epitopes for Induction of Strong Immune Using Immunoinformatics Approaches
International Journal of Peptide Research and Therapeutics (2022)
