
Abstract
The programmed death protein 1 (PD-1) and its ligand (PD-L1) represent a well-characterized immune checkpoint in cancer, effectively targeted by monoclonal antibodies that are approved for routine clinical use. The regulation of PD-L1 expression is complex, varies between different tumor types and occurs at the genetic, transcriptional and post-transcriptional levels. Copy number alterations of PD-L1 locus have been reported with varying frequency in several tumor types. At the transcriptional level, a number of transcriptional factors seem to regulate PD-L1 expression including HIF-1, STAT3, NF-κΒ, and AP-1. Activation of common oncogenic pathways such as JAK/STAT, RAS/ERK, or PI3K/AKT/MTOR, as well as treatment with cytotoxic agents have also been shown to affect tumoral PD-L1 expression. Correlative studies of clinical trials with PD-1/PD-L1 inhibitors have so far shown markedly discordant results regarding the value of PD-L1 expression as a marker of response to treatment. As the indications for immune checkpoint inhibition broaden, understanding the regulation of PD-L1 in cancer will be of utmost importance for defining its role as predictive marker but also for optimizing strategies for cancer immunotherapy. Here, we review the current knowledge of PD-L1 regulation, and its use as biomarker and as therapeutic target in cancer.
Similar content being viewed by others


Introduction
Cancer development and progression raises a strong antitumor immune response through which the immune system can eliminate cancer cells. This immunosurveillance theory describes the complex interactions between immune and cancer cells, divided in three distinct but often overlapping stages: elimination, equilibrium, and evasion. Thus, tumors can suppress immunity and escape eradication; evading immune destruction has been characterized as a hallmark of cancer [1, 2].
Programmed death protein 1 (PD-1) and its ligand (PD-L1) have been recognized as inhibitory molecules that cause impaired immune response against cancer cells. Therapeutic antibodies targeting PD-1/PD-L1 have been introduced into clinical practice, leading to better patient outcomes [3]. Immune checkpoint regulation has been under intense investigation over the last decades, however, the underlying mechanisms regulating the PD1 and PD-L1 expression are not fully understood; several oncogenic signaling pathways, epigenetic modifications, and genetic variations have been suggested. The aim of this review is to summarize the current knowledge on PD-L1 regulation and its emerging role as a target in cancer immunotherapy.
Immune surveillance: the role of PD-1/PD-L1 axis as immune checkpoint
PD-1 (CD279) is a transmembrane protein, member of the CD28 family. It is mainly expressed on activated T cells but it can also be detected in other cells such as B- and natural killer (NK) cells upon induction [4]. PD-1 has two ligands, PD-L1 (CD274, B7-H1) and PD-L2 (CD273, B7-DC), which belong to the B7-CD28 protein family [5]. PD-L1 is expressed on tumor cells but it can also be present on the surface of other cell types including T cells, B cells, dendritic cells, macrophages, mesenchymal stem cells, epithelial, endothelial cells, and as recently shown, brown adipocytes [6]. PD-L2 is typically expressed in antigen-presenting cells (APCs). PD-L1 is expressed upon stimulation of cytokine interferon-γ (IFNg), secreted by activated T cells [7, 8].
PD-L1 and PD-L2 are encoded by the CD274 and PDCD1LG2 genes, respectively, located on chromosome 9p.24.1, whereas PD-1 is encoded by the PDCD1 gene located on chromosome 2q37.3 [4].
PD-1/PD-L1 axis plays an important role in the regulation of T-cell immunity and has been also implicated in autoimmunity and infection [9]. The PD-1/PD-L1 interaction has been characterized as an “immune checkpoint” due to its impact on the orchestration of immune response against tumor antigens. Along with cytotoxic T-lymphocyte-associated protein 4 (CTLA-4, CD152), they represent immunological “brakes” that modulate T-cell activation leading to an impaired immunosurveillance.
T-cell activation involves a two signal-model; APCs require a first signal from T-cell receptor (TCR), which recognizes the antigen along with the major histocompatibility complex (MHC) presented on the surface of APC. The second signal includes the co-stimulatory interaction between CD28 on the surface of T cells and CD80 (B7.1) or CD86 (B7.2) on the surface of APC [10, 11].
The engagement of PD-1 with its ligands leads to the inhibition of T-cell activation and response, via mechanisms that include blocking of proliferation, induction of apoptosis, and regulatory T-cell differentiation and therefore immune inhibition [11]. Blocking the PD-1/PD-L1 axis with potent monoclonal antibodies may reverse the impaired anticancer immunity and thus represents an appealing target of cancer immunotherapy [12].
The genetic basis of PD-L1 expression in cancer
The genetic aberrations of the PD-L1/PD-L2 gene loci represent a key mechanism of PD-L1 expression both in solid and hematologic tumors. Studies of copy number alterations (CNAs) have been reported in several tumor types (Table 1). The highest frequencies of CNAs have been seen in squamous cell carcinomas of vulva and cervix and triple-negative breast cancer (TNBC), as well as in classical Hodgkin lymphoma (cHL) and primary mediastinal B-cell lymphoma (PMBCL). Contrary, low or absent CNAs have been reported in small and non-small cell lung cancer (NSCLC) and in diffuse large B-cell lymphomas (DLBCL). In general, copy number gains and especially amplifications are well correlated with the protein levels of PD-L1. Given the challenges in determining the protein levels of PD-L1 as detailed below, detection of CNAs is an attractive alternative for identifying patients who could benefit from treatment with checkpoint inhibitors. Table 1 summarizes the current literature of the genetic regulation of PD-L1 [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. In addition to these individual studies, a large in silico analysis of CNAs in PD-L1 has been conducted using the Cancer Genome Atlas datasets (22 cancer types, 9771 tumors). Interestingly, deletions of 9p24.1 were more common than gains in this analysis and were found mostly in melanoma and NSCLC, with gains occurring frequently in ovarian, head and neck, bladder, and cervical carcinomas [29].
Furthermore, a novel genetic regulatory mechanism of PD-L1 gene expression involving the disruption of its 3′ untranslated region (3′-UTR) has been shown in multiple tumor types including T-cell leukemia/lymphoma, DLBCL, and gastric adenocarcinoma. Through interruption of PD-L1 3′-UTR by structural variation, a deviant increase in PD-L1 transcripts occurs leading to immune escape in murine EG7-OVA cancer cells, which in turn can be reversed by PD-L1/PD-1 inhibition [30].
PD-L1 regulation via oncogenic signaling pathways
RAS/RAF/MEK/MAPK-ERK pathway
The mitogen-activated protein kinase (MAPK) pathway is crucial for various functions in normal cells, including growth and differentiation. Its role is also important in carcinogenesis because its activation leads to cancer development [31]. The ERK-MAPK pathway has been shown to regulate PD-L1 expression in different cancer types. Both pharmacologic inhibition of mitogen-activated protein kinase (MEK) and small interfering RNA (siRNA) knockdown of ERK1/2 resulted in decreased levels of PD-L1 in melanoma cells resistant to BRAF inhibition [32]. Interestingly, in TNBC cells, MEK inhibition resulted in upregulation of MHC II and PD-L1 expression both in vitro and in vivo, whereas combined MEK/PD-1 inhibition increased the effectiveness of antitumor immunity [33]. MAPK signaling pathway was also responsible for the ectopic expression of PD-L1 in v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS)-mutant NSCLC cell lines, as revealed by the decrease in PD-L1 levels after both MEK and extracellular signal-regulated MAP kinase (ERK) abrogation [34]. In another study, Toll-like receptor 4 activation resulted in upregulation of PD-L1 in bladder cancer cells. The use of both ERK and JNK inhibitors abrogated PD-L1 expression, further supporting the contribution of MAPK signaling in PD-L1 regulation [35]. Moreover, the interaction of tyrosine kinase receptor c-Met with its ligand hepatocyte growth factor (HGF) induced Ras activation. Ablation of Ras effect led to downregulation of c-Met-mediated expression of PD-L1 in renal cancer cells [36].
KRAS activation may also induce PD-L1 expression, as it resulted in stabilization of PD-L1 mRNA transcript assessed through Adenylate-uridylate-rich elements identification in its 3′-UTR in lung cancer cell lines. Additionally, MEK and Phosphoinositide 3-kinase (PI3K) inhibition led to decreased PD-L1 levels and enhanced effectiveness of antitumor immunity in vivo [37].
PI3K/PTEN/Akt/mTOR pathway
The PI3K/Akt/mTOR signaling represents another pathway that affects immune surveillance through the regulation of PD-L1. Its activation by either oncogenic PIK3CA mutations (catalytic subunit alpha of PI3K) or by loss-of-function mutations of its negative regulator, phosphatase and tensin homolog (PTEN) modulates immune responses contributing to a survival benefit of cancer cells [38]. In human gliomas, loss of PTEN and activation of PI3K pathway enhanced PD-L1 expression [39]. In TNBC, knockdown of PTEN by short hairpin RNA resulted in elevated levels of both PD-L1 protein expression and mRNA transcripts, whereas inhibition of Akt and mechanistic target of rapamycin (mTOR) decreased PD-L1 expression [40]. In a murine model of lung SCC, concurrent inactivation of PTEN and Lbk1 resulted in increased levels of PD-L1 [41]. PI3K inhibition, resulted in PD-L1 downregulation in different cancer types including renal cell carcinoma through HGF/c-Met [36], KRAS- or EGFR-mutated NSCLC [42] and melanoma [32]. Conversely, LY294002 did not abrogate PD-L1 expression in bladder cancer cells [35]. Moreover, mTOR inhibition with rapamycin reduced levels of PD-L1, both in human cell lines and in murine models of NSCLC and combined treatment with rapamycin and anti-PD-1 antibody inhibited tumor growth in mice [42].
Epidermal growth factor receptor (EGFR)
EGFR is commonly mutated in NSCLC and has been associated with PD-L1 upregulation in these tumors [43]. PD-L1 was overexpressed in EGFR-mutant murine lung cancer, whereas treatment with an anti-PD-1 antibody restrained tumor growth. Forced ectopic expression of mutant EGFR on bronchial epithelial cells resulted in PD-L1 upregulation, whereas this effect was abolished upon treatment with EGFR tyrosine kinase inhibitors [44, 45]. The EGFR-mediated regulation of PD-L1 in EGFR mutant NSCLC was dependent on MAPK pathway activation. Inhibition of ERK1/2/c-Jun resulted in reduced PD-L1 levels in PD-L1 overexpressing lung cancer cells [46]. In another more recent study, EGFR was shown to regulate the expression of PD-L1 through the activation of Interleukin-6 (IL-6)/Janus Kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) pathway in EGFR-driven NSCLC [47].
EML4-ALK
PD-L1 upregulation has been observed in patients with NSCLC harboring the anaplastic lymphoma kinase (ALK) and echinoderm microtubule-associated protein like-4 (EML4) chromosomal rearrangement. Activation of EML4-ALK was associated with increased PD-L1 expression; furthermore, treatment with either the ALK inhibitor alectinib or ALK gene silencing with siRNA abrogated this effect. Notably, PD-L1 upregulation was dependent on MAPK/ERK/MEK and PI3K/Akt signaling pathways [48]. In another study using pulmonary adenocarcinoma cell lines, EML4-ALK transcriptionally regulated PD-L1 via STAT3 and HIF-1a [49]. These studies indicate the different ways in which this chimeric protein can regulate the expression of PD-L1 and thus reveal the complexity of signaling pathways and their downstream targets. The various crosstalks in the cellular level can influence anticancer immunity and at the same time offer possible appealing therapeutic targets.
Transcriptional control of PD-L1
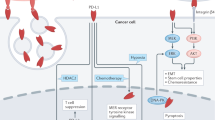
The transcriptional regulation of PD-L1 is summarized in Fig. 1.

Transcriptional and post-transcriptional control of PD-L1 in cancer. Regulation of PD-L1 is complex and occurs at different levels. Several signaling pathways are involved including RAS/RAF/MEK/MAPK-ERK and PI3K/PTEN/Akt/mTOR. Their activation by oncogenic and/or loss-of-function mutations can lead either to direct action on target genes or to the activation of transcription factors. Such molecules as STAT3, STAT1, c-Jun, HIFs, or NF-κB can shuttle into the nucleus, bind to specific sites on PD-L1 gene promoter and induce its expression. PD-L1 is also regulated post-transcriptionally by microRNAs, which bind to mRNA and lead to its translational repression or enhancement
The JAK/STAT pathway
STAT3 plays a key role in promoting cancer cell survival and proliferation, as well as creating immunosuppressive and thus pro-carcinogenic conditions in the tumor microenvironment (TME) [50]. Furthermore, STAT3 is involved in PD-L1 regulation in various cancer types. In nucleophosmin-anaplastic large-cell lymphoma kinase (NPM-ALK) positive anaplastic large-cell lymphoma (ALCL), STAT3 is activated by NPM-ALK oncoprotein through JAK3 activation, binds physically to the PD-L1 gene promoter, and induces its expression in vitro and in vivo [51]. This STAT3-mediated transcriptional regulation of PD-L1 has been recently shown in another T-cell lymphoma, namely the ALK-negative ALCL. STAT3 gene silencing led to decreased PD-L1 levels in ALK-ALCL [52] and also in KRAS-mutant NSCLC cell lines [34]. By contrast, chromatin immunoprecipitation analysis did not show active binding of STAT3 directly on the promoter of PD-L1 in melanoma cells, despite the presence of putative binding sites of STAT3 on the promoter identified in silico. Abrogation of STAT3 resulted in enhancement of PD-L1 construct activity mediated by IFNg [53]. PD-L1 was also induced by latent membrane protein-1 in Epstein–Barr virus (EBV)-associated nasopharyngeal carcinomas (NPC) through JAK3/STAT3 activation [54].
Another STAT family member, STAT1, is considered to be a tumor suppressor that reduces proliferation, induces apoptosis, and enhances cancer immunosurveillance [55]. Accumulating evidence indicates the emerging role of STAT1 in tumor growth, immune suppression, and therapeutic resistance [56]. Upon stimulation with IFNg, STAT1 activation resulted in PD-L1 upregulation and in reduction of NK-cell activity against tumor cells in multiple myeloma, acute myeloid leukemia (AML), and acute lymphoblastic leukemia (ALL) [57]. Similarly, STAT1 inhibition led to decreased PD-L1 levels in myeloma cells and thus suppressed the antitumor function of cytotoxic T cells [58]. PD-L1 upregulation was JAK2/STAT1-dependent in head and neck cancer with wild-type EGFR, whereas JAK2 inhibition resulted in both basal and EGF-mediated downregulation of PD-L1. Moreover, knockdown of STAT1 gene abolished both IFNg- and EGF-mediated upregulation of PD-L1. Of note, EGFR activation promotes phosphorylation of STAT1, which in turn binds to the promoter of PD-L1 and controls its expression [59]. Although putative binding sites for STAT1 on PD-L1 promoter have been postulated, active binding of STAT1 on PD-L1 gene promoter could not be demonstrated in melanoma cells [53].
Interferon regulatory factor 1 (IRF1) is a downstream effector of STAT1 upon IFNg stimulation. Its role is crucial in both constitutive and IFNg-mediated upregulation of PD-L1. Inhibition of IRF1 activity or expression resulted in decreased PD-L1 levels in human lung cancer cells [60]. The key role of IRF1 and interferon receptor pathway in the regulation of PD-L1 has also been implied in melanoma, where putative binding sites for IRF1 have been identified in the PD-L1 promoter and abrogation of IRF1 site resulted in reduced PD-L1 levels [53, 61]. Recently, another novel mechanism of PD-L1 regulation by DNA double-strand breaks (DSBs) was unveiled. This DSB-dependent PD-L1 upregulation was mediated by the activation of STAT1/STAT3 phosphorylation and IRF1 [62].
Hypoxia-inducible factors (HIFs)
Hypoxia signaling represents an important pathway in oncogenesis. HIF-1a and HIF-2a are the major components of a transcriptional complex, through which tumor cells adapt to hypoxic conditions. HIF stabilization leads to its binding to specific regions called hypoxia response elements (HRE) on certain gene promoters [63]. High levels of HIF-1 have been correlated with both worse outcomes and resistance to cytotoxic therapy [64]. Intriguingly, HIF-1 expression by different cellular sub-populations of the innate and adaptive immunity can modify antitumor activity by repressing the effective T-cell response and alter TME to promote tumor cell survival [63]. A recent study revealed that HIF-1α guided CD8 + T-cell migration and function, whereas its depletion on T cells resulted in increased tumor growth and impaired antitumor control [65]. One of the mechanisms by which hypoxia signaling impairs T-cell functionality is the induction of PD-L1 on myeloid-derived suppressor cells under hypoxic conditions. Indeed, HIF-1a transcriptionally regulates PD-L1 expression by binding on HRE of its promoter [66]. Furthermore, PD-L1 may be a target of HIF2a in clear cell renal cell carcinoma (ccRCC) cells in which the tumor-suppressor pVHL was abrogated. Upon deficiency of pVHL increased PD-L1 levels, associated with HIF-2a activation, were observed in vitro [67]. Similar results were obtained from ccRCC patient samples with VHL loss-of-function mutations, where a positive correlation was seen between PD-L1 expression, HIF-2a expression and VHL mutations. Of note, HIF-2a transcriptionally regulates PD-L1 by binding to the active HRE of its promoter [68]. Moreover, STAT3 can cooperate with HIF-1, but not HIF-2, in the regulation of HIF target genes in response to hypoxia. Inhibition of STAT3 expression or activity in breast and RCC cell lines reduced the expression of genes targeted by HIF-1 [69]. These findings support the idea of combining HIF-targeting therapies and immunotherapy.
The role of nuclear factor kappa B (NF-κB)
NF-κB is a master transcription factor activated in several cancer types, promoting inflammation, inhibiting apoptosis, and impairing effective antitumor immunity [70]. The NF-κB family contains seven members, with the most representative being the p65 RelA/p50. This cytoplasmic heterodimer translocates to the nucleus and acts as a transcription factor of κB upon degradation of the IκΒ-α inhibitor [71, 72]. In melanoma cells, NF-κB mediated PD-L1 overexpression induced by IFN-γ. PD-L1 upregulation by NF-κΒ was independent of STAT3 and c-Jun, whereas targeting of MAPK and PI3K signaling pathways had a minor impact on PD-L1 expression [72]. Notably, STAT3 regulates and cooperates with NF-κΒ in additional cancer types [73]. For example, PD-L1 regulation may be dependent on p65/NF-κB and mediated by LMP1 in EBV-positive NPC, as inhibition of NF-κB activity resulted in decreased PD-L1 levels [54].
The Myc oncogene
Myc plays a pivotal role in carcinogenesis by controlling cell proliferation and survival in various cell systems. Tumor regression after Myc inactivation is associated with a not fully understood immune response, as reflected by the accumulation of CD4 + T cells [74,75,76]. Furthermore, a novel role of Myc was recently revealed in the context of avoiding effective cancer immunosurveillance. Using a Tet-off MYC-dependent mouse model of T-ALL (MYC T-ALL), Casey et al. showed that Myc transcriptionally regulates PD-L1 and CD47, an inhibitory regulator of the innate immune system [77]. Moreover, forced expression of PD-L1 and CD47 upon Myc inactivation was correlated with worse antitumor immune response as indicated by the reduction of macrophages and CD4 + T cells in TME, tumor progression, and maintenance of angiogenesis and senescence [78]. Elucidating the role of Myc in the regulation of immune-mediated antitumor response, the potential crosstalks with other oncogenic pathways and the immune infiltrate in TME may pave the way for the use of immune checkpoint inhibitors in patients with Myc-overexpressing tumors [79]. A recent work on ALK-negative ALCL also supports a Myc-mediated regulation of PD-L1, as forced expression of Myc led to PD-L1 upregulation in cell lines showing low baseline levels of PD-L1. Similarly, both inhibition and silencing of Myc resulted in PD-L1 downregulation in lymphoma cells [52].
The bromodomain and extraterminal (BET) protein BRD4
BET proteins modulate gene expression through enzymes that regulate chromatin and histone modification [80]. Specifically, the BET protein BRD4 acts through RNA polymerase II by binding to the acetyl-lysine region of histones [81]. Inhibition of BRD4 by the JQ1 inhibitor decreased PD-L1 expression and tumor growth. BRD4 gene silencing also resulted in decreased PD-L1 levels in mouse models and in ovarian cancer cell lines. Notably, BRD4 transcriptionally regulated PD-L1 by binding on its promoter [82]. Similarly, in a recent study on B-cell lymphoma, BET inhibitors enhanced effective antitumor immunity through regulation of PD-L1, whereas inhibition and genetic ablation of BRD4 resulted in suppression of PD-L1 expression in a transcriptional, Myc-independent, manner. Moreover, BRD4 synergized with IRF1 to regulate PD-L1 expression induced by IFN-γ [83]. Also, another BET inhibitor (I-BET151) was shown to abrogate NF-Κβ activity in melanoma, both in vitro and in vivo, thus indirectly affecting PD-L1 expression [84].
Histone deacetylases (HDACs)
The role of the epigenetic modifiers HDACs in the modification of non-histone targets, including those participating in tumor-host interactions, has recently been investigated [85, 86]. In a study in melanoma, both inhibition and depletion of HDAC6 resulted in reduced PD-L1 levels in vitro and in vivo. PD-L1 regulation by HDAC6 was mediated by STAT3 and both HDAC6 and STAT3 were recruited to the PD-L1 gene promoter [87]. It should be noted that HDAC have pleiotropic effects within both the innate and adaptive immune response, and may thus affect PD-L1 levels via interferons [88].
The role of cell cycle
Cyclin-dependent kinases (CDKs) have a key role in cell cycle [89]. Cyclin-dependent kinase 5 (Cdk5) is a serine-threonine kinase important in central nervous system function [90] and other cellular functions [91, 92]. In a study of medulloblastoma, depletion of Cdk5 led to the upregulation of interferon regulatory factor 2 and interferon regulatory factor binding protein 2, which in turn, suppressed the expression of PD-L1. Cdk5 was thus necessary for PD-L1 upregulation after IFN-γ stimulation through STA1/IRF1 axis and its disruption led to tumor rejection in a CD4 + T-cell-dependent manner in medulloblastoma mouse models [93]. These data highlight Cdk5 as a novel target for interventions in combination with immune checkpoint blockade. Additionally, CDK4/6 inhibition has been recently shown to enhance antitumor immunity through increased T-cell cytotoxicity and Treg suppression [94]. This is discussed in detail in the post-translational regulation of PD-L1 hereunder.
The AP-1 transcription factors
c-Jun, the best known member of the AP-1 family, represents another transcription factor that is implicated in PD-L1 gene regulation. Knockdown of c-Jun resulted in decreased levels of PD-L1 in melanoma cells resistant to BRAF inhibitors [32], and co-activation of STAT3 and the subsequent formation of a transcriptional complex further enhanced these effects [95]. Similarly, combined knockdown of c-Jun and STAT3 genes in the same melanoma model showed a synergistic effect on PD-L1 downregulation [32]. Additionally, c-Jun and JUNB have been shown to bind AP-1 sites in the PD-L1 promoter in HL cells [96] and in KRAS-mutant NSCLC. In lung adenocarcinoma cell lines, the transcriptional activity was subjected to MAPK signaling pathway [34]. MAPK/AP-1 was also shown to contribute to LMP1-mediated upregulation of PD-L1 in EBV-associated NPC [54].
The ambivalent role of p53
The tumor-suppressor gene p53 has been implicated in antitumor immunity by regulating several genes involved in the immune system. Indeed, immune checkpoint regulation has been shown to represent a major target of p53 [97]. Paradoxically, activation of wild-type p53 using the small molecule Nutlin-3a resulted in increased expression of PD-L1 in human breast cancer [98] and in ALK-negative ALCL cells [52]. In p53-mutated NSCLC, downregulation of miR-34 resulted in increased PD-L1 levels [99], whereas an inverse correlation between miR-34a and PD-L1 was also confirmed in AML [100].
MicroRNAs
MicroRNAs can bind to 3′-UTR of mRNAs and lead to their degradation or translational repression [101]. MiR-513 was shown to increase PD-L1 expression in cholangiocytes [102], whereas mutation in the 3′-UTR of PD-L1 mRNA led to overexpression of the protein by preventing miR-570 binding in gastric cancer [103, 104]. On the contrary, miR-197 downregulated PD-L1 by affecting STAT3 in platinum-resistant NSCLC [105], whereas miR-138-5p was associated with decreased levels of PD-L1 in colorectal cancer (CRC) [106]. Also in CRC, miR-20b, miR-21, and miR-130b caused PD-L1 upregulation through attenuation of PTEN [107].
Post-translational regulation of PD-L1
The role of ubiquitination
In a recent study by Lim et al., a novel regulatory mechanism involving the fifth protein element of COP9 signalosome complex (CSN5), also known as Jab1, was revealed in breast cancer. CSN5 has been associated with increased proliferation, decreased apoptotic rates, and survival of cancer cells [108]. Under chronic inflammatory conditions, tumor necrosis factor alpha (TNF-α), secreted mostly by macrophages, led to PD-L1 stabilization and therefore to an immunosuppressive profile of the tumor environment [61]. The stabilization of PD-L1 by TNF-a was shown to be mediated by NF-κΒ subunit RelA/p65, which binds on the promoter of CSN5 gene and has a direct effect on its regulation. CSN5 in turn, prevents the ubiquitination of PD-L1, hinders its degradation and as a result enhances tumor escape from immunosurveillance. Indeed, CSN5 inhibition or gene silencing abolished PD-L1 expression and tumor proliferation in vivo. Curcumin, a CSN5 inhibitor, induced better responses to anti-CTLA-4 treatment in vitro, indicating the potential of combinational administration of immune checkpoint with CSN5 inhibitors [61, 109, 110]. In another in vitro study, induction of both PD-L1 ubiquitination and PD-L1 protein levels was noted upon treatment with epidermal growth factor. An increase of mono- and multiubiquitination of PD-L1 was seen, an effect that was abrogated upon inhibition of the EGFR pathway and/or ubiquitin E1 activating enzyme [111]. Furthermore, a recent study demonstrated a novel role of cyclin D-CDK4 and cullin 3-speckle-type POZ protein (SPOP) E3 ligase in regulating the expression of PD-L1. Cyclin D1-CDK4 was shown to phosphorylate SPOP and lead to ubiquitination-mediated PD-L1 destabilization. Thus, either inhibition of CDK4/6 or loss-of-function mutations of SPOP led to increased levels of PD-L1 and reduced tumor-infiltrating lymphocytes. Additionally, treatment with a CDK4/6 inhibitor and an anti-PD-1 antibody resulted in tumor regression and improved survival in vivo [112].
Lysosomal-mediated degradation
CKLF-like MARVEL transmembrane domain containing protein 6 (CMTM6) was recently identified as a novel regulator of PD-L1 [113, 114]. CMTM6—a tetraspanin protein—interacted with PD-L1 through its transmembrane domain and regulated PD-L1 expression in cancer and myeloid cells, both in vitro and in vivo [115]. Depletion of CMTM6 did not influence the CD274 transcript, but led to reduction of PD-L1 protein expression and augmentation of antitumor immunity. The mechanism of action of CMTM6 involves the avoidance of PD-L1 lysosome-mediated degradation, probably through prevention of its ubiquitination, as these two proteins are co-localized in the plasma membrane [116].
The role of glycosylation
N-glycosylation represents a crucial post-translational modification determining protein formation, functionality, and interaction with other proteins [117]. A novel association between procedure-glycosylation and ubiquitination in the regulation of PD-L1 has recently been unveiled. In basal-like breast cancer cells, N-glycosylation of PD-L1 (highly at sites N35, N192, N200, and N219) led to protein stabilization and avoidance of its degradation by 26S proteasome. In contrast, non-glycosylated forms interrelated with Glycogen synthase kinase 3 beta (GSK3β), which in turn phosphorylated PD-L1 resulting in its degradation. Inhibition of GSK3β activity augmented immune suppression by tumor cells both in vitro and in vivo. Furthermore, EGFR promoted inactivation of GSK3β, and EGFR signaling blockade reversed stabilization of PD-L1 and led to enhanced antitumor responses [118]. In another study, N-linked glycosylation of PD-L1 (gPD-L1) was shown to increase PD-L1/PD-1 interaction, and consequently immunosuppression in TNBC. Its targeting with monoclonal antibodies or drug-conjugated gPD-L1 was thus proposed as a promising target of post-translational modifications of immune checkpoints [119].
Effect of chemotherapy in PD-L1 expression
Chemotherapeutic agents, apart from their direct cytotoxic effects on cancer cells, can also modulate immune responses against tumors [120, 121]. Treatment with paclitaxel, etoposide and 5-fluorouracil induced PD-L1 expression in breast cancer cell lines in a dose-dependent manner [122]. Paclitaxel was also associated with elevated levels of PD-L1 in human CRC and hepatocellular carcinoma cell lines. This regulation was dependent on MAPK activation [123]. Likewise, cisplatin induced PD-L1 expression in hepatoma cells in ERK1/2 phosphorylation-dependent manner [124]. In another study, doxorubicin led to PD-L1 downregulation on cell surface and a simultaneous PD-L1 upregulation in the nucleus of breast cancer cells. Nuclear PD-L1 expression was accompanied by nuclear AKT phosphorylation and proved to be dependent on PI3K/AKT pathway, whereas knockdown of PD-L1 was associated with enhanced doxorubicin-mediated apoptosis [125].
Targeting immune checkpoint regulators: the era of immunotherapy in cancer
The introduction of systemic cancer immunotherapy in clinical practice significantly predates the first randomized trials of immune checkpoint inhibitors. Despite the occurrence of rare, prolonged complete remissions in patients with metastatic melanoma and ccRCC [126, 127], the use of high-dose IL-2 was restricted by the significant, often fatal adverse events and the need for intensive monitoring and experience in its administration, whereas the use of IFNg in ccRCC was characterized by its perceived low efficacy [128]. The clinical application of cancer immunotherapy had remained stagnant until the first checkpoint inhibitor received regulatory approval for use in metastatic melanoma, the CTLA-4 inhibitor ipilimumab. Ipilimumab exhibits several recurring characteristics of immunotherapy: slow induction of response, a striking disassociation between imaging-assessed objective responses and survival, which led to the introduction of immune-related response criteria [129], unique patterns of toxicity termed “immune-related adverse events” [130] and robust, durable improvements in terms of patient survival [131].
Shortly after the approval of ipilimumab the first trials of PD-1 and later PD-L1 inhibitors were published. Their results have vastly changed the treatment landscape in multiple human malignancies, adding a new category of effective and, compared with cytotoxic chemotherapy, less toxic agents to the therapeutic armamentarium. The results of the published phase 3 trials are presented in Table 2 [132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148], whereas a selection of ongoing randomized trials in an ever-expanding list of indications, both at refractory disease, as well as in earlier lines of therapy or at the adjuvant setting is presented in Table 3. The results of these trials are eagerly awaited, because there are high unmet needs in many of the indications that these agents are being tested. Of interest are also hematologic malignancies; preliminary trials report impressive response rates in otherwise refractory disease [149], believed to be driven by both the inherent role of the PD-1/PD-L1 axis in the evasion of immunosurveillance in lymphoid tumors, particularly in those with a viral etiology [150], and by the presumed significance of PDL1 and PDL2 amplification in the biology of certain neoplasms such as Hodgkin lymphoma [22]. In contrast, the recent discontinuation of the ongoing phase 3 trials in multiple myeloma due to an increased risk of death underscores the fact that better understanding of the underlying immune mechanisms is still needed.
Importantly, a new generation of clinical trials has been initiated and initial results are already available regarding a multi-faceted attempt to improve upon the efficacy of PD-1/PD-L1 inhibitors as monotherapy: their combination with CTLA-4 inhibitors, already shown to improve outcomes in metastatic melanoma [141] and pursued in other malignancies including NSCLC and SCLC; their combination with cytotoxic chemotherapy, based upon the premise of the prevention of early disease progression due to the simultaneous administration of chemotherapy and the release of neoantigens due to the cytotoxic effects of the combinatory treatment, which may potentiate the activity of PD-1 inhibitors, an approach that has shown promising results in advanced NSCLC and at the neoadjuvant setting of TNBC [151, 152]; the combination of targeted agents and PD-1 axis blockade [153], with preliminary results showing that combining immunotherapy with inhibitors of known effectors of the axis, such as CDK4/6, results in promising activity [154]; and finally, the combination with inhibitors or stimulators of modulatory molecules such as indoleamine 2,3-dioxygenase (IDO) inhibitors, because IDO is a major negative feedback pathway regulated by IFNg. Preliminary results of the IDO inhibitor epacadostat with nivolumab in a variety of tumors and with pembrolizumab in melanoma are promising and phase 3 results are eagerly awaited [155, 156].
In short, the current era of cancer immunotherapy could be characterized as the “end of the beginning”. A variety of agents is available for use in multiple indications and clinical experience is accumulating. The next phase, namely the optimization of the use of the available agents and the exploration for novel combinations, has already begun.
Immune checkpoint regulators as novel biomarkers: prognostic and predictive value
Taking into account the significant clinical efficacy of PD-1/PD-L1 blockade in a small subset of patients, the considerable costs and potential for devastating immune-related adverse events associated with the use of these inhibitors and the robust theoretical background explaining the biology of their mechanism of action, considerable efforts have been undertaken in order to identify putative predictive biomarkers. The best characterized biomarker is the immunohistochemistry (IHC)-assessed PD-L1 expression. The conflicting results of individual trials have been summarized in meta-analyses, which indicate that increased levels of PD-L1 expression are associated with an improved probability for objective response [157, 158]. Supporting these results are two recently published clinical trials in the first line of advanced NSCLC, KEYNOTE-024, and CheckMate 026. In the former, overall survival (OS) in patients selected for PD-L1 positivity ≥50% was improved with pembrolizumab compared with platinum-based chemotherapy [132]. Contrary, in the latter trial there were no OS gains in PD-L1 ≥5% patients treated with nivolumab versus chemotherapy [133]. As there are no perceived differences in the potency of these antibodies, the obvious discrepancy in the patient population could account for the different outcome. However, several observations hinder the routine selection of appropriate candidates according to PD-L1 expression. First, in addition to the modest concordance rates between the various antibodies used to assess PD-L1 expression reported in the literature, questions still remain regarding the uncontrolled pre-analytical conditions and the assay and inter-pathologist discrepancies [159], which can lead to PD-L1 status misclassifications despite the similar analytical performance of the available assays [160]. Second, PD-L1 expression exhibits significant intratumoral, intertumoral and temporal heterogeneity [161, 162], putting into question the widespread practice of assessing PD-L1 IHC expression on archival tissue. Third, as clearly shown in individual randomized trials such as the CheckMate 017 trial at the second line of lung SCC [135], characterizing patients as appropriate for anti-PD-1 therapy according to PD-L1 expression both includes patients who do not respond to treatment and also excludes potential responders. Fourth, in the aforementioned CheckMate 026 trial, nivolumab was not more effective than chemotherapy even in the subgroup of 50% or higher PD-L1 expression. As this was not a stratification factor, imbalances such as the sex of the patients could have confounded the results, implying that PD-L1 positivity by itself is not a strong predictive biomarker [133]. Finally, the association of objective response rates and PD-L1 expression in the trial-level meta-analyses is of unsure clinical importance, since checkpoint inhibitors can confer prolonged, clinically meaningful periods of disease stabilization and because their use beyond progression in patients deemed to derive clinical benefit has been found to improve outcomes in a diverse selection of solid malignancies [163,164,165].
Keeping in mind the shortcomings of PD-L1 expression, other biomarkers have been explored. Following the observation that smokers with NSCLC seem to derive improved benefit from anti-PD-1 agents [166], it was postulated that this effect may be a surrogate marker for an increased mutational load and subsequent increased neoantigen production and exposure and more effective immune response in patients chronically exposed to a strong mutagenic factor such as smoking. Indeed, mutational load has been found to be a predictive factor in NSCLC [167]. Supporting this association is the observation that mismatch repair defective, and thus hypermutated tumors, are exquisitely sensitive to PD-1 blockade [168, 169]. In addition, NSCLC harboring driver molecular aberrations such as EGFR mutations, which exhibit lesser mutational loads have been shown to be relatively resistant to immune checkpoint inhibition [170], a finding supported by a recently published meta-analysis on the prediction of response in NSCLC patients. EGFR mutant and KRAS wild-type status were associated with a lack of sensitivity to PD-1/PD-L1 inhibition, whereas clinical factors such as smoking status, histology, sex, performance status, and age did not affect the magnitude of benefit [171].
The quantitative and qualitative assessment of the host immune response has also been explored as a predictor in checkpoint inhibition. Factors such as the abundance of pre-existing CD8 (+) T cells, a restricted (clonal) TCR repertoire, a TH1-type response, increased levels of IFN-γ and IL-18 and decreased levels of IL-6, among others, have been correlated with improved responses [166, 172], but these results need to be evaluated prospectively in randomized trials. The implementation of multiparametric, high-throughput flow cytometry, and multiplex immunohistochemical staining techniques that vastly improve the T-cell population analysis [173] and of whole-exome sequencing for the evaluation of the mutational load and the presence of specific, predictive molecular alterations will aid in this respect.
On the other hand, PD-1 and PD-L1 expression both at the tissue level and on circulating tumor cells have been evaluated in a wide variety of malignancies for their prognostic impact (Table 4) [17, 21, 174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229]. The results have been thus far inconsistent among tumor types and somewhat confusing, with reports supporting both an improved and a decreased OS conferred by high expression, a phenomenon that resonates the previously mentioned shortcomings of the assessment of PD-L1. The biologic background of these observations is as of yet uncertain. Moreover, as the expansion of the indications of PD-1/PD-L1 blockade continues with the conduct and report of clinical trials, these associations could be affected due to the increasing use of these agents, making their clinical utility questionable at the moment.
Open questions for future research
Despite the progress in genetic and epigenetic regulation of PD-L1 expression, several gaps in the literature should be covered by intensive laboratory-based research. For instance, the signaling transduction pathways involved in PD-L1 regulation are only partially understood. Better understanding of the signaling mechanisms could provide the biologic rationale for combined targeted therapy with immunotherapy strategies in cancer. Furthermore, little is known about the post-translational modifications of PD-L1 protein including tyrosine or serine/threonine phosphorylation, acetylation, ubiquitination, and SUMOylation. It is also largely unknown how possible post-translational modifications not only regulate PD-L1 levels in the tumor cells, but also how they might affect its physiologic function or its interaction with the PD-1 receptor. In addition to PD-L1, the non-genetic mechanisms underlying PD-L2 expression and function in solid tumors and hematologic malignancies should be investigated, as both ligands compete for the same receptor, PD1, and therefore the relative levels of both proteins may impact certain immunotherapy approaches.
Regarding clinical practice, regulatory authorities both in Europe (European Medicine Agency), and the United States (Food and Drug Administration) have approved the use of PD-1/PD-L1 inhibitors for a variety of malignancies regardless of the presence or absence of predictive biomarkers. Exceptions include the use of pembrolizumab at the first and second line of NSCLC, which requires PD-L1 expression levels of ≥50% and ≥1% respectively, as well as the site agnostic indication for mismatch repair deficient tumors. In addition, the financial burden of the generalized use of these agents is considerable even in high-resource settings [230]. Overcoming this obstacle and achieving the personalized use of these agents requires a stepwise approach: first, taking into account the previously mentioned shortcomings of PD-L1 as a potential biomarker, it is important to retrospectively identify, in the large amount of collected tumor material from prospective studies, novel predictive biomarkers. These would ideally be prospectively validated, although the logistics of repeating single agent trials might be prohibitive. Instead, these biomarkers could form the basis of the next-generation combinatorial trials, of trials addressing the as yet unanswered question of the optimal duration of treatment or of trials in earlier disease settings where the overtreatment of already cured individuals in a massive scale could pose a significant public health burden.
Summary
Despite the clinical success of immune checkpoint inhibition in many tumors through PD-L1/PD-1 blockade, relatively little is known regarding the biology of these regulators of cancer immune surveillance. Many mechanisms have been demonstrated to regulate the expression of PD-L1 including signaling pathways, transcriptional factors, and post-transcriptional modulators. The oncogenic signaling pathways such as JAK/STAT, RAS/ERK, or PI3K/AKT/MTOR are activated by gene mutations and growth factors. At the transcriptional level, a number of transcriptional factors seem to regulate PD-L1 expression including HIF-1, STAT3, NF-κΒ, and AP-1. PD-L1 is subject to post-transcriptional regulation by several miRNAs, CSN5, CMTM6, CDK4 and possibly other, still unknown mechanisms. Better understanding of PD-L1 regulation may pave the way for combinational treatments with both immune checkpoint inhibitors and targeted therapies against kinases or transcription factors many of which are already available for clinical use.
References
Finn OJ. Cancer immunology. N Engl J Med. 2008;358:2704–15.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
La-Beck NM, Jean GW, Huynh C, Alzghari SK, Lowe DB. Immune checkpoint inhibitors: new insights and current place in cancer therapy. Pharmacotherapy. 2015;35:963–76.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
Xia Y, Jeffrey Medeiros L, Young KH. Signaling pathway and dysregulation of PD1 and its ligands in lymphoid malignancies. Biochim Biophys Acta. 2016;1865:58–71.
Ingram JR, Dougan M, Rashidian M, Knoll M, Keliher EJ, Garrett S, et al. PD-L1 is an activation-independent marker of brown adipocytes. Nat Commun. 2017;8:647.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Chikuma S. Basics of PD-1 in self-tolerance, infection, and cancer immunity. Int J Clin Oncol. 2016;21:448–55.
Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–47.
Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016;27:409–16.
Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14.
Goldmann T, Kugler C, Reinmuth N, Vollmer E, Reck M. PD-L1 copy number gain in nonsmall-cell lung cancer defines a new subset of patients for anti PD-L1 therapy. Ann Oncol. 2016;27:206–7.
George J, Saito M, Tsuta K, Iwakawa R, Shiraishi K, Scheel AH, et al. Genomic amplification of CD274 (PD-L1) in small-cell lung cancer. Clin Cancer Res. 2017;23:1220–6.
Howitt BE, Sun HH, Roemer MG, Kelley A, Chapuy B, Aviki E, et al. Genetic basis for PD-L1 expression in squamous cell carcinomas of the cervix and vulva. JAMA Oncol. 2016;2:518–22.
Guo L, Li W, Zhu X, Ling Y, Qiu T, Dong L, et al. PD-L1 expression and CD274 gene alteration in triple-negative breast cancer: implication for prognostic biomarker. Springerplus. 2016;5:805.
Barrett MT, Anderson KS, Lenkiewicz E, Andreozzi M, Cunliffe HE, Klassen CL, et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget. 2015;6:26483–93.
Straub M, Drecoll E, Pfarr N, Weichert W, Langer R, Hapfelmeier A, et al. CD274/PD-L1 gene amplification and PD-L1 protein expression are common events in squamous cell carcinoma of the oral cavity. Oncotarget. 2016;7:12024–34.
Clave S, Pijuan L, Casadevall D, Taus A, Gimeno J, Hernandez-Llodra S, et al. CD274 (PDL1) and JAK2 genomic amplifications in pulmonary squamous-cell and adenocarcinoma patients. Histopathology. 2018;72:259–69.
Georgiou K, Chen L, Berglund M, Ren W, de Miranda NF, Lisboa S, et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood. 2016;127:3026–34.
Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 genetic alterations define classical hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34:2690–7.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9.
Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–77.
Chong LC, Twa DD, Mottok A, Ben-Neriah S, Woolcock BW, Zhao Y, et al. Comprehensive characterization of programmed death ligand structural rearrangements in B-cell non-Hodgkin lymphomas. Blood. 2016;128:1206–13.
Twa DD, Chan FC, Ben-Neriah S, Woolcock BW, Mottok A, Tan KL, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. 2014;123:2062–5.
Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood. 2016;127:869–81.
Ali HR, Glont SE, Blows FM, Provenzano E, Dawson SJ, Liu B, et al. PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann Oncol. 2015;26:1488–93.
Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, et al. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget. 2015;6:5449–64.
Budczies J, Bockmayr M, Denkert C, Klauschen F, Groschel S, Darb-Esfahani S, et al. Pan-cancer analysis of copy number changes in programmed death-ligand 1 (PD-L1, CD274)—associations with gene expression, mutational load, and survival. Genes Chromosomes Cancer. 2016;55:626–39.
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. 2016;534:402–6.
Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90.
Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19:598–609.
Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22:1499–509.
Sumimoto H, Takano A, Teramoto K, Daigo Y. RAS-mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PLoS ONE. 2016;11:e0166626.
Qian Y, Deng J, Geng L, Xie H, Jiang G, Zhou L, et al. TLR4 signaling induces B7-H1 expression through MAPK pathways in bladder cancer cells. Cancer Invest. 2008;26:816–21.
Balan M, Mier y Teran E, Waaga-Gasser AM, Gasser M, Choueiri TK, Freeman G, et al. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J Biol Chem. 2015;290:8110–20.
Coelho M, Kumar M, Moore C, Diefenbacher M, Downward J. Cell intrinsic upregulation of PD-L1 through oncogenic KRAS signalling. 10th NCRI Cancer Conference: Liverpool, UK, 2014.
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88.
Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2:361–70.
Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25:590–604.
Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76:227–38.
Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol. 2014;25:1935–40.
Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63.
Rech AJ, Vonderheide RH. Dynamic interplay of oncogenes and T cells induces PD-L1 in the tumor microenvironment. Cancer Discov. 2013;3:1330–2.
Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015;10:910–23.
Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. 2016;49:1360–8.
Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. 2015;21:4014–21.
Koh J, Jang JY, Keam B, Kim S, Kim MY, Go H, et al. EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1alpha and STAT3. Oncoimmunology. 2016;5:e1108514.
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809.
Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci USA. 2008;105:20852–7.
Atsaves V, Tsesmetzis N, Chioureas D, Kis L, Leventaki V, Drakos E, et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia. 2017;31:1633–7.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201.
Fang W, Zhang J, Hong S, Zhan J, Chen N, Qin T, et al. EBV-driven LMP1 and IFN-gamma up-regulate PD-L1 in nasopharyngeal carcinoma: implications for oncotargeted therapy. Oncotarget. 2014;5:12189–202.
Koromilas AE, Sexl V. The tumor suppressor function of STAT1 in breast cancer. Jakstat. 2013;2:e23353.
Meissl K, Macho-Maschler S, Muller M, Strobl B. The good and the bad faces of STAT1 in solid tumours. Cytokine. 2017;89:12–20.
Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, et al. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015;4:e1008824.
Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110:296–304.
Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR, et al. Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFN gamma that induce PD-L1 expression in head and neck cancer. Cancer Res. 2016;76:1031–43.
Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006;580:755–62.
Grinberg-Bleyer Y, Ghosh S. A novel link between inflammation and cancer. Cancer Cell. 2016;30:829–30.
Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017;8:1751.
Labiano S, Palazon A, Melero I. Immune response regulation in the tumor microenvironment by hypoxia. Semin Oncol. 2015;42:378–86.
Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410.
Palazon A, Tyrakis PA, Macias D, Velica P, Rundqvist H, Fitzpatrick S, et al. An HIF-1alpha/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell. 2017;32:669. e665
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–90.
Ruf M, Moch H, Schraml P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer. 2016;139:396–403.
Messai Y, Gad S, Noman MZ, Le Teuff G, Couve S, Janji B, et al. Renal cell carcinoma programmed death-ligand 1, a new direct target of hypoxia-inducible factor-2 alpha, is regulated by von Hippel-Lindau gene mutation status. Eur Urol. 2016;70:623–32.
Pawlus MR, Wang L, Hu CJ. STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene. 2014;33:1670–9.
DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400.
Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012;12:121–32.
Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS ONE. 2015;10:e0123410.
Fan Y, Mao R, Yang J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. 2013;4:176–85.
Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297:102–4.
Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–7.
Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18:485–98.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–31.
Spranger S, Gajewski TF, Kline J. MYC—a thorn in the side of cancer immunity. Cell Res. 2016;26:639–40.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–31.
Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–77.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73.
Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16:2829–37.
Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18:2162–74.
Gallagher SJ, Mijatov B, Gunatilake D, Gowrishankar K, Tiffen J, James W, et al. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res. 2014;27:1126–37.
Woan KV, Lienlaf M, Perez-Villaroel P, Lee C, Cheng F, Knox T, et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: enhanced antitumor immunity and impaired cell proliferation. Mol Oncol. 2015;9:1447–57.
Woan KV, Sahakian E, Sotomayor EM, Seto E, Villagra A. Modulation of antigen-presenting cells by HDAC inhibitors: implications in autoimmunity and cancer. Immunol Cell Biol. 2012;90:55–65.
Lienlaf M, Perez-Villarroel P, Knox T, Pabon M, Sahakian E, Powers J, et al. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol Oncol. 2016;10:735–50.
Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–91.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66.
Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–59.
Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, et al. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–8.
Utreras E, Futatsugi A, Pareek TK, Kulkarni AB. Molecular roles of Cdk5 in pain signaling. Drug Discov Today Ther Strateg. 2009;6:105–11.
Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, Chirieleison SM, et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016;353:399–403.
Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471–5.
Schaefer TS, Sanders LK, Nathans D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc Natl Acad Sci USA. 1995;92:9097–101.
Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. 2012;18:1611–8.
Munoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol. 2016;16:741–50.
Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science. 2015;349:1261669.
Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 regulation by p53 via miR-34. J Natl Cancer Inst. 2016;108:djv303.
Wang X, Li J, Dong K, Lin F, Long M, Ouyang Y, et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015;27:443–52.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Gong AY, Zhou R, Hu G, Li X, Splinter PL, O’Hara SP, et al. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J Immunol. 2009;182:1325–33.
Wang W, Sun J, Li F, Li R, Gu Y, Liu C, et al. A frequent somatic mutation in CD274 3’-UTR leads to protein over-expression in gastric cancer by disrupting miR-570 binding. Hum Mutat. 2012;33:480–4.
Wang W, Li F, Mao Y, Zhou H, Sun J, Li R, et al. A miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Hum Genet. 2013;132:641–8.
Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther. 2015;23:717–27.
Zhao L, Yu H, Yi S, Peng X, Su P, Xiao Z, et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget. 2016;7:45370–84.
Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, et al. MiR-20b, -21, and -130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol. 2014;75:348–53.
Shackleford TJ, Claret FX. JAB1/CSN5: a new player in cell cycle control and cancer. Cell Div. 2010;5:26.
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–39.
Schlierf A, Altmann E, Quancard J, Jefferson AB, Assenberg R, Renatus M, et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat Commun. 2016;7:13166.
Horita H, Law A, Hong S, Middleton K. Identifying regulatory posttranslational modifications of PD-L1: a focus on monoubiquitinaton. Neoplasia. 2017;19:346–53.
Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–95.
Han W, Ding P, Xu M, Wang L, Rui M, Shi S, et al. Identification of eight genes encoding chemokine-like factor superfamily members 1-8 (CKLFSF1-8) by in silico cloning and experimental validation. Genomics. 2003;81:609–17.
Sanchez-Pulido L, Martin-Belmonte F, Valencia A, Alonso MA. MARVEL: a conserved domain involved in membrane apposition events. Trends Biochem Sci. 2002;27:599–601.
Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–5.
Visan I. CMTM6 controls PD-L1. Nat Immunol. 2017;18:1067.
Cheung JC, Reithmeier RA. Scanning N-glycosylation mutagenesis of membrane proteins. Methods. 2007;41:451–9.
Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632.
Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–201. e110
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–33.
Luo M, Fu L. The effect of chemotherapy on programmed cell death 1/programmed cell death 1 ligand axis: some chemotherapeutical drugs may finally work through immune response. Oncotarget. 2016;7:29794–803.
Zhang P, Su DM, Liang M, Fu J. Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Mol Immunol. 2008;45:1470–6.
Gong W, Song Q, Lu X, Gong W, Zhao J, Min P, et al. Paclitaxel induced B7-H1 expression in cancer cells via the MAPK pathway. J Chemother. 2011;23:295–9.
Qin X, Liu C, Zhou Y, Wang G. Cisplatin induces programmed death-1-ligand 1(PD-L1) over-expression in hepatoma H22 cells via Erk/MAPK signaling pathway. Cell Mol Biol (Noisy-Le-Grand). 2010;56(Suppl):Ol1366–1372.
Ghebeh H, Lehe C, Barhoush E, Al-Romaih K, Tulbah A, Al-Alwan M, et al. Doxorubicin downregulates cell surface B7-H1 expression and upregulates its nuclear expression in breast cancer cells: role of B7-H1 as an anti-apoptotic molecule. Breast Cancer Res. 2010;12:R48.
Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6(Suppl 1):S11–14.
Belldegrun AS, Klatte T, Shuch B, LaRochelle JC, Miller DC, Said JW, et al. Cancer-specific survival outcomes among patients treated during the cytokine era of kidney cancer (1989-2005): a benchmark for emerging targeted cancer therapies. Cancer. 2008;113:2457–63.
Coppin C, Porzsolt F, Awa A, Kumpf J, Coldman A, Wilt T. Immunotherapy for advanced renal cell cancer. Cochrane Database Syst Rev. 2005: CD001425.
Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:e143–e152.
Horvat TZ, Adel NG, Dang TO, Momtaz P, Postow MA, Callahan MK, et al. Immune-related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol. 2015;33:3193–8.
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–94.
Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–33.
Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376:2415–26.
Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–50.
Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–35.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–39.
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–65.
Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. 2017;390:1853–62.
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30.
Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–84.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34.
Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015–26.
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–13.
Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–67.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377:1919–29.
Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377:1824–35.
Powles T, Duran I, van der Heijden MS, Loriot Y, Vogelzang NJ, De Giorgi U, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2018;391:748–57.
Escudier B, Tannir NM, McDermott DF, Frontera OA, Melichar B, Plimack ER, et al. LBA5CheckMate 214: efficacy and safety of nivolumab+ipilimumab (N+I) v sunitinib (S) for treatment-naïve advanced or metastatic renal cell carcinoma (mRCC), including IMDC risk and PD-L1 expression subgroups. Ann Oncol. 2017;28:mdx440.029–mdx440.029.
Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. 2016;34:2698–704.
Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol. 2017;14:203–20.
Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17:1497–508.
Nanda R, Liu MC, Yau C, Asare S, Hylton N, Veer LVt, et al. Pembrolizumab plus standard neoadjuvant therapy for high-risk breast cancer (BC): results from I-SPY 2. J Clin Oncol. 2017;35:506–506.
Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17:286–301.
Rugo HS, Kabos P, Dickler MN, John WJ, Smith I, Lu Y, Young S, et al. A phase 1b study of abemaciclib plus pembrolizumab for patients with hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-) metastatic breast cancer (MBC). 2017 San Antonio Breast Cancer Symposium: San Antonio, Texas, 2017.
Perez R, Riese M, Lewis M, Saleh M, Daud A, Berlin J, et al. Epacadostat plus nivolumab in patients with advanced solid tumors: preliminary phase I/II results of ECHO-204. Abstract 3003. American Society of Clinical Oncology annual meeting: Chicago, Illinois, USA, 2017.
Gangadhar T, Hamid O, Smith D, Bauer T, Wasser J, Olszanski A, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Abstract 1110PD. European Society for Medical Oncology annual meeting: Copenhagen, Denmark, 2016.
Gandini S, Massi D, Mandala M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: a systematic review and meta-analysis. Crit Rev Oncol Hematol. 2016;100:88–98.
Khunger M, Hernandez A, Pasupuleti V, Rakshit S, Pennell N, Stevenson J, et al. Programmed cell death 1 (PD-1) ligand (PD-L1) expression in solid tumors as a predictive biomarker of benefit from PD-1/PD-L1 axis inhibitors: a systematic review and meta-analysis. JCO Precis Oncol. 2017;1:1–15.
Micke P, Johansson A, Westborn-Fremer A, Backman M, Djureinovic D, Patthey A, et al. PD-L1 immunohistochemistry in clinical diagnostics: inter-pathologist variability is as high as assay variability. Abstract e20637. American Society of Clinical Oncology annual meeting: Chicago, Illinois, USA, 2017.
Hirsch FR, McElhinny A, Stanforth D, Ranger-Moore J, Jansson M, Kulangara K, et al. PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the Blueprint PD-L1 IHC assay comparison project. J Thorac Oncol. 2017;12:208–22.
McLaughlin J, Han G, Schalper KA, Carvajal-Hausdorf D, Pelekanou V, Rehman J, et al. Quantitative assessment of the heterogeneity of PD-L1 expression in non-small-cell lung cancer. JAMA Oncol. 2016;2:46–54.
Casadevall D, Clave S, Taus A, Hardy-Werbin M, Rocha P, Lorenzo M, et al. Heterogeneity of tumor and immune cell PD-L1 expression and lymphocyte counts in surgical NSCLC samples. Clin Lung Cancer. 2017;18:682–91.
Gandara D, Pawel J, Sullivan R, Helland A, Han J, Aix S, et al. Impact of atezolizumab (atezo) treatment beyond disease progression (TBP) in advanced NSCLC: results from the randomized phase III OAK study. Abstract 9001. American Society of Clinical Oncology Annual Meeting: Chicago, Illinois, USA, 2017.
Escudier B, Motzer RJ, Sharma P, Wagstaff J, Plimack ER, Hammers HJ, et al. Treatment beyond progression in patients with advanced renal cell carcinoma treated with nivolumab in CheckMate 025. Eur Urol. 2017;72:368–76.
Long GV, Weber JS, Larkin J, Atkinson V, Grob JJ, Schadendorf D, et al. Nivolumab for patients with advanced melanoma treated beyond progression: analysis of 2 phase 3 clinical trials. JAMA Oncol. 2017;3:1511–19.
Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22:4585–93.
Lee CK, Man J, Lord S, Cooper W, Links M, Gebski V, et al. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non-small cell lung carcinoma: a systematic review and meta-analysis. JAMA Oncol. 2018;4:210–16.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71.
Zaritskaya L, Shurin MR, Sayers TJ, Malyguine AM. New flow cytometric assays for monitoring cell-mediated cytotoxicity. Expert Rev Vaccin. 2010;9:601–16.
Li X, Li M, Lian Z, Zhu H, Kong L, Wang P, et al. Prognostic role of programmed death ligand-1 expression in breast cancer: a systematic review and meta-analysis. Target Oncol. 2016;11:753–61.
Uhercik M, Sanders AJ, Owen S, Davies EL, Sharma AK, Jiang WG, et al. Clinical significance of PD1 and PDL1 in human breast cancer. Anticancer Res. 2017;37:4249–54.
Li X, Wetherilt CS, Krishnamurti U, Yang J, Ma Y, Styblo TM, et al. Stromal PD-L1 expression is associated with better disease-free survival in triple-negative breast cancer. Am J Clin Pathol. 2016;146:496–502.
Chen S, Wang RX, Liu Y, Yang WT, Shao ZM. PD-L1 expression of the residual tumor serves as a prognostic marker in local advanced breast cancer after neoadjuvant chemotherapy. Int J Cancer. 2017;140:1384–95.
Zhang M, Sun H, Zhao S, Wang Y, Pu H, Wang Y, et al. Expression of PD-L1 and prognosis in breast cancer: a meta-analysis. Oncotarget. 2017;8:31347–54.
Tsang JY, Au WL, Lo KY, Ni YB, Hlaing T, Hu J, et al. PD-L1 expression and tumor infiltrating PD-1+lymphocytes associated with outcome in HER2+breast cancer patients. Breast Cancer Res Treat. 2017;162:19–30.
Jung HI, Jeong D, Ji S, Ahn TS, Bae SH, Chin S, et al. Overexpression of PD-L1 and PD-L2 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res Treat. 2017;49:246–54.
Gu X, Gao XS, Xiong W, Guo W, Han L, Bai Y, et al. Increased programmed death ligand-1 expression predicts poor prognosis in hepatocellular carcinoma patients. Onco Targets Ther. 2016;9:4805–13.
Diana A, Wang LM, D’Costa Z, Allen P, Azad A, Silva MA, et al. Prognostic value, localization and correlation of PD-1/PD-L1, CD8 and FOXP3 with the desmoplastic stroma in pancreatic ductal adenocarcinoma. Oncotarget. 2016;7:40992–1004.
Dai C, Wang M, Lu J, Dai Z, Lin S, Yang P, et al. Prognostic and predictive values of PD-L1 expression in patients with digestive system cancer: a meta-analysis. Onco Targets Ther. 2017;10:3625–34.
Liu YX, Wang XS, Wang YF, Hu XC, Yan JQ, Zhang YL, et al. Prognostic significance of PD-L1 expression in patients with gastric cancer in East Asia: a meta-analysis. Onco Targets Ther. 2016;9:2649–54.
Gu L, Chen M, Guo D, Zhu H, Zhang W, Pan J, et al. PD-L1 and gastric cancer prognosis: a systematic review and meta-analysis. PLoS One. 2017;12:e0182692.
Lee KS, Kwak Y, Ahn S, Shin E, Oh HK, Kim DW, et al. Prognostic implication of CD274 (PD-L1) protein expression in tumor-infiltrating immune cells for microsatellite unstable and stable colorectal cancer. Cancer Immunol Immunother. 2017;66:927–39.
Wang H, Yao H, Li C, Liang L, Zhang Y, Shi H, et al. PD-L2 expression in colorectal cancer: independent prognostic effect and targetability by deglycosylation. Oncoimmunology. 2017;6:e1327494.
Webb JR, Milne K, Kroeger DR, Nelson BH. PD-L1 expression is associated with tumor-infiltrating T cells and favorable prognosis in high-grade serous ovarian cancer. Gynecol Oncol. 2016;141:293–302.
Gevensleben H, Dietrich D, Golletz C, Steiner S, Jung M, Thiesler T, et al. The immune checkpoint regulator PD-L1 is highly expressed in aggressive primary prostate cancer. Clin Cancer Res. 2016;22:1969–77.
Ness N, Andersen S, Khanehkenari MR, Nordbakken CV, Valkov A, Paulsen EE, et al. The prognostic role of immune checkpoint markers programmed cell death protein 1 (PD-1) and programmed death ligand 1 (PD-L1) in a large, multicenter prostate cancer cohort. Oncotarget. 2017;8:26789–801.
Nakanishi J, Wada Y, Matsumoto K, Azuma M, Kikuchi K, Ueda S. Overexpression of B7-H1 (PD-L1) significantly associates with tumor grade and postoperative prognosis in human urothelial cancers. Cancer Immunol Immunother. 2007;56:1173–82.
Erlmeier F, Weichert W, Autenrieth M, Wiedemann M, Schrader AJ, Hartmann A, et al. PD-L2: a prognostic marker in chromophobe renal cell carcinoma? Med Oncol. 2017;34:71.
Motoshima T, Komohara Y, Ma C, Dewi AK, Noguchi H, Yamada S, et al. PD-L1 expression in papillary renal cell carcinoma. BMC Urol. 2017;17:8.
Choueiri TK, Fay AP, Gray KP, Callea M, Ho TH, Albiges L, et al. PD-L1 expression in nonclear-cell renal cell carcinoma. Ann Oncol. 2014;25:2178–84.
Abbas M, Steffens S, Bellut M, Eggers H, Grosshennig A, Becker JU, et al. Intratumoral expression of programmed death ligand 1 (PD-L1) in patients with clear cell renal cell carcinoma (ccRCC). Med Oncol. 2016;33:80.
Leite KR, Reis ST, Junior JP, Zerati M, Gomes Dde O, Camara-Lopes LH, et al. PD-L1 expression in renal cell carcinoma clear cell type is related to unfavorable prognosis. Diagn Pathol. 2015;10:189.
Shin SJ, Jeon YK, Kim PJ, Cho YM, Koh J, Chung DH, et al. Clinicopathologic analysis of PD-L1 and PD-L2 expression in renal cell carcinoma: association with oncogenic proteins status. Ann Surg Oncol. 2016;23:694–702.
Lin YM, Sung WW, Hsieh MJ, Tsai SC, Lai HW, Yang SM, et al. High PD-L1 expression correlates with metastasis and poor prognosis in oral squamous cell carcinoma. PLoS ONE. 2015;10:e0142656.
Kogashiwa Y, Yasuda M, Sakurai H, Nakahira M, Sano Y, Gonda K, et al. PD-L1 expression confers better prognosis in locally advanced oral squamous cell carcinoma. Anticancer Res. 2017;37:1417–24.
Ikeda S, Okamoto T, Okano S, Umemoto Y, Tagawa T, Morodomi Y, et al. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J Thorac Oncol. 2016;11:62–71.
Chang YL, Yang CY, Huang YL, Wu CT, Yang PC. High PD-L1 expression is associated with stage IV disease and poorer overall survival in 186 cases of small cell lung cancers. Oncotarget. 2017;8:18021–30.
Tsao MS, Le Teuff G, Shepherd FA, Landais C, Hainaut P, Filipits M, et al. PD-L1 protein expression assessed by immunohistochemistry is neither prognostic nor predictive of benefit from adjuvant chemotherapy in resected non-small cell lung cancer. Ann Oncol. 2017;28:882–9.
Xia H, Shen J, Hu F, Chen S, Huang H, Xu Y, et al. PD-L1 over-expression is associated with a poor prognosis in Asian non-small cell lung cancer patients. Clin Chim Acta. 2017;469:191–4.
Takada K, Okamoto T, Toyokawa G, Kozuma Y, Matsubara T, Haratake N, et al. The expression of PD-L1 protein as a prognostic factor in lung squamous cell carcinoma. Lung Cancer. 2017;104:7–15.
Igawa S, Sato Y, Ryuge S, Ichinoe M, Katono K, Hiyoshi Y, et al. Impact of PD-L1 expression in patients with surgically resected non-small-cell lung cancer. Oncology. 2017;92:283–90.
Okita R, Maeda A, Shimizu K, Nojima Y, Saisho S, Nakata M. PD-L1 overexpression is partially regulated by EGFR/HER2 signaling and associated with poor prognosis in patients with non-small-cell lung cancer. Cancer Immunol Immunother. 2017;66:865–76.
Que Y, Xiao W, Guan YX, Liang Y, Yan SM, Chen HY, et al. PD-L1 expression is associated with FOXP3+regulatory T-cell infiltration of soft tissue sarcoma and poor patient prognosis. J Cancer. 2017;8:2018–25.
Obeid JM, Erdag G, Smolkin ME, Deacon DH, Patterson JW, Chen L, et al. PD-L1, PD-L2 and PD-1 expression in metastatic melanoma: correlation with tumor-infiltrating immune cells and clinical outcome. Oncoimmunology. 2016;5:e1235107.
Li Y, Liang L, Dai W, Cai G, Xu Y, Li X, et al. Prognostic impact of programed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor infiltrating lymphocytes in colorectal cancer. Mol Cancer. 2016;15:55.
Darb-Esfahani S, Kunze CA, Kulbe H, Sehouli J, Wienert S, Lindner J, et al. Prognostic impact of programmed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor-infiltrating lymphocytes in ovarian high grade serous carcinoma. Oncotarget. 2016;7:1486–99.
Schmidt LH, Kummel A, Gorlich D, Mohr M, Brockling S, Mikesch JH, et al. PD-1 and PD-L1 expression in NSCLC indicate a favorable prognosis in defined subgroups. PLoS ONE. 2015;10:e0136023.
Danilova L, Wang H, Sunshine J, Kaunitz GJ, Cottrell TR, Xu H, et al. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc Natl Acad Sci USA. 2016;113:E7769–E7777.
Velcheti V, Schalper KA, Carvajal DE, Anagnostou VK, Syrigos KN, Sznol M, et al. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest. 2014;94:107–16.
Jesinghaus M, Steiger K, Slotta-Huspenina J, Drecoll E, Pfarr N, Meyer P, et al. Increased intraepithelial CD3.T-lymphocytes and high PD-L1 expression on tumor cells are associated with a favorable prognosis in esophageal squamous cell carcinoma and allow prognostic immunogenic subgrouping. Oncotarget. 2017;18:46756–68.
Kim HR, Ha SJ, Hong MH, Heo SJ, Koh YW, Choi EC, et al. PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor for head and neck cancer patients. Sci Rep. 2016;6:36956.
Kiyasu J, Miyoshi H, Hirata A, Arakawa F, Ichikawa A, Niino D, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood. 2015;126:2193–201.
Ma K, Wei X, Dong D, Wu Y, Geng Q, Li E. PD-L1 and PD-1 expression correlate with prognosis in extrahepatic cholangiocarcinoma. Oncol Lett. 2017;14:250–6.
Huang Y, Zhang SD, McCrudden C, Chan KW, Lin Y, Kwok HF. The prognostic significance of PD-L1 in bladder cancer. Oncol Rep. 2015;33:3075–84.
Muenst S, Schaerli AR, Gao F, Daster S, Trella E, Droeser RA, et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat. 2014;146:15–24.
Fan Y, Ma K, Wang C, Ning J, Hu Y, Dong D, et al. Prognostic value of PD-L1 and PD-1 expression in pulmonary neuroendocrine tumors. Onco Targets Ther. 2016;9:6075–82.
Zhang Y, Kang S, Shen J, He J, Jiang L, Wang W, et al. Prognostic significance of programmed cell death 1 (PD-1) or PD-1 ligand 1 (PD-L1) expression in epithelial-originated cancer: a meta-analysis. Medicine (Baltim). 2015;94:e515.
Koh YW, Han JH, Yoon DH, Suh C, Huh J. PD-L1 expression correlates with VEGF and microvessel density in patients with uniformly treated classical Hodgkin lymphoma. Ann Hematol. 2017;96:1883–90.
Wang L, Wang H, Chen H, Wang WD, Chen XQ, Geng QR, et al. Serum levels of soluble programmed death ligand 1 predict treatment response and progression free survival in multiple myeloma. Oncotarget. 2015;6:41228–36.
Huang SY, Lin HH, Lin CW, Li CC, Yao M, Tang JL, et al. Soluble PD-L1: a biomarker to predict progression of autologous transplantation in patients with multiple myeloma. Oncotarget. 2016;7:62490–502.
Paydas S, Bagir E, Seydaoglu G, Ercolak V, Ergin M. Programmed death-1 (PD-1), programmed death-ligand 1 (PD-L1), and EBV-encoded RNA (EBER) expression in Hodgkin lymphoma. Ann Hematol. 2015;94:1545–52.
Bi XW, Wang H, Zhang WW, Wang JH, Liu WJ, Xia ZJ, et al. PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J Hematol Oncol. 2016;9:109.
Fang X, Xiu B, Yang Z, Qiu W, Zhang L, Zhang S, et al. The expression and clinical relevance of PD-1, PD-L1, and TP63 in patients with diffuse large B-cell lymphoma. Medicine (Baltim). 2017;96:e6398.
Pyo JS, Kang G, Kim JY. Prognostic role of PD-L1 in malignant solid tumors: a meta-analysis. Int J Biol Markers. 2017;32:e68–e74.
Wang Q, Liu F, Liu L. Prognostic significance of PD-L1 in solid tumor: an updated meta-analysis. Medicine (Baltim). 2017;96:e6369.
Tartari F, Santoni M, Burattini L, Mazzanti P, Onofri A, Berardi R. Economic sustainability of anti-PD-1 agents nivolumab and pembrolizumab in cancer patients: recent insights and future challenges. Cancer Treat Rev. 2016;48:20–24.
Acknowledgements
We thank Dr. Ioannis Mantas for his help with illustrative work.
Funding
This study was supported by the Swedish Cancer Society (CAN 2015/713 to TF); the Cancer Society in Stockholm (154132 to TF); The Swedish Breast Cancer Association (IZ, TF); European Society for Medical Oncology Georges Mathé Translational Research Fellowship (AM); and Hellenic Society of Medical Oncology (AM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Foukakis has received research grants (institutional) from Pfizer and Roche; personal fees from Novartis, Pfizer, Roche and UpToDate outside the submitted work. The remaining authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zerdes, I., Matikas, A., Bergh, J. et al. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene 37, 4639–4661 (2018). https://doi.org/10.1038/s41388-018-0303-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0303-3
This article is cited by
-
Investigation of the potential effects of estrogen receptor modulators on immune checkpoint molecules
Scientific Reports (2024)
-
Prognostic significance of blood-based PD-L1 analysis in patients with non-small cell lung cancer undergoing immune checkpoint inhibitor therapy: a systematic review and meta-analysis
World Journal of Surgical Oncology (2023)
-
mTORC1 upregulates B7-H3/CD276 to inhibit antitumor T cells and drive tumor immune evasion
Nature Communications (2023)
-
LncTUG1 promotes hepatocellular carcinoma immune evasion via upregulating PD-L1 expression
Scientific Reports (2023)
-
Ponatinib delays the growth of solid tumours by remodelling immunosuppressive tumour microenvironment through the inhibition of induced PD-L1 expression
British Journal of Cancer (2023)
