
Abstract
Chemotherapy, radiation therapy, as well as targeted anticancer agents can induce clinically relevant tumor-targeting immune responses, which critically rely on the antigenicity of malignant cells and their capacity to generate adjuvant signals. In particular, immunogenic cell death (ICD) is accompanied by the exposure and release of numerous damage-associated molecular patterns (DAMPs), which altogether confer a robust adjuvanticity to dying cancer cells, as they favor the recruitment and activation of antigen-presenting cells. ICD-associated DAMPs include surface-exposed calreticulin (CALR) as well as secreted ATP, annexin A1 (ANXA1), type I interferon, and high-mobility group box 1 (HMGB1). Additional hallmarks of ICD encompass the phosphorylation of eukaryotic translation initiation factor 2 subunit-α (EIF2S1, better known as eIF2α), the activation of autophagy, and a global arrest in transcription and translation. Here, we outline methodological approaches for measuring ICD markers in vitro and ex vivo for the discovery of next-generation antineoplastic agents, the development of personalized anticancer regimens, and the identification of optimal therapeutic combinations for the clinical management of cancer.
Similar content being viewed by others

Facts
-
Immunogenic cell death (ICD) can initiate adaptive immune responses, because it is accompanied by the emission of adjuvant-like signals commonly known as damage-associated molecular patterns (DAMPs).
-
Key DAMPs for cell death to be perceived as immunogenic include calreticulin, HMGB1, ATP, ANXA1, and type I IFN.
-
DAMP emission by cells undergoing ICD often relies on the activation of intracellular responses of adaptation to stress.
-
Although detecting DAMP emission from dying cells informs on the potential immunogenicity of cell death, in vivo assays are required to validate bona fide instances of ICD.
Open questions
-
Does a core set of DAMPs common to all instances of ICD exist?
-
Can we harness transcriptional signatures of pattern recognition receptor (PRR) signaling to assess DAMP emission in patient biopsies?
-
Will therapeutic strategies specifically conceived to restore the immunogenicity of cell death enter the clinical practice for cancer therapy?
Introduction
The emergence and progression of human neoplasms strongly depends on the interaction between cancer cells and their microenvironment, especially in its immunological components1,2,3,4,5,6,7. Immunosurveillance is generally mediated by type 1 CD4+ T-helper (TH1) cells and CD8+ cytotoxic T lymphocytes (CTLs), which specifically recognize antigenic epitopes emerging during malignant transformation and tumor progression8,9,10,11,12,13. Specifically, neoplastic cells can become visible to the adaptive immune system as a consequence of non-synonymous mutations in the coding region of actively expressed genes (leading to antigens that are not covered by central tolerance) or the ectopic expression of otherwise normal antigens for which central tolerance is leaky9,14,15,16,17. Thus, the (neo)antigenic profile of a tumor is a key determinant of anticancer immune responses18,19,20, as demonstrated by the fact that several solid tumors become resistant to immunotherapy with immune checkpoint inhibitors (ICIs) by acquiring defects in the antigen presentation machinery21,22,23,24,25. However, antigenicity is not sufficient for malignant cells to initiate anticancer immunity, as antigen presentation by dendritic cells (DCs) in the absence of co-stimulatory signals to T cells generally results in the establishment of immunological tolerance26,27,28,29,30. Thus, malignant cells can also escape immunosurveillance by losing their capacity to promote the recruitment and functional maturation of DCs or their precursors, a feature that is cumulatively referred to as adjuvanticity31,32,33. Although the adjuvanticity of infected cells mostly originates from microbial products commonly referred to as microbe-associated molecular patterns (MAMPs; e.g., lipopolysaccharide)34,35,36, malignant cells mediate chemotactic and immunostimulatory effects by emitting the so-called damage-associated molecular patterns (DAMPs) and secreting cytokines as they adapt or succumb to microenvironmental perturbations16,37,38.
Immunogenic cell death (ICD) represents a functionally unique response pattern that comprises the induction of organellar and cellular stress, and culminates with cell death accompanied by the exposure, active secretion, or passive release of numerous DAMPs16,37,39,40,41. The spatiotemporally defined emission of DAMPs in the course of ICD and their binding to specific pattern recognition receptors (PRRs) expressed by DCs initiates a cellular cascade that ultimately results in the activation of both innate and adaptive immune responses34,42,43,44. In line with this notion, accumulating preclinical and clinical evidence indicates that various DAMPs and DAMP-associated processes may have prognostic and predictive value for patients affected by a variety of tumors45. Moreover, there is ample evidence that treatment-driven ICD can elicit anticancer immune responses that reinforce the therapeutic effects of conventional anticancer chemotherapies and radiotherapy46,47,48,49. So far, however, only a few bona fide ICD inducers have been successfully employed in the clinic as therapeutics (Table 1)46,50. These agents may be particularly relevant to initiate anticancer immune responses that can be actioned by ICIs or other forms of immunotherapy in the context of combinatorial treatment regimens46,51,52,53,54,55, as demonstrated in some clinical56,57 and numerous preclinical58,59,60 studies. In line with this notion, numerous Food and Drug Administration-approved ICD inducers are currently being investigated in off-label oncological settings, especially in combination with ICIs or other immunotherapeutics61,62,63,64,65.
Thus, the development of methodological approaches and platforms for identifying novel ICD inducers should accelerate the development of next-generation anticancer therapeutics, ultimately improving the clinical management of a large population of oncological patients.
Main hallmarks of ICD
ICD can be induced by different stressors, including but not limited to (1) intracellular pathogens66,67,68; (2) conventional chemotherapeutics such as anthracyclines, DNA-damaging agents, and proteasomal inhibitors50,69,70,71,72; (3) targeted anticancer agents such as the tyrosine kinase inhibitor crizotinib, the epidermal growth factor receptor-specific monoclonal antibody cetuximab and poly-ADP-ribose polymerase (PARP) inhibitors59,73,74,75,76; and (4) numerous physical modalities, encompassing hypericin- and redaporfin-based photodynamic therapy, extracorporeal photochemotherapy, various forms of ionizing radiation, high hydrostatic pressure, and severe heat shock77,78,79,80,81.
DAMPs emitted in the course of ICD include endoplasmic reticulum (ER) chaperones such as calreticulin (CALR) and heat-shock proteins (HSPs), which are exposed on the cell surface, the non-histone chromatin-binding protein high-mobility group box 1 (HMGB1), the cytoplasmic protein annexin A1 (ANXA1), and the small metabolite ATP that are liberated from dying cells into the extracellular space, as well as type I interferons (IFNs) that are released upon de novo synthesis38,82,83,84. DAMPs can be recognized by both the innate and adaptive immune systems via distinct PRRs driving chemoattraction, homing, activation, and/or maturation, ultimately resulting in the cross-presentation of tumor antigens to CD8+ CTLs in the context of robust immunostimulation34,43. Other hallmarks of ICD include the phosphorylation of eukaryotic translation initiation factor 2 subunit-α (EIF2S1, better known as eIF2α), the activation of autophagy, and a global arrest in transcription and translation85,86,87,88. Importantly, not all ICD inducers activate the same stress responses and hence elicit the same molecular signals16. Thus, for instance, although autophagy is strictly required for anthracycline-driven cancer cell death to be perceived as immunogenic86, the same does not hold true for the demise of cancer cells exposed to ionizing radiation89 (Fig. 1).

Different inducers of immunogenic cell death (ICD) have been shown to elicit incompletely overlapping molecular signatures with respect to ICD biomarkers. This not only reinforces the need for the simultaneous assessment of multiple surrogate ICD biomarkers in the context of screening campaigns, but also identifies an originally unsuspected diversity in the molecular and cellular mechanisms supporting adaptive immunity downstream of danger signaling. ANXA1, annexin A1; CALR, calreticulin, CXCL10, C-X-C motif chemokine ligand 10; ECP, extracorporeal photochemotherapy; HHP, high hydrostatic pressure; HMGB1, high-mobility group box 1; IFN, interferon; IL-1β (official name: IL1B), interleukin 1 beta; IL-17 (official name: IL17), interleukin 17; PDT, photodynamic therapy.
Calreticulin
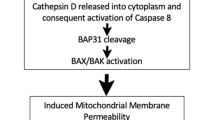
CALR exposed on the plasma membrane of malignant cells undergoing ICD serves as an “eat-me” signal that facilitates the engulfment of dying cells or their corpses by DCs or their precursors, thus providing them with an abundant source of antigenic material90,91,92,93. The molecular mechanism underlying the ICD-associated exposure of CALR include (1) the phosphorylation of eIF2α, accompanied by (2) a robust arrest of protein translation and (3) the activation of pro-apoptotic caspase 8 (CASP8), followed by the cleavage of B-cell receptor-associated protein 31 (BCAP31), the aggregation of the pro-apoptotic Bcl-2 family members BCL2-associated X protein (BAX) and BCL2-antagonist/killer 1 (BAK1) at the outer mitochondrial membrane, and (4) the vesicle-associated membrane protein 1 (VAMP1)- and synaptosome-associated protein 25 (SNAP25)-mediated anterograde transport to the Golgi apparatus and exocytosis94.
Surface-exposed CALR binds to LDL-receptor-related protein 1 (LRP1, best known as CD91), which is the main ER chaperone-sensing PRR expressed by antigen-presenting cells including DCs95,96. CD91 ligation promotes the engulfment of cellular corpses and debris by a mechanism that depends on the GTPase Rac family small GTPase 1 (RAC1)78,95,97. Consistent with the key role of CALR exposure in the perception of cell death as immunogenic, CALR knockdown by RNA interference (RNAi), CALR deletion by CRISPR/Cas9, or CALR blockade by neutralizing antibodies decreases the potency of ICD-mediated anticancer immune responses in a variety of settings79. Besides the role of CALR on the initiation of adaptive T-cell-mediated immunity downstream of ICD, we have recently demonstrated that CALR spontaneously exposed on the surface of malignant blasts from acute myeloid leukemia patients is associated with improved innate immunity as a consequence of improved interleukin 15 (IL15) trans-presentation to natural killer (NK) cells98,99. Altogether, these findings document that surface-exposed ER chaperones are important hallmarks of ICD stimulating both innate and adaptive anticancer immunity.
High-mobility group box 1
The release of HMGB1 from cancer cells undergoing ICD involves the permeabilization of both the nuclear lamina and the plasma membrane in a two-step process that enables the translocation of the protein from the nucleus to the cytoplasm, followed by its liberation into the extracellular space100,101. Extracellular HMGB1 can bind multiple PRRs expressed by myeloid cells, encompassing advanced glycosylation end-product-specific receptor (AGER, best known as RAGE) and Toll-like receptor 4 (TLR4)102,103,104,105. However, it appears that TLR4 signaling via MYD88 innate immune signal transduction adaptor (MYD88)106,107,108 is required and sufficient for cell death to be perceived as immunogenic, as demonstrated with a variety of genetic and pharmacological approaches16. In line with this notion, the knockout of HMGB1 in cancer cells and the antibody-mediated neutralization of TLR4 in the host limit therapeutically relevant immune responses (and hence disease control) driven by anthracyclines, cyclophosphamide, or oxaliplatin in preclinical in vivo models103,109. In addition, loss-of-function polymorphisms in TLR4 have been associated with unfavorable disease outcome in patients with breast cancer receiving anthracyclines as part of their clinical management103, in head and neck squamous cell carcinoma patients exposed to standard-of-care chemotherapy110, as well as in melanoma patients treated with an experimental DC-based vaccine111,112. Altogether, these findings indicate that the HMGB1-mediated activation of TLR4 is a crucial constituent of ICD-elicited immunogenicity. That said, HMGB1 release appears to be common to both immunogenic and non-immunogenic variants of cell deaths. In line with this notion, extracellular HMGB1 has been consistently used as a biomarker for plasma membrane permeabilization113. This implies that the mere detection of HMGB1 release cannot be interpreted as a reliable sign of ICD.
ATP
During the course of ICD, ATP is released in an autophagy-dependent manner through the active exocytosis of ATP-containing vesicles via pannexin channels114,115,116. Extracellular ATP operates as a prominent “find-me” signal for DC precursors and macrophages upon binding to purinergic receptor P2Y2 (P2RY2, a metabotropic receptor), thus facilitating the recruitment of myeloid cells to sites of active ICD117,118. Moreover, extracellular ATP mediates pro-inflammatory effects upon activation of the CASP1-dependent NLRP3 inflammasome and consequent secretion of mature interleukin 1 beta (IL1B, best known as IL-1β) and IL-18119,120,121. These effects, which originate from purinergic receptor P2X 7 (P2RX7, an inotropic receptor), culminate with the activation of CD8+ T cells and IL-17-producing γδ T cells119. Consistent with the importance of these events for immune responses driven by ICD, the immunogenicity of cell death is abrogated when either ATP fails to accumulate in the microenvironment of dying cancer cells or when P2RX7 or P2RY2 are absent from the myeloid compartment of the host119. Thus, overexpression of the ATP-degrading ectoenzyme ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, best known as CD39) in malignant tissues exerts potent immunosuppressive effects122,123,124. Moreover, loss-of-function polymorphisms in P2RX7 has been associated with poor clinical outcome in breast cancer patients subjected to anthracycline-based chemotherapy119. Of note, the cell death-associated secretion of ATP resembles HGMB1 release in that it can also accompany non-immunogenic variants of cell death. At least in vitro, however, normalizing extracellular ATP levels to percentage of dead cells or to the ATP plateau that can be achieved with a detergent aids the discrimination of non-immunogenic vs. immunogenic forms of cell death.
Annexin A1
The mechanisms involved in the release of ANXA1 by cancer cells succumbing to ICD remain largely obscure. However, ANXA1 appears to mediate a non-redundant role as a homing factor that governs the final approach of DCs or their precursors to malignant cells undergoing ICD125. Consistent with such a key function, malignant cells lacking Anxa1 exhibit limited sensitivity to anthracycline-based chemotherapy in vivo125. Similarly, the anticancer activity of anthracyclines in mice is amply compromised when the host lacks formyl peptide receptor 1 (FPR1), which is the main receptor for extracellular ANXA1125. Moreover, loss-of-function polymorphisms in FPR1 have been associated with poor overall survival and metastasis-free survival in breast cancer patients receiving adjuvant anthracycline-based chemotherapy125.
Type I interferon
Finally, ICD is accompanied by a robust type I IFN response, which can be driven by both RNA and DNA species126,127,128. In the former setting, the receptor is endosomal TLR3126,129, whereas in the latter scenario a key role is played by cytosolic cyclic GMP-AMP synthase (CGAS) and its signal transducer stimulator of IFN response cGAMP interactor 1 (STING1, best known as STING)89,130,131. Irrespective of the precise mechanism underlying type I IFN secretion, this cytokine mediates prominent immunostimulatory effects upon binding to homodimeric or heterodimeric receptors expressed by various immune cells36,132,133. For instance, type I IFN is known to enhance the cytotoxic functions of both CD8+ T cells and NK cells134, and promote cross-priming by DCs135,136. Moreover, type I IFN can trigger the secretion of pro-inflammatory mediators by macrophages137 and inhibit the immunosuppressive functions of CD4+CD25+FOXP3+ regulatory T cells138. Besides these direct immunostimulatory functions, type I IFN also elicits the synthesis of the C-X-C motif chemokine ligand 10 (CXCL10, a prominent chemotactic factor) by cancer cells undergoing ICD via an autocrine signaling loop126. The immunogenicity of ICD driven by anthracyclines and radiation therapy strongly relies on type I IFN signaling, as documented by the fact that therapeutic efficacy in mice is amply reduced when neoplastic lesions key components of the type I IFN response, such as Ifnar1, Ifnar2, Tlr3, Cgas, or Sting1, as well as when mice are co-treated with IFNAR1-blocking antibodies126,139,140. Along similar lines, Ifnar1−/− neoplastic cells exposed to doxorubicin in vitro lost their capacity to vaccinate syngeneic hosts against a rechallenge with living cells of the same type due their inability to prime adaptive immune responses126. That said, although acute, robust type I IFN responses have been consistently associated with immunostimulation, chronic, indolent type I IFN signaling mediates immunosuppressive effects83. Thus, caution should be employed when characterizing type I IFN responses in the context of ICD.
Irrespective of this and other caveats, DAMP-dependent adjuvanticity occupies a key position in the mechanism that governs the immunogenicity of malignant cells succumbing to ICD.
Monitoring CALR, HSPs, and the ISR
The exposure of CALR and other ER chaperones such as heat-shock protein family A (Hsp70) member 1A (HSPA1A, best known as HSP70) and heat-shock protein 90 α-family class A member 1 (HSP90AA1, best known as HSP90) on the plasma membrane of cells undergoing ICD can be monitored by several assays. The cytofluorometric detection of CALR exposure requires the use of specific anti-CALR antibodies and vital dyes such as such as 4′,6-diamidino-2-phenylindole (DAPI), propidium iodide (PI), or 7-aminoactinomycin D (7-AAD), to exclude permeabilized cells from the analysis and hence to avoid false-positive values98,99. The transgene-enforced expression of a CALR-HaloTag™ fusion protein141,142 can be also be used to specifically detect CALR exposure on (hitherto) living cells based on a cell-impermeant fluorescent HaloTag™ ligand143. However, this approach requires transgenic cell lines and hence is not suitable for ex vivo applications on freshly collected malignant cells. Surface-exposed CALR and other ER chaperones can also be detected by immunoblotting after cell surface proteins are biotinylated in pre-apoptotic cells (to avoid the detection of intracellular chaperones), followed by streptavidin-mediated precipitation78. Alternatively, fluorescence microscopy can be harnessed to monitor subcellular CALR localization, either upon immunostaining with CALR-specific antibodies, or in cells that have been engineered to express CALR in conjunction with a fluorescent moiety144,145. The latter technology is particularly advantageous for high-content screening (HCS) campaigns aimed at the identification of agents that cause CALR/HSP translocation on the plasma membrane. Retrospectively monitoring CALR/HSP expression in formalin-fixed paraffin-embedded bioptic samples from cancer patients by immunohistochemistry coupled to the evaluation of clinicopathological variables, offers a tool to estimate the impact of CALR/HSP exposure on disease progression91,146. Nevertheless, this technique is unable to precisely distinguish between the intracellular and surface-exposed pools of CALR and HSPs.
The ICD-associated exposure of CALR/HSP depends on the so-called integrated stress response (ISR), which is orchestrated around the inactivating phosphorylation of eIF2α147,148. The latter is generally catalyzed by eukaryotic translation initiation factor 2α kinase 2 (EIF2AK2, best known as PKR) and EIF2AK3 (best known as PERK), which are particularly sensitive to the accumulation of unfolded proteins within the ER149. Intriguingly, the other reticular arms of the ISR such as the splicing of X-box binding protein 1 (XBP1)150, as well as the derepression of activating transcription factor 4 (ATF4) and ATF6151,152 are not mechanistically linked to the immunogenicity of dying cancer cells, meaning that solely the phosphorylation of eIF2α constitutes a pathognomonic feature of ICD149,153. The ICD-associated phosphorylation of eIF2α can be detected by immunoblotting, flow cytometry, and immunofluorescence microscopy based on phosphoneoepitope-specific antibodies154,155,156,157,158, with the latter two approaches offering the scalability that is needed for HCS applications.
Monitoring HMGB1 release
The ICD-associated release of HMGB1 can be evaluated indirectly, upon quantification of the residual pool of intracellular HMGB1 by immunoblotting, as well as directly, upon assessment of extracellular HMGB1 levels in cell culture supernatants based on commercially available enzyme-linked immunosorbent assay (ELISA) kits80,103,143,159,160,161,162. ELISA kits are advantageous in that they offer a precise and sensitive means to quantify HMGB1 in a variety of samples including culture supernatants, sera and other biological fluids163,164,165. An alternative technological approach consists in the generation of cells expressing a green fluorescent protein (GFP)-tagged variant of HMGB1, which can be assessed by fluorescence microscopy in the presence of an appropriate nuclear counterstain, to quantify the residual pool of nuclear HMGB1160. This approach offers adequate scalability for HCS applications, but obviously cannot be employed to retrospectively investigate HMGB1 release from patient samples. Immunohistochemistry has been successfully harnessed to such aim, although (at least in some setting) a clear distinction between nuclear and cytoplasmic HMGB1 has been relatively hard to make166,167,168. Recently, the retention using selective hooks (RUSH) system169,170 has also been established as a fully automated technology with high-throughput workflow to determine the presence of DAMPs in distinct subcellular compartments. In the RUSH system, a streptavidin-NLS3 fusion protein is used as a nuclear hook to sequestrate a streptavidin-binding peptide (SBP) fused with a target and a reporter such as HMGB1 and GFP, respectively160. In this setting, the exogenous addition of biotin competitively disrupts the interaction between streptavidin-NLS3 and HMGB1-SBP-GFP to release the biosensor from its hook, hence allowing the fluorescent signal to leave the nucleus provided that an HMGB1-releasing stimulus is present160. The main advantage of the RUSH system is that it limits the amount of false-positive hits due to auto-fluorescent molecules and enables the retention of HMGB1 unless biotin is provided, constituting an interesting investigational platform to assess ICD-related processes specifically linked to HMGB1 release in the context of near-to-normal HGMB1 levels (which is not the case for RNAi- or CRISPR/Cas9-based manipulations).
Monitoring ATP secretion
In analogy to HMGB1, ICD-associated secretion of ATP can be monitored both directly, upon quantification of extracellular ATP, and indirectly, upon assessment of the residual pool of intracellular ATP (after cell lysis)86. Commercial luminescence-based assays represent the gold standard for both direct and indirect quantification of ATP levels171,172. Indeed, luciferase can catalyze the oxidation of its substrate luciferin, which is associated with light emission, only in the presence of magnesium, oxygen, and ATP173,174, which can be harnessed for quantitative assessments based on a conventional standard curve. The main disadvantage of the direct approach reflects the potential expression of ATP-degrading enzymes such as CD39 by cancer cells175,176, which may lower ATP concentrations below limit-of-detection. The indirect approach may potentially be confounded by agents that alter intracellular ATP levels in the absence of any cytotoxicity (and hence any ATP release), such as drugs targeting bioenergetic metabolism177,178,179. As an alternative to luminescence-based approaches, intracellular ATP-containing vesicles can be visualized and quantified by quinacrine180, a fluorochrome that emits in green in the presence of ATP, enabling quantitative assessment by flow cytometry and fluorescence microscopy181,182.
Monitoring the release of type I IFNs
The secretion of type I IFN from cancer cells undergoing ICD can be monitored by several assays183. In this setting, ELISA-based detection represents the gold standard approach, as it enables the quantitative assessment of type I IFN in a wide panel of biological specimens with superior sensitivity184,185,186. However, ELISAs are disadvantageous in that they cannot be harnessed to precisely identify type I IFN-producing cells within heterogeneous cell populations187,188. Such a disadvantage can be overcome by cytofluorometric tests based on intracellular staining with a type I IFN-specific (most often IFNB1-specific) antibody98. This approach can be widely used to analyze the production of type I IFN in cultured cells, as well as in primary tumor cells freshly isolated from patients91, although it is intrinsically unapt for the assessment of actual type I IFN secretion. RT-PCR and immunoblotting on cell lysates are also commonly employed to monitor type I IFN expression in cells responding to stress139,189. However, neither of these approaches can be employed to evaluate type I IFN secretion (as opposed to intracellular expression). Moreover, mRNA measurements do not formally evaluate type I IFN signaling, as transcription is not necessarily associated with translation190,191. Immunostaining based on type I IFN-specific antibodies coupled with immunohistochemistry or immunofluorescence microscopy has also been successfully employed to detect type I IFN in bioptic specimens from cancer patients and mice192,193,194,195,196,197,198. However, it is complex to discriminate between intracellular expression and secretion on these technical platforms. As an alternative to direct type I IFN measurements, genes expressed by cells exposed to type I IFN (which are commonly referred to as IFN-stimulated genes, ISGs), including MX dynamin-like GTPase 1 (MX1), have been evaluated by RT-PCR as proxies for the transcriptional response driven by IFN receptor dimers126,199. This approach overcomes several of the aforementioned limitations, although it cannot be implemented on a per cell basis. To this aim, although, biosensor cell lines expressing GFP under the control of the MX1 promoter have been engineered200. Such cells are amenable not only to cytofluorometric studies, but also to plate-based fluorescence measurements for HCS applications. A similar strategies relying on type I IFN signaling effectors has been adopted for the immunohistochemical evaluation of type I IFN activity in patient biopsies201,202.
Assessment of transcription and translation
One salient feature of ICD is the inhibition of RNA transcription85, constituting yet another feature that can be monitored in screening campaigns aimed at the identification of novel ICD inducers. Stalled RNA synthesis can be assessed in vitro by means of a chemically derivatized uridine analog that incorporates into nascent RNA and can be visualized as a fluorescent signal by click chemistry85. Alternatively, the inhibition of transcription can be accessed via the immunofluorescence microscopy-based detection of nucleolin and fibrillarin, two proteins that colocalize in the nucleus when RNA synthesis is active, yet can be detected as separate entities when transcription is stalled85. Besides laborious methods based on the incorporation of radiolabeled amino acids into nascent proteins203, translational proficiency can be assessed by polysome profiling, which is commonly based on the separation of cellular lysates on a sucrose gradient coupled to immunoblotting for ribosomal subunits on the fractions collected therefrom204,205. It may be difficult, however, to scale up polysome profiling for HCS applications. Irrespective of these and other unresolved issues, incorporating the assessment of transcriptional and translational proficiency into screening campaigns aimed at identifying novel ICD inducers may limit false positivity rate.
Discovery platform for the identification of ICD inducers
To address the need for novel ICD-inducing agents, we have built a phenotypic screening platform that incorporates many of the aforementioned assays coupled to automated epifluorescence microscopy (Fig. 2). Specifically, we employ biosensor cell lines to measure fluorescent surrogate markers for ATP release (with quinacrine), CALR exposure (using cells stably expressing CALR-GFP), type I IFN signaling (with cells expressing GFP under the control of the MX1 promoter), and HMGB1 release (in cells stably transduced with an HMGB1-GFP fusion) along with morphological traits of cell death such as the rarefaction of cells or the appearance of pyknotic nuclei206. These biosensors can be cultured in the presence of agents from large chemical collections and screened for ICD manifestations in a semi-automated manner, followed by in vitro validation experiments with alternative methods for ICD detection and additional cell lines. Finally, potential ICD inducers selected from the phenotypic screening need to be validated for their capacity to induce anticancer immune responses in vivo, in mouse models of prophylactic vaccination or therapeutic challenge. In some cases, indeed, abundant DAMP emission does not necessarily correlate with the ability of dying cells to drive anticancer immunity143 and in vivo functional assays remain the gold standard approach to identify bona fide ICD. More recently, we have used artificial intelligence to design algorithms that relate physicochemical descriptors of chemical agents with biological activity. These algorithms can predict the likelihood of distinct molecules to induce ICD, hence enabling the pre-selection of drugs with a high probability to operate as bona fide ICD inducers and hence reducing the cost of screening campaigns149,206.

The main hallmarks of immunogenic cell death (ICD) can be assessed by flow cytometry, (immuno)fluorescence microscopy, immunoblotting, or luminometry, based on a variety of different approaches. ANXA1, annexin A1; CALR, calreticulin, CXCL10, C-X-C motif chemokine ligand 10; HMGB1, high-mobility group box 1; IFN, interferon; IL-1β (official name: IL1B), interleukin 1 beta; IL-17 (official name: IL17), interleukin 17; RUSH, retention using selective hooks.
Concluding remarks
In summary, we and others have developed assays to assess ICD parameters in vitro (in cell cultures) and ex vivo (on tumor biopsies), which can be harnessed for the discovery of next-generation anticancer agents and the identification of optimal therapeutic regimens for clinical application, respectively. The construction of a multistep discovery pipeline involving artificial intelligence-driven pre-selection and a robotized workflow for the detection of surrogate ICD biomarkers enables us to implement various HCS campaigns that ultimately identified novel ICD inducers206. Some of the compounds identified with this platform have entered clinical trials, either as single agents or in combination with ICIs61,63. Similarly, novel approaches to drive ICD have been harnessed for the development of therapeutic DC-based vaccines, which are currently under clinical evaluation207,208. Moreover, the systematic assessment of ICD biomarkers such as the expression of CALR or HMGB1 on tumor biopsies may yield useful information for patient stratification (Fig. 3). That said, DAMP detection in patient samples remains particularly challenging16, as even in the case of proteins (which are considerably more stable than ATP), expression levels do not necessarily relate to emission16. At least in part, this problem could be circumvented by concomitantly assessing: (1) the intracellular levels of a specific DAMP (when possible, as for proteins) and/or the activation of the intracellular stress response that drives the emission of such DAMP (together, assessing the probability of DAMP emission); and (2) transcriptional programs driven by PRR activated by the same DAMP (as a measure of active signaling). To the best of our knowledge, however, such a combinatorial approach has not yet been undertaken.

Several high-content screening (HCS) campaigns have led to the discovery of novel immunogenic cell death (ICD) inducers that have entered clinical testing, either alone (A), or combined with immune checkpoint inhibitors (B). ICD induction is also being harnessed for the generation of dendritic cell (DC)-based vaccines for therapeutic purposes (C). Finally, biomarkers of ICD may be used to stratify patient populations and hence identify individuals with an elevated likelihood to respond to treatment and/or subjects that would benefit from strategies correcting existing defects in ICD signaling (D). HHP, high hydrostatic pressure.
On theoretical grounds, the absence or limited availability of ICD biomarkers should prompt the use of therapeutic approaches that (attempt to) compensate for the missing factors209. For instance, the absence of CALR might be compensated by the direct injection of recombinant CALR into the tumor or the administration of a CD47-blocking antibody, which neutralizes the main functional antagonist of CALR210,211. Similarly, the absence of HMGB1 might be compensated by the administration of recombinant HMGB1 itself or alternative TLR4 agonists212. These examples illustrate how the in-depth exploration of ICD-related processes and molecules might yield knowledge that may be harnessed to improve cancer therapies in a personalized, biomarker-driven manner.
References
Smyth, M. J., Dunn, G. P. & Schreiber, R. D. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 90, 1–50 (2006).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011).
Galluzzi, L., Spranger, S., Fuchs, E. & López-Soto, A. WNT signaling in cancer immunosurveillance. Trends Cell Biol. 29, 44–65 (2019).
Salmon, H., Remark, R., Gnjatic, S. & Merad, M. Host tissue determinants of tumour immunity. Nat. Rev. Cancer 19, 215–227 (2019).
Sautes-Fridman, C., Petitprez, F., Calderaro, J. & Fridman, W. H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325 (2019).
Galon, J., Angell, H. K., Bedognetti, D. & Marincola, F. M. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 39, 11–26 (2013).
Roumenina, L. T., Daugan, M. V., Petitprez, F., Sautes-Fridman, C. & Fridman, W. H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 19, 698–715 (2019).
Chalmin, F., Humblin, E., Ghiringhelli, F. & Vegran, F. Transcriptional programs underlying Cd4 T cell differentiation and functions. Int. Rev. Cell Mol. Biol. 341, 1–61 (2018).
Peng, D. et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253 (2015).
Dunn, G. P., Koebel, C. M. & Schreiber, R. D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 6, 836–848 (2006).
Rao, S., Gharib, K. & Han, A. Cancer immunosurveillance by T cells. Int Rev. Cell Mol. Biol. 342, 149–173 (2019).
Dunn, G. P., Old, L. J. & Schreiber, R. D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21, 137–148 (2004).
Ivashkiv, L. B. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 18, 545–558 (2018).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
Haen, S. P., Loffler, M. W., Rammensee, H. G. & Brossart, P. Towards new horizons: characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 17, 595–610 (2020).
Galluzzi, L. et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 8, e000337 (2020).
Smith, C. C. et al. Alternative tumour-specific antigens. Nat. Rev. Cancer 19, 465–478 (2019).
Vitale, I. et al. Mutational and antigenic landscape in tumor progression and cancer immunotherapy. Trends Cell Biol. 29, 396–416 (2019).
Hu, Z., Ott, P. A. & Wu, C. J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 18, 168–182 (2018).
Ren, Y. et al. HLA class-I and class-II restricted neoantigen loads predict overall survival in breast cancer. Oncoimmunology 9, 1744947 (2020).
Sade-Feldman, M. et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 8, 1136 (2017).
Kotsias, F., Cebrian, I. & Alloatti, A. Antigen processing and presentation. Int. Rev. Cell Mol. Biol. 348, 69–121 (2019).
Yamamoto, K. et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105 (2020).
Kalbasi, A. & Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 20, 25–39 (2020).
Lee, J. H. et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat. Commun. 11, 1897 (2020).
Lee, Y. S. & Radford, K. J. The role of dendritic cells in cancer. Int. Rev. Cell Mol. Biol. 348, 123–178 (2019).
Martinek, J., Wu, T. C., Cadena, D., Banchereau, J. & Palucka, K. Interplay between dendritic cells and cancer cells. Int. Rev. Cell Mol. Biol. 348, 179–215 (2019).
Vacchelli, E. et al. Trial watch: dendritic cell-based interventions for cancer therapy. Oncoimmunology 2, e25771 (2013).
Worbs, T., Hammerschmidt, S. I. & Forster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 17, 30–48 (2017).
Amon, L. et al. Transcriptional control of dendritic cell development and functions. Int. Rev. Cell Mol. Biol. 349, 55–151 (2019).
Krysko, O., Løve Aaes, T., Bachert, C., Vandenabeele, P. & Krysko, D. V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 4, e631 (2013).
Sprooten, J., Agostinis, P. & Garg, A. D. Type I interferons and dendritic cells in cancer immunotherapy. Int. Rev. Cell Mol. Biol. 348, 217–262 (2019).
Balan, S., Saxena, M. & Bhardwaj, N. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol. 348, 1–68 (2019).
Gong, T., Liu, L., Jiang, W. & Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 20, 95–112 (2020).
Bloes, D. A., Kretschmer, D. & Peschel, A. Enemy attraction: bacterial agonists for leukocyte chemotaxis receptors. Nat. Rev. Microbiol. 13, 95–104 (2015).
McNab, F., Mayer-Barber, K., Sher, A., Wack, A. & O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103 (2015).
Yatim, N., Cullen, S. & Albert, M. L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 17, 262–275 (2017).
Krysko, D. V. et al. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860–875 (2012).
Zhou, J. et al. Immunogenic cell death in cancer therapy: present and emerging inducers. J. Cell Mol. Med. 23, 4854–4865 (2019).
Garg, A. D. et al. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim. Biophys. Acta 1805, 53–71 (2010).
Boada-Romero, E., Martinez, J., Heckmann, B. L. & Green, D. R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 21, 398–414 (2020).
Paludan, S. R., Reinert, L. S. & Hornung, V. DNA-stimulated cell death: implications for host defence, inflammatory diseases and cancer. Nat. Rev. Immunol. 19, 141–153 (2019).
Vanpouille-Box, C., Hoffmann, J. A. & Galluzzi, L. Pharmacological modulation of nucleic acid sensors - therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 18, 845–867 (2019).
West, A. P. & Shadel, G. S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17, 363–375 (2017).
Fucikova, J. et al. Prognostic and predictive value of DAMPs and DAMP-associated processes in cancer. Front. Immunol. 6, 402 (2015).
Galluzzi, L., Humeau, J., Buque, A., Zitvogel, L. & Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 17, 725–741 (2020).
Deutsch, E., Chargari, C., Galluzzi, L. & Kroemer, G. Optimising efficacy and reducing toxicity of anticancer radioimmunotherapy. Lancet Oncol. 20, e452–e463 (2019).
Ye, W. et al. ASTX660, an antagonist of cIAP1/2 and XIAP, increases antigen processing machinery and can enhance radiation-induced immunogenic cell death in preclinical models of head and neck cancer. Oncoimmunology 9, 1710398 (2020).
Xiao, R. et al. Antagonist of cIAP1/2 and XIAP enhances anti-tumor immunity when combined with radiation and PD-1 blockade in a syngeneic model of head and neck cancer. Oncoimmunology 7, e1471440 (2018).
Pol, J. et al. Trial Watch: immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology 4, e1008866 (2015).
Hwang, W. L., Pike, L. R. G., Royce, T. J., Mahal, B. A. & Loeffler, J. S. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat. Rev. Clin. Oncol. 15, 477–494 (2018).
Galon, J. & Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019).
Ngwa, W. et al. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 18, 313–322 (2018).
Truxova, I. et al. Rationale for the combination of dendritic cell-based vaccination approaches with chemotherapy agents. Int. Rev. Cell Mol. Biol. 330, 115–156 (2017).
Brown, J. S., Sundar, R. & Lopez, J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br. J. Cancer 118, 312–324 (2018).
Antonia, S. J. et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N. Engl. J. Med. 377, 1919–1929 (2017).
Voorwerk, L. et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat. Med. 25, 920–928 (2019).
Yamazaki, T., Buqué, A., Ames, T. D. & Galluzzi, L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. Oncoimmunology 9, 1721810 (2020).
Liu, P. et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat. Commun. 10, 1486 (2019).
D’Amico, L. et al. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J. Immunother. Cancer 7, 16 (2019).
Vacchelli, E. et al. Trial Watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology 3, e27878 (2014).
Han, X. & Vesely, M. D. Stimulating T cells against cancer with agonist immunostimulatory monoclonal antibodies. Int. Rev. Cell Mol. Biol. 342, 1–25 (2019).
Tang, J. et al. Trial watch: the clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discov. 17, 854–855 (2018).
Aranda, F. et al. Trial Watch: immunostimulatory monoclonal antibodies in cancer therapy. Oncoimmunology 3, e27297 (2014).
Borrie, A. E. & Maleki Vareki, S. T lymphocyte-based cancer immunotherapeutics. Int. Rev. Cell Mol. Biol. 341, 201–276 (2018).
Sun, C. et al. Reactive oxygen species involved in CT26 immunogenic cell death induced by Clostridium difficile toxin B. Immunol. Lett. 164, 65–71 (2015).
Donnelly, O. G. et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 20, 7–15 (2013).
Bommareddy, P. K., Shettigar, M. & Kaufman, H. L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 18, 498–513 (2018).
Dudek, A. M., Garg, A. D., Krysko, D. V., De Ruysscher, D. & Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 24, 319–333 (2013).
Serrano-Del Valle, A., Anel, A., Naval, J. & Marzo, I. Immunogenic cell death and immunotherapy of multiple myeloma. Front. Cell Dev. Biol. 7, 50 (2019).
Giglio, P. et al. PKR and GCN2 stress kinases promote an ER stress-independent eIF2alpha phosphorylation responsible for calreticulin exposure in melanoma cells. Oncoimmunology 7, e1466765 (2018).
Huang, F. Y. et al. Induction of enhanced immunogenic cell death through ultrasound-controlled release of doxorubicin by liposome-microbubble complexes. Oncoimmunology 7, e1446720 (2018).
Garrido, G. et al. Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a specific antibody. J. Immunol. 187, 4954–4966 (2011).
Pozzi, C. et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat. Med. 22, 624–631 (2016).
Chabanon, R. M. et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Invest. 129, 1211–1228 (2019).
Pantelidou, C. et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 9, 722–737 (2019).
Rodriguez-Ruiz, M. E., Vitale, I., Harrington, K. J., Melero, I. & Galluzzi, L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat. Immunol. 21, 120–134 (2020).
Garg, A. D. et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 31, 1062–1079 (2012).
Tatsuno, K. et al. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis. 10, 578 (2019).
Fucikova, J. et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int. J. Cancer 135, 1165–1177 (2014).
Krombach, J. et al. Priming anti-tumor immunity by radiotherapy: dying tumor cell-derived DAMPs trigger endothelial cell activation and recruitment of myeloid cells. Oncoimmunology 8, e1523097 (2019).
Di Virgilio, F., Sarti, A. C., Falzoni, S., De Marchi, E. & Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 18, 601–618 (2018).
Vanpouille-Box, C., Demaria, S., Formenti, S. C. & Galluzzi, L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell 34, 361–378 (2018).
Rosenzweig, R., Nillegoda, N. B., Mayer, M. P. & Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 20, 665–680 (2019).
Humeau, J. et al. Inhibition of transcription by dactinomycin reveals a new characteristic of immunogenic cell stress. EMBO Mol. Med. 12, e11622 (2020).
Michaud, M. et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577 (2011).
Bezu, L. et al. eIF2α phosphorylation: a hallmark of immunogenic cell death. Oncoimmunology 7, e1431089 (2018).
Rybstein, M. D., Bravo-San Pedro, J. M., Kroemer, G. & Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 20, 243–251 (2018).
Yamazaki, T. et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 21, 1160–1171 (2020).
Poon, I. K., Lucas, C. D., Rossi, A. G. & Ravichandran, K. S. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol. 14, 166–180 (2014).
Kasikova, L. et al. Calreticulin exposure correlates with robust adaptive antitumor immunity and favorable prognosis in ovarian carcinoma patients. J. Immunother. Cancer 7, 312 (2019).
Guermonprez, P., Gerber-Ferder, Y., Vaivode, K., Bourdely, P. & Helft, J. Origin and development of classical dendritic cells. Int. Rev. Cell Mol. Biol. 349, 1–54 (2019).
Schcolnik-Cabrera, A. et al. Calreticulin in phagocytosis and cancer: opposite roles in immune response outcomes. Apoptosis 24, 245–255 (2019).
Panaretakis, T. et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 28, 578–590 (2009).
Gardai, S. J. et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334 (2005).
Gardai, S. J. et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 115, 13–23 (2003).
Byrne, J. C. et al. Bruton’s tyrosine kinase is required for apoptotic cell uptake via regulating the phosphorylation and localization of calreticulin. J. Immunol. 190, 5207–5215 (2013).
Truxova, I. et al. Calreticulin exposure on malignant blasts correlates with improved natural killer cell-mediated cytotoxicity in acute myeloid leukemia patients. Haematologica 105, 1868–1878 (2020).
Fucikova, J. et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 128, 3113–3124 (2016).
Yang, H., Wang, H., Chavan, S. S. & Andersson, U. High mobility group box protein 1 (HMGB1): the prototypical endogenous danger molecule. Mol. Med. 21(Suppl 1), S6–S12 (2015).
Zhu, X. et al. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J. Clin. Invest. 125, 1098–1110 (2015).
Park, J. S. et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 290, C917–C924 (2006).
Apetoh, L. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059 (2007).
Taguchi, A. et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 405, 354–360 (2000).
Smith, M. et al. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 7, e1526250 (2018).
Medzhitov, R. et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2, 253–258 (1998).
Gay, N. J., Symmons, M. F., Gangloff, M. & Bryant, C. E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 14, 546–558 (2014).
Vigneron, C. et al. Sepsis inhibits tumor growth in mice with cancer through Toll-like receptor 4-associated enhanced natural killer cell activity. Oncoimmunology 8, e1641391 (2019).
Nayagom, B. et al. Immunogenic cell death in a combined synergic gene- and immune-therapy against cancer. Oncoimmunology 8, e1667743 (2019).
Bergmann, C. et al. Toll-like receptor 4 single-nucleotide polymorphisms Asp299Gly and Thr399Ile in head and neck squamous cell carcinomas. J. Transl. Med. 9, 139 (2011).
Tittarelli, A. et al. Toll-like receptor 4 gene polymorphism influences dendritic cell in vitro function and clinical outcomes in vaccinated melanoma patients. Cancer Immunol. Immunother. 61, 2067–2077 (2012).
Sprooten, J. et al. Trial watch: dendritic cell vaccination for cancer immunotherapy. Oncoimmunology 8, e1638212 (2019).
Galluzzi, L. et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 16, 1093–1107 (2009).
Martins, I. et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 21, 79–91 (2014).
Anderson, C. M. & Macleod, K. F. Autophagy and cancer cell metabolism. Int. Rev. Cell Mol. Biol. 347, 145–190 (2019).
Follo, C., Cheng, Y., Richards, W. G., Bueno, R. & Broaddus, V. C. Autophagy facilitates the release of immunogenic signals following chemotherapy in 3D models of mesothelioma. Mol. Carcinog. 58, 1754–1769 (2019).
Burnstock, G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 87, 659–797 (2007).
Elliott, M. R. et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286 (2009).
Ghiringhelli, F. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178 (2009).
Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006).
Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Ohta, A. & Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414, 916–920 (2001).
Stagg, J. et al. CD73-deficient mice are resistant to carcinogenesis. Cancer Res. 72, 2190–2196 (2012).
Allard, B., Allard, D., Buisseret, L. & Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 17, 611–629 (2020).
Vacchelli, E. et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 350, 972–978 (2015).
Sistigu, A. et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 20, 1301–1309 (2014).
Rodriguez-Ruiz, M. E. et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology 8, e1655964 (2019).
Khodarev, N. N. Intracellular RNA sensing in mammalian cells: role in stress response and cancer therapies. Int. Rev. Cell Mol. Biol. 344, 31–89 (2019).
Medler, T. et al. Activating the nucleic acid-sensing machinery for anticancer immunity. Int. Rev. Cell Mol. Biol. 344, 173–214 (2019).
Hopfner, K. P. & Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 21, 501–521 (2020).
McLaughlin, M. et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 20, 203–217 (2020).
Motwani, M., Pesiridis, S. & Fitzgerald, K. A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 20, 657–674 (2019).
Kawasaki, T. & Kawai, T. Discrimination between self and non-self-nucleic acids by the innate immune system. Int. Rev. Cell Mol. Biol. 344, 1–30 (2019).
Oh, J. H. et al. Sustained type I interferon reinforces NK cell-mediated cancer immunosurveillance during chronic virus infection. Cancer Immunol. Res. 7, 584–599 (2019).
Bek, S. et al. Targeting intrinsic RIG-I signaling turns melanoma cells into type I interferon-releasing cellular antitumor vaccines. Oncoimmunology 8, e1570779 (2019).
Takeda, Y. et al. Type I interferon-independent dendritic cell priming and antitumor T cell activation induced by a Mycoplasma fermentans lipopeptide. Front. Immunol. 9, 496 (2018).
Muller, E. et al. Toll-like receptor ligands and interferon-gamma synergize for induction of antitumor M1 macrophages. Front. Immunol. 8, 1383 (2017).
Gangaplara, A. et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. 14, e1006985 (2018).
Vanpouille-Box, C. et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 8, 15618 (2017).
Deng, L. et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852 (2014).
Los, G. V. & Wood, K. The HaloTag: a novel technology for cell imaging and protein analysis. Methods Mol. Biol. 356, 195–208 (2007).
Los, G. V. et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382 (2008).
Menger, L. et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci. Transl. Med. 4, 143ra199 (2012).
Pacheco, A., Merianda, T. T., Twiss, J. L. & Gallo, G. Mechanism and role of the intra-axonal Calreticulin translation in response to axonal injury. Exp. Neurol. 323, 113072 (2020).
Gao, G., Zhu, C., Liu, E. & Nabi, I. R. Reticulon and CLIMP-63 regulate nanodomain organization of peripheral ER tubules. PLoS Biol. 17, e3000355 (2019).
Fucikova, J. et al. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res. 76, 1746–1756 (2016).
Costa-Mattioli, M. & Walter, P. The integrated stress response: from mechanism to disease. Science 368, eaat5314 (2020).
Hetz, C. & Papa, F. R. The unfolded protein response and cell fate control. Mol. Cell 69, 169–181 (2018).
Bezu, L. et al. eIF2α phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ. 25, 1375–1393 (2018).
Glimcher, L. H., Lee, A. H. & Iwakoshi, N. N. XBP-1 and the unfolded protein response (UPR). Nat. Immunol. 21, 963–965 (2020).
Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 (2012).
Rainbolt, T. K. & Frydman, J. Dynamics and clustering of IRE1alpha during ER stress. Proc. Natl Acad. Sci. USA 117, 3352–3354 (2020).
Hetz, C., Chevet, E. & Harding, H. P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719 (2013).
Harding, H. P., Zhang, Y. & Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 (1999).
van Schadewijk, A., van’t Wout, E. F., Stolk, J. & Hiemstra, P. S. A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell Stress Chaperones 17, 275–279 (2012).
Hayashi, A. et al. The role of brain-derived neurotrophic factor (BDNF)-induced XBP1 splicing during brain development. J. Biol. Chem. 282, 34525–34534 (2007).
Prischi, F., Nowak, P. R., Carrara, M. & Ali, M. M. Phosphoregulation of Ire1 RNase splicing activity. Nat. Commun. 5, 3554 (2014).
Reineke, L. C., Dougherty, J. D., Pierre, P. & Lloyd, R. E. Large G3BP-induced granules trigger eIF2α phosphorylation. Mol. Biol. Cell 23, 3499–3510 (2012).
Dong Xda, E. et al. High mobility group box I (HMGB1) release from tumor cells after treatment: implications for development of targeted chemoimmunotherapy. J. Immunother. 30, 596–606 (2007).
Zhao, L., Liu, P., Kepp, O. & Kroemer, G. Methods for measuring HMGB1 release during immunogenic cell death. Methods Enzymol. 629, 177–193 (2019).
Candolfi, M. et al. Release of HMGB1 in response to proapoptotic glioma killing strategies: efficacy and neurotoxicity. Clin. Cancer Res. 15, 4401–4414 (2009).
Bommareddy, P. K., Zloza, A., Rabkin, S. D. & Kaufman, H. L. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology 8, 1591875 (2019).
Lei, C., Geng, J. & Zhong, L. The association between plasma HMGB1 and sRAGE and clinical outcome in intracerebral hemorrhage. J. Neuroimmunol. 345, 577266 (2020).
Bruck, E. et al. Prolonged elevation of plasma HMGB1 is associated with cognitive impairment in intensive care unit survivors. Intensive Care Med. 46, 811–812 (2020).
Wang, Y. et al. Urinary MCP-1HMGB1 increased in calcium nephrolithiasis patients and the influence of hypercalciuria on the production of the two cytokines. Urolithiasis 45, 159–175 (2017).
Suzuki, Y. et al. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 72, 3967–3976 (2012).
Suren, D. et al. HMGB1 expression in urothelial carcinoma of the bladder. J. BUON 23, 1882–1886 (2018).
Mohajertehran, F., Ayatollahi, H., Khazaeni, K., Shakeri, M. T. & Mohtasham, N. Overexpression of high-mobility motor box 1 in the blood and tissues of patients with head and neck squamous cell carcinoma. Iran. J. Otorhinolaryngol. 30, 261–271 (2018).
Boncompain, G. et al. Synchronization of secretory protein traffic in populations of cells. Nat. Methods 9, 493–498 (2012).
Boncompain, G. & Perez, F. Synchronizing protein transport in the secretory pathway. Curr. Protoc. Cell Biol. Chapter 15, Unit 15.19 (2012).
Dubyak, G. R. Luciferase-assisted detection of extracellular ATP and ATP metabolites during immunogenic death of cancer cells. Methods Enzymol. 629, 81–102 (2019).
Branchini, B. R. & Southworth, T. L. A highly sensitive biosensor for ATP using a chimeric firefly luciferase. Methods Enzymol. 589, 351–364 (2017).
Lundin, A. & Thore, A. Analytical information obtainable by evaluation of the time course of firefly bioluminescence in the assay of ATP. Anal. Biochem. 66, 47–63 (1975).
Lundin, A. Use of firefly luciferase in ATP-related assays of biomass, enzymes, and metabolites. Methods Enzymol. 305, 346–370 (2000).
Nagate, Y. et al. Ectonucleotidase CD39 is highly expressed on ATLL cells and is responsible for their immunosuppressive function. Leukemia. https://doi.org/10.1038/s41375-020-0788-y (2020).
Li, X. Y. et al. Targeting CD39 in cancer reveals an extracellular ATP- and inflammasome-driven tumor immunity. Cancer Discov. 9, 1754–1773 (2019).
Patel, C. H., Leone, R. D., Horton, M. R. & Powell, J. D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 18, 669–688 (2019).
Altman, B. J., Stine, Z. E. & Dang, C. V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 619–634 (2016).
Hoang, G., Udupa, S. & Le, A. Application of metabolomics technologies toward cancer prognosis and therapy. Int. Rev. Cell Mol. Biol. 347, 191–223 (2019).
Marceau, F., Roy, C. & Bouthillier, J. Assessment of cation trapping by cellular acidic compartments. Methods Enzymol. 534, 119–131 (2014).
Forveille, S. et al. Quinacrine-mediated detection of intracellular ATP. Methods Enzymol. 629, 103–113 (2019).
Vessey, K. A., Ho, T., Jobling, A. I., Wang, A. Y. & Fletcher, E. L. Fluorescent labeling and quantification of vesicular ATP release using live cell imaging. Methods Mol. Biol. 2041, 209–221 (2020).
Borden, E. C. Interferons alpha and beta in cancer: therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 18, 219–234 (2019).
Formenti, S. C. et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 24, 1845–1851 (2018).
Becker-Merok, A., Ostli-Eilersten, G., Lester, S. & Nossent, J. Circulating interferon-alpha2 levels are increased in the majority of patients with systemic lupus erythematosus and are associated with disease activity and multiple cytokine activation. Lupus 22, 155–163 (2013).
Cherney, D. Z., Scholey, J. W., Sochett, E., Bradley, T. J. & Reich, H. N. The acute effect of clamped hyperglycemia on the urinary excretion of inflammatory cytokines/chemokines in uncomplicated type 1 diabetes: a pilot study. Diabetes Care 34, 177–180 (2011).
Leylek, R. & Idoyaga, J. The versatile plasmacytoid dendritic cell: function, heterogeneity, and plasticity. Int. Rev. Cell Mol. Biol. 349, 177–211 (2019).
Moore, E. C. et al. Nanocomplex-based TP53 gene therapy promotes anti-tumor immunity through TP53- and STING-dependent mechanisms. Oncoimmunology 7, e1404216 (2018).
Diamond, J. M. et al. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol. Res 6, 910–920 (2018).
Gry, M. et al. Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genomics 10, 365 (2009).
Liu, Y., Beyer, A. & Aebersold, R. On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550 (2016).
Downey, J. et al. RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to influenza A virus infection. PLoS Pathog. 13, e1006326 (2017).
Sarkar, M. K. et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann. Rheum. Dis. 77, 1653–1664 (2018).
Takahashi, A. et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun. 9, 1249 (2018).
Skopelja-Gardner, S. et al. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci. Rep. 10, 7908 (2020).
Demaria, O. et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl Acad. Sci. USA 112, 15408–15413 (2015).
Wu, H. et al. Mumps virus-induced innate immune responses in mouse Sertoli and Leydig cells. Sci. Rep. 6, 19507 (2016).
Zhu, W. et al. Pattern recognition receptor-initiated innate antiviral responses in mouse epididymal epithelial cells. J. Immunol. 194, 4825–4835 (2015).
Bordignon, J. et al. Expression profile of interferon stimulated genes in central nervous system of mice infected with dengue virus Type-1. Virology 377, 319–329 (2008).
Zhou, H. et al. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis. 7, e2134 (2016).
Clifford, J. L. et al. Expression of protein mediators of type I interferon signaling in human squamous cell carcinoma of the skin. Cancer Epidemiol. Biomark. Prev. 9, 993–997 (2000).
Clifford, J. L. et al. Suppression of type I interferon signaling proteins is an early event in squamous skin carcinogenesis. Clin. Cancer Res. 8, 2067–2072 (2002).
Cooke, S. K. et al. Effects of the presence and absence of amino acids on translation, signaling, and long-term depression in hippocampal slices from Fmr1 knockout mice. J. Neurochem. 151, 764–776 (2019).
Chassé, H., Boulben, S., Costache, V., Cormier, P. & Morales, J. Analysis of translation using polysome profiling. Nucleic Acids Res. 45, e15 (2017).
Coudert, L., Adjibade, P. & Mazroui, R. Analysis of translation initiation during stress conditions by polysome profiling. J. Vis. Exp. 87, 51164 (2014).
Kepp, O. et al. A fluorescent biosensor-based platform for the discovery of immunogenic cancer cell death inducers. Oncoimmunology 8, 1606665 (2019).
Han, P., Hanlon, D., Sobolev, O., Chaudhury, R. & Edelson, R. L. Ex vivo dendritic cell generation-A critical comparison of current approaches. Int. Rev. Cell Mol. Biol. 349, 251–307 (2019).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020).
Bezu, L. et al. Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 6, 187 (2015).
Feng, M. et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586 (2019).
Chao, M. P., Majeti, R. & Weissman, I. L. Programmed cell removal: a new obstacle in the road to developing cancer. Nat. Rev. Cancer 12, 58–67 (2011).
Hennessy, E. J., Parker, A. E. & O’Neill, L. A. Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307 (2010).
Acknowledgements
G.K. is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR)—Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085 and GDW20181100051), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM). L.G. is supported by a Breakthrough Level 2 grant from the US Department of Defense (DoD), Breast Cancer Research Program (BRCP) (#BC180476P1), by the 2019 Laura Ziskin Prize in Translational Research (#ZP-6177, PI: Formenti) from the Stand Up to Cancer (SU2C), by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukemia and Lymphoma Society (LLS), by a startup grant from the Dept. of Radiation Oncology at Weill Cornell Medicine (New York, US), by a Rapid Response Grant from the Functional Genomics Initiative (New York, US), by industrial collaborations with Lytix (Oslo, Norway) and Phosplatin (New York, US), and by donations from Phosplatin (New York, US), the Luke Heller TECPR2 Foundation (Boston, US), and Sotio a.s. (Prague, Czech Republic).
Author information
Authors and Affiliations
Contributions
Concept and design: J.F., O.K., and L.G. Writing, review, and/or revision of the manuscript: J.F., L.K., T.Y., G.P., P.L., L.Z., O.K., R.S., G.K., and L.G. Display item design: O.K. and G.P. All authors approve the final version of the article.
Corresponding authors
Ethics declarations
Conflict of interest
J.F. and R.S. are full-time employees of Sotio. O.K. is a scientific co-founder of Samsara Therapeutics. G.K. has been holding research contracts with Bayer Healthcare, Genentech, Glaxo Smyth Kline, Institut Mérieux, Lytix Pharma, PharmaMar, Sotio, and Vasculox. He is on the Board of Directors of the Bristol Myers Squibb Foundation France and is a scientific co-founder of everImmune, Samsara Therapeutics, and Therafast Bio. L.G. received research support from Lytix and Phosplatin, consulting fees from OmniSEQ, Astra Zeneca, Inzen, and the Luke Heller TECPR2 Foundation, and is a member of the Scientific Advisory Committee of Boehringer Ingelheim, The Longevity Labs, and OmniSEQ. All other authors have no conflicts to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Piacentini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fucikova, J., Kepp, O., Kasikova, L. et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis 11, 1013 (2020). https://doi.org/10.1038/s41419-020-03221-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03221-2
This article is cited by
-
PREX2 contributes to radiation resistance by inhibiting radiotherapy-induced tumor immunogenicity via cGAS/STING/IFNs pathway in colorectal cancer
BMC Medicine (2024)
-
The application of nanoparticles-based ferroptosis, pyroptosis and autophagy in cancer immunotherapy
Journal of Nanobiotechnology (2024)
-
Supporting gut health with medicinal cannabis in people with advanced cancer: potential benefits and challenges
British Journal of Cancer (2024)
-
Non-canonical BIM-regulated energy metabolism determines drug-induced liver necrosis
Cell Death & Differentiation (2024)
-
Dendritic cells as orchestrators of anticancer immunity and immunotherapy
Nature Reviews Clinical Oncology (2024)
