




Abstract
We assessed the clinicopathological features of 92 patients with primary Sjögren's syndrome-associated neuropathy (76 women, 16 men, 54.7 years, age at onset). The majority of patients (93%) were diagnosed with Sjögren's syndrome after neuropathic symptoms appeared. We classified these patients into seven forms of neuropathy: sensory ataxic neuropathy (n = 36), painful sensory neuropathy without sensory ataxia (n = 18), multiple mononeuropathy (n = 11), multiple cranial neuropathy (n = 5), trigeminal neuropathy (n = 15), autonomic neuropathy (n = 3) and radiculoneuropathy (n = 4), based on the predominant neuropathic symptoms. Acute or subacute onset was seen more frequently in multiple mononeuropathy and multiple cranial neuropathy, whereas chronic progression was predominant in other forms of neuropathy. Sensory symptoms without substantial motor involvement were seen predominantly in sensory ataxic, painful sensory, trigeminal and autonomic neuropathy, although the affected sensory modalities and distribution pattern varied. In contrast, motor weakness and muscle atrophy were observed in multiple mononeuropathy, multiple cranial neuropathy and radiculoneuropathy. Autonomic symptoms were often seen in all forms of neuropathy. Abnormal pupils and orthostatic hypotension were particularly frequent in sensory ataxic, painful, trigeminal and autonomic neuropathy. Unelicited somatosensory evoked potentials and spinal cord posterior column abnormalities in MRI were observed in sensory ataxic, painful and autonomic neuropathy. Sural nerve biopsy specimens (n = 55) revealed variable degrees of axon loss. Predominantly large fibre loss was observed in sensory ataxic neuropathy, whereas predominantly small fibre loss occurred in painful sensory neuropathy. Angiitis and perivascular cell invasion were seen most frequently in multiple mononeuropathy, followed by sensory ataxic neuropathy. The autopsy findings of one patient with sensory ataxic neuropathy showed severe large sensory neuron loss paralleling to dorsal root and posterior column involvement of the spinal cord, and severe sympathetic neuron loss. Degrees of neuron loss in the dorsal and sympathetic ganglion corresponded to segmental distribution of sensory and sweating impairment. Multifocal T-cell invasion was seen in the dorsal root and sympathetic ganglion, perineurial space and vessel walls in the nerve trunks. Differential therapeutic responses for corticosteroids and IVIg were seen among the neuropathic forms. These clinicopathological observations suggest that sensory ataxic, painful and perhaps trigeminal neuropathy are related to ganglioneuronopathic process, whereas multiple mononeuropathy and multiple cranial neuropathy would be more closely associated with vasculitic process.
Introduction
Primary Sjögren's syndrome is a systemic autoimmune disease characterized by xerophthalmia and xerostomia, and is associated with systemic visceral involvement, including pneumonitis, renal tubular acidosis, pancreatitis, myositis, and occasionally lymphocytic proliferation. A wide variety of neurological complications also are characteristic features of primary Sjögren's syndrome (Attwood et al., 1961; Alexander et al., 1982; Delalande et al., 2004). Peripheral neuropathy is a major neurological manifestation of Sjögren's syndrome and its aetiology has been considered to be vasculitis in the peripheral nerves, similar to that observed in other collagen diseases. In 1986 and 1990, it was demonstrated that dorsal root ganglionitis with degeneration of dorsal root ganglion neurons and mononuclear cell infiltration without vasculitis are associated with the sensory ataxic form of Sjögren's syndrome-associated neuropathy, suggesting that ganglion neurons themselves can be a target of Sjögren's syndrome (Malinow et al., 1986; Griffin et al., 1990).
Sjögren's syndrome-associated neuropathy has been shown to manifest as a variety of forms of neuropathy, including sensory ataxic neuropathy (Kennett et al., 1986; Griffin et al., 1990; Kaplan et al., 1990; Sobue et al., 1993), trigeminal neuropathy (Kaltreider et al., 1969), multiple mononeuropathy (Peyronnard et al., 1982; Molina et al., 1985), radiculoneuropathy (Gross et al., 1987; Grant et al., 1997), painful sensory neuropathy without sensory ataxia (Denislic et al., 1995; Mori et al., 2003), autonomic neuropathy with anhidrosis (Kumazawa et al., 1993; Goto et al., 2000) and multiple cranial neuropathy (Touze et al., 1999; Chu et al., 2000; Urban et al., 2001). While the wide spectrum of these neuropathies has been described in anecdotal reports or in studies of the systemic manifestations of Sjögren's syndrome (Mellgren et al., 1989; Gemignani et al., 1994; Mauch et al., 1994), the pathogenic mechanism responsible for most forms of Sjögren's syndrome-associated neuropathy remains unresolved. Furthermore, the spectrum of neuropathy and neuropathic symptoms of each form of neuropathy, particularly in the pathological and electrophysiological background, have not been well elucidated. In addition, since the prevalence of Sjögren's syndrome is growing in the elderly (Lafitte et al., 2001), Sjögren's syndrome-associated neuropathy also has become more prevalent. It is therefore necessary to re-evaluate the clinical spectrum, and pathological and electrophysiological features of Sjögren's syndrome-associated neuropathy.
In this study, we assessed the clinicopathological and electrophysiological features of a large number of patients associated with primary Sjögren's syndrome-associated neuropathy and determined the range of clinical manifestations of neuropathy.
Patients and methods
Patients
A total of 92 patients (76 women, 16 men; mean age, 59.7 years), all of Japanese descent, who fulfilled the diagnostic criteria for primary Sjögren's syndrome and who had been referred to the Hospital of Nagoya University School of Medicine and its affiliated hospitals between 1985 and 2004 were the subjects of this study. The diagnosis of primary Sjögren's syndrome was established by the criteria proposed by the Diagnostic Committee of Health and Welfare of Japan (Fujibayashi et al., 1999) and by the American-European Community (Vitali et al., 2002). These criteria included symptoms of xerophthalmia and xerostomia, objective evidence of keratoconjunctivitis such as an abnormal Schirmer's test and an abnormal Rose Bengal score, evidence of chronic lymphocytic sialoadenitis on a minor salivary grand biopsy specimen, abnormal salivary gland scintigraphy or sialography, decreased salivary flow determined by a gum test and the presence of either anti-Sjögren's syndrome A or B (anti-SS-A or SS-B) autoantibodies (Fujibayashi et al., 1999; Vitali et al., 2002). Patients with other collagen diseases, such as systemic lupus erythematosus, rheumatoid arthritis, mixed connective tissue disease, progressive systemic sclerosis, polyarteritis nodosa, polymyositis and Churg–Strauss syndrome, diagnosed by the diagnostic criteria appropriate to each condition (Anonymous, 1980; Tan et al., 1982; Arnett et al., 1988; Masi et al., 1990) and designated as having secondary Sjögren's syndrome, were excluded from this study. Patients underwent neurological examinations, blood studies, CSF studies, nerve conduction studies (NCS), sural nerve biopsies, somatosensory evoked potentials (SEPs) and spinal MRI.
A patient with the sensory ataxic form of Sjögren's syndrome-associated neuropathy who died at 88 years of age underwent autopsy and histological examination.
Assessment of neurological symptoms, activities of daily living and autonomic nerve dysfunction
Neurological examinations for somatic motor and sensory symptoms were performed by at least one neurologist. Sensory examinations were performed for light touch, pinprick, vibratory sensation and joint position sense, as well as for the presence of sensory ataxia and pain or painful dysaesthesia. Muscle strength was assessed using the Medical Research Council scale. Cranial nerve function, Romberg's sign, walking pattern, deep tendon reflexes and pathological reflexes were also assessed. Autonomic symptoms were assessed as described elsewhere.
For the assessment of clinical disability on daily life the modified Rankin scale (van Swieten et al., 1988) was used. For the assessment of autonomic nerve dysfunction, we evaluated pupil abnormalities, including presence of Adie's pupils, anisocoria and elliptic pupils, urinary disturbances, diarrhoea and constipation, hypohidrosis and anhidrosis, orthostatic hypotension and 123I-meta-iodobenzylguanidine (MIBG) cardiac accumulation. Autonomic symptoms generally were assessed by examining or interviewing patients or interviewing patients' family members, or reviewing the clinical records. Urinary symptoms were estimated by nocturnal or diurnal urinary frequency, a sensation of urgency, urinary incontinence, voiding difficulty and retention. Constipation was considered to be present if there were no stools for more than 3 days. Orthostatic hypotension was defined as a fall in systolic blood pressure of ≥30 mmHg upon standing from a recumbent position. For assessment of 123I-MIBG cardiac accumulation, 123I-MIBG 111 mBq (myoMIBG-123I for injection; Daiichi Radioisotope Laboratories Co., Tokyo, Japan) was given intravenously in Lugol's solution (200 mg iodine) to block the thyroid uptake. Cardiac MIBG uptake was expressed as a heart/ mediastinum ratio (H/M ratio) at 30 min (early scan) and 4 h (delayed scan) as described before (Watanabe et al., 2001). Thermography and quantitative sweating measurements were performed on some selected patients as previously described (Kumazawa et al., 1993).
Nerve conduction studies and somatosensory evoked potentials
Motor and sensory NCS were performed in the median, tibial and sural nerves using a standard method as described before (Sobue et al., 1989). Motor nerve conduction velocity (MCV), distal motor latency (DL) and compound muscle action potential (CMAP) were recorded for the median and tibial nerves. Sensory nerve conduction velocity (SCV) and sensory nerve action potential (SNAP) were assessed for the median and sural nerves. Control values were obtained in 191 normal volunteers (mean age ± SD, 48.7 ± 16.5 years; men : women, 97 : 94) for the median nerve, 121 (mean age ± SD, 49.9 ± 15.0; men : women, 64 : 57) for the tibial nerve and 133 (mean age ± SD, 50.6 ± 15.6; men : women, 74 : 59) for the sural nerve (Koike et al., 2001). Blink reflexes were recorded using a standard technique (Kimura, 2001).
SEPs were recorded using median nerve stimulation at the wrist (Kachi et al., 1994). Cortical (N20), cervical (N13), and Erb's point (N9) peaks were assessed by separate stimulation. Controls of the latency of SEPs were obtained in 37 normal volunteers (mean age ± SD, 38 ± 7 years).
Sural nerve biopsy and autopsy study
Sural nerve biopsies were performed in 55 patients as described previously (Sobue et al., 1989). Informed consent was established beforehand. Sural nerve biopsy specimens were examined by standard light microscopic methods and by teased fibre techniques. Specimens were divided into two portions. The first portion was fixed in 2.5% glutaraldehyde solution in 0.125 M cacodylate buffer (pH 7.4) and then embedded in an epoxy resin for morphometric and ultrastructural study. Density of myelinated fibres and morphological features were assessed in sections embedded in the epoxy resin and stained with toluidine blue using a computer-assisted image analyser (Luzex FS; Nikon, Tokyo, Japan), and densities of small and large myelinated fibres were calculated as described previously (Sobue et al., 1989; Koike et al., 2001). Some parts of specimens were processed for teased fibre study and were assessed for pathological conditions according to criteria described previously (Sobue et al., 1989; Dyck et al., 1993). For electron microscopic examination, epoxy resin-embedded specimens were processed for ultrathin sectioning. To assess the density of unmyelinated fibres, electron microscopic photomicrographs at a magnification of ×4000 were taken in random fashion to cover the ultrathin transverse section. The density of the unmyelinated fibres was estimated from the photomicrographs using a computer-assisted image analysis system.
For the autopsy study, the brain, spinal cord, sympathetic and sensory ganglia, peripheral nerve trunks, submandibular and subauricular salivary glands as well as various visceral organs were sampled systemically at the time of autopsy and examined in paraffin and epoxy-resin embedded sections.
MRI assessment of cervical spinal cord
A total of 27 patients underwent MRI of the cervical spinal cord, including the C4 level on a 1.5 T unit. We used axial T2*-weighted gradient echo images (repetition time/echo time/excitations, 700/21/3; flip angle, 20°; matrix, 256 × 256) as described previously (Yasuda et al., 1994; Sobue et al., 1995; Mori et al., 2001). MRI findings were assessed on their distributions of abnormal high intensity area in the posterior columns of the spinal cord.
Laboratory data
Routine blood tests were performed, including anti-SS-A and anti-SS-B antibodies. These autoantibodies were detected using enzyme-linked immunosorbent assay and immunoblotting [Mesacup-2 test, according to the manufacturer's instructions (MBL, Ltd. Japan)]. Alpha-fodrin, a candidate autoantigen for Sjögren's syndrome (Haneji et al., 1997) also was examined as follows. The purified recombinant N-terminal portion of alpha-fodrin and GST (glutathione-S-transferase) fusion protein (JS-1) were loaded onto 10% polyacrylamide gels and transferred to nitrocellulose membranes by electroblotting. The membranes were blocked overnight at room temperature with Tris-buffered saline containing 3% non-fat dry milk. The membranes were incubated with sera from patients with Sjögren's syndrome (1 : 200 dilution) for 4 h at room temperature. Then, bound antibodies were detected with biotinylated anti-human IgG antibodies and alkaline phosphatase-conjugated streptavidin (both from Jackson Immunoresearch, West Grove, PA) using 5-bromo-4-chloro-3-indolyl phosphate and nitro blue tetrazorium as substrates.
Statistical analysis
All statistical analyses were performed using the Mann–Whitney U-test. P-values of <0.05 were considered significant.
Results
General clinical features and classification of neuropathy
All 92 patients fulfilled the diagnostic criteria for Sjögren's syndrome (Fujibayashi et al., 1999; Vitali et al., 2002). The majority of patients (86 patients) were diagnosed as having Sjögren's syndrome after neurological symptoms developed, while only six patients were diagnosed with Sjögren's syndrome before the neurological symptoms appeared. Thus most of the patients had been followed for a neuropathy of unknown cause for a while before being diagnosed with Sjögren's syndrome. We classified these patients into seven forms of neuropathy: sensory ataxic neuropathy, painful sensory neuropathy without sensory ataxia, multiple mononeuropathy, multiple cranial neuropathy, trigeminal neuropathy, autonomic neuropathy and radiculoneuropathy, based on the predominant neuropathic symptoms. Sensory ataxic neuropathy was defined as one with sensory neuropathy predominantly manifesting as impairment of joint-position sense leading to sensory ataxia but preserved muscle power, muscle volume and motor nerve function (Sobue et al., 1993). A total of 36 patients were included in the sensory ataxic neuropathy group. Painful neuropathy without sensory ataxia (Mori et al., 2003) was another form of sensory neuropathy but with predominant involvement of superficial sensation of pain and light touch sense without or with minor impairment of deep sensation resulting in a painful sensory neuropathy without sensory ataxia. Motor function was well preserved with this neuropathy. Eighteen patients were included in this group. Eleven patients were considered to have multiple mononeuropathy. This form of neuropathy was characterized by multiple mononeuropathy mainly distributed in the distal portion of the limbs with both motor and sensory involvement. Sensory involvement generally included both superficial and deep sensation. Twenty patients were classified as having cranial neuropathy. Of the 20, 5 patients had multiple cranial neuropathy and 15 patients had isolated trigeminal neuropathy. Multiple cranial neuropathy affects multiple cranial motor and sensory nerves including the trigeminal nerve. Trigeminal neuropathy was defined as a pure sensory neuropathy restricted to the territory of the sensory trigeminal nerves. Autonomic neuropathy was characterized by predominant autonomic dysfunction. Three patients were considered to have this neuropathy. Radiculoneuropathy was defined by lesions restricted predominantly to the spinal roots or the very proximal portion of the spinal nerves. Radiculoneuropathy often mimics chronic inflammatory demyelinating polyneuropathy. Four patients were included in this category of neuropathy.
The age at first examination and the age of onset of neuropathic symptoms varied to some extent, but did not differ among the forms of neuropathy (Table 1). A female predominance was commonly observed in all of the neuropathies. Sjögren's syndrome-related symptoms also were seen at similar rates among the neuropathies. More than half of the patients had sicca syndrome, manifested by either xerophthalmia or xerostomia. Schirmer's test and the Rose Bengal test were positive in >50% of the examined patients. Almost all of the examined patients had either lymphocytic infiltration of the salivary glands, salivary gland cell destruction or both on minor salivary gland biopsy. Sialography and salivary scintigraphy also were positive in a majority of patients in each neuropathic group. Antibodies to SS-A and SS-B were present in 20–100% and 0–50% of patients in each neuropathic group, respectively. Only 13 patients were both SS-A and SS-B positive. Anti-alpha-fodrin antibodies were detected in 60–100% of patients in each neuropathic group. This positive rate was extremely high as compared with those in the control group without neuropathy (<14% positive), while it was not significantly different between neuropathic groups. Mild increases in the CSF protein concentration were seen in some of the patients examined.
Clinical features of patients with peripheral neuropathy associated with Sjögren's syndrome
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 11) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 36) | Painful (n = 18) | Multiple (n = 5) | Trigeminal (n = 15) | |||||||||||
| Age (years) | 65.2 ± 7.8 | 58.1 ± 15.9 | 59.1 ± 18.2 | 55.6 ± 12.7 | 55.6 ± 9.4 | 46.3 ± 18.0 | 57.0 ± 11.0 | |||||||
| Age of onset of neuropathy (years) | 64.9 ± 12.9 | 56.0 ± 13.8 | 58.1 ± 13.5 | 55.1 ± 14.8 | 51.7 ± 11.6 | 42.5 ± 17.4 | 49.0 ± 12.2 | |||||||
| Sex, women:men (n) | 26:10 | 16:2 | 10:1 | 4:1 | 15:0 | 2:1 | 3:1 | |||||||
| Follow-up (years) | 5.7 ± 4.6 | 3.6 ± 2.8 | 2.3 ± 1.3 | 5.2 ± 6.6 | 7.0 ± 4.1 | 1.7 ± 1.6 | 7.3 ± 3.8 | |||||||
| (range) (years) | (1–18) | (1–12) | (1–4) | (1–7) | (2–10) | (1–3) | (3–10) | |||||||
| Sjögren's syndrome | ||||||||||||||
| Dry eye: n (%)/dry mouth: n (%) | 20 (56)/23 (64) | 13 (72)/12 (67) | 7 (64)/7 (64) | 3 (60)/2 (40) | 10 (67)/10 (67) | 2 (67)/2 (67) | 2 (50)/2 (50) | |||||||
| Positive findings | ||||||||||||||
| Schirmer's test: n (%) | 27/29 (93) | 14/15 (93) | 7/8 (88) | 5/5 (100) | 8/10 (80) | 3/3 (100) | 2/4 (50) | |||||||
| Rose Bengal test: n (%) | 20/29 (69) | 11/12 (92) | 6/6 (100) | 3/5 (60) | 8/9 (89) | 2/2 (100) | ND | |||||||
| Salivary gland biopsy: n (%) | 26/28 (93) | 13/16 (81) | 8/9 (89) | 5/5 (100) | 11/11 (100) | 2/2 (100) | 4/4 (100) | |||||||
| Sialography, cintigraphy: n (%) | 9/10 (90) | 6/6 (100) | 3/3 (100) | 2/3 (67) | 8/8 (100) | 1/1 (100) | ND | |||||||
| SS-A: n (%) | 19/36 (53) | 7/18 (39) | 7/11 (64) | 1/5 (20) | 6/15 (40) | 2/3 (67) | 4/4 (100) | |||||||
| SS-B: n (%) | 4/36 (11) | 3/18 (17) | 2/11 (18) | 1/5 (20) | 0/15 (0) | 1/3 (33) | 2/4 (50) | |||||||
| Alpha-fodrin: n (%) | 14/16 (88) | 6/7 (86) | 3/5 (60) | 5/5 (100) | 2/2 (100) | 1/1 (100) | 3/3 (100) | |||||||
| CSF protein elevation: n (%) | 8/23 (35) | 3/10 (30) | 5/8 (63) | 1/3 (33) | 0/1 (0) | 0/1 (0) | 4/4 (100) | |||||||
| Associated symptoms (n) | T (4), P (2), Pa (2), L (3), Ly (1) | T (2), A (2), R (1), P (1), M (1) | T (2) | – | – | – | T (1), L (1) | |||||||
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 11) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 36) | Painful (n = 18) | Multiple (n = 5) | Trigeminal (n = 15) | |||||||||||
| Age (years) | 65.2 ± 7.8 | 58.1 ± 15.9 | 59.1 ± 18.2 | 55.6 ± 12.7 | 55.6 ± 9.4 | 46.3 ± 18.0 | 57.0 ± 11.0 | |||||||
| Age of onset of neuropathy (years) | 64.9 ± 12.9 | 56.0 ± 13.8 | 58.1 ± 13.5 | 55.1 ± 14.8 | 51.7 ± 11.6 | 42.5 ± 17.4 | 49.0 ± 12.2 | |||||||
| Sex, women:men (n) | 26:10 | 16:2 | 10:1 | 4:1 | 15:0 | 2:1 | 3:1 | |||||||
| Follow-up (years) | 5.7 ± 4.6 | 3.6 ± 2.8 | 2.3 ± 1.3 | 5.2 ± 6.6 | 7.0 ± 4.1 | 1.7 ± 1.6 | 7.3 ± 3.8 | |||||||
| (range) (years) | (1–18) | (1–12) | (1–4) | (1–7) | (2–10) | (1–3) | (3–10) | |||||||
| Sjögren's syndrome | ||||||||||||||
| Dry eye: n (%)/dry mouth: n (%) | 20 (56)/23 (64) | 13 (72)/12 (67) | 7 (64)/7 (64) | 3 (60)/2 (40) | 10 (67)/10 (67) | 2 (67)/2 (67) | 2 (50)/2 (50) | |||||||
| Positive findings | ||||||||||||||
| Schirmer's test: n (%) | 27/29 (93) | 14/15 (93) | 7/8 (88) | 5/5 (100) | 8/10 (80) | 3/3 (100) | 2/4 (50) | |||||||
| Rose Bengal test: n (%) | 20/29 (69) | 11/12 (92) | 6/6 (100) | 3/5 (60) | 8/9 (89) | 2/2 (100) | ND | |||||||
| Salivary gland biopsy: n (%) | 26/28 (93) | 13/16 (81) | 8/9 (89) | 5/5 (100) | 11/11 (100) | 2/2 (100) | 4/4 (100) | |||||||
| Sialography, cintigraphy: n (%) | 9/10 (90) | 6/6 (100) | 3/3 (100) | 2/3 (67) | 8/8 (100) | 1/1 (100) | ND | |||||||
| SS-A: n (%) | 19/36 (53) | 7/18 (39) | 7/11 (64) | 1/5 (20) | 6/15 (40) | 2/3 (67) | 4/4 (100) | |||||||
| SS-B: n (%) | 4/36 (11) | 3/18 (17) | 2/11 (18) | 1/5 (20) | 0/15 (0) | 1/3 (33) | 2/4 (50) | |||||||
| Alpha-fodrin: n (%) | 14/16 (88) | 6/7 (86) | 3/5 (60) | 5/5 (100) | 2/2 (100) | 1/1 (100) | 3/3 (100) | |||||||
| CSF protein elevation: n (%) | 8/23 (35) | 3/10 (30) | 5/8 (63) | 1/3 (33) | 0/1 (0) | 0/1 (0) | 4/4 (100) | |||||||
| Associated symptoms (n) | T (4), P (2), Pa (2), L (3), Ly (1) | T (2), A (2), R (1), P (1), M (1) | T (2) | – | – | – | T (1), L (1) | |||||||
n/n, positive patient number to all examined patient number. As for associated symptoms, T, hypothyroidism; P, interstitial pneumonia; Pa, pancreatitis; A, anaemia; M, myositis; L, liver dysfunction; Ly, lymphoma; R, renal involvement.
Clinical features of patients with peripheral neuropathy associated with Sjögren's syndrome
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 11) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 36) | Painful (n = 18) | Multiple (n = 5) | Trigeminal (n = 15) | |||||||||||
| Age (years) | 65.2 ± 7.8 | 58.1 ± 15.9 | 59.1 ± 18.2 | 55.6 ± 12.7 | 55.6 ± 9.4 | 46.3 ± 18.0 | 57.0 ± 11.0 | |||||||
| Age of onset of neuropathy (years) | 64.9 ± 12.9 | 56.0 ± 13.8 | 58.1 ± 13.5 | 55.1 ± 14.8 | 51.7 ± 11.6 | 42.5 ± 17.4 | 49.0 ± 12.2 | |||||||
| Sex, women:men (n) | 26:10 | 16:2 | 10:1 | 4:1 | 15:0 | 2:1 | 3:1 | |||||||
| Follow-up (years) | 5.7 ± 4.6 | 3.6 ± 2.8 | 2.3 ± 1.3 | 5.2 ± 6.6 | 7.0 ± 4.1 | 1.7 ± 1.6 | 7.3 ± 3.8 | |||||||
| (range) (years) | (1–18) | (1–12) | (1–4) | (1–7) | (2–10) | (1–3) | (3–10) | |||||||
| Sjögren's syndrome | ||||||||||||||
| Dry eye: n (%)/dry mouth: n (%) | 20 (56)/23 (64) | 13 (72)/12 (67) | 7 (64)/7 (64) | 3 (60)/2 (40) | 10 (67)/10 (67) | 2 (67)/2 (67) | 2 (50)/2 (50) | |||||||
| Positive findings | ||||||||||||||
| Schirmer's test: n (%) | 27/29 (93) | 14/15 (93) | 7/8 (88) | 5/5 (100) | 8/10 (80) | 3/3 (100) | 2/4 (50) | |||||||
| Rose Bengal test: n (%) | 20/29 (69) | 11/12 (92) | 6/6 (100) | 3/5 (60) | 8/9 (89) | 2/2 (100) | ND | |||||||
| Salivary gland biopsy: n (%) | 26/28 (93) | 13/16 (81) | 8/9 (89) | 5/5 (100) | 11/11 (100) | 2/2 (100) | 4/4 (100) | |||||||
| Sialography, cintigraphy: n (%) | 9/10 (90) | 6/6 (100) | 3/3 (100) | 2/3 (67) | 8/8 (100) | 1/1 (100) | ND | |||||||
| SS-A: n (%) | 19/36 (53) | 7/18 (39) | 7/11 (64) | 1/5 (20) | 6/15 (40) | 2/3 (67) | 4/4 (100) | |||||||
| SS-B: n (%) | 4/36 (11) | 3/18 (17) | 2/11 (18) | 1/5 (20) | 0/15 (0) | 1/3 (33) | 2/4 (50) | |||||||
| Alpha-fodrin: n (%) | 14/16 (88) | 6/7 (86) | 3/5 (60) | 5/5 (100) | 2/2 (100) | 1/1 (100) | 3/3 (100) | |||||||
| CSF protein elevation: n (%) | 8/23 (35) | 3/10 (30) | 5/8 (63) | 1/3 (33) | 0/1 (0) | 0/1 (0) | 4/4 (100) | |||||||
| Associated symptoms (n) | T (4), P (2), Pa (2), L (3), Ly (1) | T (2), A (2), R (1), P (1), M (1) | T (2) | – | – | – | T (1), L (1) | |||||||
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 11) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 36) | Painful (n = 18) | Multiple (n = 5) | Trigeminal (n = 15) | |||||||||||
| Age (years) | 65.2 ± 7.8 | 58.1 ± 15.9 | 59.1 ± 18.2 | 55.6 ± 12.7 | 55.6 ± 9.4 | 46.3 ± 18.0 | 57.0 ± 11.0 | |||||||
| Age of onset of neuropathy (years) | 64.9 ± 12.9 | 56.0 ± 13.8 | 58.1 ± 13.5 | 55.1 ± 14.8 | 51.7 ± 11.6 | 42.5 ± 17.4 | 49.0 ± 12.2 | |||||||
| Sex, women:men (n) | 26:10 | 16:2 | 10:1 | 4:1 | 15:0 | 2:1 | 3:1 | |||||||
| Follow-up (years) | 5.7 ± 4.6 | 3.6 ± 2.8 | 2.3 ± 1.3 | 5.2 ± 6.6 | 7.0 ± 4.1 | 1.7 ± 1.6 | 7.3 ± 3.8 | |||||||
| (range) (years) | (1–18) | (1–12) | (1–4) | (1–7) | (2–10) | (1–3) | (3–10) | |||||||
| Sjögren's syndrome | ||||||||||||||
| Dry eye: n (%)/dry mouth: n (%) | 20 (56)/23 (64) | 13 (72)/12 (67) | 7 (64)/7 (64) | 3 (60)/2 (40) | 10 (67)/10 (67) | 2 (67)/2 (67) | 2 (50)/2 (50) | |||||||
| Positive findings | ||||||||||||||
| Schirmer's test: n (%) | 27/29 (93) | 14/15 (93) | 7/8 (88) | 5/5 (100) | 8/10 (80) | 3/3 (100) | 2/4 (50) | |||||||
| Rose Bengal test: n (%) | 20/29 (69) | 11/12 (92) | 6/6 (100) | 3/5 (60) | 8/9 (89) | 2/2 (100) | ND | |||||||
| Salivary gland biopsy: n (%) | 26/28 (93) | 13/16 (81) | 8/9 (89) | 5/5 (100) | 11/11 (100) | 2/2 (100) | 4/4 (100) | |||||||
| Sialography, cintigraphy: n (%) | 9/10 (90) | 6/6 (100) | 3/3 (100) | 2/3 (67) | 8/8 (100) | 1/1 (100) | ND | |||||||
| SS-A: n (%) | 19/36 (53) | 7/18 (39) | 7/11 (64) | 1/5 (20) | 6/15 (40) | 2/3 (67) | 4/4 (100) | |||||||
| SS-B: n (%) | 4/36 (11) | 3/18 (17) | 2/11 (18) | 1/5 (20) | 0/15 (0) | 1/3 (33) | 2/4 (50) | |||||||
| Alpha-fodrin: n (%) | 14/16 (88) | 6/7 (86) | 3/5 (60) | 5/5 (100) | 2/2 (100) | 1/1 (100) | 3/3 (100) | |||||||
| CSF protein elevation: n (%) | 8/23 (35) | 3/10 (30) | 5/8 (63) | 1/3 (33) | 0/1 (0) | 0/1 (0) | 4/4 (100) | |||||||
| Associated symptoms (n) | T (4), P (2), Pa (2), L (3), Ly (1) | T (2), A (2), R (1), P (1), M (1) | T (2) | – | – | – | T (1), L (1) | |||||||
n/n, positive patient number to all examined patient number. As for associated symptoms, T, hypothyroidism; P, interstitial pneumonia; Pa, pancreatitis; A, anaemia; M, myositis; L, liver dysfunction; Ly, lymphoma; R, renal involvement.
As complicating systemic inflammatory symptoms, hypothyroidism was seen in nine patients, dyshaematopoietic anaemia in two patients, interstitial pneumonia in three patients, myositis in one patient, liver dysfunction in four patients, pancreatitis in two patients, renal involvement in one patient and lymphoma in one patient (Table 1).
Neuropathic features of each form of neuropathy
Sensory ataxic neuropathy
A total of 36 patients had this form of neuropathy (Table 2). This neuropathy was characterized by sensory ataxia due to kinaesthetic deep sensory impairment without substantial motor symptoms. The initial symptom was usually paraesthesias in the digits of the foot or hand. These paraesthesias were often unilateral, and gradually spread to the limbs, trunk and face. In three patients, the initial paraesthesia was localized to the trigeminal nerve area. The time from the onset to the development of full-blown symptoms of sensory involvement was variable among the patients, weeks to months in four patients, but usually months to years. The sensory symptoms were mostly asymmetrical, segmental or multi-focal rather than presenting as a symmetrical polyneuropathy, particularly in the progression stage. Ten patients had trigeminal nerve involvement. Muscle weakness and mild atrophy were observed in four patients. Sensory impairment was mostly deep sensory predominant with Romberg's sign and pseudoathetosis being present in all of the patients. Pain or painful dysaesthesia was present in 18 patients. A total of 10 and 20 patients showed facial and truncal sensory involvement, respectively. There was generalized areflexia in all patients. The walking pattern was characteristic of sensory ataxia. In the patients with advanced disease, they were unable to walk and were wheel-chair bound.
Neuropathic symptoms
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 11) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 36) | Painful (n = 18) | Multiple (n = 5) | Trigeminal (n = 15) | |||||||||||
| Initial symptom | ||||||||||||||
| Sensory disturbance: n (%) | 36 (100) | 3 (17) | 11 (100) | 0 (0) | 15 (100) | 0 (0) | 4 (100) | |||||||
| Pain/painful dysaesthesia: n (%) | 0 (0) | 18 (100) | 1 (9) | 1 (20) | 2 (13) | 0 (0) | 0 (0) | |||||||
| Weakness: n (%) | 0 (0) | 0 (0) | 8 (73) | 0 (0) | 0 (0) | 0 (0) | 2 (50) | |||||||
| Autonomic symptoms: n (%) | 0 (0) | 1 (6) | 0 (0) | 0 (0) | 0 (0) | 3 (100) | 0 (0) | |||||||
| Cranial nerve symptoms: n (%) | 3* (8) | 0 (0) | 1 (9)* | 5 (100) | 15 (100) | 0 (0) | 1 (25)** | |||||||
| Initial progression | ||||||||||||||
| Acute: n (%) | 0 (0) | 3 (17) | 2 (18) | 3 (60) | 0 (0) | 1 (33) | 0 (0) | |||||||
| Subacute: n (%) | 4 (11) | 1 (6) | 4 (36) | 0 (0) | 3 (20) | 0 (0) | 1 (25) | |||||||
| Chronic: n (%) | 32 (89) | 14 (78) | 5 (45) | 2 (40) | 12 (80) | 2 (67) | 3 (75) | |||||||
| Cranial nerve involvement: n (nerve) | 10 (V) | 8 (V) | 2 (V) | 2 (III), 3 (V), 2 (VI), 3 (VII), 3 (IX), 3 (X), 1(XII) | 15 (V) | 1 (V) | 1 (III) | |||||||
| Muscle weakness/atrophy: n (%) | 4 (11) | 1 (6) | 10 (91) | 0 (0) | 0 (0) | 0 (0) | 2 (50) | |||||||
| Sensory impairment: n (%) | 36 (100) | 18 (100) | 11 (100) | 3 (60) | 15 (100) | 2 (67) | 4 (100) | |||||||
| Modality | ||||||||||||||
| Deep > superficial sensation: n (%) | 33 (92) | 0 (0) | 1 (9) | 0 (0) | 0 (0) | 1 (33) | 3 (75) | |||||||
| Deep = superficial sensation: n (%) | 3 (8) | 0 (0) | 7 (64) | 0 (0) | 0 (0) | 1 (33) | 1 (25) | |||||||
| Superficial > deep sensation: n (%) | 0 (0) | 18 (100) | 3 (27) | 3 (60) | 15 (100) | 0 (0) | 0 (0) | |||||||
| Pain/painful dysaesthesia: n (%) | 18 (50) | 18 (100) | 7 (64) | 1 (20) | 2 (13) | 0 (0) | 0 (0) | |||||||
| Sensory ataxia: n (%) | 36 (100) | 2 (11) | 1 (9) | 0 (0) | 0 (0) | 2 (67) | 2 (50) | |||||||
| Distribution | ||||||||||||||
| Face: n (%) | 10 (28) | 8 (44) | 2 (18) | 3 (60) | 15 (100) | 1 (33) | 0 (0) | |||||||
| Trunk: n (%) | 20 (56) | 10 (56) | 2 (18) | 2 (40) | 0 (0) | 2 (67) | 1 (25) | |||||||
| Limbs: n (%) | 36 (100) | 18 (100) | 11 (100) | 2 (40) | 2 (13) | 2 (67) | 4 (100) | |||||||
| Areflexia: n (%) | 36 (100) | 9 (50) | 7 (64) | 0 (0) | 0 (0) | 2 (67) | 4 (100) | |||||||
| Modified Rankin scale (mean ± SD) (range) | 3.3 ± 0.8 (2–5) | 2.3 ± 0.8 (1–4) | 2.3 ± 0.8 (1–3) | – | – | 3.3 ± 1.2 (2–4) | 2.3 ± 1.3 (1–4) | |||||||
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 11) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 36) | Painful (n = 18) | Multiple (n = 5) | Trigeminal (n = 15) | |||||||||||
| Initial symptom | ||||||||||||||
| Sensory disturbance: n (%) | 36 (100) | 3 (17) | 11 (100) | 0 (0) | 15 (100) | 0 (0) | 4 (100) | |||||||
| Pain/painful dysaesthesia: n (%) | 0 (0) | 18 (100) | 1 (9) | 1 (20) | 2 (13) | 0 (0) | 0 (0) | |||||||
| Weakness: n (%) | 0 (0) | 0 (0) | 8 (73) | 0 (0) | 0 (0) | 0 (0) | 2 (50) | |||||||
| Autonomic symptoms: n (%) | 0 (0) | 1 (6) | 0 (0) | 0 (0) | 0 (0) | 3 (100) | 0 (0) | |||||||
| Cranial nerve symptoms: n (%) | 3* (8) | 0 (0) | 1 (9)* | 5 (100) | 15 (100) | 0 (0) | 1 (25)** | |||||||
| Initial progression | ||||||||||||||
| Acute: n (%) | 0 (0) | 3 (17) | 2 (18) | 3 (60) | 0 (0) | 1 (33) | 0 (0) | |||||||
| Subacute: n (%) | 4 (11) | 1 (6) | 4 (36) | 0 (0) | 3 (20) | 0 (0) | 1 (25) | |||||||
| Chronic: n (%) | 32 (89) | 14 (78) | 5 (45) | 2 (40) | 12 (80) | 2 (67) | 3 (75) | |||||||
| Cranial nerve involvement: n (nerve) | 10 (V) | 8 (V) | 2 (V) | 2 (III), 3 (V), 2 (VI), 3 (VII), 3 (IX), 3 (X), 1(XII) | 15 (V) | 1 (V) | 1 (III) | |||||||
| Muscle weakness/atrophy: n (%) | 4 (11) | 1 (6) | 10 (91) | 0 (0) | 0 (0) | 0 (0) | 2 (50) | |||||||
| Sensory impairment: n (%) | 36 (100) | 18 (100) | 11 (100) | 3 (60) | 15 (100) | 2 (67) | 4 (100) | |||||||
| Modality | ||||||||||||||
| Deep > superficial sensation: n (%) | 33 (92) | 0 (0) | 1 (9) | 0 (0) | 0 (0) | 1 (33) | 3 (75) | |||||||
| Deep = superficial sensation: n (%) | 3 (8) | 0 (0) | 7 (64) | 0 (0) | 0 (0) | 1 (33) | 1 (25) | |||||||
| Superficial > deep sensation: n (%) | 0 (0) | 18 (100) | 3 (27) | 3 (60) | 15 (100) | 0 (0) | 0 (0) | |||||||
| Pain/painful dysaesthesia: n (%) | 18 (50) | 18 (100) | 7 (64) | 1 (20) | 2 (13) | 0 (0) | 0 (0) | |||||||
| Sensory ataxia: n (%) | 36 (100) | 2 (11) | 1 (9) | 0 (0) | 0 (0) | 2 (67) | 2 (50) | |||||||
| Distribution | ||||||||||||||
| Face: n (%) | 10 (28) | 8 (44) | 2 (18) | 3 (60) | 15 (100) | 1 (33) | 0 (0) | |||||||
| Trunk: n (%) | 20 (56) | 10 (56) | 2 (18) | 2 (40) | 0 (0) | 2 (67) | 1 (25) | |||||||
| Limbs: n (%) | 36 (100) | 18 (100) | 11 (100) | 2 (40) | 2 (13) | 2 (67) | 4 (100) | |||||||
| Areflexia: n (%) | 36 (100) | 9 (50) | 7 (64) | 0 (0) | 0 (0) | 2 (67) | 4 (100) | |||||||
| Modified Rankin scale (mean ± SD) (range) | 3.3 ± 0.8 (2–5) | 2.3 ± 0.8 (1–4) | 2.3 ± 0.8 (1–3) | – | – | 3.3 ± 1.2 (2–4) | 2.3 ± 1.3 (1–4) | |||||||
Cranial nerve symptoms in initial symptom:
trigeminal nerve lesion;
, diplopia and ptosis. Modified Rankin scale: 0, asymptomatic; 1, non-disabling symptoms not interfering with lifestyle; 2, mildly disabling symptoms leading to some restrictions of lifestyle but not interfering with capacity to look after oneself; 3, moderately disabling symptoms significantly interfering with lifestyle or precluding totally independent existence; 4, moderately severe disability precluding independent existence while not requiring constant attention around the clock; 5, severe disability with total dependency requiring constant attention day and night.
Neuropathic symptoms
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 11) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 36) | Painful (n = 18) | Multiple (n = 5) | Trigeminal (n = 15) | |||||||||||
| Initial symptom | ||||||||||||||
| Sensory disturbance: n (%) | 36 (100) | 3 (17) | 11 (100) | 0 (0) | 15 (100) | 0 (0) | 4 (100) | |||||||
| Pain/painful dysaesthesia: n (%) | 0 (0) | 18 (100) | 1 (9) | 1 (20) | 2 (13) | 0 (0) | 0 (0) | |||||||
| Weakness: n (%) | 0 (0) | 0 (0) | 8 (73) | 0 (0) | 0 (0) | 0 (0) | 2 (50) | |||||||
| Autonomic symptoms: n (%) | 0 (0) | 1 (6) | 0 (0) | 0 (0) | 0 (0) | 3 (100) | 0 (0) | |||||||
| Cranial nerve symptoms: n (%) | 3* (8) | 0 (0) | 1 (9)* | 5 (100) | 15 (100) | 0 (0) | 1 (25)** | |||||||
| Initial progression | ||||||||||||||
| Acute: n (%) | 0 (0) | 3 (17) | 2 (18) | 3 (60) | 0 (0) | 1 (33) | 0 (0) | |||||||
| Subacute: n (%) | 4 (11) | 1 (6) | 4 (36) | 0 (0) | 3 (20) | 0 (0) | 1 (25) | |||||||
| Chronic: n (%) | 32 (89) | 14 (78) | 5 (45) | 2 (40) | 12 (80) | 2 (67) | 3 (75) | |||||||
| Cranial nerve involvement: n (nerve) | 10 (V) | 8 (V) | 2 (V) | 2 (III), 3 (V), 2 (VI), 3 (VII), 3 (IX), 3 (X), 1(XII) | 15 (V) | 1 (V) | 1 (III) | |||||||
| Muscle weakness/atrophy: n (%) | 4 (11) | 1 (6) | 10 (91) | 0 (0) | 0 (0) | 0 (0) | 2 (50) | |||||||
| Sensory impairment: n (%) | 36 (100) | 18 (100) | 11 (100) | 3 (60) | 15 (100) | 2 (67) | 4 (100) | |||||||
| Modality | ||||||||||||||
| Deep > superficial sensation: n (%) | 33 (92) | 0 (0) | 1 (9) | 0 (0) | 0 (0) | 1 (33) | 3 (75) | |||||||
| Deep = superficial sensation: n (%) | 3 (8) | 0 (0) | 7 (64) | 0 (0) | 0 (0) | 1 (33) | 1 (25) | |||||||
| Superficial > deep sensation: n (%) | 0 (0) | 18 (100) | 3 (27) | 3 (60) | 15 (100) | 0 (0) | 0 (0) | |||||||
| Pain/painful dysaesthesia: n (%) | 18 (50) | 18 (100) | 7 (64) | 1 (20) | 2 (13) | 0 (0) | 0 (0) | |||||||
| Sensory ataxia: n (%) | 36 (100) | 2 (11) | 1 (9) | 0 (0) | 0 (0) | 2 (67) | 2 (50) | |||||||
| Distribution | ||||||||||||||
| Face: n (%) | 10 (28) | 8 (44) | 2 (18) | 3 (60) | 15 (100) | 1 (33) | 0 (0) | |||||||
| Trunk: n (%) | 20 (56) | 10 (56) | 2 (18) | 2 (40) | 0 (0) | 2 (67) | 1 (25) | |||||||
| Limbs: n (%) | 36 (100) | 18 (100) | 11 (100) | 2 (40) | 2 (13) | 2 (67) | 4 (100) | |||||||
| Areflexia: n (%) | 36 (100) | 9 (50) | 7 (64) | 0 (0) | 0 (0) | 2 (67) | 4 (100) | |||||||
| Modified Rankin scale (mean ± SD) (range) | 3.3 ± 0.8 (2–5) | 2.3 ± 0.8 (1–4) | 2.3 ± 0.8 (1–3) | – | – | 3.3 ± 1.2 (2–4) | 2.3 ± 1.3 (1–4) | |||||||
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 11) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 36) | Painful (n = 18) | Multiple (n = 5) | Trigeminal (n = 15) | |||||||||||
| Initial symptom | ||||||||||||||
| Sensory disturbance: n (%) | 36 (100) | 3 (17) | 11 (100) | 0 (0) | 15 (100) | 0 (0) | 4 (100) | |||||||
| Pain/painful dysaesthesia: n (%) | 0 (0) | 18 (100) | 1 (9) | 1 (20) | 2 (13) | 0 (0) | 0 (0) | |||||||
| Weakness: n (%) | 0 (0) | 0 (0) | 8 (73) | 0 (0) | 0 (0) | 0 (0) | 2 (50) | |||||||
| Autonomic symptoms: n (%) | 0 (0) | 1 (6) | 0 (0) | 0 (0) | 0 (0) | 3 (100) | 0 (0) | |||||||
| Cranial nerve symptoms: n (%) | 3* (8) | 0 (0) | 1 (9)* | 5 (100) | 15 (100) | 0 (0) | 1 (25)** | |||||||
| Initial progression | ||||||||||||||
| Acute: n (%) | 0 (0) | 3 (17) | 2 (18) | 3 (60) | 0 (0) | 1 (33) | 0 (0) | |||||||
| Subacute: n (%) | 4 (11) | 1 (6) | 4 (36) | 0 (0) | 3 (20) | 0 (0) | 1 (25) | |||||||
| Chronic: n (%) | 32 (89) | 14 (78) | 5 (45) | 2 (40) | 12 (80) | 2 (67) | 3 (75) | |||||||
| Cranial nerve involvement: n (nerve) | 10 (V) | 8 (V) | 2 (V) | 2 (III), 3 (V), 2 (VI), 3 (VII), 3 (IX), 3 (X), 1(XII) | 15 (V) | 1 (V) | 1 (III) | |||||||
| Muscle weakness/atrophy: n (%) | 4 (11) | 1 (6) | 10 (91) | 0 (0) | 0 (0) | 0 (0) | 2 (50) | |||||||
| Sensory impairment: n (%) | 36 (100) | 18 (100) | 11 (100) | 3 (60) | 15 (100) | 2 (67) | 4 (100) | |||||||
| Modality | ||||||||||||||
| Deep > superficial sensation: n (%) | 33 (92) | 0 (0) | 1 (9) | 0 (0) | 0 (0) | 1 (33) | 3 (75) | |||||||
| Deep = superficial sensation: n (%) | 3 (8) | 0 (0) | 7 (64) | 0 (0) | 0 (0) | 1 (33) | 1 (25) | |||||||
| Superficial > deep sensation: n (%) | 0 (0) | 18 (100) | 3 (27) | 3 (60) | 15 (100) | 0 (0) | 0 (0) | |||||||
| Pain/painful dysaesthesia: n (%) | 18 (50) | 18 (100) | 7 (64) | 1 (20) | 2 (13) | 0 (0) | 0 (0) | |||||||
| Sensory ataxia: n (%) | 36 (100) | 2 (11) | 1 (9) | 0 (0) | 0 (0) | 2 (67) | 2 (50) | |||||||
| Distribution | ||||||||||||||
| Face: n (%) | 10 (28) | 8 (44) | 2 (18) | 3 (60) | 15 (100) | 1 (33) | 0 (0) | |||||||
| Trunk: n (%) | 20 (56) | 10 (56) | 2 (18) | 2 (40) | 0 (0) | 2 (67) | 1 (25) | |||||||
| Limbs: n (%) | 36 (100) | 18 (100) | 11 (100) | 2 (40) | 2 (13) | 2 (67) | 4 (100) | |||||||
| Areflexia: n (%) | 36 (100) | 9 (50) | 7 (64) | 0 (0) | 0 (0) | 2 (67) | 4 (100) | |||||||
| Modified Rankin scale (mean ± SD) (range) | 3.3 ± 0.8 (2–5) | 2.3 ± 0.8 (1–4) | 2.3 ± 0.8 (1–3) | – | – | 3.3 ± 1.2 (2–4) | 2.3 ± 1.3 (1–4) | |||||||
Cranial nerve symptoms in initial symptom:
trigeminal nerve lesion;
, diplopia and ptosis. Modified Rankin scale: 0, asymptomatic; 1, non-disabling symptoms not interfering with lifestyle; 2, mildly disabling symptoms leading to some restrictions of lifestyle but not interfering with capacity to look after oneself; 3, moderately disabling symptoms significantly interfering with lifestyle or precluding totally independent existence; 4, moderately severe disability precluding independent existence while not requiring constant attention around the clock; 5, severe disability with total dependency requiring constant attention day and night.
With respect to autonomic symptoms, 17 of the 30 assessed patients exhibited abnormal pupils including Adie's pupils associated with anisocoric and elliptic pupils (Table 3). Orthostatic hypotension was present in 12 patients, mostly without syncope. Hypohidrosis or anhidrosis was observed in 21 patients, often with segmental anhidrosis in the trunk (Fig. 1). Marked decreases in 123I-MIBG cardiac uptake were present in 8 of the 11 patients who were examined.
Autonomic symptoms
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 6) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 30) | Painful (n = 16) | Multiple (n = 5) | Trigeminal (n = 9) | ||||||
| Abnormal pupils: n | 17 | 3 | 0 | 1 | 3 | 3 | 0 | ||
| Orthostatic hypotension: n | 12 | 5 | 0 | 0 | 3 | 3 | 0 | ||
| Faint: n | 0 | 0 | 0 | 0 | 0 | 3 | 0 | ||
| Hypohidrosis/anhidrosis: n | 21 | 10 | 3 | 2 | 4 | 3 | 2 | ||
| Diarrhoea: n | 6 | 0 | 1 | 0 | 1 | 3 | 0 | ||
| Constipation: n | 6 | 5 | 2 | 1 | 1 | 3 | 2 | ||
| Vomiting: n | 2 | 0 | 0 | 0 | 0 | 1 | 0 | ||
| Urinary disturbance: n | 1 | 3 | 0 | 0 | 0 | 2 | 1 | ||
| Decreased uptake of 123I-MIBG: n | 8 /11 | 5 /7 | ND | ND | ND | 2 /2 | 0 /2 | ||
| Total: n | 21 /30 | 11 /16 | 3 /6 | 2 /5 | 4 /9 | 3 /3 | 2 /4 | ||
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 6) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 30) | Painful (n = 16) | Multiple (n = 5) | Trigeminal (n = 9) | ||||||
| Abnormal pupils: n | 17 | 3 | 0 | 1 | 3 | 3 | 0 | ||
| Orthostatic hypotension: n | 12 | 5 | 0 | 0 | 3 | 3 | 0 | ||
| Faint: n | 0 | 0 | 0 | 0 | 0 | 3 | 0 | ||
| Hypohidrosis/anhidrosis: n | 21 | 10 | 3 | 2 | 4 | 3 | 2 | ||
| Diarrhoea: n | 6 | 0 | 1 | 0 | 1 | 3 | 0 | ||
| Constipation: n | 6 | 5 | 2 | 1 | 1 | 3 | 2 | ||
| Vomiting: n | 2 | 0 | 0 | 0 | 0 | 1 | 0 | ||
| Urinary disturbance: n | 1 | 3 | 0 | 0 | 0 | 2 | 1 | ||
| Decreased uptake of 123I-MIBG: n | 8 /11 | 5 /7 | ND | ND | ND | 2 /2 | 0 /2 | ||
| Total: n | 21 /30 | 11 /16 | 3 /6 | 2 /5 | 4 /9 | 3 /3 | 2 /4 | ||
Decreased uptake of 123I-MIBG = the 123I-MIBG heart/mediastinum ratio (H/M ratio) of delayed scan was <1.8 (control–2 SD) (Hamada et al., 2003). n/n, positive patient number to all examined patient number; ND, not determined.
Autonomic symptoms
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 6) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 30) | Painful (n = 16) | Multiple (n = 5) | Trigeminal (n = 9) | ||||||
| Abnormal pupils: n | 17 | 3 | 0 | 1 | 3 | 3 | 0 | ||
| Orthostatic hypotension: n | 12 | 5 | 0 | 0 | 3 | 3 | 0 | ||
| Faint: n | 0 | 0 | 0 | 0 | 0 | 3 | 0 | ||
| Hypohidrosis/anhidrosis: n | 21 | 10 | 3 | 2 | 4 | 3 | 2 | ||
| Diarrhoea: n | 6 | 0 | 1 | 0 | 1 | 3 | 0 | ||
| Constipation: n | 6 | 5 | 2 | 1 | 1 | 3 | 2 | ||
| Vomiting: n | 2 | 0 | 0 | 0 | 0 | 1 | 0 | ||
| Urinary disturbance: n | 1 | 3 | 0 | 0 | 0 | 2 | 1 | ||
| Decreased uptake of 123I-MIBG: n | 8 /11 | 5 /7 | ND | ND | ND | 2 /2 | 0 /2 | ||
| Total: n | 21 /30 | 11 /16 | 3 /6 | 2 /5 | 4 /9 | 3 /3 | 2 /4 | ||
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 6) | Cranial neuropathy | Autonomic neuropathy (n = 3) | Radiculo- neuropathy (n = 4) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 30) | Painful (n = 16) | Multiple (n = 5) | Trigeminal (n = 9) | ||||||
| Abnormal pupils: n | 17 | 3 | 0 | 1 | 3 | 3 | 0 | ||
| Orthostatic hypotension: n | 12 | 5 | 0 | 0 | 3 | 3 | 0 | ||
| Faint: n | 0 | 0 | 0 | 0 | 0 | 3 | 0 | ||
| Hypohidrosis/anhidrosis: n | 21 | 10 | 3 | 2 | 4 | 3 | 2 | ||
| Diarrhoea: n | 6 | 0 | 1 | 0 | 1 | 3 | 0 | ||
| Constipation: n | 6 | 5 | 2 | 1 | 1 | 3 | 2 | ||
| Vomiting: n | 2 | 0 | 0 | 0 | 0 | 1 | 0 | ||
| Urinary disturbance: n | 1 | 3 | 0 | 0 | 0 | 2 | 1 | ||
| Decreased uptake of 123I-MIBG: n | 8 /11 | 5 /7 | ND | ND | ND | 2 /2 | 0 /2 | ||
| Total: n | 21 /30 | 11 /16 | 3 /6 | 2 /5 | 4 /9 | 3 /3 | 2 /4 | ||
Decreased uptake of 123I-MIBG = the 123I-MIBG heart/mediastinum ratio (H/M ratio) of delayed scan was <1.8 (control–2 SD) (Hamada et al., 2003). n/n, positive patient number to all examined patient number; ND, not determined.
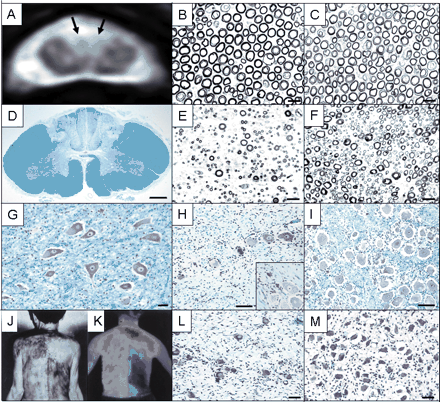
Pathological findings, MRI and sweating assessment of an autopsied patient with the sensory ataxic neuropathy. (A) Axial T2*-weighted gradient echo image of the cervical spinal cord (C4) of the patient. A high intensity area is present in the posterior column including both fasciculus cuneatus and gracilis as indicated by arrows. (B and C) Cross-section of the L4 ventral spinal root. Myelinated fibres are well preserved in the patient (B) and control (C). Scale bar = 20 µm. (D) Cross-section of the dorsal column of the cervical spinal cord. Axons are almost completely lost. Klüver–Barrera's stain. Scale bar = 1 mm. (E and F) Cross-section of the L4 dorsal spinal root. Large myelinated fibres are severely lost in the patient (E) compared with the control (F). Scale bar = 20 µm. (G) Cross-section of the L4 ventral horn. The population of the anterior horn cell is well preserved. Klüver–Barrera's stain. Scale bar = 40 µm. (H) Cross-section of the L4 dorsal root ganglion. The population of the nerve cell is decreased (H). Nageotte's nodules are occasionally seen (H). (I) Control. Klüver–Barrera's stain. Scale bar = 100 µm. (J) Thermal sweating measured by Minor's iodine–starch test in an artificial climate chamber at an ambient temperature of 40°C and 40% relative humidity. The area of anhidrosis was very distinct and distributed in a segmental manner. (K) Plain thermograms monitored by infrared thermography. Surface skin temperature was also segmental in distribution (quoted from Kumazawa et al., 1993 with permission for publication). (L) Section of the thoracic sympathetic ganglion. The population of the nerve cell bodies is decreased (L) compared with control case (M). Klüver–Barrera's stain. Scale bar = 40 µm.
With respect to nerve conduction, SNAPs in the median and sural nerves were not evoked in 61 and 50% of patients, respectively (Table 4). In contrast, CMAPs were fairly well preserved in most patients. MCV and SCV were not slowed. Temporal dispersion of the CMAPs or conduction block was not seen. SEPs were not evoked in 67, 73 and 40% of the examined patients in N20, N13 and N9, respectively (Table 4). These conduction studies indicate that axonal features were almost exclusively present in this neuropathy, and the central rami of sensory ganglion neurons were also involved in parallel.
Nerve conduction, sensory evoked potentials, and spinal cord MRI study
| Nerve conduction, SEP and MRI | Sensory neuropathy | Multiple mononeuropathy | Cranial neuropathy | Autonomic neuropathy | Radiculo- neuropathy | Controls | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic | Painful | Multiple | Trigeminal | |||||||||||||
| Nerve conduction study | n = 36 | n = 18 | n = 11 | n = 5 | n = 15 | n = 3 | n = 4 | n = 121–191 | ||||||||
| Median nerve | ||||||||||||||||
| MCV (m/s) | 53.1 ± 4.0 | 53.3 ± 33.2 | 51.3 ± 3.7 | 51.2 ± 8.1 | 54.7 ± 3.5 | 53.2 ± 0.7 | 50.5 ± 10.3 | 57.8 ± 3.7 | ||||||||
| DL (ms) | 3.7 ± 0.5 | 3.4 ± 0.2 | 3.5 ± 0.4 | 4.0 ± 0.5 | 3.9 ± 1.3 | 3.7 ± 0.6 | 4.4 ± 2.0 | 3.4 ± 0.4 | ||||||||
| CMAP (mV) | 11.1 ± 2.8 | 11.2 ± 4.0 | 10.4 ± 2.4 | 11.4 ± 3.0 | 12.7 ± 3.4 | 10.5 ± 2.7 | 5.6 ± 3.1** | 10.7 ± 3.5 | ||||||||
| SCV (m/s) | 50.0 ± 4.7 | 51.5 ± 3.5 | 49.6 ± 7.8 | 61.4 ± 1.3 | 55.1 ± 2.6 | 58.5 ± 0.3 | 50.8 ± 9.5 | 57.8 ± 4.7 | ||||||||
| SNAP ( µV) | 4.1 ± 9.0*** | 4.4 ± 6.6*** | 4.9 ± 4.7*** | 23.9 ± 1.9 | 11.3 ± 4.1 | 25.6 ± 4.0 | 22.2 ± 1.0 | 23.5 ± 8.4 | ||||||||
| N.E. (%) | 61 | 11 | 18 | 0 | 0 | 33 | 0 | 0 | ||||||||
| Tibial nerve | ||||||||||||||||
| MCV (m/s) | 43.5 ± 2.8 | 44.5 ± 3.3 | 38.6 ± 3.9 | 48.3 ± 4.2 | 43.7 ± 2.3 | 44.7 ± 1.2 | 43.5 ± 1.1 | 46.9 ± 3.5 | ||||||||
| DL (ms) | 4.8 ± 0.8 | 4.5 ± 0.4 | 5.1 ± 1.1 | 4.7 ± 1.0 | 5.1 ± 0.2 | 4.2 ± 0.9 | 6.0 ± 3.4 | 4.5 ± 0.8 | ||||||||
| CMAP (mV) | 10.5 ± 7.0 | 14.5 ± 6.8 | 6.5 ± 9.1* | 12.2 ± 9.1 | 16.4 ± 5.2 | 12.6 ± 1.0 | 9.2 ± 3.3* | 10.9 ± 3.8 | ||||||||
| Sural nerve | ||||||||||||||||
| SCV (m/s) | 45.3 ± 2.8 | 46.7 ± 6.5 | 46.4 ± 3.1 | 50.3 ± 3.5 | 53.4 ± 1.2 | 46.8 ± 0.0 | 45.0 ± 5.1 | 51.0 ± 5.1 | ||||||||
| SNAP ( µV) | 2.2 ± 3.6*** | 8.1 ± 8.1*** | 1.3 ± 1.5*** | 20.5 ± 3.5 | 22.5 ± 2.3 | 10.3 ± 1.4 | 18.3 ± 1.5 | 11.5 ± 4.7 | ||||||||
| N.E. (%) | 50 | 17 | 55 | 0 | 0 | 33 | 0 | 0 | ||||||||
| SEP | n = 15 | n = 8 | n = 3 | n = 4 | n = 37 | |||||||||||
| Median nerve stimulation | ||||||||||||||||
| N20 latency (ms) | 20.2 ± 2.1 | 20.9 ± 2.3 | ND | ND | ND | 19.5 ± 0.7 | 21.4 ± 3.2 | 18.9 ± 1.1 | ||||||||
| N.E. (%) | 67 | 0 | 33 | 0 | ||||||||||||
| N13 latency (ms) | 13.1 ± 0.8 | 14.4 ± 1.9 | ND | ND | ND | 13.1 ± 0.6 | 14.8 ± 2.6 | 12.8 ± 1.0 | ||||||||
| N.E. (%) | 73 | 0 | 33 | 0 | ||||||||||||
| N9 latency (ms) | 9.1 ± 0.9 | 9.8 ± 0.9 | ND | ND | ND | 8.9 ± 0.4 | 10.1 ± 1.2 | 8.6 ± 0.6 | ||||||||
| N.E. (%) | 40 | 13 | 33 | 0 | ||||||||||||
| MRI | n = 12 | n = 8 | n = 3 | n = 4 | ||||||||||||
| Spinal cord abnormality | 9† | 3† | ND | ND | ND | 1† | 4‡ | |||||||||
| Nerve conduction, SEP and MRI | Sensory neuropathy | Multiple mononeuropathy | Cranial neuropathy | Autonomic neuropathy | Radiculo- neuropathy | Controls | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic | Painful | Multiple | Trigeminal | |||||||||||||
| Nerve conduction study | n = 36 | n = 18 | n = 11 | n = 5 | n = 15 | n = 3 | n = 4 | n = 121–191 | ||||||||
| Median nerve | ||||||||||||||||
| MCV (m/s) | 53.1 ± 4.0 | 53.3 ± 33.2 | 51.3 ± 3.7 | 51.2 ± 8.1 | 54.7 ± 3.5 | 53.2 ± 0.7 | 50.5 ± 10.3 | 57.8 ± 3.7 | ||||||||
| DL (ms) | 3.7 ± 0.5 | 3.4 ± 0.2 | 3.5 ± 0.4 | 4.0 ± 0.5 | 3.9 ± 1.3 | 3.7 ± 0.6 | 4.4 ± 2.0 | 3.4 ± 0.4 | ||||||||
| CMAP (mV) | 11.1 ± 2.8 | 11.2 ± 4.0 | 10.4 ± 2.4 | 11.4 ± 3.0 | 12.7 ± 3.4 | 10.5 ± 2.7 | 5.6 ± 3.1** | 10.7 ± 3.5 | ||||||||
| SCV (m/s) | 50.0 ± 4.7 | 51.5 ± 3.5 | 49.6 ± 7.8 | 61.4 ± 1.3 | 55.1 ± 2.6 | 58.5 ± 0.3 | 50.8 ± 9.5 | 57.8 ± 4.7 | ||||||||
| SNAP ( µV) | 4.1 ± 9.0*** | 4.4 ± 6.6*** | 4.9 ± 4.7*** | 23.9 ± 1.9 | 11.3 ± 4.1 | 25.6 ± 4.0 | 22.2 ± 1.0 | 23.5 ± 8.4 | ||||||||
| N.E. (%) | 61 | 11 | 18 | 0 | 0 | 33 | 0 | 0 | ||||||||
| Tibial nerve | ||||||||||||||||
| MCV (m/s) | 43.5 ± 2.8 | 44.5 ± 3.3 | 38.6 ± 3.9 | 48.3 ± 4.2 | 43.7 ± 2.3 | 44.7 ± 1.2 | 43.5 ± 1.1 | 46.9 ± 3.5 | ||||||||
| DL (ms) | 4.8 ± 0.8 | 4.5 ± 0.4 | 5.1 ± 1.1 | 4.7 ± 1.0 | 5.1 ± 0.2 | 4.2 ± 0.9 | 6.0 ± 3.4 | 4.5 ± 0.8 | ||||||||
| CMAP (mV) | 10.5 ± 7.0 | 14.5 ± 6.8 | 6.5 ± 9.1* | 12.2 ± 9.1 | 16.4 ± 5.2 | 12.6 ± 1.0 | 9.2 ± 3.3* | 10.9 ± 3.8 | ||||||||
| Sural nerve | ||||||||||||||||
| SCV (m/s) | 45.3 ± 2.8 | 46.7 ± 6.5 | 46.4 ± 3.1 | 50.3 ± 3.5 | 53.4 ± 1.2 | 46.8 ± 0.0 | 45.0 ± 5.1 | 51.0 ± 5.1 | ||||||||
| SNAP ( µV) | 2.2 ± 3.6*** | 8.1 ± 8.1*** | 1.3 ± 1.5*** | 20.5 ± 3.5 | 22.5 ± 2.3 | 10.3 ± 1.4 | 18.3 ± 1.5 | 11.5 ± 4.7 | ||||||||
| N.E. (%) | 50 | 17 | 55 | 0 | 0 | 33 | 0 | 0 | ||||||||
| SEP | n = 15 | n = 8 | n = 3 | n = 4 | n = 37 | |||||||||||
| Median nerve stimulation | ||||||||||||||||
| N20 latency (ms) | 20.2 ± 2.1 | 20.9 ± 2.3 | ND | ND | ND | 19.5 ± 0.7 | 21.4 ± 3.2 | 18.9 ± 1.1 | ||||||||
| N.E. (%) | 67 | 0 | 33 | 0 | ||||||||||||
| N13 latency (ms) | 13.1 ± 0.8 | 14.4 ± 1.9 | ND | ND | ND | 13.1 ± 0.6 | 14.8 ± 2.6 | 12.8 ± 1.0 | ||||||||
| N.E. (%) | 73 | 0 | 33 | 0 | ||||||||||||
| N9 latency (ms) | 9.1 ± 0.9 | 9.8 ± 0.9 | ND | ND | ND | 8.9 ± 0.4 | 10.1 ± 1.2 | 8.6 ± 0.6 | ||||||||
| N.E. (%) | 40 | 13 | 33 | 0 | ||||||||||||
| MRI | n = 12 | n = 8 | n = 3 | n = 4 | ||||||||||||
| Spinal cord abnormality | 9† | 3† | ND | ND | ND | 1† | 4‡ | |||||||||
Values are expressed as mean ± SD. Control values are those described previously (Koike et al., 2001). MCV, motor nerve conduction velocity; DL, distal latency; CMAP, compound muscle action potentials; SCV, sensory nerve conduction velocity; SNAP, sensory nerve action potentials; N.E., not evoked; SEP, somatosensory evoked potentials; control values are from 37 conduction times.
P < 0.05,
P < 0.01,
P < 0.001 as compared with control value.
In T2* weighted gradient echo images, a high intensity area is present in the posterior column.
In T1 weighted echo images. Posterior spinal roots and cauda equina are enhanced by gadolinium.
Nerve conduction, sensory evoked potentials, and spinal cord MRI study
| Nerve conduction, SEP and MRI | Sensory neuropathy | Multiple mononeuropathy | Cranial neuropathy | Autonomic neuropathy | Radiculo- neuropathy | Controls | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic | Painful | Multiple | Trigeminal | |||||||||||||
| Nerve conduction study | n = 36 | n = 18 | n = 11 | n = 5 | n = 15 | n = 3 | n = 4 | n = 121–191 | ||||||||
| Median nerve | ||||||||||||||||
| MCV (m/s) | 53.1 ± 4.0 | 53.3 ± 33.2 | 51.3 ± 3.7 | 51.2 ± 8.1 | 54.7 ± 3.5 | 53.2 ± 0.7 | 50.5 ± 10.3 | 57.8 ± 3.7 | ||||||||
| DL (ms) | 3.7 ± 0.5 | 3.4 ± 0.2 | 3.5 ± 0.4 | 4.0 ± 0.5 | 3.9 ± 1.3 | 3.7 ± 0.6 | 4.4 ± 2.0 | 3.4 ± 0.4 | ||||||||
| CMAP (mV) | 11.1 ± 2.8 | 11.2 ± 4.0 | 10.4 ± 2.4 | 11.4 ± 3.0 | 12.7 ± 3.4 | 10.5 ± 2.7 | 5.6 ± 3.1** | 10.7 ± 3.5 | ||||||||
| SCV (m/s) | 50.0 ± 4.7 | 51.5 ± 3.5 | 49.6 ± 7.8 | 61.4 ± 1.3 | 55.1 ± 2.6 | 58.5 ± 0.3 | 50.8 ± 9.5 | 57.8 ± 4.7 | ||||||||
| SNAP ( µV) | 4.1 ± 9.0*** | 4.4 ± 6.6*** | 4.9 ± 4.7*** | 23.9 ± 1.9 | 11.3 ± 4.1 | 25.6 ± 4.0 | 22.2 ± 1.0 | 23.5 ± 8.4 | ||||||||
| N.E. (%) | 61 | 11 | 18 | 0 | 0 | 33 | 0 | 0 | ||||||||
| Tibial nerve | ||||||||||||||||
| MCV (m/s) | 43.5 ± 2.8 | 44.5 ± 3.3 | 38.6 ± 3.9 | 48.3 ± 4.2 | 43.7 ± 2.3 | 44.7 ± 1.2 | 43.5 ± 1.1 | 46.9 ± 3.5 | ||||||||
| DL (ms) | 4.8 ± 0.8 | 4.5 ± 0.4 | 5.1 ± 1.1 | 4.7 ± 1.0 | 5.1 ± 0.2 | 4.2 ± 0.9 | 6.0 ± 3.4 | 4.5 ± 0.8 | ||||||||
| CMAP (mV) | 10.5 ± 7.0 | 14.5 ± 6.8 | 6.5 ± 9.1* | 12.2 ± 9.1 | 16.4 ± 5.2 | 12.6 ± 1.0 | 9.2 ± 3.3* | 10.9 ± 3.8 | ||||||||
| Sural nerve | ||||||||||||||||
| SCV (m/s) | 45.3 ± 2.8 | 46.7 ± 6.5 | 46.4 ± 3.1 | 50.3 ± 3.5 | 53.4 ± 1.2 | 46.8 ± 0.0 | 45.0 ± 5.1 | 51.0 ± 5.1 | ||||||||
| SNAP ( µV) | 2.2 ± 3.6*** | 8.1 ± 8.1*** | 1.3 ± 1.5*** | 20.5 ± 3.5 | 22.5 ± 2.3 | 10.3 ± 1.4 | 18.3 ± 1.5 | 11.5 ± 4.7 | ||||||||
| N.E. (%) | 50 | 17 | 55 | 0 | 0 | 33 | 0 | 0 | ||||||||
| SEP | n = 15 | n = 8 | n = 3 | n = 4 | n = 37 | |||||||||||
| Median nerve stimulation | ||||||||||||||||
| N20 latency (ms) | 20.2 ± 2.1 | 20.9 ± 2.3 | ND | ND | ND | 19.5 ± 0.7 | 21.4 ± 3.2 | 18.9 ± 1.1 | ||||||||
| N.E. (%) | 67 | 0 | 33 | 0 | ||||||||||||
| N13 latency (ms) | 13.1 ± 0.8 | 14.4 ± 1.9 | ND | ND | ND | 13.1 ± 0.6 | 14.8 ± 2.6 | 12.8 ± 1.0 | ||||||||
| N.E. (%) | 73 | 0 | 33 | 0 | ||||||||||||
| N9 latency (ms) | 9.1 ± 0.9 | 9.8 ± 0.9 | ND | ND | ND | 8.9 ± 0.4 | 10.1 ± 1.2 | 8.6 ± 0.6 | ||||||||
| N.E. (%) | 40 | 13 | 33 | 0 | ||||||||||||
| MRI | n = 12 | n = 8 | n = 3 | n = 4 | ||||||||||||
| Spinal cord abnormality | 9† | 3† | ND | ND | ND | 1† | 4‡ | |||||||||
| Nerve conduction, SEP and MRI | Sensory neuropathy | Multiple mononeuropathy | Cranial neuropathy | Autonomic neuropathy | Radiculo- neuropathy | Controls | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic | Painful | Multiple | Trigeminal | |||||||||||||
| Nerve conduction study | n = 36 | n = 18 | n = 11 | n = 5 | n = 15 | n = 3 | n = 4 | n = 121–191 | ||||||||
| Median nerve | ||||||||||||||||
| MCV (m/s) | 53.1 ± 4.0 | 53.3 ± 33.2 | 51.3 ± 3.7 | 51.2 ± 8.1 | 54.7 ± 3.5 | 53.2 ± 0.7 | 50.5 ± 10.3 | 57.8 ± 3.7 | ||||||||
| DL (ms) | 3.7 ± 0.5 | 3.4 ± 0.2 | 3.5 ± 0.4 | 4.0 ± 0.5 | 3.9 ± 1.3 | 3.7 ± 0.6 | 4.4 ± 2.0 | 3.4 ± 0.4 | ||||||||
| CMAP (mV) | 11.1 ± 2.8 | 11.2 ± 4.0 | 10.4 ± 2.4 | 11.4 ± 3.0 | 12.7 ± 3.4 | 10.5 ± 2.7 | 5.6 ± 3.1** | 10.7 ± 3.5 | ||||||||
| SCV (m/s) | 50.0 ± 4.7 | 51.5 ± 3.5 | 49.6 ± 7.8 | 61.4 ± 1.3 | 55.1 ± 2.6 | 58.5 ± 0.3 | 50.8 ± 9.5 | 57.8 ± 4.7 | ||||||||
| SNAP ( µV) | 4.1 ± 9.0*** | 4.4 ± 6.6*** | 4.9 ± 4.7*** | 23.9 ± 1.9 | 11.3 ± 4.1 | 25.6 ± 4.0 | 22.2 ± 1.0 | 23.5 ± 8.4 | ||||||||
| N.E. (%) | 61 | 11 | 18 | 0 | 0 | 33 | 0 | 0 | ||||||||
| Tibial nerve | ||||||||||||||||
| MCV (m/s) | 43.5 ± 2.8 | 44.5 ± 3.3 | 38.6 ± 3.9 | 48.3 ± 4.2 | 43.7 ± 2.3 | 44.7 ± 1.2 | 43.5 ± 1.1 | 46.9 ± 3.5 | ||||||||
| DL (ms) | 4.8 ± 0.8 | 4.5 ± 0.4 | 5.1 ± 1.1 | 4.7 ± 1.0 | 5.1 ± 0.2 | 4.2 ± 0.9 | 6.0 ± 3.4 | 4.5 ± 0.8 | ||||||||
| CMAP (mV) | 10.5 ± 7.0 | 14.5 ± 6.8 | 6.5 ± 9.1* | 12.2 ± 9.1 | 16.4 ± 5.2 | 12.6 ± 1.0 | 9.2 ± 3.3* | 10.9 ± 3.8 | ||||||||
| Sural nerve | ||||||||||||||||
| SCV (m/s) | 45.3 ± 2.8 | 46.7 ± 6.5 | 46.4 ± 3.1 | 50.3 ± 3.5 | 53.4 ± 1.2 | 46.8 ± 0.0 | 45.0 ± 5.1 | 51.0 ± 5.1 | ||||||||
| SNAP ( µV) | 2.2 ± 3.6*** | 8.1 ± 8.1*** | 1.3 ± 1.5*** | 20.5 ± 3.5 | 22.5 ± 2.3 | 10.3 ± 1.4 | 18.3 ± 1.5 | 11.5 ± 4.7 | ||||||||
| N.E. (%) | 50 | 17 | 55 | 0 | 0 | 33 | 0 | 0 | ||||||||
| SEP | n = 15 | n = 8 | n = 3 | n = 4 | n = 37 | |||||||||||
| Median nerve stimulation | ||||||||||||||||
| N20 latency (ms) | 20.2 ± 2.1 | 20.9 ± 2.3 | ND | ND | ND | 19.5 ± 0.7 | 21.4 ± 3.2 | 18.9 ± 1.1 | ||||||||
| N.E. (%) | 67 | 0 | 33 | 0 | ||||||||||||
| N13 latency (ms) | 13.1 ± 0.8 | 14.4 ± 1.9 | ND | ND | ND | 13.1 ± 0.6 | 14.8 ± 2.6 | 12.8 ± 1.0 | ||||||||
| N.E. (%) | 73 | 0 | 33 | 0 | ||||||||||||
| N9 latency (ms) | 9.1 ± 0.9 | 9.8 ± 0.9 | ND | ND | ND | 8.9 ± 0.4 | 10.1 ± 1.2 | 8.6 ± 0.6 | ||||||||
| N.E. (%) | 40 | 13 | 33 | 0 | ||||||||||||
| MRI | n = 12 | n = 8 | n = 3 | n = 4 | ||||||||||||
| Spinal cord abnormality | 9† | 3† | ND | ND | ND | 1† | 4‡ | |||||||||
Values are expressed as mean ± SD. Control values are those described previously (Koike et al., 2001). MCV, motor nerve conduction velocity; DL, distal latency; CMAP, compound muscle action potentials; SCV, sensory nerve conduction velocity; SNAP, sensory nerve action potentials; N.E., not evoked; SEP, somatosensory evoked potentials; control values are from 37 conduction times.
P < 0.05,
P < 0.01,
P < 0.001 as compared with control value.
In T2* weighted gradient echo images, a high intensity area is present in the posterior column.
In T1 weighted echo images. Posterior spinal roots and cauda equina are enhanced by gadolinium.
T2*-weighted MRI demonstrated posterior column high intensity signal in 9 of the 12 examined patients (Table 4; Fig. 1). The extent of dorsal column high intensity T2* signal was well correlated with the distribution and intensity of sensory involvement and sensory ataxia, indicating the presence of central rami involvement due to sensory ganglion neuron damage (Mori et al., 2001).
Sural nerve biopsy was performed in 31 patients (Table 5). Total myelinated fibre density was variably reduced, ranging from 131 to 6918/mm2 (mean ± SD, 3287 ± 2843/mm2), and that of unmyelinated fibres was also reduced. Mean densities of large, small myelinated and unmyelinated fibres were reduced to 18, 56 and 75% of normal controls, respectively, indicating large fibre predominant loss. Axonal sprouts were not conspicuous in any case. In teased-fibre preparations, axonal degeneration was observed in 30.9 ± 36.1% of samples, while segmental demyelination was seen in only 9.7 ± 9.4% of samples, indicating that axonal changes are the predominant pathological feature. Chronic vasculitis of the arterioles in the epineurial space was seen in six patients and mild perivascular lymphocyte infiltrates in the small vessels were also seen in nine patients.
Pathological findings in the sural nerve
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 8) | Cranial neuropathy | Autonomic neuropathy (n = 2) | Radiculo- neuropathy (n = 4) | Controls (n = 7) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 31) | Painful (n = 9) | Multiple (n = 0) | Trigeminal (n = 1) | |||||||||||||
| Myelinated fibre density (no./mm2) | ||||||||||||||||
| Total | 3287 ± 2843** | 4105 ± 2260** | 1153 ± 920** | ND | 8010 | 2924 | 5985 ± 1890* | 8220 ± 614 | ||||||||
| Large | 579 ± 697*** | 2039 ± 1136* | 226 ± 262** | ND | 2994 | 1113 | 1593 ± 913* | 3150 ± 383 | ||||||||
| Small | 2878 ± 2482* | 2056 ± 1267** | 927 ± 672*** | ND | 5111 | 1811 | 4391 ± 977 | 5071 ± 397 | ||||||||
| Small/large | 13.7 ± 18.1** | 0.9 ± 0.5* | 10.3 ± 12.0* | ND | 1.7 | 2.9 | 3.1 ± 1.2 | 1.6 ± 0.2 | ||||||||
| Unmyelinated fibre density (no./mm2) | 22643 ± 9477* | 9879 ± 9203** | ND | ND | ND | 14 822 | ND | 29901 ± 2623 | ||||||||
| Segmental de-/remyelination (%) | 9.7 ± 9.4 | 10.0 ± 2.5 | 13.3 ± 13.1 | ND | 2.5 | 7.0 | 14.5 ± 9.2 | 7.2 ± 6.5 | ||||||||
| Axonal degeneration (%) | 30.9 ± 36.1** | 19.0 ± 16.1 | 61.0 ± 5.3** | ND | 0 | 12.5 | 3.5 ± 4.9 | 1.4 ± 1.6 | ||||||||
| Vasculitis: n (%) | 6 (19) | 0 (0) | 5 (63) | ND | 0 (0) | 0 (0) | 0 (0) | – | ||||||||
| Perivascular cell invasion: n (%) | 9 (29) | 1 (11) | 6 (75) | ND | 0 (0) | 0 (0) | 1 (25) | – | ||||||||
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 8) | Cranial neuropathy | Autonomic neuropathy (n = 2) | Radiculo- neuropathy (n = 4) | Controls (n = 7) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 31) | Painful (n = 9) | Multiple (n = 0) | Trigeminal (n = 1) | |||||||||||||
| Myelinated fibre density (no./mm2) | ||||||||||||||||
| Total | 3287 ± 2843** | 4105 ± 2260** | 1153 ± 920** | ND | 8010 | 2924 | 5985 ± 1890* | 8220 ± 614 | ||||||||
| Large | 579 ± 697*** | 2039 ± 1136* | 226 ± 262** | ND | 2994 | 1113 | 1593 ± 913* | 3150 ± 383 | ||||||||
| Small | 2878 ± 2482* | 2056 ± 1267** | 927 ± 672*** | ND | 5111 | 1811 | 4391 ± 977 | 5071 ± 397 | ||||||||
| Small/large | 13.7 ± 18.1** | 0.9 ± 0.5* | 10.3 ± 12.0* | ND | 1.7 | 2.9 | 3.1 ± 1.2 | 1.6 ± 0.2 | ||||||||
| Unmyelinated fibre density (no./mm2) | 22643 ± 9477* | 9879 ± 9203** | ND | ND | ND | 14 822 | ND | 29901 ± 2623 | ||||||||
| Segmental de-/remyelination (%) | 9.7 ± 9.4 | 10.0 ± 2.5 | 13.3 ± 13.1 | ND | 2.5 | 7.0 | 14.5 ± 9.2 | 7.2 ± 6.5 | ||||||||
| Axonal degeneration (%) | 30.9 ± 36.1** | 19.0 ± 16.1 | 61.0 ± 5.3** | ND | 0 | 12.5 | 3.5 ± 4.9 | 1.4 ± 1.6 | ||||||||
| Vasculitis: n (%) | 6 (19) | 0 (0) | 5 (63) | ND | 0 (0) | 0 (0) | 0 (0) | – | ||||||||
| Perivascular cell invasion: n (%) | 9 (29) | 1 (11) | 6 (75) | ND | 0 (0) | 0 (0) | 1 (25) | – | ||||||||
Values are expressed as mean ± SD. Control values (mean ± SD) were obtained from autopsy material and expressed as mean ± SD for 7 case. (Koike et al., 2004). ND, not determined; Small < 6.73 µm; large ≥ 6.73 µm in fibre diameter (Sobue et al., 1989);
P < 0.05,
P < 0.01,
P < 0.001 as compared with control values.
Pathological findings in the sural nerve
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 8) | Cranial neuropathy | Autonomic neuropathy (n = 2) | Radiculo- neuropathy (n = 4) | Controls (n = 7) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 31) | Painful (n = 9) | Multiple (n = 0) | Trigeminal (n = 1) | |||||||||||||
| Myelinated fibre density (no./mm2) | ||||||||||||||||
| Total | 3287 ± 2843** | 4105 ± 2260** | 1153 ± 920** | ND | 8010 | 2924 | 5985 ± 1890* | 8220 ± 614 | ||||||||
| Large | 579 ± 697*** | 2039 ± 1136* | 226 ± 262** | ND | 2994 | 1113 | 1593 ± 913* | 3150 ± 383 | ||||||||
| Small | 2878 ± 2482* | 2056 ± 1267** | 927 ± 672*** | ND | 5111 | 1811 | 4391 ± 977 | 5071 ± 397 | ||||||||
| Small/large | 13.7 ± 18.1** | 0.9 ± 0.5* | 10.3 ± 12.0* | ND | 1.7 | 2.9 | 3.1 ± 1.2 | 1.6 ± 0.2 | ||||||||
| Unmyelinated fibre density (no./mm2) | 22643 ± 9477* | 9879 ± 9203** | ND | ND | ND | 14 822 | ND | 29901 ± 2623 | ||||||||
| Segmental de-/remyelination (%) | 9.7 ± 9.4 | 10.0 ± 2.5 | 13.3 ± 13.1 | ND | 2.5 | 7.0 | 14.5 ± 9.2 | 7.2 ± 6.5 | ||||||||
| Axonal degeneration (%) | 30.9 ± 36.1** | 19.0 ± 16.1 | 61.0 ± 5.3** | ND | 0 | 12.5 | 3.5 ± 4.9 | 1.4 ± 1.6 | ||||||||
| Vasculitis: n (%) | 6 (19) | 0 (0) | 5 (63) | ND | 0 (0) | 0 (0) | 0 (0) | – | ||||||||
| Perivascular cell invasion: n (%) | 9 (29) | 1 (11) | 6 (75) | ND | 0 (0) | 0 (0) | 1 (25) | – | ||||||||
| Clinical features | Sensory neuropathy | Multiple mononeuropathy (n = 8) | Cranial neuropathy | Autonomic neuropathy (n = 2) | Radiculo- neuropathy (n = 4) | Controls (n = 7) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ataxic (n = 31) | Painful (n = 9) | Multiple (n = 0) | Trigeminal (n = 1) | |||||||||||||
| Myelinated fibre density (no./mm2) | ||||||||||||||||
| Total | 3287 ± 2843** | 4105 ± 2260** | 1153 ± 920** | ND | 8010 | 2924 | 5985 ± 1890* | 8220 ± 614 | ||||||||
| Large | 579 ± 697*** | 2039 ± 1136* | 226 ± 262** | ND | 2994 | 1113 | 1593 ± 913* | 3150 ± 383 | ||||||||
| Small | 2878 ± 2482* | 2056 ± 1267** | 927 ± 672*** | ND | 5111 | 1811 | 4391 ± 977 | 5071 ± 397 | ||||||||
| Small/large | 13.7 ± 18.1** | 0.9 ± 0.5* | 10.3 ± 12.0* | ND | 1.7 | 2.9 | 3.1 ± 1.2 | 1.6 ± 0.2 | ||||||||
| Unmyelinated fibre density (no./mm2) | 22643 ± 9477* | 9879 ± 9203** | ND | ND | ND | 14 822 | ND | 29901 ± 2623 | ||||||||
| Segmental de-/remyelination (%) | 9.7 ± 9.4 | 10.0 ± 2.5 | 13.3 ± 13.1 | ND | 2.5 | 7.0 | 14.5 ± 9.2 | 7.2 ± 6.5 | ||||||||
| Axonal degeneration (%) | 30.9 ± 36.1** | 19.0 ± 16.1 | 61.0 ± 5.3** | ND | 0 | 12.5 | 3.5 ± 4.9 | 1.4 ± 1.6 | ||||||||
| Vasculitis: n (%) | 6 (19) | 0 (0) | 5 (63) | ND | 0 (0) | 0 (0) | 0 (0) | – | ||||||||
| Perivascular cell invasion: n (%) | 9 (29) | 1 (11) | 6 (75) | ND | 0 (0) | 0 (0) | 1 (25) | – | ||||||||
Values are expressed as mean ± SD. Control values (mean ± SD) were obtained from autopsy material and expressed as mean ± SD for 7 case. (Koike et al., 2004). ND, not determined; Small < 6.73 µm; large ≥ 6.73 µm in fibre diameter (Sobue et al., 1989);
P < 0.05,
P < 0.01,
P < 0.001 as compared with control values.
Painful sensory neuropathy without sensory ataxia
A total of 18 patients had this form of neuropathy (Table 2). The initial symptoms were painful dysaesthesia in the most distal portions of the limbs, usually in unilateral limbs. In three patients, the initial progression was acute, occurring in days, and painful dysaesthesias were present over the entire body, including the trunk and face. In a majority of patients, spread of the dysaesthesias was chronic, occurring over months to years. The trigeminal nerve was involved in eight patients. Sensory impairment was relatively predominant with respect to superficial sensation of pain, temperature and light touch, and was associated with pain or painful dysaesthesia. Deep sensation was relatively well preserved, and motor function also was preserved. However, mild sensory ataxia in the limbs was seen in two patients. The face and trunk were involved in 8 and 10 patients, respectively, and segmental in distribution in some patients. In contrast to the sensory ataxic form, deep tendon reflexes were fairly well preserved in half of the patients. Seven patients could not walk because of severe pain.
Eleven patients showed symptoms consistent with autonomic neuropathy (Table 3). Abnormal pupils, including Adie's pupils and elliptic pupils, were seen in three patients. Orthostatic hypotension and hypohidrosis or anhidrosis were present in 5 and 10 patients. Segmental distribution of anhidrosis was often seen in the trunk. A severe decrease in 123I-MIBG cardiac uptake was seen in five of the seven examined patients. These results suggest that autonomic nerves are also widely involved in this form of neuropathy.
In contrast to sensory ataxic neuropathy, unelicited SNAPs were present in only 11 and 17% of median and sural nerves, respectively (Table 4). SCV was well preserved. MCV showed no slowing and CMAPs were well preserved. Cortical (N20) and cervical (N13) SEPs were elicited in all of the examined patients and Erb's point (N9) SEP was not elicited in only one patient examined.
T2*-MRI of the spinal cord showed minimal high intensity signal in the posterior column in three out of the eight patients studied (Table 4). The extent of high intensity signal in these patients was relatively small compared with those seen with sensory ataxic neuropathy. The sural nerve biopsy specimen in nine patients mostly showed small fibre loss (Table 5). Mean densities of large, small myelinated and unmyelinated fibres were reduced to 65, 41 and 33% of normal control, respectively, indicating small fibre predominant loss. Axonal sprouts were essentially absent. In teased-fibre preparations, axonal degeneration was seen in 19.0 ± 16.1% of fibres, predominantly in the small-diameter fibres. Perivascular cell invasion was also present in one patient.
These relatively well preserved SNAPs and SEPs, and mild T2* posterior column abnormalities on MRI as well as the predominant decrease in small myelinated and unmyelinated fibres in the sural nerve suggest that small sensory neurons are predominantly impaired and large diameter sensory neurons are fairly well preserved in this form of neuropathy.
Patients were followed-up for 1–12 years. Deep sensory impairment developed in three patients over nine years. They showed sensory ataxia in the legs and fingers. Other patients showed persistent painful sensory neuropathy with a gradual extension of the distribution of the neuropathy, without sensory ataxia.
Multiple mononeuropathy
A total of 11 patients showed a form of multiple mononeuropathy (Table 2). The initial symptom of neuropathy was the acute onset of a tingling sensation or painful dysaesthesia in the distal portion of the limbs. Subsequently, motor and sensory symptoms episodically occurred and extended to the distribution of a multiple mononeuropathy pattern mostly restricted to the limbs. Initial progression was acute or subacute in half of the patients. Trigeminal nerves and truncal intercostal nerves were involved in only two patients, respectively. Impairment during one episode subsequently disappeared, and another area of sensory impairment developed in some patients. Sensory impairment involved all modalities of both superficial and deep sensation. Muscle weakness was evident in the involved limbs, but sensory symptoms were generally more pronounced. Perinuclear antineutrophil cytoplasmic antibody (p-ANCA) and cryoglobulins were negative in all the patients examined. Systemic autonomic symptoms were relatively rare (Table 3). CMAPs and SNAPs in the involved nerves were markedly reduced (Table 4). Both large and small myelinated fibres were markedly depleted with prominent active axonal degeneration in the sural nerves. The most prominent histological feature was the frequent occurrence of vasculitic lesions associated with perivascular cellular invasions (Table 5).
Multiple cranial neuropathy
Five patients had multiple cranial neuropathy (Table 2). Involvement of the cranial nerves was bilateral VII nerve involvement in one patient, recurrent III and VI nerve involvement in one patient, III, V, VI, VII, IX and X nerve involvement in one patient, V, IX and X nerve involvement in one patient, and V, VII, IX, X and XII nerve involvement in one patient. Abnormal pupils were seen in one patient (Table 3). Three patients had acute onset of the neuropathy. With respect to extra-cranial symptoms, painful dysaesthesia in the limbs was detected in the initial phase in one patient, and truncal and limb sensory impairment developed in two patients during the follow-up. All patients had cranial motor nerve involvement in spite of the fact that the extent and degree of cranial nerve involvement was variable among the patients.
Trigeminal neuropathy
A total of 15 patients had a pure sensory trigeminal neuropathy (Table 2). Nine patients had unilateral involvement and six had bilateral involvement. Numbness or paraesthesia restricted to the trigeminal nerve region was the characteristic feature. Appreciation of pin prick and soft touch was diminished in the trigeminal nerve region, and dysaesthesia was present. Dysaesthesia of the tongue was present in one patient. Motor symptoms referable to trigeminal nerve involvement were not seen. The progression of these symptoms was indolent in most patients. Sensory disturbances in the limbs were seen in two patients. Pupillary abnormalities were seen in three patients, and orthostatic hypotension and hypohidrosis were observed in three and four patients, respectively (Table 3). There were no marked abnormalities in the routine nerve conduction of the limbs (Table 4). Blink reflex tests were performed in three patients with unilateral involvement, which confirmed trigeminal nerve involvement on the affected side (data not shown). Nerve biopsy was obtained from one patient, the findings of which were normal (Table 5).
Autonomic neuropathy
Three patients had predominant and severe autonomic symptoms and were designated as autonomic neuropathy (Tables 2 and 3). All three patients showed Adie's pupils and all patients also showed severe orthostatic hypotension with syncope. Hypohidrosis or anhidrosis also was present in the trunk and all four limbs. All patients developed abdominal pain, constipation and diarrhoea. Cardiac 123I-MIBG uptake was reduced in two patients examined. Lack of plasma norepinephrine increase in response to standing and hypersensitive blood pressure increase beyond 25 mmHg in response to low concentration of norepinephrine infusion at 3 µg/min were seen in two patients examined. These observations suggest that peripheral sympathetic nervous system was severely involved in this form of neuropathy. Limb and truncal sensory impairment was present with sensory ataxia, but without motor involvement. These symptoms appeared chronically. The SNAPs and SEPs were unelicited and high intensity MRI signal in the posterior column of the spinal cord was seen in one patient (Table 4). A moderate reduction in the myelinated and unmyelinated fibre populations was seen in the sural nerve (Table 5).
Radiculoneuropathy
Four patients had this form of neuropathy (Table 2). All patients had chronic sensorimotor polyradiculoneuropathy with progressive sensory impairment and muscle weakness. The sensory disturbance was in a glove and stocking pattern in all of the patients, with an associated sensory ataxia in three patients. Apparent muscle weakness was seen in two patients. Autonomic symptoms were generally absent, except for constipation, hypohidrosis and urinary disturbances (Table 3). The CSF protein concentration was elevated, ranging from 98 to 146 mg/dl, without pleocytosis. F-wave abnormalities, poor occurrence and prolonged latencies, were present in all patients, while motor and sensory nerve conductions were almost normal, except in one patient with mild elongated distal latency and decreased conduction velocities in the median and tibial nerves (Table 4). This nerve conduction feature was unusual in chronic inflammatory demyelinating polyradiculoneuropathy. SEPs were also substantially prolonged. MRI of the lumbar spine showed abnormal gadolinium enhancement predominantly of the dorsal spinal roots and cauda equine, in all four patients. Sural nerve biopsy showed variable degrees of myelinated fibre loss with minor to moderate demyelinating changes in all patients (Table 5). These clincopathological features suggest that the primary lesion in these patients is in the spinal nerve roots or most proximal nerve trunks, consistent with an inflammatory radiculoneuropathy.
Overlapping clinical features among the neuropathic forms
Each neuropathic form had principal and predominant clinical features characterizing each individual neuropathic form, while the clinical symptoms overlapped to some extent with each other. Sensory ataxic neuropathy frequently had painful features, autonomic symptoms and trigeminal nerve involvement. Painful sensory neuropathy also had autonomic and trigeminal nerve involvement, as well as sensory ataxic features. Multiple mononeuropathy had painful and sensory ataxic features. Trigeminal neuropathy had autonomic and painful features. Multiple cranial neuropathy had some degree of trigeminal, painful and autonomic features. Autonomic neuropathy also had sensory ataxic and trigeminal nerve involvement. These overlapping symptoms were the common features in the present analysis, while overlapping symptoms occurred during the long-standing clinical course. For instance, some patients with painful sensory neuropathy without sensory ataxia later developed sensory ataxia, or alternatively, patients with sensory ataxic neuropathy often developed painful dysaesthetic features during the clinical course. These overlapping clinical features strongly suggest that each individual neuropathic form is not the absolute clinical entity, but these individual forms share a common underlying pathological process.
Findings in an autopsied patient with the sensory ataxic form of neuropathy
An 88-year-old woman with the sensory ataxic form of neuropathy was examined at the time of autopsy. She had numbness on the right side of her face since the age of 64 years, and developed unsteadiness of gait and pseudoathetosis in the fingers at 71 years of age. She was diagnosed as having Sjögren's syndrome at the age of 71. Severe sensory ataxia in the limbs was present. A marked segmental distribution of sensory impairment, particularly with respect to a deep sensation and anhidrosis, was noted (Kumazawa et al., 1993) in the limbs and trunk (Fig. 1). Severe orthostatic hypotension, with a decrease of up to 70 mmHg in systolic pressure, and marked decrease in cardiac MIBG uptake was present. T2*-high intensity signal lesions in the spinal dorsal column were observed (Fig. 1). Respiratory failure due to pneumonia was the cause of death. The autopsy was performed 5 h postmortem.
The population of sensory ganglion neurons was severely, but variably, diminished among the spinal segments; 45% of the control value in the C5, 37% in the Th11 and 26% in the L4 segments (Fig. 1). Nageotte's nodules (Fig. 1) and mild cell infiltrations that contained mainly T-cells were seen (Fig. 2). The large sensory ganglion neurons were diminished predominantly. Myelinated fibre density in the dorsal spinal roots was also variably diminished among the spinal segments; 48% of the control value in the C5, 42% in the Th11 and 22% in the L4 segments (Fig. 1). The large myelinated fibres also were depleted predominantly. The extent of fibre loss in the dorsal spinal roots correlated well with the corresponding dorsal root ganglion cell population. The spinal dorsal column fibre population was also markedly depleted (Fig. 1). These observations strongly suggest that ganglioneuronitis affecting the sensory neurons is the major pathological process. The sympathetic ganglion cells also were severely, but variably, diminished among the segments (23–51%), with mild T-cell invasion (Fig. 1). These segmental variations in the extent of sensory ganglion neuron involvement and sympathetic ganglion neuron involvement seem to correspond to segmental variation of sensory and sweat impairments seen in this patient (Fig. 1). These clinicopathological correlates also may support the view that the major responsible lesion is of sensory and sympathetic neurons. The myelinated fibres in the sciatic, median and tibial nerves in the proximal portion of these nerve trunks showed a remarkable multifocal patchy distribution of myelinated fibre loss, present mainly in the large diameter fibres. The sural nerve revealed loss of large myelinated fibres with active axonal degeneration. Multifocal and disseminated perivascular T-cell infiltrations were seen in the endoneurial and perineurial space of the peripheral nerve trunks (Fig. 2), although the extent of cell invasion was mild. Features of arterial vasculitis, mostly in the post-active state, were seen throughout the peripheral nerve trunks (Fig. 2). Examination of the skeletal muscles showed an almost normal appearance and spinal motor neurons and ventral roots also were normal in appearance and in population (Fig. 1). Submandibular and subauricular salivary glands had T-cell invasion and acinar cell destruction (Fig. 2). Relatively mild inflammatory cell invasion in this patient may be due to the extensive therapies including prednisone.
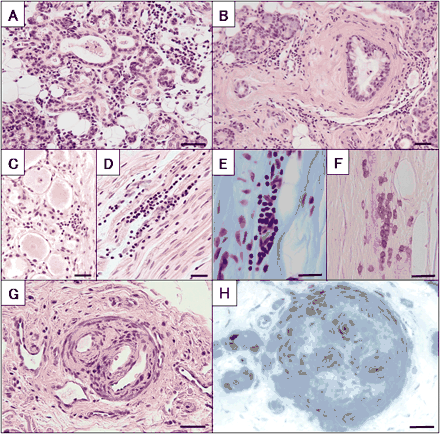
Inflammatory aspects of the autopsied patient. (A and B) Lymphocytic infiltration at parotid gland (A) and submandibular gland (B). Haematoxylin-eosin stain. Scale bar = 40 µm. (C and D) Lymphocytic infiltration at the L4 dorsal root ganglions on axial section (C), and on longitudinal section (D). Haematoxylin-eosin stain. Scale bar = 20 µm. (E and F) Longitudinal section of the median nerve. Perivenular lymphocytic infiltrates in the endoneurium. Klüver–Barrera's stain (E) and UCHL-1 positive cells (F). Scale bar = 20 µm. (G and H) Chronic vasculitis in the perineurial space. (G) Median nerve. Haematoxylin-eosin stain. (H) Sural nerve. Toluidine blue stain. Scale bar = 20 µm.
Therapeutic profiles for individual neuropathic forms
Corticosteroids (prednisone, 1 mg/kg/day) and intravenous immunoglobulin (IVIG) (400 mg/kg for 5 days) were prescribed for some of the patients (Table 6). Definite improvement in the modified Rankin scale measurement or in sensory impairments, including pain and painful dysaesthesias, after treatment was considered a favourable response (Table 6). Presence or absence of favourable response was evaluated ∼1 month after treatment.
Therapeutic profiles in prednisone- and IVIG-treated patients
| Neuropathic form | Prednisone | IVIG | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treated patients (n) | Favourable response (n) | No response (n) | % Response | Treated patients (n) | Favourable response (n) | No response (n) | % Response | |||||||||
| Sensory neuropathy | ||||||||||||||||
| Ataxic* | 22 | 4 | 18 | 18 | 13 | 3 | 10 | 23 | ||||||||
| Painful** | 6 | 1 | 5 | 17 | 3 | 2 | 1 | 67 | ||||||||
| Multiple mononeuropathy* | 11 | 8 | 3 | 73 | 1 | 0 | 1 | 0 | ||||||||
| Cranial neuropathy | ||||||||||||||||
| Multiple*** | 4 | 3 | 1 | 75 | 0 | ND | ND | ND | ||||||||
| Trigeminal*** | 3 | 1 | 2 | 33 | 0 | ND | ND | ND | ||||||||
| Autonomic neuropathy† | 2 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | ||||||||
| Radiculoneuropathy* | 3 | 0 | 3 | 0 | 4 | 4 | 0 | 100 | ||||||||
| Total | 51 | 17 | 34 | 33 | 22 | 9 | 13 | 41 | ||||||||
| Neuropathic form | Prednisone | IVIG | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treated patients (n) | Favourable response (n) | No response (n) | % Response | Treated patients (n) | Favourable response (n) | No response (n) | % Response | |||||||||
| Sensory neuropathy | ||||||||||||||||
| Ataxic* | 22 | 4 | 18 | 18 | 13 | 3 | 10 | 23 | ||||||||
| Painful** | 6 | 1 | 5 | 17 | 3 | 2 | 1 | 67 | ||||||||
| Multiple mononeuropathy* | 11 | 8 | 3 | 73 | 1 | 0 | 1 | 0 | ||||||||
| Cranial neuropathy | ||||||||||||||||
| Multiple*** | 4 | 3 | 1 | 75 | 0 | ND | ND | ND | ||||||||
| Trigeminal*** | 3 | 1 | 2 | 33 | 0 | ND | ND | ND | ||||||||
| Autonomic neuropathy† | 2 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | ||||||||
| Radiculoneuropathy* | 3 | 0 | 3 | 0 | 4 | 4 | 0 | 100 | ||||||||
| Total | 51 | 17 | 34 | 33 | 22 | 9 | 13 | 41 | ||||||||
IVIG, intravenous immunoglobulin therapy; ND, not determined.
Favourable response:
For sensory ataxic neuropathy, multiple mononeuropathy and radiculoneuropathy, positive therapeutic response with reduction of one or more points of the modified Rankin scale.
For painful neuropathy, positive therapeutic response with three or more reduction of Visual Analogue Scale (VAS) rating for pain, ranging from 0 = no pain to 10 = maximal pain intensity.
For cranial neuropathy, therapeutic response was assessed for the improvement of the symptoms of each cranial nerve. Favourable response was designated as definite subjective and objective improvement.
As for autonomic neuropathy, autonomic symptoms did not show a definite favourable response to the therapy.
Therapeutic profiles in prednisone- and IVIG-treated patients
| Neuropathic form | Prednisone | IVIG | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treated patients (n) | Favourable response (n) | No response (n) | % Response | Treated patients (n) | Favourable response (n) | No response (n) | % Response | |||||||||
| Sensory neuropathy | ||||||||||||||||
| Ataxic* | 22 | 4 | 18 | 18 | 13 | 3 | 10 | 23 | ||||||||
| Painful** | 6 | 1 | 5 | 17 | 3 | 2 | 1 | 67 | ||||||||
| Multiple mononeuropathy* | 11 | 8 | 3 | 73 | 1 | 0 | 1 | 0 | ||||||||
| Cranial neuropathy | ||||||||||||||||
| Multiple*** | 4 | 3 | 1 | 75 | 0 | ND | ND | ND | ||||||||
| Trigeminal*** | 3 | 1 | 2 | 33 | 0 | ND | ND | ND | ||||||||
| Autonomic neuropathy† | 2 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | ||||||||
| Radiculoneuropathy* | 3 | 0 | 3 | 0 | 4 | 4 | 0 | 100 | ||||||||
| Total | 51 | 17 | 34 | 33 | 22 | 9 | 13 | 41 | ||||||||
| Neuropathic form | Prednisone | IVIG | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treated patients (n) | Favourable response (n) | No response (n) | % Response | Treated patients (n) | Favourable response (n) | No response (n) | % Response | |||||||||
| Sensory neuropathy | ||||||||||||||||
| Ataxic* | 22 | 4 | 18 | 18 | 13 | 3 | 10 | 23 | ||||||||
| Painful** | 6 | 1 | 5 | 17 | 3 | 2 | 1 | 67 | ||||||||
| Multiple mononeuropathy* | 11 | 8 | 3 | 73 | 1 | 0 | 1 | 0 | ||||||||
| Cranial neuropathy | ||||||||||||||||
| Multiple*** | 4 | 3 | 1 | 75 | 0 | ND | ND | ND | ||||||||
| Trigeminal*** | 3 | 1 | 2 | 33 | 0 | ND | ND | ND | ||||||||
| Autonomic neuropathy† | 2 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | ||||||||
| Radiculoneuropathy* | 3 | 0 | 3 | 0 | 4 | 4 | 0 | 100 | ||||||||
| Total | 51 | 17 | 34 | 33 | 22 | 9 | 13 | 41 | ||||||||
IVIG, intravenous immunoglobulin therapy; ND, not determined.
Favourable response:
For sensory ataxic neuropathy, multiple mononeuropathy and radiculoneuropathy, positive therapeutic response with reduction of one or more points of the modified Rankin scale.
For painful neuropathy, positive therapeutic response with three or more reduction of Visual Analogue Scale (VAS) rating for pain, ranging from 0 = no pain to 10 = maximal pain intensity.
For cranial neuropathy, therapeutic response was assessed for the improvement of the symptoms of each cranial nerve. Favourable response was designated as definite subjective and objective improvement.
As for autonomic neuropathy, autonomic symptoms did not show a definite favourable response to the therapy.
Multiple mononeuropathy and multiple cranial neuropathy showed the most favourable response to corticosteroid therapy. Sensory ataxic neuropathy showed a favourable response to corticosteroid treatment in only 18% of the patients (Table 6). The rate of favourable response to IVIG therapy for radiculoneuropathy, painful sensory neuropathy and sensory ataxic neuropathy was 100, 67 and 23%, respectively, although the number of patients treated was limited (Table 6). This suggests that the rate of favourable response to corticosteroid or IVIG therapy was different among the neuropathic form, probably reflecting the underlying pathology. However, these favourable therapeutic responses were rather short-lived. In the long-term follow-up, these patients ultimately showed progression of symptoms.
Discussion
Underlying pathological features in each form of neuropathy
In this study, we assessed that Sjögren's syndrome-associated neuropathy has a broad clinical spectrum, including sensory ataxic neuropathy, painful sensory neuropathy without sensory ataxia, multiple mononeuropathy, multiple cranial neuropathy, trigeminal neuropathy, autonomic neuropathy and radiculoneuropathy. Here, we discuss the pathological background underlying several forms of neuropathy. Sensory ganglion cell destruction associated with lymphocytic infiltration detected by dorsal root ganglion biopsy provided direct proof that ganglioneuronitis is responsible for lesions in the sensory ataxic form of neuropathy (Malinow et al., 1986; Griffin et al., 1990). Most of our patients with sensory ataxic neuropathy had lesions of the central rami as well as the peripheral rami of the sensory neurons, as assessed by low amplitude or unelicitable SEPs and SNAPs, dorsal spinal column T2*-high intensity signal lesions and segmental sensory impairment. Furthermore, the autopsy findings of a patient with the sensory ataxic form had severe depletion of large-sized sensory ganglion neurons accompanied by T-cell invasion, which strongly support this view. In addition, substantial preservation of motor nerve function and a lack of axonal sprouting with large axon loss in the sural nerve biopsy specimens also support the view that the sensory neurons are primarily affected.
In contrast, in the painful sensory neuropathy form without sensory ataxia there is predominantly superficial sensory impairment, well preserved motor nerve function and small axon loss with relative preservation of large axons. SEPs are relatively well preserved compared with the sensory ataxic form, but T2*-high intensity signal lesions in the dorsal column of the spinal cord were observed, although the extent of high intensity was smaller than those in sensory ataxic neuropathy. Lack of axonal sprouts in the sural nerve biopsy also argues against the presence of a primary axonal lesion. We did not perform histological examination of the dorsal root ganglion. However, based on our clinical, laboratory, and electrophysiological data, we can speculate that this form of neuropathy is another form of sensory ganglioneuronopathy that affects small ganglion neurons. Some patients with painful sensory neuropathy eventually developed sensory ataxia due to the impairment of deep kinaesthetic sensation during long-term follow-up, although its distribution was restricted. Alternatively, some of the patients with sensory ataxic neuropathy had impairment of superficial sensation with painful dysaesthesias. These overlapping symptoms, observed in these two forms of neuropathy, may also support the hypothesis that these two neuropathies are part of a spectrum of disorders with a similar pathology.
The pathological basis of trigeminal neuropathy is not known. However, isolated sensory deficits along the territory of the trigeminal nerve are characteristic, and motor nerve dysfunction, even trigeminal motor dysfunction, is not present. Furthermore, pure sensory trigeminal neuropathy is occasionally the initial symptom of the sensory ataxic form of neuropathy or can present as one of the subsequent symptoms of sensory ataxic and painful sensory neuropathies. In addition, nature of autonomic symptoms such as pupillary abnormality and orthostatic hypotension, and highly chronic initial progression pattern in trigeminal neuropathy are similarly shared with those in sensory ataxic and painful neuropathy forms. These clinical features would suggest that trigeminal neuropathy is a cranial nerve version of sensory ganglionopathy, although further evidence is necessary to confirm this hypothesis.
In contrast to sensory ataxic neuropathy, painful sensory neuropathy and trigeminal neuropathy, multiple mononeuropathy and multiple cranial neuropathy often include motor nerve involvement with predominantly acute and subacute onset. Motor nerve involvement can be assessed accurately using the electrophysiological findings in these forms of neuropathy. In some patients with multiple mononeuropathy, evidence of motor nerve denervation on EMG or NCS can be detected. These observations suggest that this form of neuropathy represents a combined sensory and motor neuropathy, rather than an isolated sensory neuropathy. Furthermore, multiple cranial neuropathy and multiple mononeuropathy are not seen in the sensory ataxic and painful sensory forms of neuropathy, suggesting that these neuropathies are distinct from the sensory neuropathies. Vasculitis in the small arteries or arterioles in a sural nerve biopsy were detected in five out of eight patients with multiple mononeuropathy, the frequency of which was significantly higher than those in other forms of sensory neuropathies. Based on these observations, vasculitis and subsequent axonopathy might be the aetiology of multiple mononeuropathy, and possibly of multiple cranial neuropathy.
With respect to the pathological basis of Sjögren's syndrome-associated neuropathy, the autopsy findings of the patient with the sensory ataxic form suggest that there may be a continuous spectrum of pathological processes among the different forms of neuropathy. Sensory and autonomic ganglionitis accompanied by T-cell invasion was present in this patient, while disseminated vasculitis and perivascular T-cell infiltration were also present throughout the peripheral nerve trunks. The patients in whom the ganglionitis process was predominant, as in this autopsied patient, will present with the sensory ataxic form. In contrast, if the vasculitic process in the nerve trunk predominates, the patient would show the features of multiple mononeuropathy, including motor symptoms rather than symptoms of sensory or autonomic ganglionitis. We need further histological studies to confirm these findings, while we may speculate that sensory ganglionopathic lesions would contribute more profoundly to the sensory ataxic, painful sensory and trigeminal neuropathy forms, and vasculitic lesions would result in the multiple mononeuropathy and possibly multiple cranial neuropathy forms.
Neuropathy and other non-sicca symptomatic manifestations of Sjögren's syndrome
The striking feature was that the clinical manifestations of neuropathy preceded the development of sicca syndrome or laboratory findings consistent with Sjögren's syndrome in most patients. Thus, in most patients, neuropathy developed first and then the diagnosis of Sjögren's syndrome was made up to 12 years later, well in agreement with previous studies from our group and other groups (Sobue et al., 1993; Grant et al., 1997; Mori et al., 2001, 2003). This chronological sequence is true for all forms of neuropathy, but is more characteristic in the ganglionitis-related neuropathy forms, such as sensory ataxic and painful sensory neuropathy. Extraneural symptoms, such as pancreatitis and interstitial pneumonia, also can precede the clinical manifestations of Sjögren's syndrome (Garcia-Carrasco et al., 2002). These observations strongly suggest that neural tissues, particularly dorsal root sensory ganglion cells and probably autonomic ganglion cells, are the primary targets in Sjögren's syndrome in addition to the salivary and lacrimal glands (Greenspan et al., 1974), and visceral organs including the pancreas, lung, and thyroid (Swigris et al., 2002).
Antigens primarily responsible for the Sjögren's syndrome, which could be universally present among the target tissues, have been investigated. Whether alpha-fodrin antibody is specific to Sjögren's syndrome or not has been debated (de Seze et al., 2004; Ruffatti et al., 2004), but alpha-fodrin has still been proposed as a candidate antigen (Haneji et al., 1997). We examined anti-alpha-fodrin antibodies in the serum of patients from the present study and found that this antibody is elevated in patients with Sjögren's syndrome-associated neuropathy. However, increases in this antibody were also observed in other types of neuropathy (data not shown) suggesting that this antibody is a candidate marker for Sjögren's syndrome, but its specificity needs to be assessed further. Additional antigens responsible for Sjögren's syndrome that are expressed in all of the target organs need to be identified.
We still do not know why the neuropathic symptoms precede the manifestations of sicca symptoms and other characteristic features in the Sjögren's syndrome-associated neuropathy patients. One possible situation would be that the patients with neuropathic symptoms as the initial symptom would first be referred to a neurology clinic rather than to a rheumatology clinic, while in the case of patients with sicca syndrome they would be referred to a rheumatology clinic. In the case of patients presenting with pancreatitis as the initial symptom, these patients tended to be referred to the gastroenterology clinic rather than to rheumatology clinic. The low prevalence of anti SS-A and SS-B antibodies in our neuropathic patients may also contribute to the earlier occurrence of neuropathies before the diagnosis of Sjögren's syndrome. Taken together, the current diagnostic criteria for Sjögren's syndrome based on the sicca syndrome may need to be re-evaluated.
Autonomic symptoms and the autonomic neuropathy form
Autonomic symptoms are widely present in Sjögren's syndrome-associated neuropathy, particularly in the sensory ataxic, painful sensory and autonomic neuropathy form. Autonomic symptoms may be attributed to a different pathologic cause, such as autonomic ganglioneuronitis and peripheral autonomic nerve involvement due to direct T-cell attack of the nerves or ischaemia due to vasculitis. The findings from the autopsied patient, including the loss of sympathetic ganglion neurons associated with T-cell invasion, strongly support the view that autonomic ganglion cells are primarily involved, in a fashion similar to the involvement of sensory ganglion cells. In this patient, the segmental distribution of anhidrosis and skin temperature changes corresponded to the segmental variation in the extent of autonomic ganglion cell involvement, also supporting the hypothesis that the primary lesions in autonomic ganglion cells are responsible for autonomic symptoms (Fig. 1). Two of three autonomic neuropathy patients also had sensory ataxia, suggesting that the autonomic ganglionopathy has a similar aetiology as sensory ataxic neuropathy. The presence of Adie's pupils, which is often associated with Sjögren's syndrome-associated neuropathy, is also probably attributable to ciliary ganglion cell involvement (Waterschoot et al., 1991), although further histological assessment is needed. The degree of orthostatic hypotension, anhidrosis, constipation and loss of 123I-MIBG uptake were unexpectedly severe when the autonomic system was involved. Autonomic symptoms in Sjögren's syndrome-associated neuropathy are generally considered mild in their manifestations compared with the sensory symptoms (Wright et al., 1999). Our three patients with autonomic neuropathy were exceptions, since the autonomic symptoms, including bowel dysfunction, were extremely prominent symptoms, suggesting that severe autonomic neuropathy can be present in the spectrum of neuropathies associated with Sjögren's syndrome (Goto et al., 2000; Sakakibara et al., 2004). The present observations suggest that autonomic symptoms are one of the major symptoms in this neuropathy.
Therapeutic approach to Sjögren's syndrome-associated neuropathy
Corticosteroids and immunosuppressants have been employed for the treatment of Sjögren's syndrome, resulting in improvement of non-neuropathic Sjögren's syndrome-associated symptoms, such as sicca syndrome and pneumonitis (Zandbelt et al., 2001; Swigris et al., 2002).
For the therapy of neuropathy associated with Sjögren's syndrome, corticosteroids (Griffin et al., 1990; Noguchi et al., 2003), immunosuppressants (Griffin et al., 1990), plasmapheresis (Chen et al., 2001), d-penicillamine (Asahina et al., 1998), infliximab (Caroyer et al., 2002) and immunoglobulin (Molina et al., 1996; Pascual et al., 1998; Takahashi et al., 2003) administration have been reported anecdotally and suggest a favourable therapeutic response. In the present study, a favourable response to treatment was assessed in an open manner, and both standard corticosteroid and IVIG treatment had similar frequencies of favourable response. Based on the limited number of patients treated, there may be marked differences in the rates of favourable therapeutic response among the neuropathic forms, reflecting major differences in the causes of neuropathy. Corticosteroid therapy is likely a good candidate for multiple mononeuropathy and multiple cranial neuropathy, and favourable improvement may be seen in the painful dysaesthesias of the painful sensory neuropathy and radiculoneuropathy forms with IVIG therapy. Although these symptomatic therapeutic responses were seen in certain patients, overall progression of the neuropathic symptoms as well as of Sjögren's syndrome itself occurred. The findings of this study suggest that IVIG and corticosteroids may be efficacious in treating the neuropathic symptoms of Sjögren's syndrome, although these favourable responses were only seen in certain subpopulations of patients. Randomized controlled studies are needed to assess the efficacy of these treatments for neuropathic symptoms of Sjögren's syndrome.
The authors are grateful to Dr Hayashi (Department of Oral Molecular Pathology, The University of Tokushima Graduate School of Dentistry, Japan) for providing us with alpha-fodrin vector, and information on alpha-fodrin. The authors would like to express their deep appreciation to Dr Tsutomu Yanagi (Department of Neurology, Nagoya Daini Red Cross Hospital, Japan), Dr Eiichiro Mukai (Department of Neurology, Nagoya Medical Center, Japan), Drs Kazuhiro Imamura and Yuichi Kawagashira (Department of Neurology, Okazaki City Hospital, Japan), Dr Shigehisa Mitake (Department of Neurology, Tosei General Hospital, Japan), Dr Kenji Mokuno (Department of Neurology, Toyohashi Municipal Hospital, Japan) and Dr Yukihiko Matsuoka (Department of Neurology, Higashi Nagoya Hospital, Japan) for their support in the clinical evaluation of patients and for referring the patients for this study. The authors also are grateful for the expert pathologic technical assistance given by Dr Nozomi Hishikawa and Ms Sugiko Yokoi (Department of Neurology, Nagoya University Graduate School of Medicine, Japan). This study was partly supported by the grants from the Ministry of Health, Labor, and Welfare, Japan.
References
Alexander EL, Provost TT, Stevens MB, Alexander GE. Neurologic complications of primary Sjögren's syndrome.
Anonymous. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee.
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis.
Asahina M, Kuwabara S, Asahina M, Nakajima M, Hattori T. d-penicillamine treatment for chronic sensory ataxic neuropathy associated with Sjögren's syndrome.
Caroyer JM, Manto MU, Steinfeld SD. Severe sensory neuronopathy responsive to infliximab in primary Sjögren's syndrome.
Chen WH, Yeh JH, Chiu HC. Plasmapheresis in the treatment of ataxic sensory neuropathy associated with Sjögren's syndrome.
Chu K, Kang DW, Song YW, Yoon BW. Trochlear nerve palsy in Sjögren's syndrome.
Delalande S, de Seze J, Fauchais AL, Hachulla E, Stojkovic T, Ferriby D, et al. Neurologic manifestations in primary Sjögren syndrome: a study of 82 patients.
Denislic M, Meh D, Popovic M, Kos-Golja M. Small nerve fibre dysfunction in a patient with Sjögren's syndrome. Neurophysiological and morphological confirmation.
de Seze J, Dubucquoi S, Fauchais AL, Hachulla E, Matthias T, Lefranc D, et al. Autoantibodies against alpha-fodrin in Sjögren's syndrome with neurological manifestations.
Dyck PJ, Giannini C, Lais A. Pathologic alterations of nerves. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, editors. Peripheral neuropathy. 3rd ed. Philadelphia: WB Saunders;
Fujibayashi T, Sugai S, Miyasaka N, Toujou T, Miyawaki S, Ichikawa Y, et al. Revised Japanese diagnostic criteria for Sjögren's syndrome. Annual Report of Research Committee for Immune Disease. Tokyo: Japanese Ministry of Health and Welfare;
Garcia-Carrasco M, Ramos-Casals M, Rosas J, Pallares L, Calvo-Alen J, Cervera R, et al. Primary Sjögren's syndrome: clinical and immunologic disease patterns in a cohort of 400 patients.
Gemignani F, Marbini A, Pavesi G, Di Vittorio S, Manganelli P, Cenacchi G, et al. Peripheral neuropathy associated with primary Sjögren's syndrome.
Goto H, Matsuo H, Fukudome T, Shibuya N, Ohnishi A, Nakamura H. Chronic autonomic neuropathy in a patient with primary Sjögren's syndrome.
Grant IA, Hunder GG, Homburger HA, Dyck PJ. Peripheral neuropathy associated with sicca complex.
Greenspan JS, Daniels TE, Talal N, Sylvester RA. The histopathology of Sjögren's syndrome in labial salivary gland biopsies.
Griffin JW, Cornblath DR, Alexander E, Campbell J, Low PA, Bird S, et al. Ataxic sensory neuropathy and dorsal root ganglionitis associated with Sjögren's syndrome.
Gross M. Chronic relapsing inflammatory polyneuropathy complicating sicca syndrome.
Hamada K, Hirayama M, Watanabe H, Kobayashi R, Ito H, Ieda T, et al. Onset age and severity of motor impairment are associated with reduction of myocardial 123I-MIBG uptake in Parkinson's disease.
Haneji N, Nakamura T, Takio K, Yanagi K, Higashiyama H, Saito I, et al. Identification of alpha-fodrin as a candidate autoantigen in primary Sjögren's syndrome.
Kachi T, Sobue G, Yamamoto M, Igata A. Sensory conduction study in chronic sensory ataxic neuropathy.
Kaltreider HB, Talal N. The neuropathy of Sjögren's syndrome. Trigeminal nerve involvement.
Kaplan JG, Rosenberg R, Reinitz E, Buchbinder S, Schaumburg HH. Invited review: peripheral neuropathy in Sjögren's syndrome.
Kennett RP, Harding AE. Peripheral neuropathy associated with the sicca syndrome.
Kimura J. The blink reflex. In: Kimura J, editor. Electrodiagnosis in diseases of nerve and muscle. Principles and practice. New York: Oxford University Press;
Koike H, Mori K, Misu K, Hattori N, Ito H, Hirayama M, et al. Painful alcoholic polyneuropathy with predominant small-fiber loss and normal thiamine status.
Koike H, Misu K, Sugiura M, Iijima M, Mori K, Yamamoto M, et al. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy.
Kumazawa K, Sobue G, Yamamoto K, Mitsuma T. Segmental anhidrosis in the spinal dermatomes in Sjögren's syndrome-associated neuropathy.
Lafitte C, Amoura Z, Cacoub P, Pradat-Diehl P, Picq C, Salachas F, et al. Neurological complications of primary Sjögren's syndrome.
Malinow K, Yannakakis GD, Glusman SM, Edlow DW, Griffin J, Pestronk A, et al. Subacute sensory neuronopathy secondary to dorsal root ganglionitis in primary Sjögren's syndrome.
Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg–Strauss syndrome (allergic granulomatosis and angiitis).
Mauch E, Volk C, Kratzsch G, Krapf H, Kornhuber HH, Laufen H, et al. Neurological and neuropsychiatric dysfunction in primary Sjögren's syndrome.
Mellgren SI, Conn DL, Stevens JC, Dyck PJ. Peripheral neuropathy in primary Sjögren's syndrome.
Molina JA, Benito-Leon J, Bermejo F, Jimenez-Jimenez FJ, Olivan J. Intravenous immunoglobulin therapy in sensory neuropathy associated with Sjögren's syndrome.
Molina R, Provost TT, Alexander EL. Peripheral inflammatory vascular disease in Sjögren's syndrome. Association with nervous system complications.
Mori K, Koike H, Misu K, Hattori N, Ichimura M, Sobue G. Spinal cord magnetic resonance imaging demonstrates sensory neuronal involvement and clinical severity in neuronopathy associated with Sjögren's syndrome.
Mori K, Iijima M, Sugiura M, Koike H, Hattori N, Ito H, et al. Sjögren's syndrome associated painful sensory neuropathy without sensory ataxia.
Noguchi Y, Tsuchiyama T, Matsumoto T, Fujigasaki H, Inaba A, Yokota T, et al. Two distinct types of neuropathy associated with Sjögren's syndrome developed in one patient. The importance of the selection of an appropriate therapeutic regimen.
Pascual J, Cid C, Berciano J. High-dose IV immunoglobulin for peripheral neuropathy associated with Sjögren's syndrome.
Peyronnard JM, Charron L, Beaudet F, Couture F. Vasculitic neuropathy in rheumatoid disease and Sjögren's syndrome.
Ruffatti A, Ostuni P, Grypiotis P, Botsios C, Tonello M, Grava C, et al. Sensitivity and specificity for primary Sjögren's syndrome of IgA and IgG anti-alpha-fodrin antibodies detected by ELISA.
Sakakibara R, Hirano S, Asahina M, Sawai S, Nemoto Y, Hiraga A, et al. Primary Sjögren's syndrome presenting with generalized autonomic failure.
Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study.
Sobue G, Yasuda T, Kachi T, Sakakibara T, Mitsuma T. Chronic progressive sensory ataxic neuropathy: clinicopathological features of idiopathic and Sjögren's syndrome-associated cases.
Sobue G, Yasuda T, Kumazawa K, Yamamoto K, Mitsuma T. MRI demonstrates dorsal column involvement of the spinal cord in Sjögren's syndrome-associated neuropathy.
Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid interstitial pneumonia: a narrative review.
Takahashi Y, Takata T, Hoshino M, Sakurai M, Kanazawa I. Benefit of IVIG for long-standing ataxic sensory neuronopathy with Sjögren's syndrome. IV immunoglobulin.
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus.
Touze E, Blanche P, Zuber M. A 35-year history of recurrent multiple cranial neuropathy due to primary Sjögren's syndrome.
Urban PP, Keilmann A, Teichmann EM, Hopf HC. Sensory neuropathy of the trigeminal, glossopharyngeal, and vagal nerves in Sjögren's syndrome.
van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver agreement for the assessment of handicap in stroke patients.
Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjögren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group.
Watanabe H, Ieda T, Katayama T, Takeda A, Aiba I, Doyu M, et al. Cardiac 123I-meta-iodobenzylguanidine (MIBG) uptake in dementia with Lewy bodies: comparison with Alzheimer's disease.
Waterschoot MP, Guerit JM, Lambert M, de Barsy T. Bilateral tonic pupils and polyneuropathy in Sjögren's syndrome: a common pathophysiological mechanism?
Wright RA, Grant IA, Low PA. Autonomic neuropathy associated with sicca complex.
Yasuda T, Sobue G, Hirose Y, Mimura M, Yanagi T. MR of acute autonomic and sensory neuropathy.
Author notes
1Department of Neurology, Nagoya University Graduate School of Medicine, 2Department of Neurology, Higashi Nagoya Hospital, Nagoya, 3Department of Neurology, Hokkaido University School of Medicine, Sapporo, Japan, 4Rehabilitation Center, Kitasato University School of Medicine, Kanagawa, Japan, 5Department of Microbiology and Immunology, Kanazawa Medical Collage, Kanazawa, Japan and 6Department of Neuropathology, Institute for Medical Science of Aging, Aichi Medical University, Japan


{kind=link}
{kind=link}