





Abstract
Previous studies have not completely clarified the precise defect that characterizes B cell development in aged animals. The question of which developmental mechanism is actually deficient in aging remains controversial. The goal of this study was to elucidate the effects of aging on bone marrow B cell population dynamics. We used mathematical modeling to predict the outcome of the different possible effects, and then compared these predictions to experimental data, to find the most plausible effects. Our model shows that the three main differences between B cell development in young and old mice are a decrease in the maximum number of cells in the pre-B compartment and increases in the rate of transition from cycling pre-B cells to resting pre-B cells and in the fractions of static cells included in the immature B cell subset.
Introduction
The immune system's function deteriorates with aging. Age-associated thymic involution is associated with the decreased production of naive T lymphocytes, an altered T cell repertoire and a decreased production of T cell factors (1–3). The number of blood B lymphocytes secreting Ig increases with age (4). However, the frequency and concentration of auto-antibodies increase with age, as do the occurrences of clonal B cell expansions and mAbs (5). The age-associated dysregulation of humoral immunity is associated with changes in the antibody repertoire. These alterations shift the antibody specificity from foreign to self-antigens, the isotypes from IgG to IgM, the affinity to antigens from high to low affinity and the source from antibodies produced by CD5− B cells to those produced by CD5+ B cells (1–3). The age-associated decline in the generation of naive B cells is not associated with macroscopic or microscopic ‘involution’ of the bone marrow since myeloid cells, erythroid cells and platelets continue to be produced throughout life (4).
The age-related changes in B cell development in the bone marrow are rather subtle. Consistent with the ∼90% reduction in common lymphocyte progenitor numbers in 20-month-old mice, the frequency and the number of pro-B cells are also reduced substantially with age (6). This observation disagrees with earlier studies indicating modest or no significant decreases in the pro-B cell compartment in old mice (7, 8). The reason for this discrepancy may be the strategies used to identify and count pro-B cells. Pro-B cells in the bone marrow are CD43+B220lo, and cells with this phenotype also include non-B lineage cells. The report concluding that there is a diminution in pro-B cell numbers with age excluded the latter cells from analysis (6), whereas the earlier two reports analyzed the total CD43+B220+ population (7, 8). Pre-B lymphocytes CD43−B220+, the most numerous of bone marrow B cell precursors, have been reported to decrease in number by 60–90% during aging (5, 9). Thus, the defect in B cell development appears to affect the transition of Hardy stage C (pro-B cells) to Hardy stage D (pre-B cells). The numbers of pre-B cells generated in vitro from pro-B cells were reduced by ∼20% in cultures of pro-B cells from old compared with young mice (10). Furthermore, kinetic studies of B cell regeneration revealed that pre-B cells and splenic B cell numbers were reduced by ∼3-fold in old compared with young recipients of bone marrow cells from young mice (11).
The transition from pro-B cells to pre-B cells depends on the surface expression of the pre-B cell receptor (BCR) and decreases with age (7–8, 12) because of impaired VDJ heavy chain gene recombination (13). Extrinsic factors in the bone marrow that decline with age are largely responsible for the less efficient V(D)J recombination in pro-B cells and diminished progression to the pre-B cell stage (14). Pre-B cells that do not express the pre-BCR do not survive. Evidence for this fact is the age-associated increase in the percentage of bone marrow pre-B cells that are undergoing apoptosis (10). Pre-BCR signaling stimulates the expression of the anti-apoptotic BCL-XL gene product that is critical for pre-B cell survival (15–18). The decreased numbers of pre-B lymphocytes from old mice express lower amounts of BCL-XL mRNA and protein (10).
Fewer pre-B and mature B cells are generated from pro-B cells derived from old compared with young mice when pro-B cells are cultured with stromal cells (10, 19). This defect is at least in part attributable to the impaired ability of bone marrow stromal cells to support B cell generation. Extrinsic factors that influence B cell precursor development include IL-7 produced by bone marrow stromal cells as well as IL-16 and TCP17, a 17-kDa protein secreted by activated CD8 T cells (5). The IL-7-mediated increase in the number of pre-B cells was greater when IL-7 was added to bone marrow cultures from old compared with young mice. This suggested that the production of and/or the response to IL-7 is impaired in old mice. However, extrinsic effects alone cannot explain decreases in the number of B lineage cells with age because pro-B cells exposed in vitro to non-limiting amounts of IL-7 still proliferate less than their young counterparts (6, 20). Pro-B cells from old mice express the IL-7R and common chains in normal levels (20), so age-related defects in IL-7 responsiveness might instead be due to aberrations in IL-7-signaling pathways. If this were the case, it could help to explain defects in V-DJ joining in pro-B cells, which is an IL-7-dependent event (21).
Despite the decreased number of recent bone marrow B cell emigrants in the periphery, the number of peripheral B lymphocytes does not diminish with age (7–8, 11, 19). This is due both to the capacity of B lymphocytes for peripheral self-renewal and to the increased life span of B lymphocytes in old mice (8, 22). Although the steady-state number of peripheral B cells does not decline with age, the impaired export of B cells from the bone marrow in old mice influences the rate of regeneration of splenic B cell number and diversity. Both old and young recipients do ultimately maintain the same number of splenic B lymphocytes. However, the diversity of the regenerated peripheral B cell repertoire is reduced in old compared with young mice (5). As the bone marrow is the only source of a diverse population of B cells, aging may be associated with a decreased production of a diverse population of peripheral B lymphocytes by the bone marrow.
Previous studies have not completely clarified the precise defect(s) that characterize B cell development in aged animals. The question of which developmental mechanism is actually deficient in aging remains controversial: Johnson et al. (7) have not found a decreased production of B cells in the bone marrow as reported by Klinman et al. (8). In order to understand the effects of aging on B cell development, we used in the present study the combination of published experimental data on B cell populations in aged mice (7) and our mathematical model of B cell populations in the bone marrow (23), aiming to discover which parameters of the developmental process change with age. The model enumerates cells in the main bone marrow B cell subsets and the fractions of 5-bromo-2-deoxyuridine (BrdU)-labeled cells in each subset, based on their rates of differentiation, proliferation and death. Aging may cause a decrease in proliferation rates, an increase in death rates or changes in differentiation rates—and BrdU-labeling studies alone cannot distinguish between these possibilities. Neither do linear estimates of the decrease in cell production, based on the labeling curve, give the correct estimates. Only fitting a non-linear mathematical model to the experimental data, as we have done in the present study, may reveal which of the above-mentioned parameter changes occur in aged mice, and estimate the magnitude of the changes. The results point at three main changes: the rate of transition from cycling pre-B cells to resting pre-B cells increases by ∼50%, the carrying capacity of the pre-B compartment—that is, the maximum number of cells of this type that the bone marrow can support (explained below)—is reduced by ∼30% and the fractions of static cells, which must be included in the immature B cell subset in order to explain why labeling does not reach 100% even in a long-term steady state, are increased 5-fold. Thus, the modeling settles the controversy between the apparently conflicting experimental results: the overall numbers of cells produced are lower in older mice but the proliferation rate is unchanged, as explained below.
Methods
Data for model fitting
To form a more accurate account of age-related changes in the population dynamics of the B cell lineage, researchers used BrdU labeling to assess the kinetics of B cell development in young and old animals. Johnson et al. (7) looked at early kinetics (12 h to 11 days) of the bone marrow B cell populations. They examined a large number of mice (up to 12) at each time point, providing a statistical basis for their conclusions in spite of the mouse-to-mouse variability in conducting aging studies. They found an overall increase in the number of bone marrow cells in old animals as compared with young (Table 1). Despite a significant decrease in the percentages of pro- and immature B cells in old mice, there was no significant decrease in the actual numbers of pro- and immature B cells as compared with young mice due to the increase in the overall cellularity. The number of B220hi mature recirculating bone marrow B cells was increased in old animals, accounting for ∼10% of the increase in total bone marrow cellularity. In both young and old mice, the pro-B cell compartment, which contains the rapidly proliferating compartment of developing B cells, was fully labeled within 1 ± 2 days of BrdU administration. The pre-B cell compartment in both age groups was fully labeled by day 3. The number of pre-B cells was significantly decreased by ∼30% in old mice. The immature B cells of young mice reached maximum BrdU labeling at ∼95% by day 7, while in old mice maximum labeling plateaued at 75% of the total immature B cell compartment after 7 days.
Numbers of cells at each stage of development in the bone marrow of 2- and 22-month-old mice (7) and the steady-state cell numbers obtained in our simulations (×106)
B cell subset | Number of cells (×106) | ||
|---|---|---|---|
| Young | Old | ||
| Total number of cells | 45.8 ± 1.6 | 58.7 ± 2.4 | |
| Pro-B | 2.7 ± 0.2 | 2.5 ± 0.2 | |
| Pre-B | 5.0 ± 0.2 | 3.6 ± 0.3** | |
| Immature | 2.7 ± 0.2 | 2.3 ± 0.1 | |
| Mature recirculating | 1.8 ± 0.1 | 3.1 ± 0.2 ** | |
| Simulation results | |||
| Pro-B | 2.6 ± 0.1 | 2.31 ± 0.06 | |
| Pre-B | 4.89 ± 0.05 | 3.39 ± 0.07 | |
| Immature | 2.56 ± 0.04 | 2.2 ± 0.2 | |
B cell subset | Number of cells (×106) | ||
|---|---|---|---|
| Young | Old | ||
| Total number of cells | 45.8 ± 1.6 | 58.7 ± 2.4 | |
| Pro-B | 2.7 ± 0.2 | 2.5 ± 0.2 | |
| Pre-B | 5.0 ± 0.2 | 3.6 ± 0.3** | |
| Immature | 2.7 ± 0.2 | 2.3 ± 0.1 | |
| Mature recirculating | 1.8 ± 0.1 | 3.1 ± 0.2 ** | |
| Simulation results | |||
| Pro-B | 2.6 ± 0.1 | 2.31 ± 0.06 | |
| Pre-B | 4.89 ± 0.05 | 3.39 ± 0.07 | |
| Immature | 2.56 ± 0.04 | 2.2 ± 0.2 | |
The experimental results represent the mean and the standard deviation of 56 young and 51 old mice. (** indicates P < 0.0001; it should be noted that in terms of percentages out of the total bone marrow or spleen nucleated cell numbers, all these populations were significantly decreased.) The simulation results represent the mean and standard deviation of at least 29 000 runs, in which parameters were varied in the ranges shown in Table 2.
Numbers of cells at each stage of development in the bone marrow of 2- and 22-month-old mice (7) and the steady-state cell numbers obtained in our simulations (×106)
B cell subset | Number of cells (×106) | ||
|---|---|---|---|
| Young | Old | ||
| Total number of cells | 45.8 ± 1.6 | 58.7 ± 2.4 | |
| Pro-B | 2.7 ± 0.2 | 2.5 ± 0.2 | |
| Pre-B | 5.0 ± 0.2 | 3.6 ± 0.3** | |
| Immature | 2.7 ± 0.2 | 2.3 ± 0.1 | |
| Mature recirculating | 1.8 ± 0.1 | 3.1 ± 0.2 ** | |
| Simulation results | |||
| Pro-B | 2.6 ± 0.1 | 2.31 ± 0.06 | |
| Pre-B | 4.89 ± 0.05 | 3.39 ± 0.07 | |
| Immature | 2.56 ± 0.04 | 2.2 ± 0.2 | |
B cell subset | Number of cells (×106) | ||
|---|---|---|---|
| Young | Old | ||
| Total number of cells | 45.8 ± 1.6 | 58.7 ± 2.4 | |
| Pro-B | 2.7 ± 0.2 | 2.5 ± 0.2 | |
| Pre-B | 5.0 ± 0.2 | 3.6 ± 0.3** | |
| Immature | 2.7 ± 0.2 | 2.3 ± 0.1 | |
| Mature recirculating | 1.8 ± 0.1 | 3.1 ± 0.2 ** | |
| Simulation results | |||
| Pro-B | 2.6 ± 0.1 | 2.31 ± 0.06 | |
| Pre-B | 4.89 ± 0.05 | 3.39 ± 0.07 | |
| Immature | 2.56 ± 0.04 | 2.2 ± 0.2 | |
The experimental results represent the mean and the standard deviation of 56 young and 51 old mice. (** indicates P < 0.0001; it should be noted that in terms of percentages out of the total bone marrow or spleen nucleated cell numbers, all these populations were significantly decreased.) The simulation results represent the mean and standard deviation of at least 29 000 runs, in which parameters were varied in the ranges shown in Table 2.
Mathematical model of B cell populations in the bone marrow
We implemented our model of B cell development (23) by fitting it to published experimental data from 2- and 22-month-old mice (7). The model is based on differential equations representing developing B lymphocyte subsets. This addresses the issue of intercellular variability by representing each compartment as a ‘pool’ of cells, where we fix only the probability (per unit time) that a cell would leave this pool by using a population rate of transition. Cell division and death are similarly treated as probabilistic events by using population rates. A schematic representation of the model is presented in Fig. 1. Using this model, we have in previous studies shown that (i) the labeling data (23) are incompatible with B cell development being a synchronous, conveyor-belt-type process, and that (ii) B cell development may not be completely unidirectional, because our results support the possibility of a phenotypic ‘reflux’ from the immature to the pre-B cell subsets.
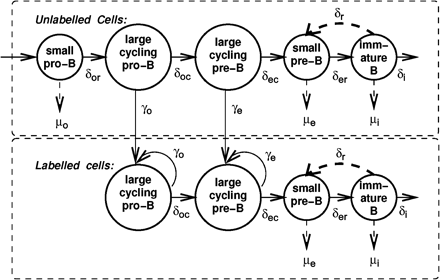
Model of developing B cell populations including BrdU labeling. All dividing cells are assumed to be labeled. Cell subsets and parameters represented in our model are shown (see Methods for details).
Our model deals with three populations: pro-B, pre-B and immature B cells, with cell numbers in these subsets represented by Bo, Be and Bi, respectively. However, previous experimental observations distinguish between small, non-cycling cells and large, cycling cells in both the pro-B and pre-B compartments, where the transition from pro-B to pre-B occurs while the cells are cycling. Hence, we break the pro-B and pre-B subsets into two subsets each: Bor for small, resting pro-B cells (Hardy's fractions A through C), and Boc for large, cycling pro-B cells (part of Hardy's fraction C′); similarly, Bec for large, cycling pre-B cells (the remainder of fraction C′), and Ber for small, resting pre-B cells (fraction D). For the sake of comparison with our experimental results, however, in the graphs presented in this paper, all pro-B cells were grouped together, and similarly, all pre-B cells were grouped together into one compartment.
The input of stem cells into the pro-B compartment is denoted by s (for ‘source’) in units of cells per time unit. We chose the time unit for which parameter values were defined to be 6 h, which is a minimum estimate for the cell-cycle time of bone marrow cells (24) and is smaller than the minimal time interval between experimental measurements, which was 12 h (7). All other parameters are rate parameters, defined as fractions of cell subsets per time unit. The rates of differentiation between subsets are represented by δX, with X representing the compartment the cells are differentiating from. Proliferation (population growth) is assumed to occur only in the late pro-B compartment (with the rate denoted by γo) and the early pre-B compartment (with rate γe).
Proliferation of developing B cells is known to be limited by the finite space and resources (e.g. contact with the stroma, growth factors, nutrients) in the bone marrow (25, 26). That is, for each proliferating subset, there is a finite ‘carrying capacity’, defined as the number of cells in that compartment for which the corresponding population growth rate becomes zero. Hence, the term for proliferation in each compartment is multiplied by a logistic growth-limiting factor: 1 − Bo/Ko for pro-B cells and 1 − Be/Ke for pre-B cells, with Bo = Bor + Boc and Be = Bec + Ber. In these terms, Ko and Ke denote the carrying capacities of the pro- and pre-B compartments, respectively. Other functional forms for population growth were investigated in our previous work (23), and the logistic form shown above was found to give the best fit. The separate carrying capacities for the pro-B and pre-B compartments are based on the different requirements for survival of these two cell populations—stromal cell contact and growth factors for pro-B cells and only IL-7 for pre-B cells, as discussed in (23). While our model is, to the best of our knowledge, the only mathematical model presented so far for the population dynamics of developing B lymphocytes, and the current study is the first to address the effect of aging by searching which parameters of the mathematical model change with age, much work has been done on modeling the population dynamics of developing T lymphocytes, which are similar in many aspects to developing B lymphocytes (27–32). For developing T lymphocytes, the logistic model has also been found to be the most appropriate, hence it is not surprising that the growth limitations are similar for B lymphocytes.
Cell death is assumed in our model to occur only in the non-proliferating cell subsets because proliferation, gene rearrangement and selection occur in distinct stages, and cell death usually occurs only as a result of failure in the latter two processes. The corresponding population mortality rates are denoted by μo, μe and μi for Bor, Ber and Bi, respectively.
The equations were integrated using a simple C program. In the simulations, we first ran the model without labeling for 100 time units (which is long enough for cell numbers to arrive at their steady states), then with labeling for 28 units (168 h, as in the experiments) and then again without labeling for the remaining time. The latter part of each run (that is, the part in which there is no labeling) simulates the decay of the number of labeled cells over time, under the assumption that this decay is only due to cell death or migration, neglecting any possible reduction of the numbers of all detectable labeled due to cell divisions. The decay of labeling was not measured in the experiments, hence our simulations predict what the dynamics of decay would be. This suggests an additional way to test our model, beyond fitting to existing data. The total numbers of cells in both compartments, and the fractions of cells in each compartment, exhibit the same behavior as in the basic model because we assume that labeling does not change the kinetics of B cell development.
Model parameters for old and young mice and parameter fitting to the data
In choosing parameter ranges for exploration in the simulations of our model, we adhered to the following guidelines. (i) The parameters should be in the experimentally observed orders of magnitude, if published information is available. While these estimates (where available) are usually not given in units of population rates, so that interpretation of most of these data depends on the model used, they were useful in suggesting the appropriate ranges for some of the parameters. (ii) The steady-state values obtained using these parameters should be in agreement with our experimental observations on both the total numbers and the composition of bone marrow B cells. Any parameter set which did not conform to this criterion was rejected. (iii) The time of arrival to the steady state should also conform with the experimental data. (iv) The fraction of labeled cells in each of the three sub-populations should be within the error bars of the experimental observation.
The parameter space defined by criterion (i) was thoroughly searched in simulations, varying each parameter in intervals of 0.01 and covering all possible combinations of parameter values, as in (23). The data were insufficient to give confidence limits on all parameters, hence parameter sets were considered to have a good fit to the experimental data according to criteria (ii)–(iv) above. In Table 2, we give the ranges for each parameter alone; however, since the acceptable parameter sets do not cover this whole sub-space, further relationsips between parameter values are also discussed (Fig. 5 and text below). For the sake of drawing conclusions from the data, only parameters for which there was no overlap between the ranges for young and old mice were considered to show a meaningful change.
Parameter value ranges that gave, in our simulations, the total cell numbers and population composition within the experimental ranges
Subset | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pro-B | Pre-B | Immature B | |||||||
| Resting | Cycling | Cycling | Resting | ||||||
| Young mice | |||||||||
| Entry rate (cell number × 104) | 1–10 | ||||||||
| Proliferation rate | 0.3–1.4 | 0.9–1.3 | |||||||
| Carrying capacity (cell number × 106) | 2–3 | 5.3–5.5 | |||||||
| Death rate | 0–1 | 0.1–1 | 0.00–0.19 | ||||||
| Output (differentiation) rate | 0.1–1 | 0.1–0.3 | 0.2–0.25 | 0.5–1.2 | 0.01–0.19 | ||||
| Reflux rate | 0–0.25 | ||||||||
| Static cells (cell number × 105) | 1 | ||||||||
| Old mice | |||||||||
| Entry rate (cell number × 104) | 1–10 | ||||||||
| Proliferation rate | 0.1–1.4 | 0.5–1.2 | |||||||
| Carrying capacity (cell number × 106) | 2.4–3 | 3.5–4 | |||||||
| Death rate | 0–1 | 0.1–1 | 0.00–0.2 | ||||||
| Output (differentiation) rate | 0.1–1 | 0.1–0.3 | 0.3–0.4 | 0.5–1.15 | 0.01–0.2 | ||||
| Reflux rate | 0–0.25 | ||||||||
| Static cells (cell number × 105) | 5 | ||||||||
Subset | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pro-B | Pre-B | Immature B | |||||||
| Resting | Cycling | Cycling | Resting | ||||||
| Young mice | |||||||||
| Entry rate (cell number × 104) | 1–10 | ||||||||
| Proliferation rate | 0.3–1.4 | 0.9–1.3 | |||||||
| Carrying capacity (cell number × 106) | 2–3 | 5.3–5.5 | |||||||
| Death rate | 0–1 | 0.1–1 | 0.00–0.19 | ||||||
| Output (differentiation) rate | 0.1–1 | 0.1–0.3 | 0.2–0.25 | 0.5–1.2 | 0.01–0.19 | ||||
| Reflux rate | 0–0.25 | ||||||||
| Static cells (cell number × 105) | 1 | ||||||||
| Old mice | |||||||||
| Entry rate (cell number × 104) | 1–10 | ||||||||
| Proliferation rate | 0.1–1.4 | 0.5–1.2 | |||||||
| Carrying capacity (cell number × 106) | 2.4–3 | 3.5–4 | |||||||
| Death rate | 0–1 | 0.1–1 | 0.00–0.2 | ||||||
| Output (differentiation) rate | 0.1–1 | 0.1–0.3 | 0.3–0.4 | 0.5–1.15 | 0.01–0.2 | ||||
| Reflux rate | 0–0.25 | ||||||||
| Static cells (cell number × 105) | 5 | ||||||||
All rates are per a time unit representing 6 h. The ranges in bold are those that differed most between young and old mice.
Parameter value ranges that gave, in our simulations, the total cell numbers and population composition within the experimental ranges
Subset | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pro-B | Pre-B | Immature B | |||||||
| Resting | Cycling | Cycling | Resting | ||||||
| Young mice | |||||||||
| Entry rate (cell number × 104) | 1–10 | ||||||||
| Proliferation rate | 0.3–1.4 | 0.9–1.3 | |||||||
| Carrying capacity (cell number × 106) | 2–3 | 5.3–5.5 | |||||||
| Death rate | 0–1 | 0.1–1 | 0.00–0.19 | ||||||
| Output (differentiation) rate | 0.1–1 | 0.1–0.3 | 0.2–0.25 | 0.5–1.2 | 0.01–0.19 | ||||
| Reflux rate | 0–0.25 | ||||||||
| Static cells (cell number × 105) | 1 | ||||||||
| Old mice | |||||||||
| Entry rate (cell number × 104) | 1–10 | ||||||||
| Proliferation rate | 0.1–1.4 | 0.5–1.2 | |||||||
| Carrying capacity (cell number × 106) | 2.4–3 | 3.5–4 | |||||||
| Death rate | 0–1 | 0.1–1 | 0.00–0.2 | ||||||
| Output (differentiation) rate | 0.1–1 | 0.1–0.3 | 0.3–0.4 | 0.5–1.15 | 0.01–0.2 | ||||
| Reflux rate | 0–0.25 | ||||||||
| Static cells (cell number × 105) | 5 | ||||||||
Subset | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pro-B | Pre-B | Immature B | |||||||
| Resting | Cycling | Cycling | Resting | ||||||
| Young mice | |||||||||
| Entry rate (cell number × 104) | 1–10 | ||||||||
| Proliferation rate | 0.3–1.4 | 0.9–1.3 | |||||||
| Carrying capacity (cell number × 106) | 2–3 | 5.3–5.5 | |||||||
| Death rate | 0–1 | 0.1–1 | 0.00–0.19 | ||||||
| Output (differentiation) rate | 0.1–1 | 0.1–0.3 | 0.2–0.25 | 0.5–1.2 | 0.01–0.19 | ||||
| Reflux rate | 0–0.25 | ||||||||
| Static cells (cell number × 105) | 1 | ||||||||
| Old mice | |||||||||
| Entry rate (cell number × 104) | 1–10 | ||||||||
| Proliferation rate | 0.1–1.4 | 0.5–1.2 | |||||||
| Carrying capacity (cell number × 106) | 2.4–3 | 3.5–4 | |||||||
| Death rate | 0–1 | 0.1–1 | 0.00–0.2 | ||||||
| Output (differentiation) rate | 0.1–1 | 0.1–0.3 | 0.3–0.4 | 0.5–1.15 | 0.01–0.2 | ||||
| Reflux rate | 0–0.25 | ||||||||
| Static cells (cell number × 105) | 5 | ||||||||
All rates are per a time unit representing 6 h. The ranges in bold are those that differed most between young and old mice.
Results
The experimental data of BrdU-labeling kinetics we used here (7) reach much higher fractions of labeling than the data we used in our previous study (23). The two data sets differ because Witte et al. measured over 28 days, which is more than sufficient for the labeling fractions to reach their steady state, while the data in (23) was measured only over 60 h, which was not sufficient for the labeling fractions to reach their steady state. Hence, here we fit our model to the experimental data from the Witte group (7). These data show higher BrdU-labeling kinetics of the pro- and pre-B cell subsets. As a result, a simulation based on the data in (23) brings the steady-state labeling fractions of the immature subset to higher levels than those in the experimental data (7) from the Witte group (Fig. 2). In order to solve this problem, we included in the simulation a subset of static non-dividing, non-dying cells that have the same phenotype and hence are included within the immature subset, following the suggested explanation in (7). The parameter sets for young and old mice which gave the best fit to the experimental data are given in Table 2.
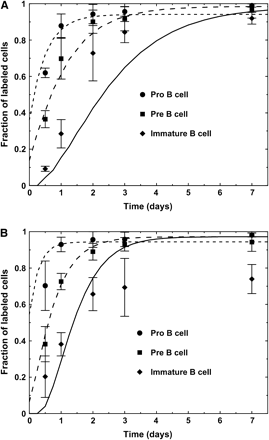
Labeling kinetics obtained by the simulation of the original model, which did not include static cells in the immature compartment. Simulation results (lines) are presented along with the experimental results (symbols with error bars). (A) Young mice. (B) Old mice.
The simulated steady-state total cell numbers at each stage of development in the bone marrow of young and old mice are within the experimentally observed ranges. The simulated B cell populations have reached these steady-state numbers within reasonable times (shown in Fig. 3 for young and old mice), and the steady-state cell numbers for both young and old mice are presented in Table 1.
![Steady-state cell numbers obtained in a simulation of the bone marrow population model which fits well with Witte's data. (A) Young mice, (B) old mice. The parameters used are the following, which are within the ranges given in Table 2; the superscripts y and o denote parameters for young and old mouse bone marrow, respectively, and ‘/tu’ means per simulation time unit (of 6 h): Sy = 50 000 cells/tu, So = 80 000 cells/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(K_{\mathrm{o}}^{\mathrm{y}}\) \end{document} = \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(K_{\mathrm{o}}^{\mathrm{o}}\) \end{document} = 3 × 106 cells, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(K_{\mathrm{e}}^{\mathrm{y}}\) \end{document} = 5.2 × 106 cells, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(K_{\mathrm{e}}^{\mathrm{o}}\) \end{document} = 3.6 × 106 cells, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\mu}}_{\mathrm{o}}^{\mathrm{y}}\) \end{document} = 0.8/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\mu}}_{\mathrm{o}}^{\mathrm{o}}\) \end{document} = 0.7/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{or}}^{\mathrm{y}}\) \end{document} = 0.95/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{or}}^{\mathrm{o}}\) \end{document} = 1.0/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\gamma}}_{\mathrm{o}}^{\mathrm{y}}\) \end{document} = \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\gamma}}_{\mathrm{o}}^{\mathrm{o}}\) \end{document} = 1.2/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{oc}}^{\mathrm{y}}\) \end{document} = 0.2/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{oc}}^{\mathrm{o}}\) \end{document} = 0.3/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\gamma}}_{\mathrm{e}}^{\mathrm{y}}\) \end{document} = 1.3/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\gamma}}_{\mathrm{e}}^{\mathrm{o}}\) \end{document} = 0.5/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{ec}}^{\mathrm{y}}\) \end{document} = 0.2/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{ec}}^{\mathrm{o}}\) \end{document} = 0.3/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\mu}}_{\mathrm{e}}^{\mathrm{y}}\) \end{document} = 0.7/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\mu}}_{\mathrm{e}}^{\mathrm{o}}\) \end{document} = 0.1/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{er}}^{\mathrm{y}}\) \end{document} = 1.0/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{er}}^{\mathrm{o}}\) \end{document} = 1.1/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\mu}}_{\mathrm{i}}^{\mathrm{y}}\) \end{document} = 0.08/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\mu}}_{\mathrm{i}}^{\mathrm{o}}\) \end{document} = 0.2/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{i}}^{\mathrm{y}}\) \end{document} = 0.1/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{i}}^{\mathrm{o}}\) \end{document} = 0.2/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{r}}^{\mathrm{y}}\) \end{document} = 0.05/tu, \batchmode \documentclass[fleqn,10pt,legalpaper]{article} \usepackage{amssymb} \usepackage{amsfonts} \usepackage{amsmath} \pagestyle{empty} \begin{document} \(\mathrm{{\delta}}_{\mathrm{r}}^{\mathrm{o}}\) \end{document} = 0.03/tu.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/intimm/18/1/10.1093/intimm/dxh346/2/m_intimmdxh346f03_lw.gif?Expires=1716503112&Signature=4z11a30FoWZyAq17-44Qzv7B-s-BM7ZYUi~NVJEkLUht5l3zBcG0umoJRW1BVocpE0PP31ovH3Wgs9jSSxmEb0oS9n4tWKwR8Yfiq~Z3xnjAbtdf0WvWr-UDlXyZ31ew3kRb6ZTKro9udgJctruR8nPQc8cnr8BzMIxsVNib1EFUiLWOlIsn5Uwhc1Mwm3QhRQvJEnTmZCMkXB0QnJJ7mU6~miR2NIpJuJs0kJ~hn4txvi8txapP-IbbOM07xy76m2exmzq03TYXwRtyh16~CZBZdcebpdIokKc67evGt5G~tiY8FFi4f3lepGbQEjZWtoVVBKSHaZ2BErX93t9lRA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Steady-state cell numbers obtained in a simulation of the bone marrow population model which fits well with Witte's data. (A) Young mice, (B) old mice. The parameters used are the following, which are within the ranges given in Table 2; the superscripts y and o denote parameters for young and old mouse bone marrow, respectively, and ‘/tu’ means per simulation time unit (of 6 h): Sy = 50 000 cells/tu, So = 80 000 cells/tu,
Simulated labeling kinetics obtained with our best-fit set of parameters are given in Fig. 4(A and B), and the post-labeling decay of BrdU-labeled cell fractions are given in Fig. 4(C and D).
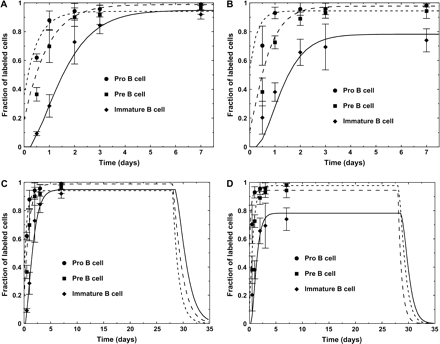
(A and B) Labeling kinetics of young (A) and old (B) mice obtained by the simulation of the model which fits to the experimental results. The parameters used are the ones given in Table 2. Simulation results (lines) are presented along with the experimental results (symbols with error bars). (C and D) Simulation of the kinetics in young (C) and old (D) mice, extended up to 35 days after the start of labeling, to show the predicted course of labeling decay.
From our simulation results it is evident that the number of hypothetical static cells, which we had to include in the immature B cell subset, is higher in old mice than in young mice (Table 2). The parameters that change most significantly with age are the differentiation rate from cycling pre-B cells to resting pre-B cells, which increases by ∼50%, and the pre-B cell-carrying capacity, which is reduced (Table 2 and Fig. 5A). Cell proliferation rates are in overlapping ranges for both young and old bone marrow B cells. However, the range of proliferation rates for young mice includes higher values than the range of proliferation rates for old mice (Fig. 5B and C).
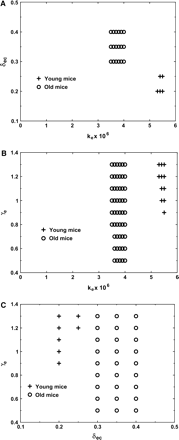
(A) The different pairs of Ke and δec values, (B) the different pairs of Ke and γe values and (C) the different pairs of δec and γe values, which gave the best fit in old and young mice. The regions covered by each parameter pair are actually contiguous.
Moreover, old mice have a lower fraction of proliferating cells in the pre-B cell subset in each generation as more cells differentiate to the resting state at each generation. Young mice also have a higher pre-B cell-carrying capacity, which allows this subset to expand to higher cell numbers. Note that the ranges are given here for each parameter separately, hence not all values in the range given for one parameter necessarily give a good fit when used with all values in the ranges of other parameters. This is clearly illustrated in Fig. 5.
Discussion
The goal of this study was to elucidate the effects of aging on bone marrow B cell population dynamics. We used mathematical modeling to predict the outcome of the different possible effects, such as slowing of division and/or differentiation in each compartment, changes in the carrying capacity or increase in the rates of cell death, and then compared these predictions to experimental data, to find the most plausible effects. Mathematical modeling was essential, as BrdU-labeling studies alone cannot distinguish between these possibilities, nor do linear estimates of the decrease in cell production, based on the labeling curve, give the correct estimates. The results point at three main differences between the parameters governing developing B cell population dynamics in young and old mice: the rate of transition from cycling pre-B cells to resting pre-B cells increases by ∼50%, the carrying capacity of the pre-B compartment is reduced by ∼30% and the fractions of static cells, which must be included in the immature B cell subset in order to explain why labeling does not reach 100% even in a long-term steady state, are increased 5-fold. These differences are discussed below.
The simulations show that there is a larger rate of transition from proliferating to resting cells in old bone marrow pre-B cells, as represented by the parameter δec attaining higher values for young mice. On the single-cell level, after each division, the daughter cells can either continue to proliferate or stop proliferating. As the animal gets older, the probability that any given cell will stop proliferating may increase, due to the accumulation of genetic defects. This would lead to an increase of the δec parameter on the population level (Fig. 6). This assumption is in line with the conclusions of previous studies (27–31), in which data on T cells from old or young mouse thymus growing in fetal thymic organ cultures were mathematically analyzed. In those studies, it was concluded that old donor-derived thymocytes probably perform a lower number of sequential divisions prior to final differentiation than young donor-derived thymocytes. Thus, we suggest that the transition from cycling cells to resting cells in both T and B lymphocytes is accelerated due to decreased capacity to resume proliferation in cells from old mice, and not a decreased rate of proliferation in the cycling compartment.
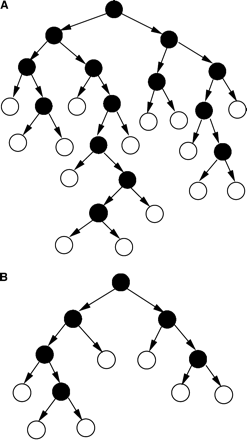
A cell lineage, starting from one-seeded cell. A cell in the lineage either divides or moves to the resting phase. Once a cell stops dividing, it remains in the resting phase. (A) Young, (B) old mice. Filled circles denote cycling cells, empty circles denote resting cells.
We have also shown here that the carrying capacity of the pre-B cell sub-population is lower in old mice than in young mice. This suggests that the space and resources, such as contact with the bone marrow stroma and the availability of growth factors and nutrients, are lower in old animals. Our results thus support previous studies that suggested that the production of and/or the response to IL-7 was impaired in old mice (6, 10, 19–22). Thus, the model solves the apparent contradiction between the two experimental studies (7, 8) by showing that production of B cells in the bone marrow is indeed decreased in the pre-B stage, and the following developmental compartments do not quite compensate for this decrease. However, the decreased numbers of cells produced are lower in older mice due to changes in differentiation rates and the carrying capacity in the pre-B compartment, while the proliferation rate itself is unchanged.
Johnson et al. (7) proposed that the static cells included in the immature compartment occur due to increased longevity within the immature compartment, such that their incidence increases with age (7), and estimated that between 20–25% of the immature cell population belongs to this subset. Our analysis agrees with this suggestion, as we find that the magnitude of this subset is ∼4% of young and ∼23% of old immature B cells, and the increase in terms of absolute cell numbers is ∼5-fold. However, since there are no specific data on this cell subset, whose existence could only be inferred from the data on other cell populations, we could not evaluate the life span of these cells, and simply assumed that their numbers are in steady state. Cell longevity may increase due to defects in the apoptotic signaling pathways, and hence lower numbers of cells die by apoptosis. This age-related defect would lead to fewer immature B cells remaining sensitive to selection against self-antigen, which would in turn increase the production of both autoreactive and functionally defective B cells. It is interesting that, despite the increase in the number of static cells in the immature B cell compartment, there is no change in the death rate, differentiation rate and reflux rate of the non-static cells in this compartment (Table 1). This supports the suggestion in (7) that the production potential of the non-static immature B cell population in aged mice remains high, although the population itself is smaller. This contrasts with the conclusions of Kline et al. (8), who argue that the production rate of new IgM+ cells in the bone marrow is drastically diminished.
This study exemplifies the use of mathematical modeling and computer simulation, not only in analyzing experimental results but also in suggesting lines of further investigation. In this case, modeling has suggested that measurements will focus on the changes in the mechanisms of signaling and cycling as a function of age.
Transmitting editor: I. Pecht
The authors are grateful to P. Witte for sharing the experimental data, to M. Cancro for critical reading of the manuscript and to H. Edelman for help in the manuscript preparation. This work was supported in part by Israel Science Foundation grant number 759/01-1 and The Yigal Alon Fellowship (to R.M.), and indirectly by a Human Frontiers Science Program grant and a Swedish Foundation for Strategic Research grant funding the Strategic Research Center for studies on Integrative Recognition in the Immune System, Karolinska Institute, Stockholm, Sweden (supporting R.M.).
References
Douek, D. C., McFarland, R. D., Keiser, P. H. et al.
LeMaoult, J., Messaoudi, I., Manavalan, J. S. et al.
Szabo, P., Zhao, K., Kirman, I. et al.
Zhao, K. S., Wang, Y. F., Gueret, R. and Weksler, M. E.
Weksler, M. E. and Goodhardt, M.
Miller, J. P. and Allman, D.
Johnson, K. M., Owen, K. and Witte, P. L.
Kline, G. H., Hayden, T. A. and Klinman, N. R.
Stephan, R. P., Sanders, V. M. and Witte, P. L.
Kirman, I., Zhao, K., Wang, Y., Szabo, P., Telford, W. and Weksler, M. E.
Li, F., Jin, F., Freitas, A., Szabo, P. and Weksier, M. E.
Cancro, M. P. and Smith, S. H.
Szabo, P., Shen, S., Telford, W. and Weksler, M. E.
Labrie, J. E., III, Sah, A. P., Allman, D. M., Cancro, M. P. and Gerstein, R. M.
Lanvin, O., Guglielmi, P., Fuentes, V. C. et al.
Uang, P. I., Morefield, S., Liu, C. Y., Chen, S., Harlan, J. M. and Willerford, D. M.
Nagaoka, H., Takahashi, Y., Hayashi, R. et al.
Grillot, D. A., Merino, R., Pena, J. C. et al.
Stephan, R. P, Reilly, C. R. and Witte, P. L.
Stephan, R. P., Lill-Elghanian, D. A. and Witte, P. L.
Corcoran, A. E., Riddell, A., Krooshoop, D. and Venkitaraman, A. R.
Sprent, J., Schaefer, M., Hurd, C. and Ron, Y.
Mehr, R., Shahaf, G., Sah, A. and Cancro, M.
Ubezio, P., Tagliabue, G., Schechter, B. and Agur, Z.
Rolink, A., Winkler, T. H., Melchers, F. and Andersson, J.
Agenes, F., Rosado, M. and Freitas, A. A.
Mehr, R., Abel, L., Ubezio, P., Globerson, A. and Agur, Z.
Mehr, R., Globerson, A. and Perelson, A.
Mehr, R., Perelson, A., Fridkis-Hareli, M. and Globerson, A.
Mehr, R., Perelson, A., Fridkis-Hareli, M. and Globerson, A.
Mehr, R., Perelson, A. S., Fridkis-Hareli, M. and Globerson, A.
Mehr, R., Ubezio, P., Kukulansky, T. and Globerson, A.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}