 Mattias Carlsten
Mattias Carlsten Richard W. Childs
Richard W. Childs- Hematology Branch, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD, USA
Given their rapid and efficient capacity to recognize and kill tumor cells, natural killer (NK) cells represent a unique immune cell to genetically reprogram in an effort to improve the outcome of cell-based cancer immunotherapy. However, technical and biological challenges associated with gene delivery into NK cells have significantly tempered this approach. Recent advances in viral transduction and electroporation have now allowed detailed characterization of genetically modified NK cells and provided a better understanding for how these cells can be utilized in the clinic to optimize their capacity to induce tumor regression in vivo. Improving NK cell persistence in vivo via autocrine IL-2 and IL-15 stimulation, enhancing tumor targeting by silencing inhibitory NK cell receptors such as NKG2A, and redirecting tumor killing via chimeric antigen receptors, all represent approaches that hold promise in preclinical studies. This review focuses on available methods for genetic reprograming of NK cells and the advantages and challenges associated with each method. It also gives an overview of strategies for genetic reprograming of NK cells that have been evaluated to date and an outlook on how these strategies may be best utilized in clinical protocols. With the recent advances in our understanding of the complex biological networks that regulate the ability of NK cells to target and kill tumors in vivo, we foresee genetic engineering as an obligatory pathway required to exploit the full potential of NK-cell based immunotherapy in the clinic.
Introduction
Natural killer (NK) cells are immune cells primarily found in the blood, liver, spleen, bone marrow, and to a lesser extent, in lymph nodes (1). They were initially identified based on their ability to lyse tumor cells without a need for priming (2–5). NK cells are now known to play an important role in host immunity against both cancers and certain viral infections (6–8).
NK cells can mediate cytotoxicity via multiple distinct mechanisms. Degranulation is the most studied cytotoxicity pathway, where NK cells release cytotoxic granules upon contact with the target. Cytotoxicity via this pathway is dictated by a balance of signals from an array of germline encoded activation and inhibitory cell surface receptors. Most activation receptors need simultaneous co-stimulation by other activation receptors to trigger NK cell cytotoxicity (9). One exception from this rule is the Fc receptor CD16, which alone can trigger NK cell degranulation against antibody-coated target cells via antibody-dependent cellular cytotoxicity (ADCC) (9). Other routes by which NK cells can kill targets are the death receptor pathways TRAIL/TRAIL-R and Fas/FasL. Instead of triggering release of cytotoxic granules, death receptor pathways prompt apoptosis via caspase activation in target cells.
More than a decade has passed since initial reports established the anti-tumor potential of NK cells in patients with cancer. These studies showed that haplo-identical donor NK cells could prevent relapse in acute myeloid leukemia (AML) following hematopoietic stem cell transplantation (HSCT) and that adoptively infused mature donor NK cells could induce remission in AML patients (6, 10). Despite this revelation, doubts remain about the true therapeutic potential of NK cells in cancer immunotherapy. In contrast to therapy utilizing T cells, enthusiasm for NK cell-based immunotherapy has been tempered by uncertainties about their in vivo persistence, and doubts regarding their ability to migrate to tumor tissues following adoptive infusions. Although recent data have shown CMV reactivation reduces the risk for AML relapse following HSCT (11) potentially caused by CMV-induced NK cells cross-reacting with AML cells, NK cells, unlike T-cells, lack antigen specificity, further tempering enthusiasm for their use as immune effectors in cellular therapy.
Genetic manipulation of NK cells to improve their persistence, cytotoxicity, tumor targeting capacity, and ability to home to disease sites in vivo holds potential to advance the efficacy of NK cell-based cancer immunotherapy. However, until relatively recently, the genetic manipulation of NK cells has proven to be challenging. Viral transduction, successfully used for T cells, has been associated with low levels of transgene expression and unfavorable effects on cell viability when used with NK cells. Recent optimization of viral transduction and the establishment of electroporation technologies for efficient gene transfection have revived the enthusiasm for studies evaluating genetic modification of NK cells. Investigators around the world are now exploring the potential of multiple different NK cell modalities to genetically reprogram with the overall aim of further improving upon their capacity to kill tumors in cancer patients. One example of how this technique can be utilized is to introduce genes into NK cells coding for gamma-cytokines (IL-2 and IL-15) to induce independence from the obligate need of exogenous cytokines for proper in vivo persistence and expansion post infusion. This and similar strategies may further improve the efficacy of NK cell-based immunotherapy, as tumor regression following adoptive NK cell infusions in AML patients has been reported to be dependent on their ability to expand in vivo (6), while being limited by regulatory T cells also mobilized following exogenous cytokine administration (12, 13). The introduction of chimeric antigen receptors (CARs) and the down-regulation of inhibitory NK cell receptors such as NKG2A are additional examples of specific genetic manipulations that can be utilized to improve the outcome of adoptive NK cell immunotherapy.
Given their rapid and efficient method of recognizing tumor cells, NK cells represent a unique immune cell to genetically reprogram in an effort to improve the outcome of cell-based cancer immunotherapy. This review focuses on methods for introducing transgenes into NK cells and the advantages and limitations of such strategies. It also gives an overview of strategies for genetic reprograming of NK cells that have been evaluated to date and an outlook on how these specific strategies may be best utilized in clinic to maximize the anti-tumor potential of NK-cell based immunotherapy.
Methods and Challenges with Genetic Manipulation of NK Cells: Viral Transduction Versus Transfection
Genetic manipulation of T cells has successfully been used in both preclinical and clinical research (14). In contrast, studies on genetically engineered NK cells have historically been limited by poor efficacy of transgene delivery and substantial procedure-associated NK cell apoptosis. In this section, we discuss available approaches for gene delivery into NK cells, characterizing how each approach developed over time while highlighting the positive and negative aspects of each method (Box 1).

Box 1. Pros and Cons for Methods of Genetic Modification of NK Cells.
Viral Transduction
The reduced efficacy of viral transduction of NK cells compared to T cells may in part be related to the innate properties that characterize NK cells. Innate immune receptors, such as pattern recognition receptors that recognize foreign genomic material, are likely involved in triggering apoptosis of NK cells following viral transduction (15). Best results from studies of viral transduction of NK cells have been achieved using either NK cell lines or primary NK cells that have undergone expansion ex vivo (Table 1). In contrast, viral transduction of primary resting human NK cells typically results in substantially lower transduction efficiencies. Most studies on viral transduction of NK cells have utilized retro- and lentiviral vectors. Although adenoviral- and vaccinia virus vectors have been utilized for transduction of NK cells, their use has been limited and they will not be discussed further in this review.
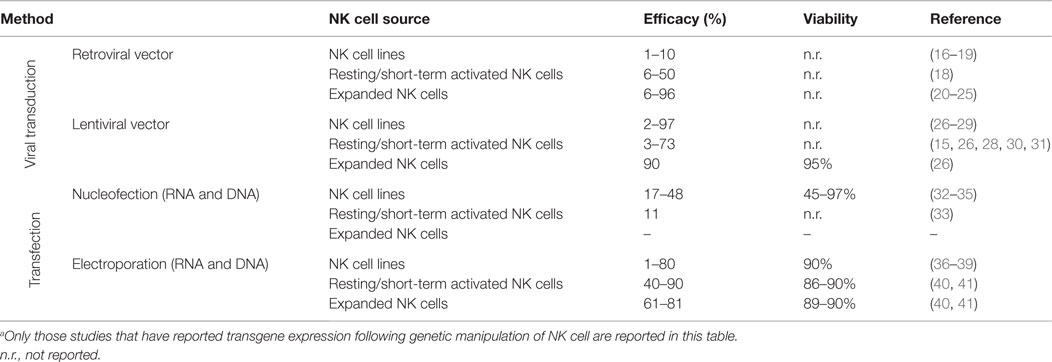
Table 1. Overview of techniques used to genetically modify NK cells with reported gene delivery efficacies and effect on cell viability.a
Retroviral vectors were the first viral vectors used to genetically modify NK cells. The first report on retroviral transduction of NK cells was published in the late 1990s and focused on genetic manipulation of the NK cell line NK-92 (16). This study reported a transduction efficacy of only 2–3%. Optimization of retroviral transduction approaches over the past decade has resulted in higher transduction efficiencies, especially when used with human NK cells that have undergone ex vivo expansion (Table 1). A recent report showed that retroviral transduction of ex vivo expanded NK cells with genes coding for either IL-15 or membrane bound IL-15 (mbIL-15) resulted in an average 69 and 71% transduction efficiency, respectively (25). Although retroviral transduction of NK cells has been reported to not alter the function, phenotype, and proliferative capacity of NK cells (20, 23), their viability following retroviral transduction has rarely been reported. A significant deleterious impact on the viability of primary NK cells undergoing retroviral transduction may preclude utilizing this approach in a clinical setting. Further, retroviral transduction also requires active cell division, impeding the use of this method with primary non-activated NK cells. This limitation is less important when retroviral transduction is utilized with NK cell lines such as NK-92 that have continuous and unlimited proliferation capacity. However, as discussed later in this review, it is important to note that this NK cell line does have phenotypic and function differences from primary human NK cells, which may have therapeutic implications for clinical therapy.
Lentiviral Vectors
More recently, studies evaluating transduction of NK cells using lentiviral vectors have been pursued. In contrast to transduction with retro- and adenoviral vectors, lentiviral vectors can incorporate transgenes into the genome of non-dividing cells. Further, lentiviral vectors allow for gene modification of NK cells without altering their phenotypic and functional properties as occurs following stimulation with i.e., cytokines. The first report on the successful use of lentiviral vectors for genetic modification of NK cells was performed in primary murine NK cells (42), with subsequent studies establishing that lentiviral transduction of human NK cells could also be achieved (Table 1). Although most studies have reported lentiviral transduction of NK cell lines with efficiencies of 15–40% (27, 28), the efficiency highly varies from only a few percent to nearly 100%, and in some cases, multiple rounds of transduction are required (26, 29). Recent data indicate that transduction efficiencies of primary human NK cells can be increased by drug-induced inhibition of intracellular innate immune receptors in NK cells (15). Unfortunately, and similar to studies utilizing retroviral transduction, the viability of NK cells after lentiviral transduction has rarely been reported. Using an optimized protocol, our lab has achieved a maximum transgene expression in up to 60% of ex vivo expanded NK cells 3 days after lentiviral transduction with GFP without incurring any deleterious effects on NK cell viability, phenotype, or function (Personal communication, R. Childs).
In summary, viral transduction of NK cells results in variable transduction efficacies and may require multiple rounds of transduction and/or post transduction cell enrichment to achieve acceptable transgene expression. Further, viral associated cell death and the need for post-transduction enrichment may compromise the clinical utility of this approach. Finally, although the risk may be low, the possibility of viral-induced insertional mutagenesis and immunogenicity (43, 44) occurring post transduction must be considered when utilizing this methodology in the clinic. Nevertheless, viral transduction of NK cells does achieve stable transgene expression which, depending on how the NK cell is being genetically modified, might be required to induce a durable and long-term clinical response.
Transfection
Compared to viral transduction, transfection of NK cells appears to be associated with lower degrees of apoptosis, less inter-individual and inter-experimental variability, with transgene delivery efficiency being completely independent of cellular division. In most cases, this approach results in a more rapid albeit transient expression of the transgene as compared to viral transduction where genes must first be incorporated into the cellular genome before expression can occur. Gene transfer using transfection can be achieved by either electroporation (including nucleofection) or lipofection. Since the latter has been used only in a few studies (45), this review will focus on strategies utilizing the electroporation approach.
Electroporation is a method where genetic material is delivered into cells following a short electric pulse that temporarily induces small pores in the cell membrane, allowing charged molecules such as DNA and RNA to move into the cell. This technology was first used with NK cell lines in the late 1990s (32, 36–38, 46, 47) and more recently has been used to genetically manipulate primary NK cells to express CARs (35, 39, 48) or cytokines for autocrine growth stimulation (49). With technological advances and the use of mRNA instead of cDNA, transfection efficiencies have increased dramatically, reaching up to 90% or more while having only a minimal deleterious effect on cell viability (Table 1). Remarkably, using mRNA electroporation, transfection efficiencies of 80–90% can be achieved in not only ex vivo expanded cells but also in primary resting (non-cytokine activated) human NK cells (40). Despite this remarkable advance, a detailed characterization on the effects of electroporation on the phenotype, function, and proliferative capacity of NK cells following electroporation has yet to be published.
As electroporation does not involve viral vectors, its use in the preclinical and clinical setting is associated with less regulatory issues. Also, as indicated above, electroporation most often leads to transient transgene expression, which may be viewed favorably from a safety perspective when new transgenes with unknown potential toxicities are being explored in early clinical trials. Regimens that use DNA electroporation technology have been employed to generate stable transgene expressing cells. Although the efficacy of this approach is typically lower than that achieved with viral transduction, it may be improved if combined with targeted integration techniques that avoid random integration in inactive heterochromatin regions. Such strategies also reduce the risk for off-target effects, including gene silencing due to random integration in active genes and integration in hot-spots that may trigger malignant transformation. With advantages in design of guiding RNAs and by having better on-target specificity compared to other gene editing technologies such as Zink-Finger nuclease (ZFN) and the transcription activator-like effector nucleases (TALEN) technologies, the recently developed clustered regularly interspaced short palindromic repeats (CRISPR) technique has rapidly become a popular tool for targeted gene integration (50). The CRISPR/Cas9 system induces permanent modifications at specific sites of the genome via double-strand breaks (DSBs), and can be used to integrate new genes at specific sites via homology-directed recombination (50). Although only moderate degrees of genome integration are currently being achieved with this technique today, the CRISPR/Cas9 system could be used to produce stably transduced NK cells by gene editing of primary NK cells prior to their ex vivo expansion.
Gene Modification Strategies Aimed at Improving the Efficacy of NK Cell-Based Cancer Immunotherapy
With new advances in the field, genetic manipulation of NK cells has opened up possibilities to study many different pathways involved in NK cell tumor targeting and the ability to genetically modify NK cells to improve their tumor cytotoxicity. Here, we will discuss reported gene modification strategies that can improve in vivo persistence and expansion, tumor tissue migration, and the tumor targeting capacity of adoptively infused NK cells (Figure 1, Table 2 and Box 2).
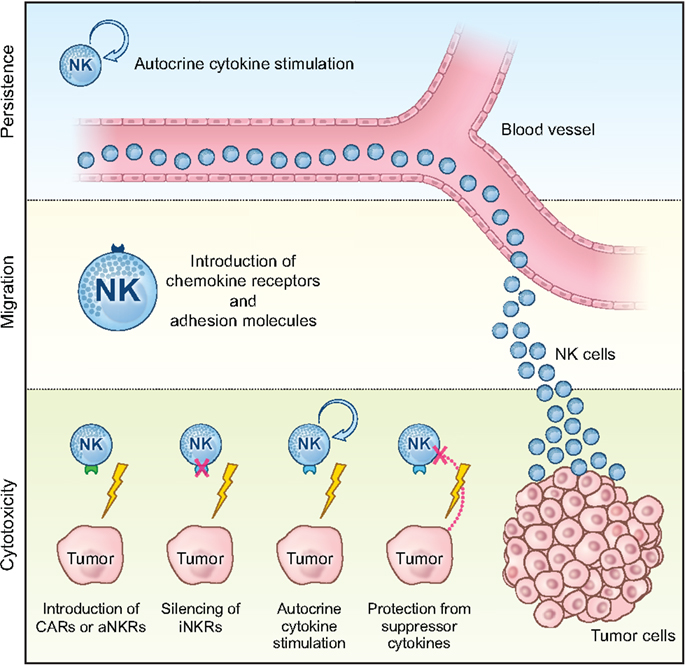
Figure 1. Schematic overview of how genetic manipulation can be can be used to improve the efficacy of NK cell-based cancer immunotherapy in the clinic. Genetic engineering of NK cells to promote persistence and expansion by autocrine cytokine stimulation, migration to the tumor tissue via introduction of receptors involved in cellular homing (i.e., chemokine receptors and adhesion molecules), as well as bolstering their anti-tumor cytotoxicity via introduction of CARs or activating NK cell receptors (aNKRs) or via silencing of inhibitory NK cell receptors (iNKRs), protection from suppressive cytokines in the tumor environment, and boosted function via autocrine cytokine stimulation.
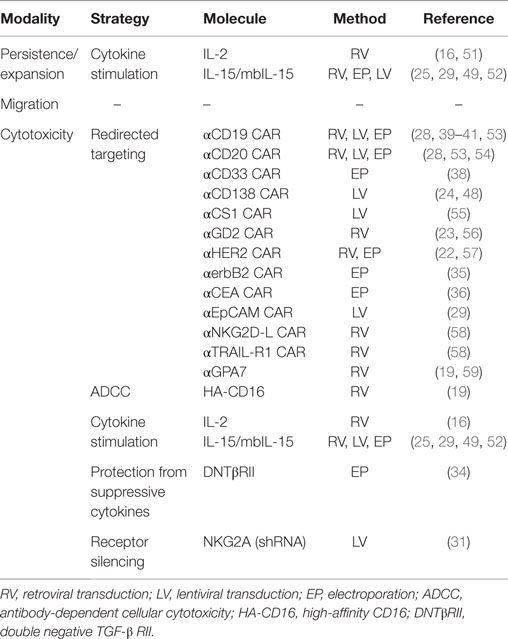
Table 2. Overview of strategies evaluated for improving the anti-tumor efficacy of primary human NK cells and NK cell lines in vitro and in preclinical animal models.

Box 2. Examples of NK Cell Modalities to Gene Manipulate for Improved Clinical Efficacy.
Strategies to Improve Persistence and Expansion of Infused NK Cells
In vivo persistence and expansion of infused NK cells have been shown critical for inducing tumor regression following adoptive NK cell infusion (6). Using retroviral transduction of the IL-2 gene into NK-92 cells, Nagashima et al. were able to propagate this NK cell line for up to 5 months in vitro without the addition of exogenous cytokines (16). Further, IL-2-expressing NK-92 cells where shown to also have enhanced tumor cytotoxicity compared to non-transduced parental NK-92 cells that were stimulated with exogenous IL-2. In line with these in vitro findings, these genetically modified cells showed improved in vivo persistence and anti-tumor responses when infused into tumor-bearing mice. Similar data with IL-2 gene delivery in expanded NK cells were reported by Konstantinidis et al. (51). As observed with IL-2 transduced NK-92 cells, retroviral transduction of ex vivo expanded NK cells with the mbIL15 gene also dramatically increased their survival in vitro; median cell recovery was 85% for mbIL-15 NK cells after 7 days in culture without IL-2, whereas mock-transduced NK cells were essentially undetectable (25). Hence, the strategy of introducing genes coding for gamma-cytokines to improve in vivo NK cell persistence and expansion following infusion independent of exogenous cytokine administration appears promising.
Strategies to Enhance Migration of Infused NK Cells
Proper tumor tissue homing of infused NK cells is a prerequisite for their ability to induce tumor regression. However, studies characterizing the in vivo migration capacity of adoptively infused NK cells have been largely overlooked (60). Recent evidence suggests non-expanded and expanded NK cells have different migration patterns when infused into animal models (61). Moreover, using trogocytosis to transfer premade cell surface molecules from a feeder cell line to NK cells, Somanshi et al. have shown that migration of infused NK cells can be redirected by equipping them with the lymph node homing receptor CCR7 (62). Despite these data, no study has so far used gene modification techniques to actively direct infused NK cells to selected organs. Based on data from Somanshi et al., we have been able to use mRNA transfection to genetically engineer NK cells with the CCR7 receptor to improve their migration toward one of its ligands CCL19 (Carlsten M., Manuscript in preparation, April 2015). Other strategies may involve utilizing chemokine receptors, such as CXCR3 to improve NK cell migration to inflamed tissues, such as those infiltrated with metastatic tumors (63).
Strategies to Increase Tumor Cytotoxicity by Infused NK Cells
The majority of reports on expression of transgenes in NK cells have characterized the effects of CARs in NK cell lines, expanded NK cells, and primary non-expanded NK cells (Table 2). CARs are engineered receptors that have the extracellular specificity of an antibody combined with potent intracellular signaling adaptors such as CD3ζ, CD28 and/or 4-1BB. Importantly, these receptors do not require stimulation through co-receptors to trigger robust anti-tumor cytotoxicity. The recent breakthrough success of anti-CD19 CAR T cell therapy in patients with B cell malignancies has stimulated the research community to develop and investigate a wide array of CARs against multiple different epitopes expressed on numerous tumor types (64). Several of these CARs have been explored in NK cells (Table 2). CD19 and CD20 specific CARs against B cell malignancies (39–41, 53, 54), and CARs targeting CD33 on leukemia cells (38), CS1 and CD138 on myeloma cells (24, 48, 55), GD2 on neuroblastoma cells (23, 56), Her2/Neu and erbB2 on breast cancer cells (22, 35), carcinoembryonic antigen (CEA) on colon cancers (36), EpCAM on epithelial tumors (29), GPA7 on melanoma (59), NKG2D ligand on leukemia and solid tumors, and TRAIL-R1 on various tumor targets (58) have all been shown to have the capacity to redirect NK cell cytotoxicity against their target antigens. The majority of these studies have used viral vectors to transduce CARs into the NK cell, albeit electroporation has also been used in a few studies (Table 2).
Based on clinical data showing superior response rates in rituximab-treated lymphoma patients homozygous for the high-affinity CD16-158V polymorphism (HA-CD16) compared to those who carry the low-affinity CD16-158F (LA-CD16) polymorphism (65, 66), several groups have recently addressed whether introduction of the HA-CD16 gene into NK cells lacking this polymorphism can be used as a strategy to augment ADCC against tumors. This approach has appeal as only a minority of patients is homozygous for HA-CD16 (67). Moreover, in contrast to CAR NK cells, infusions of NK cells genetically modified to express HA-CD16 may be used to improve the outcome of virtually any malignancy for which there is an FDA approved IgG1 antibody, without the expectation for any severe off target side-effects. In vitro experiments conducted by Binyamin and colleagues showed significantly improved cytotoxicity against a rituximab-coated B lymphoma cell line following stable transduction of the CD16 negative NK-92 cell line with HA-CD16 compared to NK-92 cells were equipped with LA-CD16 (19). Recently, our group explored a similar approach, where ex vivo expanded NK cells from CD16-158F/F (LA-CD16) donors were found to have substantially augmented ADCC following electroporation with mRNA coding for the HA-CD16 (68). These data suggest the addition of the HA-CD16 gene to patient NK cells that already express endogenous CD16 can be used to augment their ability to induce ADCC, and that this approach could be used as a strategy to improve the efficacy of antibody-based therapies for cancer patients.
Introduction of genes that render NK cells insensitive to suppressive cytokines such as TGF-β, thereby preserving their cytotoxicity, has also been studied. Yang et al. generated an NK-92 cell line resistant to the suppressive effects of TGF-β by genetically modifying them to express the dominant negative mutant form of TGF-β type II receptor (DNTβRII) on their surface (34). Adoptive transfer of these TGF-β insensitive NK-92 cells in lung cancer-bearing mice was associated with increased levels of IFN-γ released from the infused cells and resulted in increased survival rates compared to mice treated with wild-type NK-92.
Genetic reprograming of NK cells may also be directed to achieve specific protein silencing with the aim of improving tumor targeting by circumventing NK cell inhibitory signals induced upon interaction with tumor cells. Initial studies have focused on the use of shRNA technology for this purpose. In this context, shRNAs expressed inside cells are processed by the Dicer endonuclease complex to generate double-stranded small interfering RNAs that prevent translation of their target mRNAs (69), shRNAs have been used successfully to knock-down expression of the HLA-E-binding inhibitory NK cell receptor NKG2A (31). Using an inducible vector in IL-2 activated NK cells, Figueiredo et al. observed a 40% increased killing capacity against the HLA-E expressing cell line K562 HLA-E. Using a similar approach with the NK cell line NKL, our group observed increased killing capacity of HLA-E expressing 721.221 cells in vitro and in a preclinical mouse model (70). Further details on protocols for shRNA-mediated protein silencing in NK cells can be found in Purdy et al. (71). To date, studies utilizing CRISPR, ZFN, or TALEN to genetically modify NK cells to silence their inhibitory receptors for the same purpose of enhancing the anti-tumor capacity of NK cells have not yet been reported.
In conclusion, an array of gene modification strategies for NK cells has now been reported. Several of them hold promise for improving clinical responses of NK cell-based cancer immunotherapy. However, to date, few have been translated into clinical studies. The following section will discuss how these strategies can be incorporated in clinical NK cell cancer immunotherapy.
Considerations for the Development of Clinical Protocols using Genetically Engineered NK Cells
Challenges associated with genetic manipulation of NK cells have significantly delayed the debut of this strategy in clinical cancer therapy. While recently initiated trials (NCT00995137 and NCT01974479) exploiting the role for CAR19-expressing ex vivo expanded NK cells in patients with B cell malignancies will give us a first insight into the potential of this approach; further optimization of clinical compliant methods for genetic modifications of NK cells is needed to exploit the full clinical potential of this approach. Moreover, additional research on the multiple aspects of NK cell tumor targeting that could be modified with this technique is needed. Although clinical responses following infusion of NK cells may be further improved by simply augmenting their tumor targeting capacity, studies evaluating the potential of this technology to improve the persistence of infused cells as well as avenues to promote proper NK cell migration and homing to the tumor tissue are also warranted (Figure 1).
Genetic engineering of NK cells to make them cytokine independent and thereby improve persistence, while boosting their cytotoxic capacity, may be one avenue to further explore. The advantage with this approach would be that exogenous cytokines would be unnecessary following NK cell infusion, which may reduce the risk of mobilizing regulatory T cells that directly suppress NK cell cytotoxicity (13). Challenges with taking this approach to a clinical context include the risk of inducing a cytokine release syndrome due to massive and unregulated NK cell proliferation. This approach also comes with the potential risk of inducing malignant transformation of the NK cells due to permanent autocrine growth stimulation, as have been observed for IL-2 engineered T cells (72). However, such scenarios may be avoided if genes coding for IL-2 or IL-15 are only temporarily introduced via mRNA electroporation of NK cells. Should stable transgene expression be required to induce proper tumor regression, an alternative strategy to prevent runaway NK cell proliferation would be to introduce an inducible suicide gene in the modified cells (73).
Migration to the tumor tissue is another aspect governing proper tumor targeting. This aspect has been largely overlooked and could potentially improve clinical outcome if infused NK cells are redirected to the tumor site instead of circulating non-specifically into mostly non-tumor-bearing tissues. No studies aimed at improving the in vivo homing of infused gene engineered NK cells have yet been published.
As discussed above, numerous strategies for redirecting or boosting NK cell tumor killing in vitro have been explored. Introduction of CARs represent the most studied and developed approach that has recently reached clinical evaluation (Table 2). Expression of the high-affinity CD16 may soon also be tested in a clinical setting as this approach can be combined with already clinically available monoclonal antibodies that target an array of antigens expressed on a variety of different tumor types. Bolstering NK cell cytotoxicity via autocrine cytokine stimulation or via silencing of inhibitory NK cell receptors will likely require additional evaluation in preclinical animal models before they can be incorporated in clinical protocols. Once all these strategies are fully characterized pre-clinically, they may be combined to further improve the full anti-tumor potential of adoptively transferred NK cells. For instance, introduction of a CAR while simultaneously silencing the NKG2A inhibitory receptor may represent one such future approach. One can also consider adding autocrine cytokine stimulation to further improve cytotoxicity while simultaneously supporting their in vivo persistence. As NK cell degranulation is regulated by a balance of activating and inhibitory signals from well-defined cell surface receptors, it may also be possible to add CARs or other activation receptors together with selected receptors that mediate inhibition via ligands that are expressed on normal tissues (and not tumor cells), thereby giving genetically reprogramed NK cells an additional layer of target specificity. However, many additional preclinical studies will be needed before these approaches can reach clinic.
The choice of method for genetic reprograming of NK cells is another important factor that needs to be considered when taking genetically engineered NK cells to clinical evaluation. Viral transduction has the advantage of stable expression; however, as mentioned above, viral transduction of NK cells, especially primary cells, does not always lead to a satisfactory level of transgene expression and may require multiple rounds of transduction followed by selection of transgene positive cells. Moreover, proper expression of transgenes induced by viral transduction can take days, which may be of disadvantage since the lifespan of an NK cell may be relatively short following adoptive transfer (i.e., weeks). Future studies are warranted to better understand if multiple infusions of transfected NK cells can compensate transient transgene expression or if stable transgene expression is a prerequisite for inducing clinical responses following adoptive transfer of genetically engineered NK cells. Studies are also needed to fully understand the lifespan of NK cells, particularly those that have undergone ex vivo manipulation.
The optimal method for genetic manipulation of NK cells to be used in a clinical trial may also depend on what NK cell preparation is used (Box 3). The advantage with NK cell lines is that they can be utilized as an off-the-shelf product stably transduced to express the gene or genes of interest. They may also be long-lived if given the proper cytokine support. However, the downside with using NK cell lines, like NK-92, is the requirement for irradiation (10 Gy) prior to infusion to avoid the risk of engrafting cells that are potentially tumorigenic in vivo (74). Moreover, patients treated with infusions of NK cell lines would also need moderate to high level of preconditioning to suppress host immunity to avoid rejection of these allogeneic cells. Moreover, infusion of allogeneic cells can raise humoral immunity and lead to adaptive T-cell immune responses specifically against alloantigens, precluding repeated infusions even with the use of preconditioning. Similar allo-reactivity can be induced with the use of primary allogeneic NK cell infusions. The use of autologous NK cells circumvents these risks and precludes the need for preconditioning. The potential draw back with using autologous NK cells is that efficient tumor targeting can be prevented by inhibitory KIR interactions with self-HLA. A potential advantage with using an NK cell line versus primary NK cells is that large numbers of NK cells from the NK cell line can be infused, whereas the number of primary cells available for infusion are typically much more limited. However, this limitation has recently been circumvented by a number of highly efficient methods to expand primary NK cells ex vivo for clinical infusion (60). Ideally, infusion of autologous gene-modified NK cells can be used to avoid the rejection risk and the prerequisite for preconditioning. One approach to overcome limitations of autologous NK cell inactivation via self-HLA is to genetically modify these effectors to silence inhibitory self-HLA binding receptors, such as NKG2A and KIRs, which alone or in combination with for instance CARs, can improve the tumor targeting capacity of NK cells in the autologous setting.

Box 3. NK Cell Source for Adoptive NK Cell Cancer Immunotherapy.
Concluding Remarks
Anti-tumor antibodies and CAR T cells have established immunotherapy as a viable treatment option for patients with cancer. Given their rapid and efficient method of recognizing tumor cells, NK cells represent a unique immune cell to genetically reprogram in an effort to improve the outcome of cell-based cancer immunotherapy. However, technical and biological challenges associated with gene delivery into NK cells have significantly tempered this approach. Viral transduction of NK cells initially resulted in low transgene delivery efficiencies that often required multiple rounds of transduction and/or cellular enrichment to achieve acceptable numbers of transgene expressing cells. Nevertheless, recent improvements in retro- and lentiviral transduction of NK cells have led to a flurry of preclinical studies on gene engineered NK cells. A number of studies have also shown that NK cells can be genetically reprogramed using mRNA electroporation. In contrast to viral transduction, this approach offers high transfection efficiencies without compromising their viability and does not require high-level biosafety laboratories. Although promising preclinical data on mRNA electroporated NK cells have emerged recently, concerns have been raised regarding the clinical utility of this approach as it only results in transient transgene expression.
Recently initiated clinical trials will soon give insight into the potential effectiveness of cell-based cancer immunotherapy strategies that utilize genetically modified NK cells. Nevertheless, further optimization of both viral transduction and electroporation of NK cells is still needed before this approach can be fully exploited in the clinic. With the recent advances in our understanding of the complex biological networks that regulate the capacity of NK cells to target and kill tumors in vivo, and with rapid developments in clinically compliant techniques to genetically manipulate NK cells, we foresee genetic engineering as an obligatory pathway to exploit the full potential of adoptive NK cell immunotherapy in patients with cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge all the members of the NHLBI Laboratory of Transplantation Immunotherapy, the NHLBI Division of Intramural Research (DIR), the Swedish Research Council, the Dean R. O’Neill and Edward Rancic Memorial Cancer Research Fellowship for their many contributions and support for original research described in this manuscript.
References
1. Gregoire C, Chasson L, Luci C, Tomasello E, Geissmann F, Vivier E, et al. The trafficking of natural killer cells. Immunol Rev (2007) 220:169–82. doi: 10.1111/j.1600-065X.2007.00563.x
2. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer (1975) 16:230–9. doi:10.1002/ijc.2910160204
3. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer (1975) 16:216–29. doi:10.1002/ijc.2910160204
4. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol (1975) 5:117–21. doi:10.1002/eji.1830050208
5. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5:112–7. doi:10.1002/eji.1830050208
6. Miller JS, Soignier Y, Panoskaltsis-Mortari A, Mcnearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105:3051–7. doi:10.1182/blood-2004-07-2974
7. Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol (2012) 189:5082–8. doi:10.4049/jimmunol.1201964
8. Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood (2012) 119:2665–74. doi:10.1182/blood-2011-10-386995
9. Bryceson YT, Ljunggren HG, Long EO. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood (2009) 114:2657–66. doi:10.1182/blood-2009-01-201632
10. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295:2097–100. doi:10.1126/science.1068440
11. Elmaagacli AH, Steckel NK, Koldehoff M, Hegerfeldt Y, Trenschel R, Ditschkowski M, et al. Early human cytomegalovirus replication after transplantation is associated with a decreased relapse risk: evidence for a putative virus-versus-leukemia effect in acute myeloid leukemia patients. Blood (2011) 118:1402–12. doi:10.1182/blood-2010-08-304121
12. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, Mckenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood (2014) 123:3855–63. doi:10.1182/blood-2013-10-532531
13. Ito S, Bollard CM, Carlsten M, Melenhorst JJ, Biancotto A, Wang E, et al. Ultra-low dose interleukin-2 promotes immune-modulating function of regulatory T cells and natural killer cells in healthy volunteers. Mol Ther (2014) 22:1388–95. doi:10.1038/mt.2014.50
14. Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med (2014) 65:333–47. doi:10.1146/annurev-med-060512-150254
15. Sutlu T, Nystrom S, Gilljam M, Stellan B, Applequist SE, Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther (2012) 23:1090–100. doi:10.1089/hum.2012.080
16. Nagashima S, Mailliard R, Kashii Y, Reichert TE, Herberman RB, Robbins P, et al. Stable transduction of the interleukin-2 gene into human natural killer cell lines and their phenotypic and functional characterization in vitro and in vivo. Blood (1998) 91:3850–61.
17. Yusa S, Catina TL, Campbell KS. SHP-1- and phosphotyrosine-independent inhibitory signaling by a killer cell Ig-like receptor cytoplasmic domain in human NK cells. J Immunol (2002) 168:5047–57. doi:10.4049/jimmunol.168.10.5047
18. Becknell B, Trotta R, Yu J, Ding W, Mao HC, Hughes T, et al. Efficient infection of human natural killer cells with an EBV/retroviral hybrid vector. J Immunol Methods (2005) 296:115–23. doi:10.1016/j.jim.2004.11.012
19. Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol (2008) 180:6392–401. doi:10.4049/jimmunol.180.9.6392
20. Guven H, Konstantinidis KV, Alici E, Aints A, Abedi-Valugerdi M, Christensson B, et al. Efficient gene transfer into primary human natural killer cells by retroviral transduction. Exp Hematol (2005) 33:1320–8. doi:10.1016/j.exphem.2005.07.006
21. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood (2005) 106:376–83. doi:10.1182/blood-2004-12-4797
22. Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, Seliger B, et al. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc Natl Acad Sci U S A (2008) 105:17481–6. doi:10.1073/pnas.0804788105
23. Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res (2009) 15:4857–66. doi:10.1158/1078-0432.CCR-08-2810
24. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res (2013) 73:1777–86. doi:10.1158/0008-5472.CAN-12-3558
25. Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood (2014) 124:1081–8. doi:10.1182/blood-2014-02-556837
26. Micucci F, Zingoni A, Piccoli M, Frati L, Santoni A, Galandrini R. High-efficient lentiviral vector-mediated gene transfer into primary human NK cells. Exp Hematol (2006) 34:1344–52. doi:10.1016/j.exphem.2006.06.001
27. Savan R, Chan T, Young HA. Lentiviral gene transduction in human and mouse NK cell lines. Methods Mol Biol (2010) 612:209–21. doi:10.1007/978-1-60761-362-6_14
28. Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA, et al. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma (2012) 53:958–65. doi:10.3109/10428194.2011.634048
29. Sahm C, Schonfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother (2012) 61:1451–61. doi:10.1007/s00262-012-1212-x
30. Baeriswyl V, Wodnar-Filipowicz A, Kalberer CP. The effect of silencing NKG2D through RNA interference on receptor functions in interleukin-2-activated human natural killer cells. Haematologica (2006) 91:1538–41.
31. Figueiredo C, Seltsam A, Blasczyk R. Permanent silencing of NKG2A expression for cell-based therapeutics. J Mol Med (Berl) (2009) 87:199–210. doi:10.1007/s00109-008-0417-0
32. Maasho K, Marusina A, Reynolds NM, Coligan JE, Borrego F. Efficient gene transfer into the human natural killer cell line, NKL, using the amaxa nucleofection system. J Immunol Methods (2004) 284:133–40. doi:10.1016/j.jim.2003.10.010
33. Schoenberg K, Trompeter HI, Uhrberg M. Delivery of DNA into natural killer cells for immunotherapy. Methods Mol Biol (2008) 423:165–72. doi:10.1007/978-1-59745-194-9_11
34. Yang B, Liu H, Shi W, Wang Z, Sun S, Zhang G, et al. Blocking transforming growth factor-beta signaling pathway augments antitumor effect of adoptive NK-92 cell therapy. Int Immunopharmacol (2013) 17:198–204. doi:10.1016/j.intimp.2013.06.003
35. Liu H, Yang B, Sun T, Lin L, Hu Y, Deng M, et al. Specific growth inhibition of ErbB2expressing human breast cancer cells by genetically modified NK92 cells. Oncol Rep (2015) 33:95–102. doi:10.3892/or.2014.3548
36. Schirrmann T, Pecher G. Human natural killer cell line modified with a chimeric immunoglobulin T-cell receptor gene leads to tumor growth inhibition in vivo. Cancer Gene Ther (2002) 9:390–8. doi:10.1038/sj.cgt.7700453
37. Grund EM, Muise-Helmericks RC. Cost efficient and effective gene transfer into the human natural killer cell line, NK92. J Immunol Methods (2005) 296:31–6. doi:10.1016/j.jim.2004.10.008
38. Schirrmann T, Pecher G. Specific targeting of CD33(+) leukemia cells by a natural killer cell line modified with a chimeric receptor. Leuk Res (2005) 29:301–6. doi:10.1016/j.leukres.2004.07.005
39. Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res (2009) 33:1255–9. doi:10.1016/j.leukres.2008.11.024
40. Li L, Liu LN, Feller S, Allen C, Shivakumar R, Fratantoni J, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther (2010) 17:147–54. doi:10.1038/cgt.2009.61
41. Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy (2012) 14:830–40. doi:10.3109/14653249.2012.671519
42. Tran J, Kung SK. Lentiviral vectors mediate stable and efficient gene delivery into primary murine natural killer cells. Mol Ther (2007) 15:1331–9. doi:10.1038/sj.mt.6300184
43. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet (2003) 4:346–58. doi:10.1038/nrg1066
44. Biasco L, Baricordi C, Aiuti A. Retroviral integrations in gene therapy trials. Mol Ther (2012) 20:709–16. doi:10.1038/mt.2011.289
45. Zhang J, Sun R, Wei H, Zhang J, Tian Z. Characterization of stem cell factor gene-modified human natural killer cell line, NK-92 cells: implication in NK cell-based adoptive cellular immunotherapy. Oncol Rep (2004) 11:1097–106. doi:10.3892/or.11.5.1097
46. Liu JH, Wei S, Blanchard DK, Djeu JY. Restoration of lytic function in a human natural killer cell line by gene transfection. Cell Immunol (1994) 156:24–35. doi:10.1006/cimm.1994.1150
47. Daldrup-Link HE, Meier R, Rudelius M, Piontek G, Piert M, Metz S, et al. In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur Radiol (2005) 15:4–13. doi:10.1007/s00330-004-2526-7
48. Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol (2014) 8:297–310. doi:10.1016/j.molonc.2013.12.001
49. Jiang W, Zhang J, Tian Z. Functional characterization of interleukin-15 gene transduction into the human natural killer cell line NKL. Cytotherapy (2008) 10:265–74. doi:10.1080/14653240801965156
50. Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol (2014) 32:347–55. doi:10.1038/nbt.2842
51. Konstantinidis KV, Alici E, Aints A, Christensson B, Ljunggren HG, Dilber MS. Targeting IL-2 to the endoplasmic reticulum confines autocrine growth stimulation to NK-92 cells. Exp Hematol (2005) 33:159–64. doi:10.1016/j.exphem.2004.11.003
52. Jiang W, Zhang C, Tian Z, Zhang J. hIL-15 gene-modified human natural killer cells (NKL-IL15) augments the anti-human hepatocellular carcinoma effect in vivo. Immunobiology (2014) 219:547–53. doi:10.1016/j.imbio.2014.03.007
53. Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology (2013) 2:e26527. doi:10.4161/onci.26527
54. Muller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann HG, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother (2008) 57:411–23. doi:10.1007/s00262-007-0383-3
55. Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia (2014) 28:917–27. doi:10.1038/leu.2013.279
56. Esser R, Muller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med (2012) 16:569–81. doi:10.1111/j.1582-4934.2011.01343.x
57. Lee JM, Yoon SH, Kim HS, Kim SY, Sohn HJ, Oh ST, et al. Direct and indirect antitumor effects by human peripheral blood lymphocytes expressing both chimeric immune receptor and interleukin-2 in ovarian cancer xenograft model. Cancer Gene Ther (2010) 17:742–50. doi:10.1038/cgt.2010.30
58. Kobayashi E, Kishi H, Ozawa T, Hamana H, Nakagawa H, Jin A, et al. A chimeric antigen receptor for TRAIL-receptor 1 induces apoptosis in various types of tumor cells. Biochem Biophys Res Commun (2014) 453:798–803. doi:10.1016/j.bbrc.2014.10.024
59. Zhang G, Liu R, Zhu X, Wang L, Ma J, Han H, et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol Cell Biol (2013) 91:615–24. doi:10.1038/icb.2013.45
60. Childs RW, Berg M. Bringing natural killer cells to the clinic: ex vivo manipulation. Hematology Am Soc Hematol Educ Program (2013) 2013:234–46. doi:10.1182/asheducation-2013.1.234
61. Miller JS, Rooney CM, Curtsinger J, Mcelmurry R, Mccullar V, Verneris MR, et al. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: implications for clinical therapy. Biol Blood Marrow Transplant (2014) 20:1252–7. doi:10.1016/j.bbmt.2014.05.004
62. Somanchi SS, Somanchi A, Cooper LJ, Lee DA. Engineering lymph node homing of ex vivo-expanded human natural killer cells via trogocytosis of the chemokine receptor CCR7. Blood (2012) 119:5164–72. doi:10.1182/blood-2011-11-389924
63. Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother (2015) 64:225–35. doi:10.1007/s00262-014-1629-5
64. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev (2014) 257:107–26. doi:10.1111/imr.12131
65. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood (2002) 99:754–8. doi:10.1182/blood.V99.3.754
66. Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol (2003) 21:3940–7. doi:10.1200/JCO.2003.05.013
67. Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest (1997) 100:1059–70. doi:10.1172/JCI119616
68. Carlsten M, Li L, Su S, Berg M, Reger R, Madhusudan P, et al. Clinical-grade mRNA electroporation of NK cells: a novel and highly efficient method to genetically reprogram human NK cells for cancer immunotherapy. ASH Annual Meeting Abstracts: San Francisco (2014).
69. Tijsterman M, Ketting RF, Plasterk RH. The genetics of RNA silencing. Annu Rev Genet (2002) 36:489–519. doi:10.1146/annurev.genet.36.043002.091619
70. Furutani E, Su S, Smith A, Berg M, Childs R. siRNA inactivation of the inhibitory receptor NKG2A augments the anti-tumor effects of adoptively transferred NK cells in tumor-bearing hosts. ASH Annual Meeting Abstracts: Orlando (2010).
71. Purdy AK, Campbell KS. Introduction of shRNAs into human NK-like cell lines with retrovirus. Methods Mol Biol (2010) 612:223–31. doi:10.1007/978-1-60761-362-6_15
72. Karasuyama H, Tohyama N, Tada T. Autocrine growth and tumorigenicity of interleukin 2-dependent helper T cells transfected with IL-2 gene. J Exp Med (1989) 169:13–25. doi:10.1084/jem.169.1.13
73. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol (2014) 5:254. doi:10.3389/fphar.2014.00254
Keywords: NK cells, genetic manipulation, viral transduction, electroporation, cancer immunotherapy
Citation: Carlsten M and Childs RW (2015) Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front. Immunol. 6:266. doi: 10.3389/fimmu.2015.00266
Received: 19 March 2015; Accepted: 13 May 2015;
Published: 10 June 2015
Edited by:
Francisco Borrego, Cruces University Hospital, SpainReviewed by:
Claudia Rossig, University Children’s Hospital Münster, GermanyAntonio Di Stasi, University of Alabama at Birmingham, USA
Copyright: © 2015 Carlsten and Childs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mattias Carlsten, Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, 10 Center Drive (MSC 1230), Building 10-CRC/3E-5332, Bethesda, MD 20892, USA, mattias.carlsten@nih.gov