 Ryan J. Martinez
Ryan J. Martinez Brian D. Evavold
Brian D. Evavold- Department of Microbiology and Immunology, Emory University, Atlanta, GA, USA
Kinetic and biophysical parameters of T cell receptor (TCR) and peptide:MHC (pMHC) interaction define intrinsic factors required for T cell activation and differentiation. Although receptor ligand kinetics are somewhat cumbersome to assess experimentally, TCR:pMHC affinity has been shown to predict peripheral T cell functionality and potential for forming memory. Multimeric forms of pMHC monomers have often been used to provide an indirect readout of higher affinity T cells due to their availability and ease of use while allowing simultaneous definition of other functional and phenotypic characteristics. However, multimeric pMHC reagents have introduced a bias that underestimates the lower affinity components contained in the highly diverse TCR repertoires of all polyclonal T cell responses. Advances in the identification of lower affinity cells have led to the examination of these cells and their contribution to the immune response. In this review, we discuss the identification of high- vs. low-affinity T cells as well as their attributed signaling and functional differences. Lastly, mechanisms are discussed that maintain a diverse range of low- and high-affinity T cells.
Introduction
The T cell immune response is composed of diverse sets of T cell receptors with a normally distributed range of affinities for pMHC (1, 2). Upon antigen recognition, the biophysical characteristics of the TCR:pMHC interaction will drive signaling, division, and differentiation (3, 4). Current hypotheses suggest the highest affinity T cells have a competitive advantage during the immune response, based on the assumption that they would receive stronger and more prolonged activation signals than T cells with lower affinity interactions (5–7). However, evidence is emerging to suggest the lack of skewing toward the highest affinity T cell repertoire, instead of proposing a distribution of affinities and maintenance of diversity throughout the immune response (8, 9). This T cell diversity has been shown to be important for homeostasis of the immune system, but the process of maintaining affinity diversity is unclear. We further discuss factors that support and favor the survival of lower affinity T cells and, therefore, highlight the important contribution of low-affinity T cells and a broad affinity distribution in the immune response.
Measurement of TCR:pMHC Affinity
Affinity is defined as the probability of a receptor (TCR)–ligand (pMHC) interaction and is one of the most commonly used measurements to predict the T cell response to antigen. As with all receptor–ligand interactions, affinity, or the association constant (Ka) is derived from the on-rate (kon) and off-rate (koff) of receptor–ligand equilibrium and is calculated independently of concentrations of ligand and receptor (affinity = Ka = kon/koff) (Table 1) (3, 10). The affinity of an interaction is not its bond strength, or the force needed to break the bond, though these terms are often incorrectly used interchangeably. An example of this distinction can be seen in the avidin/biotin system as this receptor–ligand pair has one of the highest affinity interactions measured, but the non-covalent interactions can be easily broken, signifying lower bond strength (11). Along with TCR:pMHC affinity, the dissociation constant (Kd, inverse of Ka) and half-life [τ1/2 = ln(2)/koff] can also be derived. The combination of these measurements have been used to describe T cell activation and differentiation models (kinetic proofreading, optimal dwell time, etc.), but not all methods of biophysical measurements generate similar results (3, 12, 13). Therefore, further understanding of measurement systems is necessary.
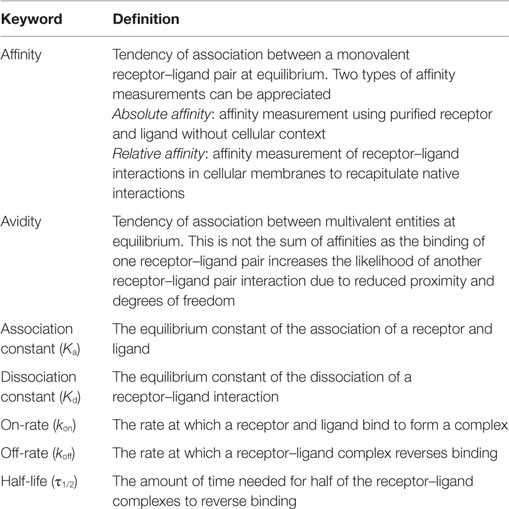
Table 1. Definitions of biophysical parameters of TCR:pMHC interaction.
Affinity is commonly measured using purified reactants in solution by surface plasmon resonance (SPR), where free ligand is flowed over receptors fixed to a surface. This absolute affinity measurement occurs in three dimensions (3D) and allows for the definition of protein interactions in their simplest, purified form with no influence from outside forces. However, the increased degrees of freedom that occur in solution may not be the optimal method to accurately assess interactions between receptor and ligands that occur in two opposing plasma membranes. A better replicate of in vivo interactions between proteins at the membrane surface can be accomplished using two-dimensional (2D) receptor–ligand binding techniques, such as flow chamber assays, thermal fluctuation assays, single molecule FRET, Zhu–Golan plots, contact area FRAP, and the adhesion frequency assay (3). Currently, the focus of our lab has been the use of the two-dimensional micropipette adhesion frequency assay (2D-MP), a measurement of the relative 2D affinity of the receptor–ligand interaction on opposing membranes (14). This 2D affinity is termed a relative affinity because it is dependent on the context in which it was measured, whereas 3D methods generate an absolute affinity measurement while ignoring all other cellular participants. This distinction of relative and absolute affinity will be discussed in a later section. When 2D and 3D affinity TCR measurements are compared, an increased affinity with an associated decreased koff can be appreciated (12, 13, 15, 16). Attempts to correlate affinity values generated by 2D and 3D methods have been achieved with little success as the parameters controlling relative 2D affinity are still unknown (12). Importantly, the relative affinity measured by 2D-MP better correlates with functional responses than 3D methods and refers to the affinity in the proper cellular context (12, 15).
The advent of recombinant pMHC tetramer reagents has allowed for the identification of antigen-specific T cells and the subsequent use of these reagents for indirect assessment of biophysical interactions of TCR:pMHC. The binding of the tetramer reagent is dependent on valency to increase its avidity as monomeric pMHC complexes do not attach well to TCR (17, 18). This lack of monomer interaction with TCR is most likely due to the reliance of pMHC tetramer staining on higher affinity interactions (8, 9). The koff and kon for each arm of the pMHC tetramer binding to TCRs are known to reflect avidity interactions, with the binding of one pMHC monomer arm enhancing the kon of the subsequent monomer arm and reducing the koff of the entire reagent (19). The use of pMHC tetramer to measure koff, kon, and τ1/2 assumes that the amount of pMHC tetramer bound to a cell is directly proportional to the affinity of that cell, with more tetramer bound to higher affinity cells than to lower affinity T cells (6, 9, 19, 20). However, this assumption may not always yield a direct correlation, with many groups demonstrating tetramer binding intensity does not equate to functional responses or SPR measurements (21–24). One possible explanation for discrepancies with SPR is that the cellular membrane can affect tetramer binding. Another possibility for these discrepancies is that TCR density affects binding because tetramer relies on avidity interactions. While many have normalized the TCR to pMHC concentrations on each cell (18, 25, 26), others do not account for the number of TCRs expressed at the cell surface (21, 27, 28). The effect of TCR density can be appreciated as the analysis of the tetramer+ populations reveals lower TCR expression as they exhibit only 20–40% of the TCR density compared to the bulk T cell population (unpublished data). This indicates tetramer+ T cells may have different TCR levels than the remaining T cell population but it is unknown if this is a cause or an effect of being a tetramer binder.
The measurement of TCR:pMHC affinity by 2D-MP is an extremely sensitive method that follows first-order kinetics and is dependent upon T cell intrinsic factors (3). Measured TCR affinities can be altered when reagents are used to change lipid composition and actin cytoskeleton (12). Adjustments of the membrane and supporting scaffolding should change 2D affinity as the characteristics of the opposing membranes during receptor–ligand interactions are fundamental for the measurement of relative 2D affinities. Much of the sensitivity of the 2D-MP assay comes from the flexibility of the red blood cell (RBC) membrane, which can be distended by the formation of a single TCR:pMHC bond (3, 29). As biotinylated pMHC is bound to the RBC through streptavidin interactions, clustering and valency of ligand could play a role in binding. Varying the concentration of pMHC on the RBC surface does not change the calculated affinity of activated T cells, signifying concentration does not affect the 2D measurements (12, 14). In experiments altering the valency of pMHC on the RBC through use of mutant streptavidin, no changes in 2D affinity were noted (12). This is in contrast to pMHC tetramers, which rely on both concentration and valency of the reagent to measure antigen specificity (30). Together, this suggests concentration and valency do not play a role in the measurement of affinity by 2D-MP. In addition, it demonstrates the micropipette values are a measure of affinity and not avidity.
An important distinction between CD4 and CD8 T cells is the contribution of coreceptor to the overall strength of binding between pMHC and TCR (18, 31). CD4 and CD8 coreceptors are thought to stabilize TCR:pMHC bonds while also recruiting Lck to the TCR complex for the initiation of the downstream signaling cascade (32, 33). Intriguingly, CD8 has a higher affinity for its coreceptor than CD4, though both interactions are of weaker affinity (13, 32). For CD8 T cells, coreceptor contributes to the binding of TCR to pMHC when assessed by pMHC tetramer, 2D-MP, and SPR (31, 34–36). The removal of CD8 contribution leads to decreased avidity and functional responses (37–39). In addition, the binding of the lowest affinity T cells are the most affected by the loss of CD8, signifying CD8 helps to increase the likelihood of low-affinity TCR:pMHC interaction and signaling (15, 37). In contrast, the role of CD4 is very different as there is little to no contribution to the TCR:pMHC interaction as measured by 2D-MP, pMHC tetramer, and SPR (13, 32). This is not to say CD4 is not important in functional responses as CD4 is required to recruit Lck for optimal initiation of T cell signaling (17, 40, 41), but at least under the conditions used in tetramer and 2D assays, there is little contribution to biophysical parameters (13). Importantly, detection of CD4 T cells by pMHCII tetramer would be expected to miss more of the lower affinity TCRs due to the lack of coreceptor contribution as compared to pMHCI tetramer and CD8 T cells. These differences in CD4 and CD8 coreceptor binding impact the use of tetramers to count antigen-specific T cells as well as measuring the kinetic rates of the TCR:pMHC interaction.
Detection of Antigen-Specific T Cells
The identification of T cells is important as the biophysical TCR:pMHC interactions discussed are correlated with functional responses. The current gold standard for identifying antigen-specific T cells are using either pMHC tetramer reagents or readouts of functional responses, but both are sub-optimal in identifying the true number of antigen-specific T cells (42–45). For example, not all T cells of the same specificity make cytokine upon stimulation as demonstrated by the use of TCR-Tg T cells (46, 47). Further, a CD4 T population has a number of distinct fates with associated effector functions, such that any one cytokine will underestimate the total number of antigen-specific cells. Interestingly, the number of pMHC tetramer+ T cells sometimes equates to the number of cytokine-producing cells, even though not all T cells will produce the target cytokine (27, 43, 45). For the most part, intracellular staining for cytokines is incompatible with tetramer staining making it unclear if cytokine-producing cells overlap with tetramer binders, or if they are distinct populations. To more accurately identify the number of antigen-specific T cells, groups have used activation markers, such as CD11a, LFA-1, or CD49d, that are upregulated on antigen-specific T cells after infection (43, 48). When these cells are quantitated using these functional cell-surface markers, they far outnumber the number of tetramer or cytokine-producing cells (43, 48). The 2D-MP further corroborates the underestimation of antigen-specific T cells by pMHC tetramers (8, 9). When compared to the total population of antigen-specific T cells, pMHC tetramer+ T cells make up only the highest affinity population (9). Most T cell repertoires have a normally distributed TCR:pMHC affinity range, with the rarest cells being the highest and lowest affinity. Based on this data, tetramer+ cells are above average in affinity and would, therefore, only make up a fraction of the total T cells in an immune response (4). This would be especially apparent for CD4 T cells since the coreceptor does not aid in tetramer avidity.
The 2D-MP assay is currently the most sensitive available to capture the entire affinity range of a T cell repertoire and not just the highest affinity T cells. To perform the 2D-MP assay, single T cells are randomly chosen for affinity measurements from a purified population of cells. As this is a sampling process that is time intensive, it is important to understand the numbers of cells needed for analysis to reflect representative data of the entire population. To address how many cells need to be measured to find the average affinity of an antigen-specific polyclonal population, we have combined previously published measurements of a polyclonal T cell population for a single antigen (MOG38–49:I-Ab) and performed random sampling experiments (Figure 1). When noting the moving average from 10 repetitions of the randomly sampled MOG-specific T cell affinities, it is apparent that the sampling affinity often reaches the average affinity rapidly, i.e., within 10 binding pairs. By the time, 16 binding T cells have been analyzed, the average affinity measured is ±5% of the affinity of the entire pooled population. This measurement is for a polyclonal population with a 2,000-fold range in affinity, meaning a repertoire with considerably less diversity (TCR-Tg or tetramer+ T cells) needs even fewer points measured to identify the average affinity. Therefore, to measure the average affinity of a normally distributed polyclonal repertoire only 10–16 T cell affinities need to be measured to be within 5% of the average affinity of the repertoire.
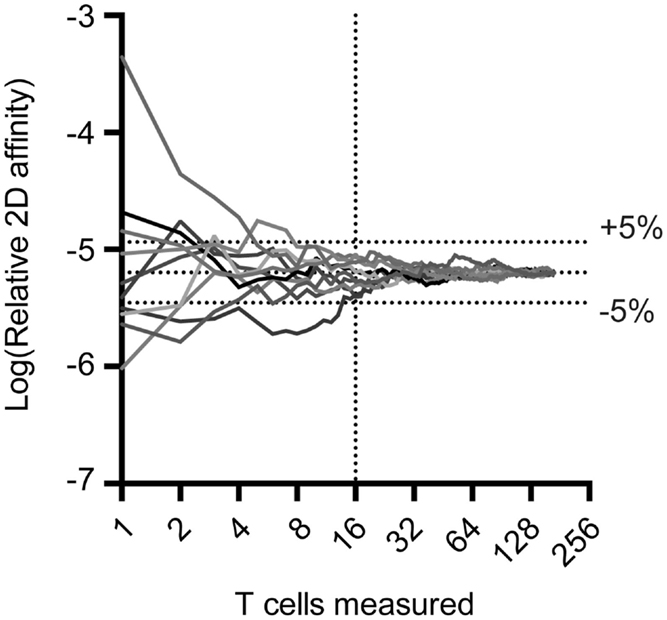
Figure 1. Random sampling of measured TCR:pMHC reveals rapid approach to average affinity for an entire population. Previous 2D affinity measurements of MOG-specific CD4 T cells were combined and randomly arranged. A moving average was calculated and graphed as a function of the number of cells sampled. Cells were then randomly rearranged and moving average calculations were performed nine more times to generate the curves illustrated.
Signaling in Low-Affinity T Cells
In both kinetic-segregation and proofreading models, the binding of TCR:pMHC is key to activation, with the koff and kon and τ1/2 controlling the strength of signal the T cell receives (49–53). Higher affinity interactions with prolonged τ1/2 have an increased likelihood of forming a stable conjugate and triggering the T cell signaling cascade, though an optimal τ1/2 most likely exists due to the hypothesized need for serial engagement (10, 50, 53). Using this logic, the lowest affinity T cells should have a reduced probability of initiating T cell signaling, but these cells do efficiently propagate the signaling cascade as low-affinity T cells do expand, and differentiate during the immune response (8, 9, 48). Thus, mechanisms must exist that allow for low-affinity T cells, with the reduced probability of initial TCR:pMHC bond formation, to receive sufficient signals to compete with high-affinity counterparts.
Developing thymocytes discriminate positive- and negative-selecting signals through both qualitative and quantitative signaling pathways. Qualitatively, studies have suggested that reduced but sustained Erk signaling is necessary for positive selection, while strong Erk, p38, and Jnk activation are necessary for negative selection (54–57). In positive selection, the protein Themis reduces TCR signaling by recruiting the phosphatase SHP-1 to inhibit Lck activation and reduces strong, transient, Erk activation, a property associated with negative selection (57, 58). Without Themis, low-affinity pMHC can produce strong agonist signals resulting in increased negative selection (58). This is of interest as negative regulatory mechanisms must be preventing low-affinity ligands from being selected like high-affinity ligands. Currently, it is unknown what controls the function of Themis or how it discriminates between high- and low-affinity pMHC to ultimately regulate downstream signals. One potential quantitative mechanism for discriminating high- and low-affinity pMHC was found in thymocyte negative selection. During negative selection, thymocytes bind to pMHC and scan coreceptors (CD4 and CD8) to find one coupled to Lck (59). CD8 coreceptors on thymocytes have a lower amount of coupled Lck than CD4 coreceptor, so a longer TCR:pMHC τ1/2 is necessary for CD8 T cell negative selection (59). If a TCR:pMHC bond is maintained during the duration for a coreceptor-Lck conjugate to be found, the T cell is determined to be high affinity and will undergo Bim-dependent apoptosis (60). These differences in signaling between positive and negative selection demonstrate how similar stimuli (pMHC) can generate distinct results depending on the TCR interaction parameters.
Naive CD4 and CD8 T cells demonstrate a distribution of reactivity to multiple foreign antigens that is established during thymic selection on self-antigens. In both naïve CD4 and CD8 T cells, multiple studies have been able to identify heterogeneity in reactivity of T cells for self- and foreign-pMHC (26, 61–64). These ranges of reactivity relate to markers of self-pMHC stimulation (CD5, Nur77, basal TCRζ phosphorylation) (26, 61, 64, 65) as well as indicators of activation in response to foreign-pMHC (ERK phosphorylation, IL-2 production) (64). Conflicting experimental evidence exists when attempting to correlate self-reactivity with foreign-reactivity (26, 61, 64). Some have demonstrated that CD5 positively correlates with foreign TCR reactivity (measured by pMHC tetramer and function) (26, 61), while others have demonstrated CD5 expression can be altered without changes to TCR affinity as measured by SPR (64). Nonetheless, based on the TCR affinity for self-pMHC, the immune system is able to diversify the reactivity of signaling machinery by controlling the basal phosphatase and kinase activity.
The first step of signaling after TCR triggering is phosphorylation of CD3 and TCRζ intracellular chains by Lck and the recruitment of the kinase Zap70. Phosphorylation of Zap70 by Lck propagates the initial signals and causes downstream activation of the MAP kinase (Erk), NF-κB, and calcineurin pathways (66). To inhibit signaling cascades, phosphatases, such as SHP-1/2 and CD45, prevent the sustained activation of kinases (66). For low-affinity TCR:pMHC interactions, the peak of T cell signaling molecules (Erk, Jun, and Ras) are delayed or absent and TCR signaling is decreased (58, 62, 67). This late and reduced activation of Erk in low-affinity T cells allows for the sustained recruitment and activity of SHP-1, decreasing the activation of Lck and reducing the T cell signaling response (58, 67, 68). Another phosphatase, PTPN22, has also been shown to inhibit Erk phosphorylation in T cells stimulated with low-affinity pMHC (69). It is surprising that even with these negative signaling events, low-affinity TCRs are able to signal and even upregulate activation genes CD69 and CD25 to similar levels as higher affinity T cells (7, 67). Similar to thymocyte selection, this demonstrates peripheral T cells have mechanisms to detect biophysical TCR interactions that can then be translated to signaling cascades as well as possess compensatory mechanisms to enhance low-affinity TCR interactions or even decrease higher affinity T cell interactions (70). Potentially, force generation by TCRs could be a mechanism by which low-affinity T cells are able to increase bond τ1/2 and receive similar signals as high-affinity counterparts (71–73). Conversely, it may be that different pathways are induced by low-affinity TCR:pMHC binding that are currently not understood. Overall, low- and high-affinity T cells use similar mechanisms and machinery to signal, but the outcomes seem to vary in their response.
Functionality of Low-Affinity T Cells
The development and function of T cells has been highly associated with TCR:pMHC affinity, with high- and low-affinity T cell interactions often performing different roles. Thymocyte selection is dependent upon TCR:pMHC affinity interactions, with positive selection requiring low-affinity interactions and negative selection deleting thymocytes greater than a threshold affinity (1, 74). The number of antigen-specific T cells for a single epitope is tightly controlled by central tolerance and, therefore, by TCRs affinity for self-pMHC presented in the thymus (75–79). The initial numbers of DP thymocytes for a single epitope are defined by the size of their positively selecting niche of self-peptides and further culled by their reactivity to self-antigen via negative selection (27, 63, 79–81). Work suggests positive selection is important in the generation of a functional T cell repertoire as alterations in the size of the selecting niche can cause T cell dysfunction or increased reactivity to antigen (63, 82). Analysis of the autoimmune prone NOD mouse has suggested its positive selecting niche is reduced, causing competition for positive selection survival signals and enriching for T cells that have a higher affinity TCR:self-pMHC interaction (81). This reduced positive selection niche and concurrent increase in TCR affinity will not be fully compensated by negative selection as negative selection is not as effective as once believed (63, 81, 83–85). The precursor frequency of a given antigen-specific T cell repertoire reproducibly generated by thymocyte selection is important as antigens with higher precursor frequencies can reach peak numbers more rapidly and provide better immune protection (27). Antigen-specific T cell repertoires with lower precursor frequencies will have a greater fold expansion, but will still not outnumber the higher precursor frequency repertoires (86). Based on this functional data, one could assume that the repertoires with larger precursor frequency have a higher affinity for pMHC as this would mean they have undergone less negative selection. Recent work supports this hypothesis as epitopes with lower precursor frequency demonstrate more similarity to mouse self-peptides as well as reduced pMHCII tetramer binding (27). Therefore, a balance exists between positive and negative selection and their preference to favor low- and high-affinity interactions between pMHC and TCR will ultimately determine the numbers and affinity of antigen-specific T cells.
All the advantages of TCRs with high affinity for pMHC would suggest they can easily outcompete the lower affinity T cells (5), but this does not seem to be occurring when the full affinity repertoire is analyzed (8, 9, 48). One caveat to many model systems is the focus on a single TCR and the use of different APLs to model the fate of polyclonal T cells in response to lower affinity antigens (7, 59, 87–89). In TCR-Tg mice, each T cell undergoes similar thymic selection mechanisms, and therefore possesses similar reactivity to antigen. Groups have shown different repertoires undergo different TCR selection and peripheral tolerance mechanisms, including negative selection, agonist selection, clonal diversion, and inhibitory molecule upregulation (65, 70, 74, 90, 91). For example, every OT-I TCR-Tg T cell will be positively and negatively selected by the same self-antigens, while in a polyclonal system, T cells with a range of affinities will recognize the OT-I cognate peptide (SIINFEKL) and will be positively and negatively selected by numerous different peptides. In the polyclonal setting, there will be a range of affinities for SIINFEKL and an accompanying range of tolerance mechanisms to control responses to this antigen. Therefore, a single TCR binding to different affinity pMHC complexes during selection is not the same as a polyclonal set of different TCRs binding to a set of pMHC. This distinction between effects at a clonal as opposed to a polyclonal level could alter functionality and impact interpretation on the role of lower affinity ligands during an immune response.
After primary antigen exposure and triggering of signaling cascades, division of CD4 and CD8 T cells will cause 100- to 1,000-fold expansion (61, 86, 92). Interestingly, low-affinity T cells are easily detectable throughout the response, signifying they are capable of expanding as well as high-affinity T cells (9, 48). Higher affinity CD8 TCR interactions cause asymmetric division that is associated with increased functionality of the proximal daughter cells (89). These CD8 T cells have similar initial rates of division, but eventually the highest affinity T cells maintain division while the lower affinity T cells begin to contract (7, 89). The contraction of lower affinity T cell is not due to increased death or lack of memory formation as a similar frequency of low-affinity pMHC primed CD8 T cells differentiates into memory T cells (88, 89). Along with differences in division rates, the migration kinetics of T cells are controlled by affinity, with the lower affinity APL stimulated T cells demonstrating increased numbers of TCR-Tg T cells in the blood at earlier time points (7). Studies in high- and low-affinity CD4 T cells demonstrates the time to the first division of high-affinity CD4 T cells is much faster than low-affinity cells, though after several divisions, the low-affinity cells reach the same absolute number as high-affinity T cells (67). Together, these data demonstrate low- and high-affinity T cells behave similarly during initial expansion, but most likely have roles in the immune response at distinct times and locations.
Evidence demonstrates T cell affinity controls effector and memory differentiation of antigen-specific populations. Using the OT-I CD8 T cell APL system, groups have demonstrated low-affinity priming generates a greater frequency of Eomes+ memory T cells (88). It was determined that TGF-βR expression, a negative regulator of T cells, is not downregulated in low-affinity T cell responses, creating a balance of the generation (IL-12R) and ablation (TGF-βR) of memory T cells (88). In CD4 T cells, TCR:pMHC affinity has been correlated with memory (6, 93, 94), T helper subset (TH1 vs. TH2), and T follicular helper (TFH) differentiation (92, 93, 95, 96) as well as prevention of exhaustion by chronic antigen exposure (97). Lower affinity TCR interactions have been shown to be biased to generate long-lived memory cells (6, 93, 94), while for TH1 vs. TH2 differentiation, greater strength of TCR stimulation increases the likelihood of TH1 differentiation (95, 96). For TFH cells, increased and decreased TCR:pMHC affinity has been correlated with differentiation, thought to be due to TCR-dependent IL-2/IL-2R alterations (28, 92, 93). The finding that TFH cells can differentiate from TCRs with low and high-affinity TCR:pMHC interactions is perplexing, but demonstrates that active mechanisms maintain the differentiation diversity of the low and high-affinity T cells. Nonetheless, comparing and contrasting the findings of CD4 and CD8 T cells demonstrates the complexity of each system, but also demonstrates unique roles and pathways for high- and low-affinity TCR:pMHC interactions to presumably maintain functional diversity.
The activation and regulation of metabolic pathways is essential for the initiation and maintenance of the immune response (98–101). In CD8 T cells, the TCR:pMHC interaction controls initial metabolic reprograming by upregulating IRF4 and Myc in a TCR:pMHC affinity-dependent manner (101). The transcription factors Myc and IRF4 coordinate the switch from fatty acid oxidation to aerobic glycolysis, which is essential for maintenance of the immune response (99, 101). Low-affinity TCR interactions led to less Myc and/or IRF4 expression, reducing the uptake of metabolic intermediates and changing the amount of T cell death during the response (99, 101). Therefore, TCR:pMHC affinity is necessary for instructing metabolic reprograming and a generating a greater functional response, but it is still unclear what function affinity-based metabolic reprograming plays for the differentiation and maintenance of low-affinity T cells.
Maintenance of Affinity Diversity
The maintenance of clonotype diversity in the immune system is essential for the health of the organism (102). By maintaining clonotype diversity, TCR affinity diversity is also preserved, with a single epitope being recognized by multiple T cells to create a normally distributed TCR:pMHC affinity population. For each epitope, a range of T cell precursors exist, whose frequency is controlled by central tolerance mechanisms (27, 103). As the preimmune frequency for a single epitope approaches 0, the capacity of the repertoire to protect the host from this epitope diminishes (90, 104). This theoretical lower limit of T cells that can effectively protect the host is defined as a protecton and is dependent upon the size of the organism and the migration velocity of T cells (105). Larger organisms need a larger protecton because their bodies have a greater volume to patrol and protect against pathogen dissemination.
Along with the size of the organism, the protecton is dependent upon the amount of cross-reactivity between TCRs. To derive the level of cross-reactivity inherent to an individual’s TCRs, the entire number of antigen-specific T cells inclusive of lower affinity ones needs to be defined. Recent studies have identified T cells with cross-reactivity using multiple high-affinity-dependent methodologies, such as pMHC tetramers, but the rules regulating cross-reactivity are still being formulated (27, 104, 106, 107). Previous studies have suggested that the highest affinity T cells are the most cross-reactive as these cells have been shown to accept the most degeneracy in TCR:pMHC interaction and still function (108–110). However, single T cells can have both increased and decreased functional responses to peptides, meaning that just because a TCR binds to one pMHC with lower affinity, it cannot bind to another pMHC with higher affinity (106). Theoretically, a single TCR should possess a range of affinity for peptides presented by MHC, meaning that cross-reactivity is not unique to only high- or low-affinity TCRs. Groups have identified low-affinity T cells during the immune response, signifying these cells must be represented in the naïve mouse, yet these lower affinity T cells are currently not being included in the calculations (8, 9, 48). If inclusion of lower affinity T cells leads to a greater number of T cells in an antigen-specific repertoire, then the amount of cross-reactivity would correspondingly change (104). Therefore, protecton size and cross-reactivity calculations may be inaccurate due to the exclusion of these cells.
Low-affinity T cells are effective immune mediators and can have dominant roles in the immune response under specific conditions (62, 88, 111–113). When T cell clones are compared for their ability to cause autoimmunity, combat infection, or prevent tumors, low-affinity T cells are often comparable in accomplishing these tasks (22, 62, 97, 112, 113). For example, when a lower affinity CD8 T cell clone specific for an influenza antigen is transferred into a mouse expressing the antigen in a tumor, little immune response occurs (113). However, when this mouse is infected with influenza and/or given CD4 T cell help, the low-affinity T cells can respond with enhanced function (113). Along with influenza, groups have demonstrated adjuvants, such as CFA, MPL, and Listeria monocytogenes, can generate a larger population of low-affinity T cells (6, 112, 114, 115). Besides adjuvants, the form of antigen can control low-affinity T cell expansion as the use of protein antigen has been demonstrated to recruit more low-affinity T cells into the immune response (94). Why antigen and adjuvant influence the affinity diversity of the T cell response is still unclear, though these factors point to the type of antigen presenting cells (APCs) as a possible manipulator of low- and high-affinity T cell skewing. This suggests high- and low-affinity T cells may compete for TCR signaling, but mechanism, such as antigen processing and presentation, may maintain and influence the affinity diversity.
Alteration of APC by using different adjuvants or forms of antigen is one way to potentially alter T cell diversity, but can diversity of affinity be regulated in a T cell intrinsic fashion? As previously mentioned, the 2D affinity measurement by 2D-MP is a relative affinity, dependent unknown factors as well as the contact area restricting the receptor and ligand interactions. Our work reveals slightly different affinities (<10-fold) for thymocytes, peripheral naïve TCR-Tg T cells, and activated T cells demonstrating that the context of the membrane environments plays a role [(13) unpublished data]. Thymocytes, naïve, and activated T cells are different sizes, which may alter contact area between the T cell and RBC during the 2D-MP measurement. At the macroscopic level, the contact area difference between thymocytes and naïve T cells seems negligible, but in fact could result in differences as lymphocytes contain excess membranes, which is stored in ruffles and protrusions that could change during development and activation state (116). If there are differences in the ruffling of the membrane along with size differences, the membrane surface area in contact containing the TCR and pMHC could vary between different populations of cells. Of note, within a given population of cells, the surface area would be similar allowing for accurate affinity measures. These effects on membrane surface would also be predicted to influence lymphocyte function in vivo, which could be why 2D affinity so accurately predicts the level of functional response.
In addition, 2D affinities are dependent on the local membrane structure that is maintained by the actin cytoskeleton and controlled by the membrane lipid composition. During TCR activation, the actin cytoskeleton is remodeled (117). This cytoskeleton change could alter the 2D-MP affinity as the integrity of the membrane structure and orientation of the surface proteins will also fundamentally change. Inhibition of actin polymerization has been demonstrated to reduce 2D affinity as well as functional responses (12, 13). The actin inhibitors again demonstrate how 2D-affinity measurements accurately readout the functionality of the TCR interaction with pMHC (12, 15). A T cell could manipulate its 2D affinity through changes in the cytoskeletal support and protein attachment to actin. Alternative 2D affinity could be regulated to a degree by alterations in lipid content as CD4 T cell subsets contain differential organization of lipids (118, 119). Lipid composition has not been studied in relation to 2D affinity, but lipid order and organization has been demonstrated to be important in T cell functional responses, implicating the affinity may be different (118, 119).
Other surface proteins could influence the ability of the TCR to interact with pMHC and the 2D affinity. In the kinetic-segregation model of T cell activation, the size of the CD45 molecule regulates the interaction of TCR:pMHC, with its exclusion from the synapse a necessary step to initiate the T cell signaling cascade (51, 52). T cell expression of a smaller isoform of CD45 would reduce steric hindrance and could increase the TCR:pMHC affinity measured by 2D-MP. Therefore, when the 2D affinity is calculated in the context of the cellular membrane, multiple T cell intrinsic factors can tune its measured value. In vivo, this fine-tuning of 2D affinity could be envisioned as a mechanism allowing for small alterations in the likelihood of TCR engagement (affinity for pMHC) while maintaining the diversity of the immune response.
Summary
Evidence demonstrates lower affinity T cells most likely have overlapping and distinct roles when compared to T cells with higher affinity interactions. Low-affinity T cells use much of the same signaling machinery for generating an immune response, but also must possess unique pathways or factors to sustain function and prevent excessive negative regulation during signaling. During differentiation, low-affinity T cells can again be found to have shared and unique roles when compared to higher affinity T cells. Low and high-affinity T cells must function together to efficiently generate a complete immune response and maintain the diversity of TCR affinity to efficiently protect the host.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Rakieb Andargachew, Dennis Neeld, Lori Blanchfield and Anna Kersh for helpful discussion.This work was supported by NIH grant T32 AI007610, RO1 NS071518, and RO1 AI110113 to BE and F31 NS086130 to RM.
References
1. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14:377–91. doi: 10.1038/nri3667
2. Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol (2012) 4:a006957. doi:10.1101/cshperspect.a006957
3. Zhu C, Jiang N, Huang J, Zarnitsyna VI, Evavold BD. Insights from in situ analysis of TCR-pMHC recognition: response of an interaction network. Immunol Rev (2013) 251:49–64. doi:10.1111/imr.12016
4. Blanchfield JL, Shorter SK, Evavold BD. Monitoring the dynamics of T cell clonal diversity using recombinant peptide: MHC technology. Front Immunol (2013) 4:170. doi:10.3389/fimmu.2013.00170
5. Busch DH, Pamer EG. T cell affinity maturation by selective expansion during infection. J Exp Med (1999) 189:701–10. doi:10.1084/jem.189.4.701
6. Malherbe L, Hausl C, Teyton L, McHeyzer-Williams MG. Clonal selection of helper T cells is determined by an affinity threshold with no further skewing of TCR binding properties. Immunity (2004) 21:669–79. doi:10.1016/j.immuni.2004.09.008
7. Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature (2009) 458:211–4. doi:10.1038/nature07657
8. Kersh AE, Edwards LJ, Evavold BD. Progression of relapsing-remitting demyelinating disease does not require increased TCR affinity or epitope spread. J Immunol (2014) 193:4429–38. doi:10.4049/jimmunol.1401456
9. Sabatino JJ, Huang J, Zhu C, Evavold BD. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med (2011) 208:81–90. doi:10.1084/jem.20101574
10. Stone JD, Kranz DM. Role of T cell receptor affinity in the efficacy and specificity of adoptive T cell therapies. Front Immunol (2013) 4:244. doi:10.3389/fimmu.2013.00244
11. Kuo SC, Lauffenburger DA. Relationship between receptor/ligand binding affinity and adhesion strength. Biophys J (1993) 65:2191–200. doi:10.1016/S0006-3495(93)81277-3
12. Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, et al. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature (2010) 464:932–6. doi:10.1038/nature08944
13. Huppa JB, Axmann M, Mörtelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature (2010) 463:963–7. doi:10.1038/nature08746
14. Chesla SE, Selvaraj P, Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys J (1998) 75:1553–72. doi:10.1016/S0006-3495(98)74074-3
15. Liu B, Zhong S, Malecek K, Johnson LA, Rosenberg SA, Zhu C, et al. 2D TCR-pMHC-CD8 kinetics determines T-cell responses in a self-antigen-specific TCR system. Eur J Immunol (2014) 44:239–50. doi:10.1002/eji.201343774
16. Adams JJ, Narayanan S, Liu B, Birnbaum ME, Kruse AC, Bowerman NA, et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity (2011) 35:681–93. doi:10.1016/j.immuni.2011.09.013
17. Hamad AR, O’Herrin SM, Lebowitz MS, Srikrishnan A, Bieler J, Schneck J, et al. Potent T cell activation with dimeric peptide-major histocompatibility complex class II ligand: the role of CD4 coreceptor. J Exp Med (1998) 188:1633–40. doi:10.1084/jem.188.9.1633
18. Crawford F, Kozono H, White J, Marrack P, Kappler J. Detection of antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity (1998) 8:675–82. doi:10.1016/S1074-7613(00)80572-5
19. Stone JD, Artyomov MN, Chervin AS, Chakraborty AK, Eisen HN, Kranz DM. Interaction of streptavidin-based peptide-MHC oligomers (tetramers) with cell-surface TCRs. J Immunol (2011) 187:6281–90. doi:10.4049/jimmunol.1101734
20. Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science (1996) 274:94–6. doi:10.1126/science.274.5284.94
21. Kim C, Wilson T, Fischer KF, Williams MA. Sustained interactions between T cell receptors and antigens promote the differentiation of CD4+ memory T cells. Immunity (2013) 39:508–20. doi:10.1016/j.immuni.2013.08.033
22. Dougan SK, Dougan M, Kim J, Turner JA, Ogata S, Cho H-I, et al. Transnuclear TRP1-specific CD8 T cells with high or low affinity TCRs show equivalent anti-tumor activity. Cancer Immunol Res (2013) 1:99–111. doi:10.1158/2326-6066.CIR-13-0047
23. al-Ramadi BK, Jelonek MT, Boyd LF, Margulies DH, Bothwell AL. Lack of strict correlation of functional sensitization with the apparent affinity of MHC/peptide complexes for the TCR. J Immunol (1995) 155:662–73.
24. Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A (2013) 110:6973–8. doi:10.1073/pnas.1221609110
25. Derby MA, Wang J, Margulies DH, Berzofsky JA. Two intermediate-avidity cytotoxic T lymphocyte clones with a disparity between functional avidity and MHC tetramer staining. Int Immunol (2001) 13:817–24. doi:10.1093/intimm/13.6.817
26. Mandl JN, Monteiro JP, Vrisekoop N, Germain RN. T cell-positive selection uses self-ligand binding strength to optimize repertoire recognition of foreign antigens. Immunity (2013) 38:263–74. doi:10.1016/j.immuni.2012.09.011
27. Nelson RW, Beisang D, Tubo NJ, Dileepan T, Wiesner DL, Nielsen K, et al. T cell receptor cross-reactivity between similar foreign and self peptides influences naïve cell population size and autoimmunity. Immunity (2014) 42(1):95–107. doi:10.1016/j.immuni.2014.12.022
28. Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol (2009) 10:375–84. doi:10.1038/ni.1704
29. Puech PH, Nevoltris D, Robert P, Limozin L, Boyer C, Bongrand P. Force measurements of TCR/pMHC recognition at T cell surface. PLoS One (2011) 6(7):e22344. doi:10.1371/journal.pone.0022344
30. Chervin AS, Stone JD, Holler PD, Bai A, Chen J, Eisen HN, et al. The impact of TCR-binding properties and antigen presentation format on T cell responsiveness. J Immunol (2009) 183:1166–78. doi:10.4049/jimmunol.0900054
31. Daniels MA, Jameson SC. Critical role for CD8 in T cell receptor binding and activation by peptide/major histocompatibility complex multimers. J Exp Med (2000) 191:335–46. doi:10.1084/jem.191.2.335
32. Artyomov MN, Lis M, Devadas S, Davis MM, Chakraborty AK. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc Natl Acad Sci U S A (2010) 107:16916–21. doi:10.1073/pnas.1010568107
33. Wooldridge L, van den Berg HA, Glick M, Gostick E, Laugel B, Hutchinson SL, et al. Interaction between the CD8 coreceptor and major histocompatibility complex class I stabilizes T cell receptor-antigen complexes at the cell surface. J Biol Chem (2005) 280:27491–501. doi:10.1074/jbc.M500555200
34. Jiang N, Huang J, Edwards LJ, Liu B, Zhang Y, Beal CD, et al. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity (2011) 34:13–23. doi:10.1016/j.immuni.2010.12.017
35. Garcia KC, Scott CA, Brunmark A, Carbone FR, Peterson PA, Wilson IA, et al. CD8 enhances formation of stable T-cell receptor/MHC class I molecule complexes. Nature (1996) 384:356–8. doi:10.1038/384577a0
36. Wyer JR, Willcox BE, Gao GF, Gerth UC, Davis SJ, Bell JI, et al. T cell receptor and coreceptor CD8 alphaalpha bind peptide-MHC independently and with distinct kinetics. Immunity (1999) 10:219–25. doi:10.1016/S1074-7613(00)80022-9
37. Slifka MK, Whitton JL. Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol (2001) 2:711–7. doi:10.1038/90650
38. Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity (2003) 18:255–64. doi:10.1016/S1074-7613(03)00019-0
39. Laugel B, van den Berg HA, Gostick E, Cole DK, Wooldridge L, Boulter J, et al. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J Biol Chem (2007) 282:23799–810. doi:10.1074/jbc.M700976200
40. Harding S, Lipp P, Alexander DR. A therapeutic CD4 monoclonal antibody inhibits TCR-zeta chain phosphorylation, zeta-associated protein of 70-kDa Tyr319 phosphorylation, and TCR internalization in primary human T cells. J Immunol (2002) 169:230–8. doi:10.4049/jimmunol.169.1.230
41. Pullar CE, Morris PJ, Wood KJ. Altered proximal T-cell receptor signalling events in mouse CD4+ T cells in the presence of anti-CD4 monoclonal antibodies: evidence for reduced phosphorylation of Zap-70 and LAT. Scand J Immunol (2003) 57:333–41. doi:10.1046/j.1365-3083.2003.01241.x
42. Kamperschroer C, Quinn DG. Quantification of epitope-specific MHC class-II-restricted T cells following lymphocytic choriomeningitis virus infection. Cell Immunol (1999) 193:134–46. doi:10.1006/cimm.1999.1458
43. Harrington LE, Most Rv RV, Whitton JL, Ahmed R. Recombinant vaccinia virus-induced T-cell immunity: quantitation of the response to the virus vector and the foreign epitope. J Virol (2002) 76:3329–37. doi:10.1128/JVI.76.7.3329-3337.2002
44. Varga SM, Welsh RM. Detection of a high frequency of virus-specific CD4+ T cells during acute infection with lymphocytic choriomeningitis virus. J Immunol (1998) 161:3215–8.
45. Murali-Krishna K, Altman JD, Suresh M, Sourdive DJD, Zajac AJ, Miller JD, et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity (1998) 8:177–87. doi:10.1016/S1074-7613(00)80470-7
46. Williams MA, Ravkov EV, Bevan MJ. Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity (2008) 28:533–45. doi:10.1016/j.immuni.2008.02.014
47. Martinez RJ, Zhang N, Thomas SR, Nandiwada SL, Jenkins MK, Binstadt BA, et al. Arthritogenic self-reactive CD4+ T cells acquire an FR4hiCD73hi anergic state in the presence of Foxp3+ regulatory T cells. J Immunol (2012) 188:170–81. doi:10.4049/jimmunol.1101311
48. McDermott DS, Varga SM. Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J Immunol (2011) 187:5568–76. doi:10.4049/jimmunol.1102104
49. Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science (1996) 273:104–6. doi:10.1126/science.273.5271.104
50. Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature (1995) 375:148–51. doi:10.1038/375148a0
51. Davis SJ, van der Merwe PA. The kinetic-segregation model: TCR triggering and beyond. Nat Immunol (2006) 7:803–9. doi:10.1038/ni1369
52. Anton van der Merwe P, Davis SJ, Shaw AS, Dustin ML. Cytoskeletal polarization and redistribution of cell-surface molecules during T cell antigen recognition. Semin Immunol (2000) 12:5–21. doi:10.1006/smim.2000.0203
53. Kalergis AM, Boucheron N, Doucey MA, Palmieri E, Goyarts EC, Vegh Z, et al. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat Immunol (2001) 2:229–34. doi:10.1038/85286
54. Sugawara T, Moriguchi T, Nishida E, Takahama Y. Differential roles of ERK and p38 MAP kinase pathways in positive and negative selection of T lymphocytes. Immunity (1998) 9:565–74. doi:10.1016/S1074-7613(00)80639-1
55. Mariathasan S, Zakarian A, Bouchard D, Michie AM, Zuniga-Pflucker JC, Ohashi PS. Duration and strength of extracellular signal-regulated kinase signals are altered during positive versus negative thymocyte selection. J Immunol (2001) 167:4966–73. doi:10.4049/jimmunol.167.9.4966
56. Fischer AM, Katayama CD, Pagès G, Pouysségur J, Hedrick SM. The role of Erk1 and Erk2 in multiple stages of T cell development. Immunity (2005) 23:431–43. doi:10.1016/j.immuni.2005.08.013
57. McNeil LK, Starr TK, Hogquist KA. A requirement for sustained ERK signaling during thymocyte positive selection in vivo. Proc Natl Acad Sci U S A (2005) 102:13574–9. doi:10.1073/pnas.0505110102
58. Fu G, Casas J, Rigaud S, Rybakin V, Lambolez F, Brzostek J, et al. Themis sets the signal threshold for positive and negative selection in T-cell development. Nature (2013) 504:441–5. doi:10.1038/nature12718
59. Stepanek O, Prabhakar AS, Osswald C, King CG, Bulek A, Naeher D, et al. Coreceptor scanning by the T cell receptor provides a mechanism for T cell tolerance. Cell (2014) 159:333–45. doi:10.1016/j.cell.2014.08.042
60. Stritesky GL, Xing Y, Erickson JR, Kalekar LA, Wang X, Mueller DL, et al. Murine thymic selection quantified using a unique method to capture deleted T cells. Proc Natl Acad Sci U S A (2013) 110:4679–84. doi:10.1073/pnas.1217532110
61. Fulton RB, Hamilton SE, Xing Y, Best JA, Goldrath AW, Hogquist KA, et al. The TCR’s sensitivity to self peptide–MHC dictates the ability of naive CD8+ T cells to respond to foreign antigens. Nat Immunol (2014) 16:107–17. doi:10.1038/ni.3043
62. Bettini M, Blanchfield L, Castellaw A, Zhang Q, Nakayama M, Smeltzer MP, et al. TCR affinity and tolerance mechanisms converge to shape T cell diabetogenic potential. J Immunol (2014) 193:571–9. doi:10.4049/jimmunol.1400043
63. Xing Y, Jameson SC, Hogquist KA. Thymoproteasome subunit-β5T generates peptide-MHC complexes specialized for positive selection. Proc Natl Acad Sci U S A (2013) 110:6979–84. doi:10.1073/pnas.1222244110
64. Persaud SP, Parker CR, Lo W-L, Weber KS, Allen PM. Intrinsic CD4+ T cell sensitivity and response to a pathogen are set and sustained by avidity for thymic and peripheral complexes of self peptide and MHC. Nat Immunol (2014) 15:266–74. doi:10.1038/ni.2822
65. Hogquist KA, Jameson SC. The self-obsession of T cells: how TCR signaling thresholds affect fate “decisions” and effector function. Nat Immunol (2014) 15:815–23. doi:10.1038/ni.2938
66. Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol (2013) 13:257–69. doi:10.1038/nri3403
67. Rosenthal KM, Edwards LJ, Sabatino JJ, Hood JD, Wasserman HA, Zhu C, et al. Low 2-dimensional CD4 T cell receptor affinity for myelin sets in motion delayed response kinetics. PLoS One (2012) 7:e32562. doi:10.1371/journal.pone.0032562
68. Stefanová I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol (2003) 4:248–54. doi:10.1038/ni895
69. Salmond RJ, Brownlie RJ, Morrison VL, Zamoyska R. The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat Immunol (2014) 15:875–83. doi:10.1038/ni.2958
70. Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, et al. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest (2013) 123:1044–65. doi:10.1172/JCI65325
71. Liu B, Chen W, Evavold BD, Zhu C. Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell (2014) 157:357–68. doi:10.1016/j.cell.2014.02.053
72. Kumar D, Feng Y, Mallis RJ, Li X, Keskin DB, Hussey RE, et al. Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc Natl Acad Sci U S A (2015) 112:1517–22. doi:10.1073/pnas.1424829112
73. Husson J, Chemin K, Bohineust A, Hivroz C, Henry N. Force generation upon T cell receptor engagement. PLoS One (2011) 6:e19680. doi:10.1371/journal.pone.0019680
74. Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu Rev Immunol (2012) 30:95–114. doi:10.1146/annurev-immunol-020711-075035
75. Jenkins MK, Moon JJ. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J Immunol (2012) 188:4135–40. doi:10.4049/jimmunol.1102661
76. Chu HH, Moon JJ, Takada K, Pepper M, Molitor JA, Schacker TW, et al. Positive selection optimizes the number and function of MHCII-restricted CD4+ T cell clones in the naive polyclonal repertoire. Proc Natl Acad Sci U S A (2009) 106:11241–5. doi:10.1073/pnas.0902015106
77. Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity (2007) 27:203–13. doi:10.1016/j.immuni.2007.07.007
78. Jenkins MK, Chu HH, McLachlan JB, Moon JJ. On the composition of the preimmune repertoire of T cells specific for peptide-major histocompatibility complex ligands. Annu Rev Immunol (2010) 28:275–94. doi:10.1146/annurev-immunol-030409-101253
79. Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell (1994) 76:17–27. doi:10.1016/0092-8674(94)90169-4
80. Merkenschlager M, Benoist C, Mathis D. Evidence for a single-niche model of positive selection. Proc Natl Acad Sci U S A (1994) 91:11694–8. doi:10.1073/pnas.91.24.11694
81. Mingueneau M, Jiang W, Feuerer M, Mathis D, Benoist C. Thymic negative selection is functional in NOD mice. J Exp Med (2012) 209:623–37. doi:10.1084/jem.20112593
82. Ignatowicz L, Kappler J, Marrack P. The repertoire of T cells shaped by a single MHC/peptide ligand. Cell (1996) 84:521–9. doi:10.1016/S0092-8674(00)81028-4
83. Taniguchi RT, DeVoss JJ, Moon JJ, Sidney J, Sette A, Jenkins MK, et al. Detection of an autoreactive T-cell population within the polyclonal repertoire that undergoes distinct autoimmune regulator (Aire)-mediated selection. Proc Natl Acad Sci U S A (2012) 109(20):7847–52. doi:10.1073/pnas.1120607109
84. Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol (2003) 4:350–4. doi:10.1038/ni906
85. Yu W, Jiang N, Ebert PJR, Kidd BA, Muller S, Lund PJ, et al. Clonal deletion prunes but does not eliminate self-specific alpha beta CD8+ T lymphocytes. Immunity (2015) 42:929–41. doi:10.1016/j.immuni.2015.05.001
86. Quiel J, Caucheteux S, Laurence A, Singh NJ, Bocharov G, Ben-Sasson SZ, et al. Antigen-stimulated CD4 T-cell expansion is inversely and log-linearly related to precursor number. Proc Natl Acad Sci U S A (2011) 108:3312–7. doi:10.1073/pnas.1018525108
87. Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity (2006) 25:261–70. doi:10.1016/j.immuni.2006.06.009
88. Knudson K, Goplen N, Cunningham C, Daniels M, Teixeiro E. Low-affinity T cells are programmed to maintain normal primary responses but are impaired in their recall to low-affinity ligands. Cell Rep (2013) 4:554–65. doi:10.1016/j.celrep.2013.07.008
89. King CG, Koehli S, Hausmann B, Schmaler M, Palmer E. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity (2013) 37:709–20. doi:10.1016/j.immuni.2012.06.021
90. Vrisekoop N, Monteiro JP, Mandl JN, Germain RN. Revisiting thymic positive selection and the mature T cell repertoire for antigen. Immunity (2014) 41:181–90. doi:10.1016/j.immuni.2014.07.007
91. Lee H-M, Bautista JL, Scott-Browne J, Mohan JF, Hsieh C-S. A broad range of self-reactivity drives thymic regulatory T cell selection to limit responses to self. Immunity (2012) 37:475–86. doi:10.1016/j.immuni.2012.07.009
92. Tubo NJ, Pagán AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single naive CD4(+) T cells from a diverse repertoire produce different effector cell types during infection. Cell (2013) 153:785–96. doi:10.1016/j.cell.2013.04.007
93. Keck S, Schmaler M, Ganter S, Wyss L, Oberle S, Huseby ES, et al. Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc Natl Acad Sci U S A (2014) 111:14852–7. doi:10.1073/pnas.1403271111
94. Baumgartner CK, Yagita H, Malherbe LP. A TCR affinity threshold regulates memory CD4 T cell differentiation following vaccination. J Immunol (2012) 189:2309–17. doi:10.4049/jimmunol.1200453
95. Van Panhuys N, Klauschen F, Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization in vivo. Immunity (2014) 41(1):63–74. doi:10.1016/j.immuni.2014.06.003
96. Brogdon JL, Leitenberg D, Bottomly K. The potency of tcr signaling differentially regulates NFATc/p activity and early IL-4 transcription in naive CD4+ T cells. J Immunol (2002) 168:3825–32. doi:10.4049/jimmunol.168.8.3825
97. Caserta S, Kleczkowska J, Mondino A, Zamoyska R. Reduced functional avidity promotes central and effector memory CD4 T cell responses to tumor-associated antigens. J Immunol (2010) 185:6545–54. doi:10.4049/jimmunol.1001867
98. O’Sullivan D, van der Windt GJW, Huang SC-C, Curtis JD, Chang C-H, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41:75–88. doi:10.1016/j.immuni.2014.06.005
99. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi:10.1016/j.immuni.2011.09.021
100. Van der Windt GJW, O’Sullivan D, Everts B, Huang SC-C, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A (2013) 110:14336–41. doi:10.1073/pnas.1221740110
101. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol (2013) 14:1155–65. doi:10.1038/ni.2710
102. Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol (2004) 4:123–32. doi:10.1038/nri1292
103. Moon JJ, Chu HH, Hataye J, Pagán AJ, Pepper M, McLachlan JB, et al. Tracking epitope-specific T cells. Nat Protoc (2009) 4:565–81. doi:10.1038/nprot.2009.9
104. Zarnitsyna VI, Evavold BD, Schoettle LN, Blattman JN, Antia R. Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front Immunol (2013) 4:485. doi:10.3389/fimmu.2013.00485
105. Langman RE, Cohn M. The E-T (elephant-tadpole) paradox necessitates the concept of a unit of B-cell function: the protection. Mol Immunol (1987) 24:675–97. doi:10.1016/0161-5890(87)90050-2
106. Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem (2012) 287:1168–77. doi:10.1074/jbc.M111.289488
107. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell (2014) 157:1073–87. doi:10.1016/j.cell.2014.03.047
108. Holler PD, Lim AR, Cho BK, Rund LA, Kranz DM. CD8(-) T cell transfectants that express a high affinity T cell receptor exhibit enhanced peptide-dependent activation. J Exp Med (2001) 194:1043–52. doi:10.1084/jem.194.8.1043
109. Donermeyer DL, Weber KS, Kranz DM, Allen PM. The study of high-affinity TCRs reveals duality in T cell recognition of antigen: specificity and degeneracy. J Immunol (2006) 177:6911–9. doi:10.4049/jimmunol.177.10.6911
110. Holler PD, Chlewicki LK, Kranz DM. TCRs with high affinity for foreign pMHC show self-reactivity. Nat Immunol (2003) 4:55–62. doi:10.1038/ni863
111. O’Sullivan JA, Zloza A, Kohlhapp FJ, Moore TV, Lacek AT, Dulin NO, et al. Priming with very low-affinity peptide ligands gives rise to CD8+ T-cell effectors with enhanced function but with greater susceptibility to transforming growth factor (TGF)β-mediated suppression. Cancer Immunol Immunother (2011) 60:1543–51. doi:10.1007/s00262-011-1043-1
112. Khan N, Cobbold M, Cummerson J, Moss PAH. Persistent viral infection in humans can drive high frequency low-affinity T-cell expansions. Immunology (2010) 131:537–48. doi:10.1111/j.1365-2567.2010.03326.x
113. Lyman MA, Nugent CT, Marquardt KL, Biggs JA, Pamer EG, Sherman LA. The fate of low affinity tumor-specific CD8+ T cells in tumor-bearing mice. J Immunol (2005) 174:2563–72. doi:10.4049/jimmunol.174.5.2563
114. Rees W, Bender J, Teague TK, Kedl RM, Crawford F, Marrack P, et al. An inverse relationship between T cell receptor affinity and antigen dose during CD4(+) T cell responses in vivo and in vitro. Proc Natl Acad Sci U S A (1999) 96:9781–6. doi:10.1073/pnas.96.17.9781
115. Srinivasan A, Foley J, McSorley SJ. Massive number of antigen-specific CD4 T cells during vaccination with live attenuated Salmonella causes interclonal competition. J Immunol (2004) 172:6884–93. doi:10.4049/jimmunol.172.11.6884
116. Majstoravich S, Zhang J, Nicholson-Dykstra S, Linder S, Friedrich W, Siminovitch KA, et al. Lymphocyte microvilli are dynamic, actin-dependent structures that do not require Wiskott-Aldrich syndrome protein (WASp) for their morphology. Blood (2004) 104:1396–403. doi:10.1182/blood-2004-02-0437
117. Sechi AS, Wehland J. Interplay between TCR signalling and actin cytoskeleton dynamics. Trends Immunol (2004) 25:257–65. doi:10.1016/j.it.2004.03.003
118. Miguel L, Owen DM, Lim C, Liebig C, Evans J, Magee AI, et al. Primary human CD4+ T cells have diverse levels of membrane lipid order that correlate with their function. J Immunol (2011) 186:3505–16. doi:10.4049/jimmunol.1002980
Keywords: TCR affinity, 2D assays, tetramers, T cells, T cell diversity
Citation: Martinez RJ and Evavold BD (2015) Lower affinity T cells are critical components and active participants of the immune response. Front. Immunol. 6:468. doi: 10.3389/fimmu.2015.00468
Received: 04 July 2015; Accepted: 28 August 2015;
Published: 10 September 2015
Edited by:
Eric Huseby, University of Massachusetts Medical School, USAReviewed by:
Tomasz Zal, University of Texas MD Anderson Cancer Center, USABrian M. Baker, University of Notre Dame, USA
Copyright: © 2015 Martinez and Evavold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian D. Evavold, Department of Microbiology and Immunology, Emory University, 1510 Clifton Road NE, Room 3127, Atlanta, GA 30322, USA, bevavol@emory.edu