 Nadège Bercovici
Nadège Bercovici Marion V. Guérin1,2,3
Marion V. Guérin1,2,3 Alain Trautmann
Alain Trautmann Emmanuel Donnadieu
Emmanuel Donnadieu- 1INSERM, U1016, Institut Cochin, Paris, France
- 2CNRS, UMR8104, Paris, France
- 3Université Paris Descartes, Sorbonne Paris Cité, Paris, France
It is well established that tumor-associated macrophages (TAM) found in most advanced tumors have a pro-tumoral role. In this context, TAM limit the activity of tumor-infiltrating lymphocytes (TIL), and a number of mechanisms have been described including a trapping in the stroma, impeding TIL to reach malignant cells. Based on these results, a number of therapeutic approaches have been designed to deplete TAM. However, during tumor regression induced by immunotherapeutic treatments, recent studies revealed that TAM can switch from pro-tumoral to anti-tumoral and actively cooperate with TIL. Here, we will review the two faces of TAM in their interaction with TIL. We will summarize how they can inhibit T cell activities in growing tumors, and how they may also, together with T cells, successfully contribute to tumor eradication after an appropriate stimulation. Finally, we will discuss current promising therapies combining TAM reprogramming with T cell-based immunotherapy.
Introduction
Macrophages are amongst the most versatile cells in the body. Resident macrophages are abundant in all tissues where, like microglia in the brain or Kuppfer cells in the liver, these “pro-tissular macrophages” contribute to optimize the functioning of the tissue in which they are, by maintaining it clean and preventing an unnecessary inflammation (1). Besides, following an appropriate stimulation, e.g., following an infection, macrophages may be key contributors to immune responses (2). They participate to a variety of functions, primarily as effector cells to eliminate the invading bodies but also to drive an acute inflammation, to promote the recruitment of other immune cells as well as to present antigens to T cells. The switch from the pro-tissular, anti-inflammatory state to the pro-immune, inflammatory one, may take place within a few minutes. This is what happens to subcapsular macrophages when they detect the arrival of pathogens in the lymph node subcapsular sinus (3). This switch may take hours or days, when it involves the recruitment of blood monocytes, followed by their appropriate differentiation in the tissue. Even though the distinction between pro-tissular and pro-immune macrophages shares similarities with the M2/M1 distinction, we favor the idea that the most important difference between these two macrophage subtypes is functional rather than phenotypic.
In advanced tumors, macrophages favor tumor growth and are associated with a bad outcome in most cancers. Therefore, tumor-associated macrophages (TAM) are usually considered as simply “pro-tumoral”. This has not always been the case. In the 1990s, a potential role of macrophages for cancer treatment has been a popular idea and this concept has begun to emerge. Indeed, in sensitized tumors, macrophages may be anti-tumoral, with the modulation of some gene expression (4).
We will summarize here some specific consequences of the functioning of macrophages in progressing tumors, in which their dominant role is pro-tumoral and immunosuppressive. In particular, we will focus on the mechanisms by which TAM limit TIL from reaching tumor cells. We will continue by considering how one can favor the switch of TAM to pro-immune cells exerting an anti-tumoral action. For these two TAM faces, our focus will be on positive or negative interactions between TAM and TIL, as summarized in the Figure 1.
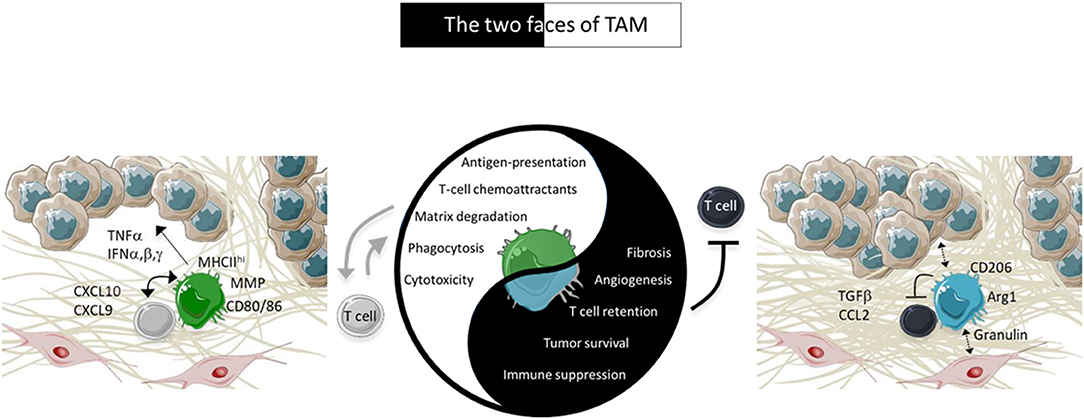
Figure 1. Characteristics of TAM in tumors. In advanced tumors, TAM are pro-tissular and promote tumor growth by several ways including by trapping TIL in the stroma. However, under appropriate activation, TAM positively cooperate with TIL to induce tumor regression. The pro-tissular and pro-immune TAM subtypes are present in two different environments depicted in the figure. Pro-tissular TAM reside in a mesenchymal environment enriched in a dense ECM network and TGFβ. Conversely, pro-immune TAM are distributed in an inflammatory milieu enriched in type I IFN and T-cell chemoattractants.
TAM Inhibit T Cell Activities in Progressing Tumors
TAM can promote tumor growth by a variety of mechanisms that include tumor cell proliferation, metastasis, angiogenesis and inhibition of T cell anti-tumoral activities. A considerable number of excellent reviews have been published on the various ways in which TAM contribute to tumor growth [for instance see (5)]. Yet, the mechanisms by which TAM negatively control T cells are not completely understood and we would like to focus on those related to intratumoral T cell migration.
TAM Impair T Cell Migration Within Tumors
Our team has recently shown that, in untreated progressing tumors, TAM have a detrimental impact on TIL ability to migrate within tumors and contact malignant cells (6). By using an experimental system based on thick slices made from fresh tumor biopsies combined with fluorescent imaging microscopy, we evidenced the presence of stable conjugates formed between TAM and CD3 T cells in the stroma of human lung tumors as illustrated in the Figure 2A. If such interactions do not result in T cell activation, macrophages could contribute to sequestering lymphocytes away from tumor cells (6). Remarkably, in mouse mammary tumor models we found that the depletion of TAM with pexidartinib, an inhibitor of the colony stimulating factor 1 receptor (CSF1R), increased the motility of TIL and their ability to reach tumor cells. This is consistent with data obtained in a mouse model of pancreatic carcinoma but with a CD8 T cell-macrophage trapping process that occurs outside the tumor (7). Whether a similar mechanism also affects CD4 T cells is unknown for the moment. In murine lymph nodes, macrophages were shown to sequester γδT cells unable to recirculate in the blood (8).
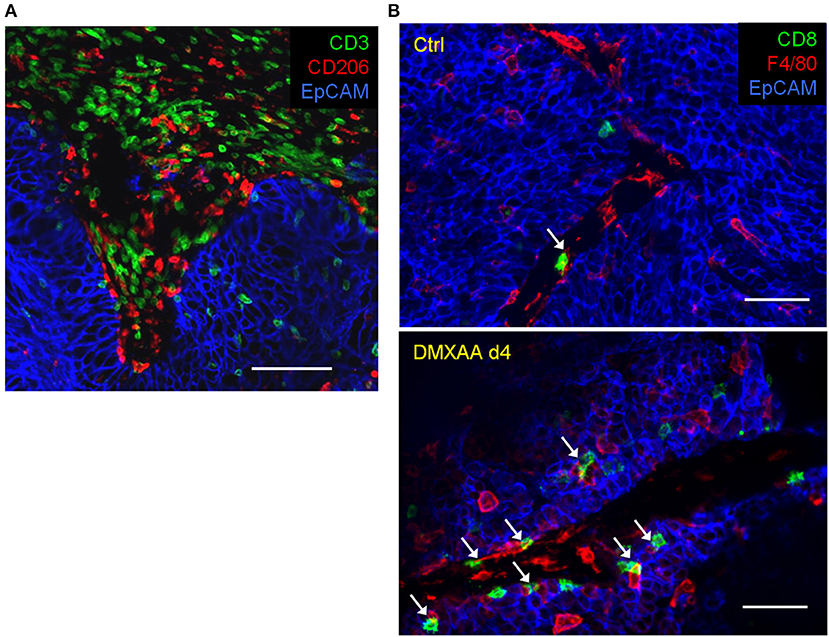
Figure 2. Within tumors, T cells and macrophages are often in contact. (A) In a human lung tumor, numerous CD3 T cells are potentially interacting with macrophages (stained by CD206). Bar, 100 μm. (B) Interactions between TIL and TAM in mammary carcinoma mouse tumors (MMTV-PyMT) before and after 4 days (4d) of treatment with the STING agonist DMXAA. Of note, the immunotherapeutic agent induces a massive recruitment of CD8 T cells and macrophages (stained by F4/80) with many contacts between both cell types. White arrows indicate TAM-T cells contacts. Bar 50 μm.
Altogether, these data suggest that TAM participate to the exclusion of TIL from the vicinity of cancer cells which is considered to be a major hurdle for T cell-based anti-tumor immunotherapy.
Mechanisms Underlying Blockade of T Cell Migration by TAM
The mechanisms by which TAM prevent CD8 T cells from reaching tumor cells is not known at the moment. We favor an adhesion process between both cell types triggered by an antigen recognition which by itself is insufficient to trigger full T cell activation. This would be in line with data showing antigen-dependent interactions between CD8 T cells and myeloid cells in a spontaneous mammary carcinoma murine model (9). However, the nature of the adhesion molecules involved in such cell-cell conjugates needs to be further investigated.
An effect of TAM on environmental factors controlling the motility of T cells cannot be ruled out. Studies performed over the last few years have provided evidence for a role of the structure of the tissue and the presence of chemokines in regulating the migration of T cells (10). By tracking T cells in fresh human lung tumor slices, we reported an important role of chemokines produced by tumor cells in the ability of T cells to infiltrate tumor islets (11). Such chemokines contribute to a low grade chronic inflammation. In mice harboring mammary tumors, we found that the depletion of TAM resulted in more inflammatory chemokines, such as CCL2 and CXCL10, which are likely to enhance the entry of T cells into the tumor and their intratumoral migration (6). The reason of an enhanced production of chemokines upon TAM depletion is not known for the moment. One possibility is that TAM could participate to the degradation and/or inactivation (e.g., nitration) of inflammatory chemokines, a process reported to occur in murine tumors (12).
A hallmark of advanced tumors is the development of a fibrosis characterized by an excessive accumulation of collagen I, likely to favor tumor progression and prevent antitumor T cell functions by limiting lymphocytes from migrating and contacting tumor cells, as we have previously demonstrated (13). Thus, a dense extracellular matrix (ECM) made by activated carcinoma-associated fibroblasts (CAF) might be responsible for the excluded T cell profile observed in various human carcinomas. The cells and elements that are susceptible to enhance collagen I production by CAF include macrophages. In many physiological situations like breast development, macrophages actively participate to the construction of the tissue (14, 15). In addition, the number of pro-tissular macrophages parallels the amount of tissue fibrosis in many human tumors. For example in colorectal tumors and in melanoma, a mesenchymal signature, associated with a bad outcomes and resistance of PD-1 therapy, are characterized by genes involved in extracellular matrix remodeling, angiogenesis, wound healing and TAM suggesting that pro-tissular macrophages and CAF are part of a similar environment (16–18). Evidence obtained in mouse models of colon cancer and pancreatic ductal adenocarcinoma indicates a role of TAM in ECM production within the tumor suggesting that TAM could indirectly inhibit T cell migration through the construction of a dense stroma (19, 20). TAM can fine-tune fibrosis by depositing and/or remodeling the ECM (20) but indirect effects through cross-talks with CAF are also envisioned (Figure 1). In that context, a recent study demonstrates that TAM activate CAF to produce excessive amount of the ECM, excluding T cells from tumor cells, through the secretion of granulin, a growth factor belonging to the epithelin family (21).
Macrophage-Depletion Strategies May Potentiate Anti-tumor T-Cell Therapies
The aforementioned studies demonstrating a negative impact of TAM on T cells fostered the development of strategies combining pro-tissular macrophage-depletion with approaches that boost T cells. In preclinical mouse tumor models, the depletion of TAM has been combined with T cell-based therapies, both anti-PD-1 and adoptive T cell transfer, which results in enhanced efficacy of the immunotherapy treatment (22–26). For example, we have shown that a macrophage-depletion strategy through CSF1R inhibition, which by itself has a minor effect on the tumor growth, also improved the efficacy of an anti-PD-1 treatment (6).
Based on these results, several therapeutic applications to impair TAM recruitment or survival are either entering or have entered clinical trials (27). CSF1R inhibitors are currently being tested, the most advanced being the small-molecule Pexidartinib (28). However, CSF1R inhibitors have shown very limited antitumor effects in patients as single agents, suggesting the need to combine these inhibitors with other approaches, including immune checkpoint inhibitors. Such combination strategies are ongoing in a number of solid malignancies (NCT02452424, NCT02713529). Macrophages also use the CCL2/CCR2 axis to enter into tumors. Thus, anti-CCR2 approaches are being developed to reduce the number of immunosuppressive macrophages into solid malignancies (29). In addition, chemotherapeutic agents (e.g., gemcitabine, cyclophosphamide, trabectedin), although not specific to TAM, have been shown to deplete myeloid cells (30–32).
Toward Strategies Favoring an Anti-tumoral Role of TAM
There is increasing evidence that an appropriate activation of macrophages, rather than their depletion, would drastically potentiate an anti-tumor immune response. Macrophages appropriately stimulated by TLR ligands or after abundant cell death, be it induced by radiotherapy, chemotherapy or other means, are key players of an acute inflammation, with numerous consequences. First, inflammatory macrophages release chemokines, leading to the recruitment of innate immune cells and T cells (33). Another major feature of acute inflammation is the activation of the tumor vasculature which controls T cell extravasation (34, 35). In addition, activated macrophages can attack and reduce the density of the intratumoral ECM (36), thus facilitating TIL mobility in the tumor microenvironment. Finally, macrophages are the most abundant cells in tumors, after tumor cells themselves, which constitute a major asset to propagate de novo inflammatory process in the tumor microenvironment. A careful analysis of the various TAM subsets infiltrating human tumors revealed in addition that a high density of anti-tumoral TAM correlated with a favorable prognosis (37).
Historically, various clinical trials were initiated, for instance with the injection of activated macrophages (38) or cytokines and microbial derived molecules (39–43) aiming at activating macrophages directly in vivo. Anti-tumoral effects of such macrophage activators have been reported, but such approaches have been globally disappointing. The conclusion to be drawn from this scientific period is that targeting only macrophages cannot induce a systematic tumor regression.
These provisional failures and the very fast increase of T cell knowledge led, in the following years, to an almost complete oversight of anti-tumoral capabilities of macrophages. However, recent advances in myeloid cell biology are putting macrophages back into play, as we will discuss.
Inhibition of Pro-tumoral TAM Orientation
The inhibition of molecules that contribute to a pro-tumoral TAM orientation represents an interesting strategy. Here we will focus on TGFβ, PI3Kγ, and some HDAC (histone deacetylases) which will nicely illustrate our point.
TGFβ Inhibition
The major effect of TGFβ, in large tumors in particular, is to resolve inflammation, to facilitate wound healing and to contribute to immunosuppression. TGFβ does it in various ways. In tumors, TAM are both a source and a target of TGFβ, which is involved in a positive feed-back loop for stabilizing the pro-tumoral TAM phenotype. In parallel, TGFβ promotes the activation of fibroblasts while it inhibits the expression of several molecules necessary for the cytotoxic activity of T cells. Therefore, this molecule appeared to be a central mediator in the TAM/CAF/TIL axis. In the literature on erythropoiesis or on fibrotic diseases, one can find that an anti-TGFβ treatment could be done, in principle, by reducing the concentration of circulating TGFβ with anti-TGFβ antibodies or with TGFβ ligand traps (44), or with pharmacological blockers of TGFβ signaling, such as SB431542 (45). Importantly for the activity of anti-tumor T cells, the combination of TGFβ blockade and anti-PDL1 has been shown to decrease the activity of fibroblasts and the density of ECM relieving the exclusion of T cells from malignant cells (46, 47). Another major effect of TGFβ is the inhibition of IFNβ signaling as we have recently shown in murine spontaneous tumors (48). Type I IFN are central in the initiation of T-cell responses and this finding could be of major importance for combined anti-tumor treatments, in which an anti-TGFβ would not be active on its own, but only for amplifying the potency of T cells. An anti-TGFβ treatment could modify the TAM phenotype and sensitivity to STING agonist within 3 days in this spontaneous tumor model (48).
Class II HDAC Inhibition
Class IIa HDAC, which includes HDAC4, act quite differently from other HDAC. A specific Class IIa HDAC inhibitor, which appears to have no effect on T cells, is able to induce an inflammatory state by promoting the infiltration of phagocytic and immunostimulatory CD40+ TAM, resulting in an anti-tumoral action (49). Thus, specific inhibitors of class IIa HDAC might prove to be of great interest in combined anti-tumoral therapies. Note that, in different instances, TGFβ effects appear to be mediated by HDAC4 (48, 50, 51). Thus, the two molecules may use common pathways to maintain an immunosuppressive, anti-inflammatory macrophage activity.
PI3Kγ Inhibition
PI3Kγ is a PI3K activated by chemokine receptors (52). In myeloid cells, PI3Kγ is important both for the recruitment of monocytes/macrophages and neutrophils (53), and for the resolution of inflammation, in particular with macrophages phagocyting apoptotic neutrophils. In macrophages, the dominant role of PI3Kγ is the resolution of inflammation and immunosuppression (54). Under these conditions, specific inhibitors of PI3Kγ (55, 56) could obviously be of major interest in combined anti-tumoral therapies.
Note that a recent study has evidenced the role of the scavenger receptor clever-1 in TAM pro-tumoral activities. In mouse tumor models, clever-1 blockade leads to macrophage repolarization that become immunostimulatory enhancing T cell responses against tumors (57). A phase I clinical trial with a blocking anti-clever-1 antibody is currently ongoing in various solid tumors (NCT03733990).
Activation of Anti-tumoral TAM Activities
We would like to shed light on two main pathways promoting an anti-tumoral TAM activity. One involves the CD40 pathway, the other one the induction of type I IFN.
The CD40L-CD40 pathway may be activated by CD40 agonists or with cells that express CD40L as it is the case with activated TIL or NKT (58). In a mouse model of pancreatic cancer, the density of the ECM was shown to be reduced after an anti-CD40 agonist treatment, through the activation of matrix metalloproteinases production by TAM, which may facilitate the motility of T cells (59). Further work from the same group indicates that a CD40 agonist triggers the release of IFNγ and CCL2 responsible for both the recruitment of monocytes/macrophages into the tumor and their polarization toward ECM-degrading cells (60).
Even if anti-CD40 antibodies may not only target TAM and DCs, but also other CD40-expressing cells such as endothelial cells, in combination with gemcitabine, CD40 agonists have already been shown to induce clinical responses in patients with surgically incurable pancreatic cancer (59). This CD40-dependent TAM activation is more efficient when combined with T cell activation (61), or with TLR9 stimulation (62).
Type I IFN has also been shown to enhance anti-tumor activities of myeloid cells. The release of IFNα/β in tumors can be achieved by irradiation (63), some chemotherapeutic agents (64) but more efficiently by a direct activation of the STING (Stimulator of Interferon Genes) molecule. TLR ligands, such as CpG, may also result in the production of IFNα/β by TAM. We have recently shown that this type I IFN contribution to anti-tumoral treatments may be strongly inhibited by TGFβ that accumulates abundantly in spontaneous tumors (48). TGFβ inhibition may therefore be an important element of an efficient combined treatment stimulating anti-tumor activity of TAM.
Overall, the balance toward anti-tumor activity of TAM maybe switched ON if one aims at inhibiting their pro-tissular activity while favoring their pro-immune activity. Various clinical trials are ongoing with such macrophages targeting agents (65).
The duration of an increased anti-tumoral activity of TAM is a question that warrants further investigations. Recruited macrophages with high cytotoxic and phagocytic activities were found to accumulate between 4 and 8 days following treatments (30, 33, 66). Thereafter, factors of the tumor microenvironment, such as VEGF, have been shown to influence the reconstitution of the TAM compartment (30), and to promote tumor outgrowth. The persistence of anti-tumor activity of macrophages may also depend on their interactions with anti-tumor T cells as will be discussed below.
Activated TAM Cooperate With TIL for a Global Anti-tumoral Activity
An increasing number of reports lead to the conclusion that T cells and TAM can cooperate to fight tumors. We have shown that activated TAM were necessary to reject transplanted tumors after therapeutic vaccination (66) or STING /type I IFN activation (33). Importantly, STING exerts an anti-tumoral activity involving both TAM and T cells (33, 48) with a key role exerted by IFNα/β production by TAM. In such an acute inflammatory context, the depletion of TAM drastically reduced the production of T-cell chemoattractants and the accumulation of CD8 T cells in tumors. Thus, TAM can either favor or prevent intra-tumoral T cell infiltration, depending on whether an inflammatory/immune response has been triggered or not. In addition, a positive feed-back may be observed, with T cells amplifying the activity of immunostimulatory TAM (66). This demonstrates that TAM and anti-tumoral CD8 TIL can work in synergy to reject tumors following an appropriate stimulation.
As a matter of fact, TAM-T cells positive interactions have been observed in various settings, but were rarely put at the forefront. Such positive interactions have been reported after intratumoral injection of TLR3 or TLR9 agonists (67, 68), after the use of checkpoints blockers anti-PD1/anti-CTLA4 (69), or after the adoptive transfer of tumor-infiltrating T cells (70) as well as chimeric antigen receptor (CAR) T cells (71). Table 1 summarizes clinical trials in which a combination of drugs targeting TAM and TIL has been evaluated.
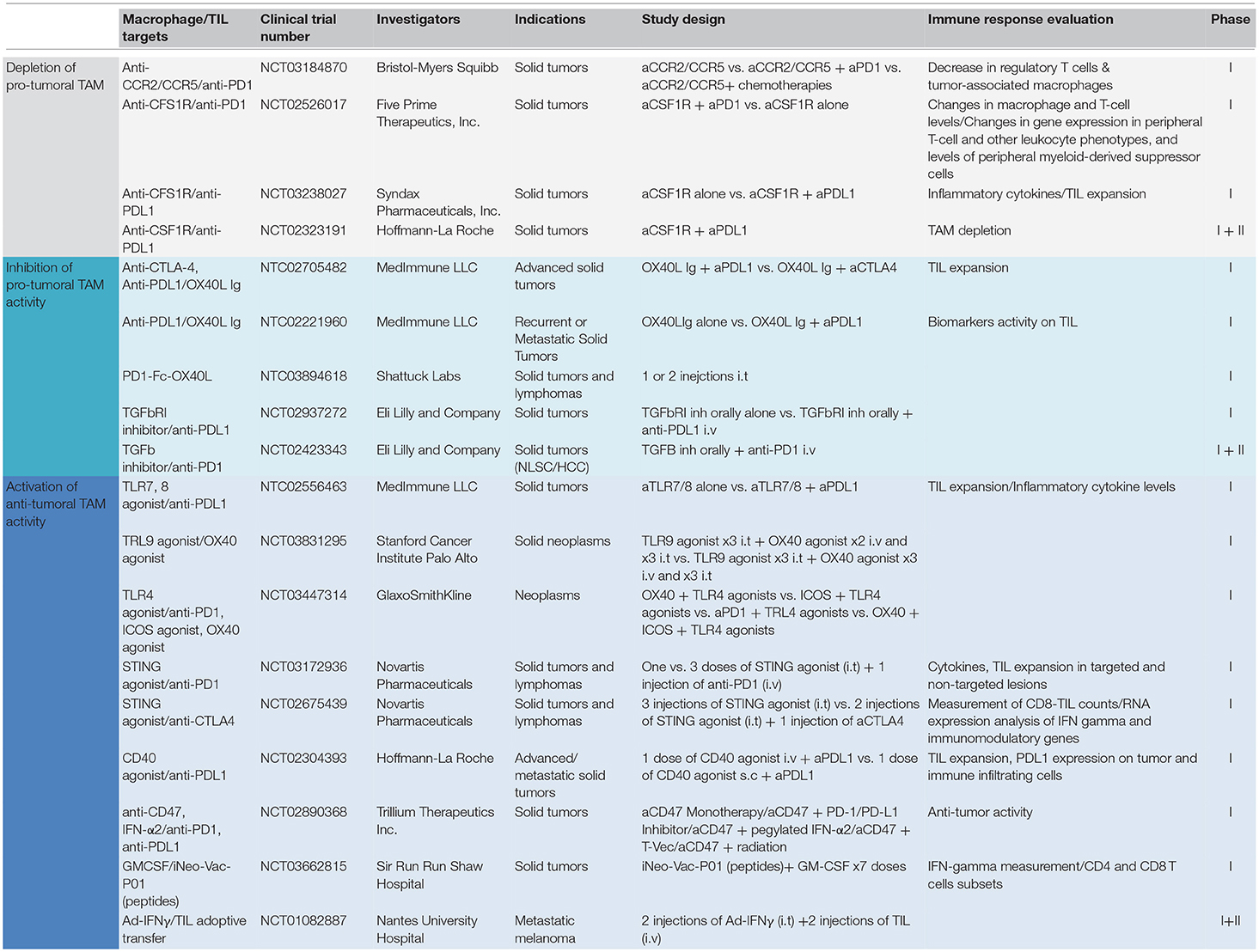
Table 1. Ongoing clinical trials targeting TAM and TIL in solid tumors.
Consequences of TAM-T Cell Cooperation
After facilitating the entry of T cells in sensitized tumors, TAM can interact closely with T cells as illustrated in Figure 2B and present tumor antigens, and thus reactivate them (72). The importance of such a reactivation may be illustrated by the fact that MHC class I expression on tumor infiltrating myeloid cells is strikingly crucial for the rejection of B16 tumor cells by adoptively transferred tumor-specific CD8 T cells (73).
Thus, TAM may help T cells, but reciprocally, T cells can contribute to macrophage activation, and the release of IFNγ seems determinant in this process (66). For instance, the anti-tumor potency of some adoptively transferred T cells was shown to rely on the IFNγ-dependent activation of TAM (74).
Another consequence of such a TAM repolarization is the acquisition of effector functions by these activated cells. Indeed, appropriately activated TAM can phagocyte and engulf tumor cells (75, 76). Additionally, they can kill tumor cells, as shown by several groups (67, 68), including ours (66). TAM, endowed with cytotoxic and cytostatic activities, can kill malignant cells by TNFα secretion (66, 77), NO (74) and sometimes on TRAIL (78).
Conclusion
The common point of view that TAM are pro-tumoral cells is only correct in one specific situation: that of growing tumors. We have recalled that TAM could play such a role in different ways, including by trapping T cells in the tumor stroma and by reducing their mobility and therefore their capacity to reach cancer cells.
However, we have also discussed that TAM, when appropriately stimulated, have the capacity to cooperate with T cells for an anti-tumoral action. Despite the well-established importance of such a cooperation in anti-infectious immune responses, its importance in anti-tumoral responses has been too often neglected. First, 30 years ago, it has been neglected by those who attempted to treat cancer by only stimulating the innate immune system. More recently, the importance of the TAM-T cell cooperation has again been ignored by those who considered that, for anti-tumoral immune responses, T cells were the good guys and macrophages the bad ones.
We have shown here that an efficient strategy should aim at stimulating both T cells and TAM so as to promote their cooperation. This cooperation is not just about a help provided by TAM to T cells: the two cell types may be both helpers for the other, and final effectors against the tumor. Treatments aiming at stimulating only one cell type (only T cells or only macrophages) should be systematically replaced by well-thought combined treatments for stimulating both of them. In particular, T cell-focused treatments with checkpoint inhibitors or CAR T cells would greatly benefit being combined with activators of pro-immune TAM activities such as CD40 agonists, STING activators or otherof IFNα inducers, and with inhibitors of pro-tissular TAM functions, i.e., inhibitors ofTGFβ or class IIa HDAC. The only caveat we would put on it is the potential immune-related adverse effects that may follow efficient tumor rejection. But taking into account that such triple combinations have the greatest chances to promote efficient anti-tumoral therapies would be a fundamental step forward.
Author Contributions
ED, AT, and NB conceived the article, wrote the first version of the manuscript, and integrated inputs from MG. MG designed the Table 1 with the contribution of NB and the Figure 1 with the contribution of AT. ED and AT designed the Figure 2. All authors approved the final version of the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was granted by Worldwide Cancer Research, the Plan Cancer (Tumor Heterogeneity and Ecosystem Program), the Comité de Paris de La Ligue Contre le Cancer, the Cancer Research for Personalized Medicine (CARPEM), CNRS, INSERM, and University Paris-Descartes. MG was granted by the Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche. We are grateful to E. Peranzoni and IMAG'IC facility of the Cochin Institute for the Figure 2.
Abbreviations
CAF, carcinoma-associated fibroblasts; CAR, chimeric antigen receptor; CSF1R, colony stimulating factor 1 receptor; ECM, extracellular matrix; HDAC, histone deacetylase; IFN, interferon; PI3K, phosphatidylinositol 3-kinase; STING, stimulator of interferon genes; TAM, tumor-associated macrophages; TIL, tumor-infiltrating lymphocytes; TGFβ, transforming growth factor beta.
References
1. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
2. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
3. Sagoo P, Garcia Z, Breart B, Lemaitre F, Michonneau D, Albert ML, et al. In vivo imaging of inflammasome activation reveals a subcapsular macrophage burst response that mobilizes innate and adaptive immunity. Nat Med. (2016) 22:64–71. doi: 10.1038/nm.4016
4. Guerriero JL. Macrophages: the road less traveled, changing anticancer therapy. Trends Mol Med. (2018) 24:472–89. doi: 10.1016/j.molmed.2018.03.006
5. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
6. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci USA. (2018) 115:E4041–E50. doi: 10.1073/pnas.1720948115
7. Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology. (2015) 149:201–10. doi: 10.1053/j.gastro.2015.04.010
8. Audemard-Verger A, Riviere M, Durand A, Peranzoni E, Guichard V, Hamon P, et al. Macrophages induce long-term trapping of gammadelta T cells with innate-like properties within secondary lymphoid organs in the steady state. J Immunol. (2017) 199:1998–2007. doi: 10.4049/jimmunol.1700430
9. Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, et al. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. (2012) 21:402–17. doi: 10.1016/j.ccr.2012.01.008
10. Krummel MF, Bartumeus F, Gerard A. T cell migration, search strategies and mechanisms. Nat Rev Immunol. (2016) 16:193–201. doi: 10.1038/nri.2015.16
11. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Investigat. (2012) 122:899–910. doi: 10.1172/JCI45817
12. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
13. Peranzoni E, Rivas-Caicedo A, Bougherara H, Salmon H, Donnadieu E. Positive and negative influence of the matrix architecture on antitumor immune surveillance. Cell Mol Life Sci. (2013) 70:4431–48. doi: 10.1007/s00018-013-1339-8
14. Ingman WV, Wyckoff J, Gouon-Evans V, Condeelis J, Pollard JW. Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev Dyn. (2006) 235:3222–9. doi: 10.1002/dvdy.20972
15. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
16. Becht E, de Reynies A, Giraldo NA, Pilati C, Buttard B, Lacroix L, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. (2016) 22:4057–66. doi: 10.1158/1078-0432.CCR-15-2879
17. Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. (2015) 47:320–9. doi: 10.1038/ng.3225
18. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. (2016) 165:35–44. doi: 10.1016/j.cell.2016.02.065
19. Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M, et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med. (2016) 213:2315–31. doi: 10.1084/jem.20151193
20. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:323–38 e6. doi: 10.1016/j.immuni.2017.07.014
21. Quaranta V, Rainer C, Nielsen SR, Raymant ML, Ahmed MS, Engle DD, et al. Macrophage-derived granulin drives resistance to immune checkpoint inhibition in metastatic pancreatic cancer. Cancer Res. (2018) 78:4253–69. doi: 10.1158/0008-5472.CAN-17-3876
22. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
23. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. (2014) 74:153–61. doi: 10.1158/0008-5472.CAN-13-1816
24. Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci Transl Med. (2018) 10:436. doi: 10.1126/scitranslmed.aan3311
25. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology. (2013) 2:e26968. doi: 10.4161/onci.26968
26. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
27. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
28. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. (2017) 5:53. doi: 10.1186/s40425-017-0257-y
29. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. (2016) 17:651–62. doi: 10.1016/S1470-2045(16)00078-4
30. Loyher PL, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. (2018) 215:2536–53. doi: 10.1084/jem.20180534
31. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. (2005) 11:6713–21. doi: 10.1158/1078-0432.CCR-05-0883
32. Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. (2005) 65:2964–71. doi: 10.1158/0008-5472.CAN-04-4037
33. Weiss JM, Guerin MV, Regnier F, Renault G, Galy-Fauroux I, Vimeux L, et al. The STING agonist DMXAA triggers a cooperation between T lymphocytes and myeloid cells that leads to tumor regression. Oncoimmunology. (2017) 6:e1346765. doi: 10.1080/2162402X.2017.1346765
34. Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. (2002) 62:1462–70.
35. Sektioglu IM, Carretero R, Bender N, Bogdan C, Garbi N, Umansky V, et al. Macrophage-derived nitric oxide initiates T-cell diapedesis and tumor rejection. Oncoimmunology. (2016) 5:e1204506. doi: 10.1080/2162402X.2016.1204506
36. Long KB, Beatty GL. Harnessing the antitumor potential of macrophages for cancer immunotherapy. Oncoimmunology. (2013) 2:e26860. doi: 10.4161/onci.26860
37. Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
38. Pages F, Lebel-Binay S, Vieillefond A, Deneux L, Cambillau M, Soubrane O, et al. Local immunostimulation induced by intravesical administration of autologous interferon-gamma-activated macrophages in patients with superficial bladder cancer. Clin Exp Immunol. (2002) 127:303–9. doi: 10.1046/j.1365-2249.2002.01766.x
39. Axelrod RS, Havas HF, Murasko DM, Bushnell B, Guan CF. Effect of the mixed bacterial vaccine on the immune response of patients with non-small cell lung cancer and refractory malignancies. Cancer. (1988) 61:2219–30.
40. Engelhardt R, Mackensen A, Galanos C. Phase I trial of intravenously administered endotoxin (Salmonella abortus equi) in cancer patients. Cancer Res. (1991) 51:2524–30.
41. Klimp AH, de Vries EG, Scherphof GL, Daemen T. A potential role of macrophage activation in the treatment of cancer. Crit Rev Oncol. (2002) 44:143–61. doi: 10.1016/S1040-8428(01)00203-7
42. Nardin A, Lefebvre ML, Labroquere K, Faure O, Abastado JP. Liposomal muramyl tripeptide phosphatidylethanolamine: targeting and activating macrophages for adjuvant treatment of osteosarcoma. Curr Cancer Drug Targets. (2006) 6:123–33. doi: 10.2174/156800906776056473
43. Tang ZY, Zhou HY, Zhao G, Chai LM, Zhou M, Lu JZ, et al. Preliminary result of mixed bacterial vaccine as adjuvant treatment of hepatocellular carcinoma. Med Oncol Tumor Pharmacother. (1991) 8:23–8.
44. Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. (2014) 20:408–14. doi: 10.1038/nm.3512
45. Farzaneh M, Derakhshan Z, Hallajzadeh J, Sarani NH, Nejabatdoust A, Khoshnam SE. Suppression of TGF-beta and ERK signaling pathways as a new strategy to provide rodent and non-rodent pluripotent stem cells. Curr Stem Cell Res Ther. (2019) 14:466–73. doi: 10.2174/1871527318666190314110529
46. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
47. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
48. Guerin M, Regnier F, Feuillet V, Vimeux L, Weiss JM, Guilbert T, et al. TGFbeta blocks STING-induced IFN release and tumor rejection in spontaneous mammary tumors. BioRxiv. (2018). doi: 10.1101/336990
49. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. (2017) 543:428–32. doi: 10.1038/nature21409
50. Glenisson W, Castronovo V, Waltregny D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim Biophys Acta. (2007) 1773:1572–82. doi: 10.1016/j.bbamcr.2007.05.016
51. Guo W, Saito S, Sanchez CG, Zhuang Y, Gongora Rosero RE, Shan B, et al. TGF-beta1 stimulates HDAC4 nucleus-to-cytoplasm translocation and NADPH oxidase 4-derived reactive oxygen species in normal human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L936–L44. doi: 10.1152/ajplung.00256.2016
52. Solinas G, Becattini B. The role of PI3Kgamma in metabolism and macrophage activation. Oncotarget. (2017) 8:106145–6. doi: 10.18632/oncotarget.22068
53. Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. (2000) 287:1049–53. doi: 10.1126/science.287.5455.1049
54. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. (2016) 539:437–42. doi: 10.1038/nature19834
55. Evans CA, Liu T, Lescarbeau A, Nair SJ, Grenier L, Pradeilles JA, et al. Discovery of a selective phosphoinositide-3-kinase (PI3K)-gamma inhibitor (IPI-549) as an immuno-oncology clinical candidate. ACS Med Chem Lett. (2016) 7:862–7. doi: 10.1021/acsmedchemlett.6b00238
56. Pemberton N, Mogemark M, Arlbrandt S, Bold P, Cox RJ, Gardelli C, et al. Discovery of highly isoform selective orally bioavailable phosphoinositide 3-kinase (PI3K)-gamma inhibitors. J Med Chem. (2018) 61:5435–41. doi: 10.1021/acs.jmedchem.8b00447
57. Viitala M, Virtakoivu R, Tadayon S, Rannikko J, Jalkanen S, Hollmen M. Immunotherapeutic blockade of macrophage clever-1 reactivates the CD8(+) T-cell response against immunosuppressive tumors. Clin Cancer Res. (2019) 25:3289–303. doi: 10.1158/1078-0432.CCR-18-3016
58. Cortesi F, Delfanti G, Grilli A, Calcinotto A, Gorini F, Pucci F, et al. Bimodal CD40/Fas-dependent crosstalk between iNKT cells and tumor-associated macrophages impairs prostate cancer progression. Cell Rep. (2018) 22:3006–20. doi: 10.1016/j.celrep.2018.02.058
59. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. (2011) 331:1612–6. doi: 10.1126/science.1198443
60. Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNgamma and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. (2016) 6:400–13. doi: 10.1158/2159-8290.CD-15-1032
61. Jackaman C, Lew AM, Zhan Y, Allan JE, Koloska B, Graham PT, et al. Deliberately provoking local inflammation drives tumors to become their own protective vaccine site. Int Immunol. (2008) 20:1467–79. doi: 10.1093/intimm/dxn104
62. Jensen JL, Rakhmilevich A, Heninger E, Broman AT, Hope C, Phan F, et al. Tumoricidal effects of macrophage-activating immunotherapy in a murine model of relapsed/refractory multiple myeloma. Cancer Immunol Res. (2015) 3:881–90. doi: 10.1158/2326-6066.CIR-15-0025-T
63. Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell. (2018) 34:361–78. doi: 10.1016/j.ccell.2018.05.013
64. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. (2014) 20:1301–9. doi: 10.1038/nm.3708
65. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. (2019) 40:310–27. doi: 10.1016/j.it.2019.02.003
66. Thoreau M, Penny HL, Tan K, Regnier F, Weiss JM, Lee B, et al. Vaccine-induced tumor regression requires a dynamic cooperation between T cells and myeloid cells at the tumor site. Oncotarget. (2015) 6:27832–46. doi: 10.18632/oncotarget.4940
67. Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci USA. (2012) 109:2066–71. doi: 10.1073/pnas.1113099109
68. Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol. (2012) 188:1592–9. doi: 10.4049/jimmunol.1101304
69. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell. (2018) 175:1443. doi: 10.1016/j.cell.2018.11.003
70. Hollenbaugh JA, Dutton RW. IFN-gamma regulates donor CD8 T cell expansion, migration, and leads to apoptosis of cells of a solid tumor. J Immunol. (2006) 177:3004–11. doi: 10.4049/jimmunol.177.5.3004
71. Textor A, Listopad JJ, Wuhrmann LL, Perez C, Kruschinski A, Chmielewski M, et al. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNgamma. Cancer Res. (2014) 74:6796–805. doi: 10.1158/0008-5472.CAN-14-0079
72. Ma Y, Mattarollo SR, Adjemian S, Yang H, Aymeric L, Hannani D, et al. CCL2/CCR2-dependent recruitment of functional antigen-presenting cells into tumors upon chemotherapy. Cancer Res. (2014) 74:436–45. doi: 10.1158/0008-5472.CAN-13-1265
73. Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Investigat. (2011) 121:4746–57. doi: 10.1172/JCI58814
74. Vicetti Miguel RD, Cherpes TL, Watson LJ, McKenna KC. CTL induction of tumoricidal nitric oxide production by intratumoral macrophages is critical for tumor elimination. J Immunol. (2010) 185:6706–18. doi: 10.4049/jimmunol.0903411
75. Bougherara H, Nemati F, Nicolas A, Massonnet G, Pugniere M, Ngo C, et al. The humanized anti-human AMHRII mAb 3C23K exerts an anti-tumor activity against human ovarian cancer through tumor-associated macrophages. Oncotarget. (2017) 8:99950–65. doi: 10.18632/oncotarget.21556
76. Montalvao F, Garcia Z, Celli S, Breart B, Deguine J, Van Rooijen N, et al. The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. J Clin Investigat. (2013) 123:5098–103. doi: 10.1172/JCI70972
77. Zhang B, Karrison T, Rowley DA, Schreiber H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Investigat. (2008) 118:1398–404. doi: 10.1172/JCI33522
Keywords: cancer, macrophages, T cells, immunotherapy, migration, inflammation
Citation: Bercovici N, Guérin MV, Trautmann A and Donnadieu E (2019) The Remarkable Plasticity of Macrophages: A Chance to Fight Cancer. Front. Immunol. 10:1563. doi: 10.3389/fimmu.2019.01563
Received: 18 March 2019; Accepted: 24 June 2019;
Published: 12 July 2019.
Edited by:
Maija Hollmén, University of Turku, FinlandReviewed by:
Paola Allavena, Milan University, ItalyReno Debets, Erasmus University Rotterdam, Netherlands
Copyright © 2019 Bercovici, Guérin, Trautmann and Donnadieu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emmanuel Donnadieu, emmanuel.donnadieu@inserm.fr