 Jil M. Jubel
Jil M. Jubel Zachary R. Barbati
Zachary R. Barbati Christof Burger1
Christof Burger1 Frank A. Schildberg
Frank A. Schildberg- 1Clinic for Orthopedics and Trauma Surgery, University Hospital Bonn, Bonn, Germany
- 2Tufts University School of Medicine, Boston, MA, United States
PD-1 as an immune checkpoint molecule down-regulates T cell activity during immune responses in order to prevent autoimmune tissue damage. In chronic infections or tumors, lasting antigen-exposure leads to permanent PD-1 expression that can limit immune-mediated clearance of pathogens or degenerated cells. Blocking PD-1 can enhance T cell function; in cancer treatment PD-1 blockade is already used as a successful therapy. However, the role of PD-1 expression and blocking in the context of acute and chronic infections is less defined. Building on its success in cancer therapy leads to the hypothesis that blocking PD-1 in infectious diseases is also beneficial in acute or chronic infections. This review will focus on the role of PD-1 expression in acute and chronic infections with virus, bacteria, and parasites, with a particular focus on recent studies regarding PD-1 blockade in infectious diseases.
Introduction
T lymphocytes play an important role both in defending the body against pathogenic microbes as well as maintaining tolerance to self. These antagonistic functions are orchestrated by different T cell subsets, which include the effector T cells (Teff), such as cytotoxic T cells (CTLs) or helper T cells (Th), and regulatory T cells (Treg). Effector T cells play an important role in acquired immunity, whereas regulatory T cells are pivotal for the development of tolerance to self and attenuating the immune response in order to reduce collateral tissue damage at the site of inflammation. As a consequence, the dynamic ratios of various T cell subsets (e.g., the Teff:Treg ratio) are important for maintaining effective immunity and tolerance to self (1). The antagonistic functions are orchestrated by several costimulatory receptors that provide stimulatory (costimulatory) and inhibitory (coinhibitory) signals to T cells, thereby modulating T cell activation and effector responses to foreign or self-antigen. Signaling through costimulatory and coinhibitory receptors only occurs secondary to successful ligation of the T cell receptor (TCR) with its cognate major histocompatibility complex (MHC). Thus, costimulation has become regarded as a second signal to T cells, providing them with additional information about the local microenvironment and inflammatory state of the host.
During infection, peptide antigens from microbes are presented on MHC complexes (peptide-MHC) to naive T cells by professional antigen-presenting cells (APCs). Importantly, in order for T cells to recognize their cognate peptide-MHC complex, they express one of two co-receptors, CD4 or CD8. CD4+ T cells recognize MHC class II molecules and are responsible for orchestrating the immune response against extracellular pathogens, such as bacteria or helminths, by providing “help” to B cells and by activating innate effector cells, such as macrophages and NK cells. In contrast, CD8+ T cells recognize MHC class I molecules and function as cytotoxic cells that target virally infected or malignant cells. After a T cell receives both primary and secondary signals via the TCR:MHC complex and through costimulation, the T cell is now considered activated and can differentiate into effector-type and memory cells (Figure 1). Costimulatory and coinhibitory receptors expressed on the surface of T cells participate in modulating T cell differentiation and thus the immune response [reviewed by (2)]. Signaling through these receptors provides information to the T cell about the local environment as well as alter T cell survival, differentiation potential, and metabolism (3–7). The coinhibitory receptor Programmed Cell Death-1 (PD-1) plays an important immunoregulatory role by reducing initial T cell activation, fine-tuning T cell differentiation and effector functions, and by contributing to the development of immunological memory (8–11). The inhibitory effects of PD-1 are mediated by engagement with its ligands PD-L1 and PD-L2 (12, 13). It is accepted that PD-1 signaling through T cells limits immune-mediated tissue damage during infection. Seeing as the PD-1 pathway also serves to limit the activation of self-reactive T cells, this signaling pathway plays a role in reducing the risk for autoimmunity and immunopathology (11).
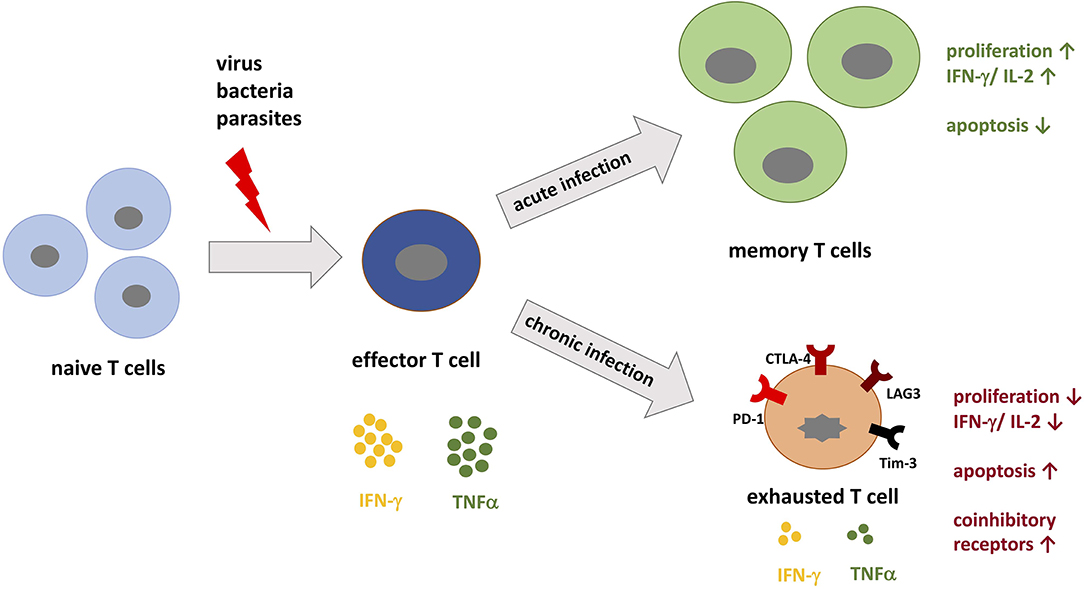
Figure 1. T cell differentiation during acute vs. chronic infections. When pathogenic peptides are presented by major histocompatibility complex (MHC) to naive T cells during infection, corresponding T cells are activated and differentiate into effector T cells specific for the pathogen. During acute infection, the immune system is able to specifically target the pathogen and eradicate it. During this process, memory T cells develop and confer immunological memory against recurrent infection. In contrast, during chronic infection the persistent load of pathogenic peptides leads to permanent stimulation of T cells, which promotes T cell exhaustion. T cell exhaustion is in part defined by the upregulation of coinhibitory receptors such as PD-1, LAG-3, and Tim-3. These receptors inhibit T cells by decreasing IL-2 production, proliferation, and the threshold for apoptosis.
Given the immunoregulatory role that PD-1 plays during inflammation, both foreign microorganisms and transformed host cells have evolved mechanisms to exploit the PD-1 pathway to evade host defenses (14, 15). Thus, examining PD-1 pathway during infection in both mice and humans has been an area of intensive research. In this review, we will cover what is known about the role of the PD-1 pathway during infectious disease. While the PD-1 pathway is an important player in regulating the activation and function of T cells at numerous points during T cell development, activation, and effector phases, here we will focus primarily on the role that the PD-1 pathway has on T cell effector responses during infection. We will also comment on the therapeutic role that PD-1 blockade may have in the context of acute and chronic infection with pathogens that are resistant to treatment. The term PD-1 blockade is a broad term that includes various mechanisms of pathway inhibition and will be used herein after to describe the use of either blocking antibodies against PD-1 or PD-L1, unless otherwise specified. Specifically, we will summarize what is known about the PD-1 pathway in the context of both acute and chronic viral infections, bacterial infections, and comment on its role during sepsis.
What Is PD-1?
PD-1 exists as a monomeric surface glycoprotein and can be recruited to the TCR signalosome during TCR ligation with peptide-MHC complexes (16, 17). It is encoded by the PDCD1 gene, which contains five exons. Exon 1 encodes a signal sequence, exon 2 encodes an IgV-like domain, and exon 3 encodes a stalk and transmembrane domain. Exons 4 and 5 encode the cytoplasmic domain, which together contain an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) (18, 19). PD-1 is inducibly expressed on CD4+ and CD8+ T cells, natural killer (NK) cells, natural killer T (NKT) cells, B cells, macrophages, and some dendritic cell (DC) subsets. T cell receptor (TCR) signaling and some cytokines, such as interleukin-2 (IL-2), interleukin-7 (IL-7), and type I interferons (IFNs), can all induce PD-1 expression on T cells (20). PD-1 expression is specifically upregulated on activated T cells and its expression can be upregulated within 24 h depending on the strength or concentration of the aforementioned stimuli. Thus, PD-1 can serve as a marker of T cell activation both in vitro and in vivo. The kinetics of PD-1 expression vary between acute and chronic infection. For example, PD-1 expression on antigen-specific T cells is transient in the context of acute infection (i.e., there is a temporary antigen load). However, in the context of chronic infection (i.e., a continuous antigen burden), PD-1 expression is sustained (21, 22). Continuous PD-1 expression on antigen-specific T cells in the context of chronic inflammation positively correlates with T cell dysfunction (21–23). These dysfunctional T cells are said to be “exhausted” (Figure 1). However, studies have found that PD-1 is necessary for the reinvigoration of exhausted T cells. Moreover, the absence of PD-1 leads to terminally exhausted T cells that cannot be recovered (14, 24). Thus, PD-1 helps avert the terminal exhaustion of T cells. Exhausted T (Tex) cells represent a heterogeneous population of antigen-specific T cells, whereby the expression of additional coinhibitory receptors and unique transcriptomes contribute to their overall dysfunctional state (25). The varying levels of other coinhibitory receptors further act to modulate T cell function in the context of acute and chronic infections (22).
The inhibitory effects of PD-1 are mediated by engagement with its ligands PD-L1 and PD-L2 (8, 12). While PD-L2 is inducibly expressed by dendritic cells (DCs), macrophages, and cultured bone marrow-derived mast cells (2), PD-L1 is constitutively expressed on the aforementioned cell types, B cells, and mesenchymal stem cells (26). Additionally, PD-L1 is expressed by a variety of non-hematopoietic cells, such as hepatocytes, vascular endothelial cells, epithelial cells, myocytes, pancreatic islet cells, and astrocytes. Of note, there is a remarkable degree of PD-L1 expression at sites of immune privilege, such as the placenta and the eye (27, 28). While there is overlap between PD-1 activation via PD-L1 and PD-L2, there is evidence to support that they independently regulate T cell responses. For example, research supports PD-L2 as the predominant regulator of CD8+ T cell proliferation and for PD-L1 as an important player for the induction of peripheral Treg cells (see below) (25, 29).
PD-1 modulates T cell activity by a number of mechanisms. Engagement of PD-1 with PD-1 ligands has been shown to inhibit proximal TCR signaling and downstream responses (Figure 2). Data collected from in vitro studies suggest that PD-1 engagement accomplishes this regulatory activity by multiple mechanisms. Engagement of PD-1 ligands with PD-1 leads to tyrosine phosphorylation of the cytoplasmic tail of PD-1 and the subsequent recruitment of the phosphatase SHP-2, a protein tyrosine phosphatase (PTP). PTPs function to dephosphorylate kinases and as a consequence, the positive signals downstream TCR and CD28 activation become antagonized. SHP-2 has been shown to directly attenuate TCR signaling by reducing phosphorylation of the Zap70/CD3ε signalosome (11, 30, 31). The downstream effects of PD-1 signaling include inhibition of AKT, phosphoinositide 3-kinase (PI3K), extracellular-signal regulated kinase (ERK), and phosphoinositide phospholipase C-γ (PLCγ) and regulation of the cell cycle leading to decreased IFN-γ/IL-2 production, reduced proliferation potential, and increased risk for apoptosis (3, 16, 26, 31). Additionally, PD-1 signaling alters T cell metabolism by inhibiting glycolysis and by promoting lipolysis and fatty acid oxidation (32, 33).
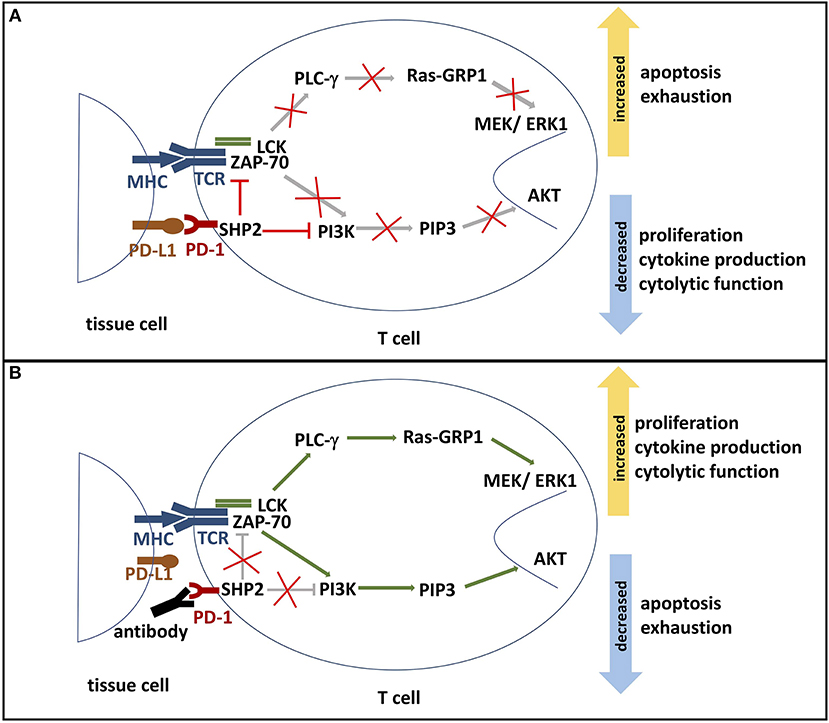
Figure 2. (A) PD-1 signaling pathway. The binding of PD-L1 or PD-L2 to its receptor PD-1 results in the phosphorylation of PD-1's ITSM and ITIM tyrosine motifs, which are located on its cytoplasmic domain. Phosphorylation leads to the recruitment of protein tyrosine phosphatases, such as SHP2. SHP2 subsequently inhibits two important pathways: One, it competes with kinases to prevent the activation of PI3K by phosphorylation. This inhibits phosphorylation of PIP2 to PIP3, thereby inhibiting Akt activation. Deactivation of serine-threonine kinase Akt reduces T cell proliferation, increases apoptosis, and promotes T cell exhaustion. Effector functions such as cytokine production and cytolytic function are also reduced. Two, SHP2 inhibits the Ras-MEK-ERK pathway. Dephosphorylation of ZAP-70 and LCK antagonize the positive downstream effects of the MHC-TCR pathway, leading to deactivation of PLC-γ, Ras-GRP1 and MEK/ERK1. ERK1 normally activates transcription factors that induce T cell proliferation and differentiation. Thus, decreased ERK1 activation reduces proliferation and differentiation potential. (B) Blockade of PD-1. In the presence of a PD-1 blocking antibody, the engagement of PD-1 and its ligands is inhibited. Consequently, SHP2 is not activated and neither PI3K/Akt pathway nor Ras-MEK-ERK pathway are repressed. Activated AKT and ERK support T cell cytokine production, proliferation, and differentiation. Furthermore, PD-1 blockade reduces T cell exhaustion and the rate of apoptosis. ITSM, immunoreceptor tyrosine-based switch motif; ITIM: immunoreceptor tyrosine-based inhibition motif; SHP2, Src homology region 2 domain-containing phosphatase 2; PI3K, phosphoinositide 3-kinase; PIP2, phosphoinositide-3,4-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; Ras, rat sarcoma; MEK, MAK-/ERK-kinase; ERK1, extracellular-signal regulated kinases 1; Zap-70, zeta-chain-associated protein kinase 70; LCK, lymphocyte-specific protein tyrosine kinase; PLC-γ, Phosphoinositide phospholipase C-γ.
Together, the downstream effect of PD-1 signaling serves to modulate T cell activation and effector function in the context of infection. Murine models of PD-1 deficiency are associated with lethal immunopathology during acute infection. Immunopathology is associated with high levels of systemic cytokines, endothelial cell death, and local tissue damage (21, 34). These data support the role for the PD-1 pathway in limiting the pro-inflammatory immune response during infection and suggest that the PD-1 pathway contributes to immune cell contraction after infection. Additionally, the PD-1 pathway plays an important role in regulating tolerance to self. In murine models, blocking the PD-1 pathway via genetic knock-down or through the administration of blocking antibodies increases the risk for developing autoimmune dilated cardiomyopathy and experimental autoimmune encephalomyelitis (35). Additionally, transgenic mice that express PD-1 with a mutant ITIM motif develop lupus-like autoimmune diseases (36, 37). In humans, single-nucleotide polymorphisms (SNP) of the PDCD1 gene have been connected with different autoimmune diseases, such as systemic lupus erythematosus (38). Whether SNPs are correlative or causative is not yet identified (11).
Inhibitory signals, like PD-L1 and PD-L2, control induction and maintenance of tolerance to self-antigens through the PD-1 pathway (12, 37). One mechanism by which the PD-1 pathway may regulate auto-reactivity is through the induction of regulatory T (Treg) cells in peripheral circulation. This is in contrast to natural Treg cells, which are centrally derived through thymic selection and express the transcription factor forkhead box P3 (FoxP3), a key transcriptional regulator of the Treg cell phenotype. Naive CD4+ T cells that enter peripheral circulation can be induced to express FoxP3 through the engagement of both the TCR and PD-1 on its surface (38). Both natural and induced Treg cells function to suppress the immune response by producing immune modulatory molecules, such as the anti-inflammatory cytokines transforming growth factor-β (TGF-β) and interleukin-10 (IL-10) (15, 39). Activated Treg cells express higher levels of PD-1, which is thought to contribute to Treg cell activity. Consistent with this, blocking PD-1 leads to decreased suppressive activity of Treg cells in vivo (26). The induction of peripheral Treg cells is thought to reduce both the activation and effector activity of self-reactive CD4+ and CD8+ T cells. Together, these data underscore the complexity of PD-1 engagement and the unique roles that the PD-1 pathway plays in the context of infection and autoimmunity. It is thus thought that the PD-1 pathway works to reduce the activation of self-reactive T cells as well as modulate the degree of activation and the effector activity of antigen-specific T cells.
The role of inhibiting the PD-1 pathway has had profound impact on tumor biology and cancer immunobiology. PD-L1 expressing tumor cells are able to escape both detection and elimination by tumor-specific CD8+ T cells (40, 41). Furthermore, PD-L1 expressing tumor cells both induce T cell exhaustion of tumor-specific T cells as well as induce tumor-specific Treg cells (42, 43). The impact of PD-1 blockade is so profound that it has already been translated to therapy to treat some forms of cancer despite the pathway's recent discovery. PD-1 blockade is an example of checkpoint inhibition, whereby natural immune checkpoints are therapeutically inhibited thereby permitting disease-specific T cell activity (11). Monoclonal blocking antibodies against PD-1 and PD-L1 are now used to treat advanced melanoma, non-small cell lung cancer, and head and neck squamous cell cancer (44). Checkpoint blockade increases the effector function of tumor-specific CD8+ T cells, which exert powerful natural defenses against some forms of malignancy. Where PD-1 blockade alone is not sufficient, combination therapies targeting more than one checkpoint have proven to reinvigorate tumor-specific T cells and reduce or even completely eliminate the tumor burden in some patients (29). Despite these promising results, it's important to note that some patients developed severe immune-related adverse events, such as colitis, hepatitis and skin disorders. These results underscore the need to better understand why some patients benefit and other patients suffer from checkpoint blockade (11, 29). Interestingly, the impact that PD-1 blockade has on viral and bacterial infection is less well-defined. Here, we will give an overview of what is known about PD-1 blockade in the context of acute and chronic infections caused by notable pathogens.
What Is Known About the PD-1 Pathway in Chronic Viral Infection?
During the course of a chronic viral infection there is a continuous burden of viral antigen. As a consequence, antigen-specific T cells are continually stimulated during the course of infection. This causes antigen-specific T cells to enter a state called T cell exhaustion (21–23). Exhausted T cells loose effector functions, such as reduced production of IL-2 and other cytokines and a reduction in proliferative and cytotoxic capacity (Figure 1). Another important indicator of T cell exhaustion is the upregulation of additional coinhibitory receptors (1, 29). The phenotypic characterization of Tex cells was born out of murine studies aimed at understanding the differences between T cell responses during acute and chronic infections with lymphocytic choriomeningitis virus (LCMV). In these seminal experiments, antigen-specific T cells taken from mice infected with a chronic strain of LCMV exhibited higher and more persistent levels of PD-1 expression when compared to antigen-specific T cells taken from mice infected with an acute strain of LCMV (22). Two different populations of antigen-specific dysfunctional T cells were identified by their expression level of PD-1. In the population of T cells where PD-1 expression was intermediate, T cell effector function could be recovered by blocking PD-1. This is in contrast to the population of T cells that expressed high levels of PD-1. PD-1 blockade could not rescue effector function of these T cells and thus were considered to be terminally exhausted (14, 45). Importantly, Blackburn et al. demonstrate that blocking the PD-1 pathway could functionally reverse T cell exhaustion and improve control over viral infection by increased T cell proliferation, killing capacity of CD8+ T cells and cytokine production (11, 22). This finding demonstrated an important role for PD-1 signaling in restraining ongoing responses in the context of chronic or persistent viral infection (15, 21). Combined blockade of both PD-1 and the coinhibitory molecule LAG-3 provided even better results when compared to blocking PD-1 alone, supporting the role of additional coinhibitory molecules in T cell exhaustion (15).
In humans, PD-1 expression on antigen-specific T cells and T cell exhaustion have been documented in numerous chronic infections, including infections with the hepatitis B virus (HBV), hepatitis C virus (HCV), and human immunodeficiency virus (HIV) (46–48). There are an estimated 257 million people chronically infected with HBV and more than 800,000 people die each year due to complications such as cirrhosis and liver cancer (49). While hepatitis B is easily prevented by vaccination, established infections are often resistant to treatment, thereby leaving infected individuals on life-long antiviral treatment. In chronic infection with HBV, PD-1 expression on HBV-specific T cells is increased (46, 47, 50, 51). Not surprisingly, in these studies PD-1 expression negatively correlates with CD8+ T cell responses (52). Research also suggests that the expression of PD-1 on HBV-specific CD8+ T cells at the earliest time points of infection positively correlate with serum ALT levels, thereby suggesting that early PD-1 expression in patients with chronic hepatitis B may serve as a biomarker for liver damage (53, 54). Newer studies also demonstrate similar results when examining PD-1 expression on HBV-specific CD4+ T cells (54).
Efforts have been made to examine how blocking the PD-1 pathway would impact the course of the infectious disease. The results and efficacy of PD-1 blockade vary considerably across studies and between pathogens (Table 1). In the context of chronic infection with HBV, PD-1 blockade increases T cell proliferation and the production of interferon-γ (IFN-γ) and IL-2 by HBV-specific CD8+ T cells taken from both the peripheral blood and livers of patients with chronic hepatitis B (55). While PD-1 blockade restored CD8+ T cell activity in this study, another study using HBV-specific T cells taken from the peripheral blood of chronic hepatitis B paints a different picture. Tang et al. demonstrate that blocking the PD-1 pathway more significantly altered CD4+ T cell function rather than CD8+ T cell function. Specifically, they report enhanced IFN-γ, IL-2 and TNFα expression by treated CD4+ T cells when compared to CD4+ T cells treated with an isotype control, as well as increased expression of the transcription factor T-bet and reduced expression of the transcription factor GATA-3 (54, 56). Differences in studies as such could potentially be resolved by better defining the HBV-specific T cell population. For example, one could imagine different responses to PD-1 blockade when more than one population of exhausted antigen-specific T cells exist (e.g., one population that expresses multiple coinhibitory receptors and a second population that predominantly expresses PD-1). Furthermore, differences in the virulence of the viral strain and variability between the populations being studied may also contribute to the degree of T cell exhaustion in the context of a chronic infection. Indeed, Tang et al. report that HBV-specific T cells express additional coinhibitory receptors, for example Tim-3 and LAG-3 (54, 71) in the context of chronic viral infection. It's plausible that PD-1 blockade may exert different effects on the CD4+ and CD8+ T cell compartments depending on the time of treatment (early vs. late) or on the degree of T cell exhaustion as measured by the number of coinhibitory receptors expressed by HBV-specific T cells. Using combination therapy to block two or more coinhibitory pathways may better serve T cell exhaustion in the context of chronic infection with HBV. However, additional work is needed to prove the efficacy of combination therapy. Importantly, while checkpoint inhibition might enhance viral clearance in patients with chronic hepatitis B, there is a risk that reactivation of subsets of exhausted T cells may increase tissue immunopathology (72).
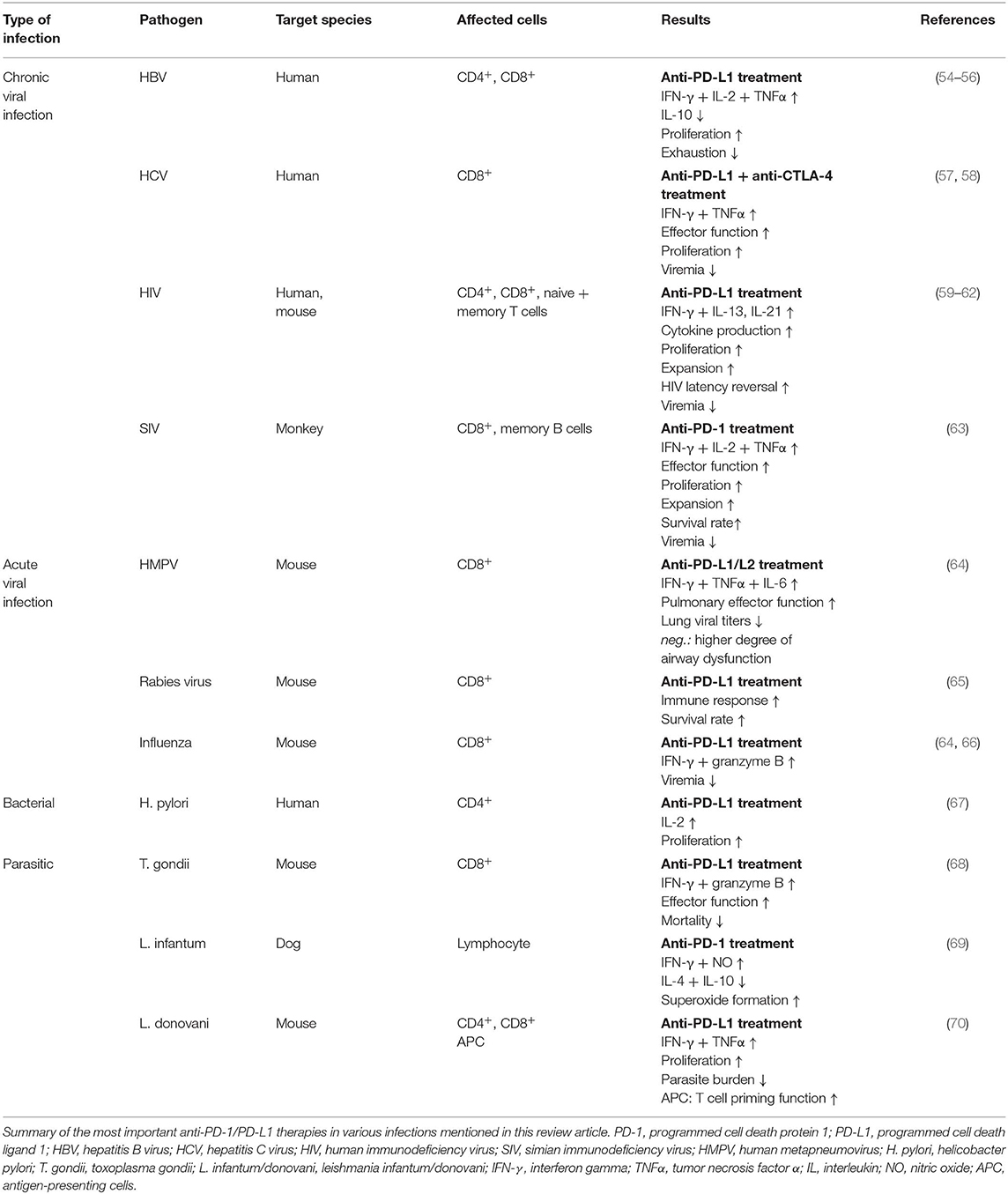
Table 1. Summary of PD-1 blockade in various infectious diseases.
Studies examining patients with chronic hepatitis C virus (HCV) also have varying results. HCV-specific CD8+ T cells have been shown to express PD-1 in several studies (57, 58, 73, 74). Ex vivo analysis of CD8+ T cells taken from patients with both acute and chronic HCV show that PD-1 expression on HCV-specific CD8+ T cells directly correlates with functional impairment. Interestingly, data from this same study reveal that blocking the PD-1 pathway restores the effector function of peripheral HCV-specific CD8+ T cells but not on HCV-specific CD8+ T cells taken from the liver (58). Importantly, cells taken from the liver were more highly positive for PD-1 than those taken from the periphery, suggesting that these cells may be more “exhausted” than those circulating in the periphery (58). These data point to differences in compartmentalization and the tissue microenvironment being important factors to consider when using PD-1 expression as a marker for T cell exhaustion and when predicting responsiveness to PD-1 blocking therapy. These results support the need to better define populations of antigen-specific T cells in the context of chronic infection when considering the impact that PD-1 blockade could have on the course of an infection (75, 76). Indeed, Gardiner et al. address the fact that the degree of T cell exhaustion (i.e., the degree of PD-1 expression and the number of additional coinhibitory receptors expressed) may confound the efficacy of PD-1 blockade. Ex vivo experiments done on PD-1+ HCV-specific CD8 T cells taken from the livers of patients chronically infected with HCV show that combined blockade against CTLA-4 and PD-1 restored T cell function whereas blockade against PD-1 or CTLA-4 alone did not (57) (Table 1).
In a randomized, double-blind, placebo-controlled study assessing the efficacy of a single dose of anti-PD-1 monoclonal antibody for the treatment of chronic hepatitis C in human patients, a protocol-defined clinical response (HCV RNA reductions of more than 0.5 log10 IU/mL were observed on 2 or more consecutive visits) was observed in five out of 45 patients. Three out of 20 patients treated with the highest administered dose (10 mg/kg) experienced >4 log10 IU/mL reductions in HCV RNA at the end of a 12-weeks, and two patients achieved HCV RNA below the lower limit of quantitation (74). While these data are promising, these numbers may not be suitable for justifying the cost of PD-1 blockade for the treatment of chronic hepatitis C given the efficacy of direct acting antivirals (e.g., Sofosbuvir or Ledipasvir) for treating both acute and chronic infections (77, 78). However, these data provide us with an important proof of concept that warrants further investigation for the use of PD-1 blockade to treat chronic infections. In line with the work done in the context of chronic hepatitis B, it is important for investigators to better define the T cell populations that respond to PD-1 blocking therapy in order to establish criteria that can be used to target patients that may benefit from immunotherapy.
Infection with HIV has a significant burden on the world population. Worldwide, there are over 37.9 million HIV-infected people and 1 million people die each year due to complications (79). Dysfunctional HIV-specific T and B cell responses are one of the primary reasons why patients living with HIV exhibit diminished immune control over the course of infection (80). Chronic HIV infection is characterized by continuous viral replication in the majority of HIV infected persons, which if left untreated, invariably leads to disease progression and acquired immunodeficiency syndrome (AIDS). Exceptions do exist; elite controllers are individuals that can control viral infection in the absence of therapy. Unsurprisingly, PD-1 expression on HIV-specific CD4+ and CD8+ T cells is associated with T cell exhaustion and disease progression (59, 81, 82). Additionally, in comparison to healthy controls, HIV-infected individuals have a higher frequency of Treg cells (83–85). PD-1 expression correlated positively with additional markers of disease progression such as viral load and reduced CD4+ T cell numbers (86). While today's antiretroviral therapy (ART) permits individuals living with HIV to lead relatively normal and healthful lives, these drugs do not eliminate the virus and patients must remain on antiretroviral therapy for the rest of their lives (11).
CD4+ T cells constitute the major cell type infected by HIV. The death and loss of functional CD4+ T cells causes AIDS in these individuals. Immuno-phenotyping CD4+ T cells taken from HIV-positive patients receiving suppressive ART reveals that these cells express PD-1, TIGIT and LAG-3 alone or in combination (48, 59, 87, 88). The population of CD4+ T cells that coexpressed all three coinhibitory receptors positively correlated both with the size of the HIV reservoir and the amount of integrated HIV DNA. These authors also demonstrate that memory CD4+ T cells expressing coinhibitory receptors contribute to the pool of HIV genomes during ART. Thus, it's possible that blocking coinhibitory receptors, such as PD-1, may target latently infected cells in virally suppressed individuals and possibly facilitate viral eradication (60, 72, 88). Indeed, ex vivo studies performed on CD4+ T cells taken from ART-suppressed patients treated with PD-1 blockade potentiates HIV latency reversal. Importantly, another study has revealed that in both aviremic and ART-suppressed patients the most important source of virus comes from PD-1+ Th cells (60, 72).
It has been shown that the number of HIV-specific CD4+ T cells correlate with control of viremia (89). This is consistent with data that has shown that the depletion of CD4+ T cells leads to a loss of CTL activity. The loss of CTL activity is caused most directly be the loss of CD4+ T helper cells and their many subsets (e.g., Th1, Th2, and T follicular cells). Additional work has shown that in vitro blockade of the PD-1 pathway restores HIV-specific CD4+ T cell proliferation and enhances HIV-specific CD4+ T cells ability to produce T helper cell subset specific cytokines, such as IFN-γ (Th1), IL-13 (Th2), and IL-21 (TFH) (59) (Table 1). Blockade also restored cytokine secretion in both CD4+ T cells that expressed intermediate and high levels of PD-1. When comparing HIV-specific CD4+ and CD8+ T cells, CD4+ T cells expressed lower levels of the inhibitory receptors CD160 and 2B4, thereby demonstrating key differences in immunoregulation between CD4+ and CD8+ T cells during chronic infection with HIV (59, 90). Together, these data show that the PD-1 pathway impairs multiple subsets of HIV-specific T helper cell responses (90). Studies performed in humanized BLT mice infected with HIV report similar results. Mice treated with PD-1 blockade exhibit improved CD4+ T cell levels and reduced viremia when compared to controls (61) (Table 1).
PD-1 blockade has also been shown to have an impact on antigen-specific CD8+ T cells. In similar experiments using humanized BLT mice, HIV-infected mice treated with PD-1 pathway blocking antibodies reduced viremia by a 45-fold reduction when compared to control animals 4-weeks post-treatment. The treatment group also exhibited an increase in the number of HIV-specific CD8+ T cells (62). Investigation of PD-1 blockade in the context of chronic simian immunodeficiency virus (SIV) in macaque monkeys yielded similar results. In these experiments, the authors demonstrate that PD-1 blockade causes SIV-specific CD8+ T cells to respond by both rapid expansion and improved functionality. Importantly, the authors investigated responses both in the blood and the gut and saw enhanced T cell immunity in both compartments. The investigators also measured improved memory B cell responses and increased levels of SIV specific antibody. Treated animals exhibited significant reductions in viremia and prolonged survival when compared to untreated animals (63) (Table 1). Despite these promising results, one should be aware of the systemic effects of the immunotherapy treatments like PD-1 blockade. At present, there is one case of HBV reactivation in a HIV-infected patient treated with a PD-1 inhibitor (76).
Given these data, it has been postulated that impaired CD4+ T cell help to B cells and B cell dysfunction may help explain the low frequency of broadly neutralizing antibodies in HIV-infected individuals. Additional work has also been done to investigate the level of expression of PD-1 on the surface on virus-specific B cells during chronic infection with HIV. These data demonstrate that during chronic infection with HIV, virus-specific B cells respond to antigen poorly and that this poor response can be relieved by suppressing the PD-1 pathway (91). In addition, there is evidence that exhausted B cells in HIV express other coinhibitory receptors. Blocking these receptors in addition to PD-1 could enhance the immune response (15). Additional work also suggests that chronic infection with HIV also dampens the activity of T follicular helper cells (TFH) (70), which are necessary for the proper formation of germinal centers that facilitate the development of high-affinity antibodies by B cells (92). Other studies have reported, that in highly viremic HIV infected children the rate of memory TFH was lower compared to controls. It was shown, that a high rate of memory TFH was associated with elevated ratio of resting memory B cells (93). Together, these results demonstrate that blocking the PD-1 pathway can enhance both the cellular and humoral immune response during infection with pathogenic immunodeficiency viruses (15). Despite these promising results, one should be aware of the other coinhibitory receptors that play an important role during chronic infections. More research is needed to understand the correlation of all these receptors and their different roles in T cell exhaustion.
Acute Viral Infection and the PD-1 Pathway
The pathobiology of acute viral infection differs significantly from that of chronic infection. During acute infection the immune system is highly activated by virus particles. At the end of the infection, the virus is eliminated, and memory cells have been differentiated. The patient shows acquired immunity. Conversely, in chronic infections the virus is not eliminated and the continuously released virus particles result in T cell exhaustion (Figure 1). It is not yet clear if T cell exhaustion arises during acute infections (29). PD-1 upregulation has been noted in the context of acute viral infection with vaccinia virus, influenza virus, rabies in mice, and hepatitis A virus (HAV), HBV, and HCV in humans (21, 53, 94, 95). During the acute phase of infection, virus-specific T cells respond to activation with increased expression of PD-1 among other markers of activation. Additionally, the virus upregulates PD-L1 on different cell types either directly through pattern recognition receptor signaling or indirectly by prompting the release of inflammatory cytokines (96). Studies that examine the functional role of the PD-1 pathway during acute infection suggest that PD-1 is involved in protecting against lethal immunopathology (37, 97). For example, during the acute phase of infection with the chronic strain of LCMV, PD-1-deficient mice die from lethal immunopathology within 1 week after infection (21, 34). The severe immunopathology is characterized by high levels of systemic cytokines and pulmonary pathology as a result of endothelial cell death (34). In contrast, PD-1-deficient mice infected with the acute strain of LCMV responded similarly to infection when compared to wild type animals (21). These results suggest two important ideas regarding the role of the PD-1 pathway during acute infection. First, they highlight the capacity of certain strains of virus to establish a chronic infection by subverting immune clearance during the acute phase of infection by taking advantage of the immunosuppressive properties of the PD-1 pathway. Second, they underscore the importance of the PD-1 pathway in dampening the immune response during the acute phase of persistent infections to protect against severe immunopathology.
Studies exploring respiratory viral infections in mice using influenza virus (IAV) and human metapneumovirus (HMPV) also support the role for the PD-1 pathway in reducing acute phase immunopathology (64, 98, 99). In these experiments, anti-PD-L1 treatment of mice infected with HMPV resulted in improved pulmonary CD8+ T cell function and reduced lung viral titers within 1-week post-infection. Anti-PD-L1 treatment was associated with increased levels of the pro-inflammatory cytokines IFN-γ, TNFα, and IL-6. While exacerbated lung pathology was not observed in mice treated with anti-PD-L1 when compared to mice treated with an isotype control, the mice treated with anti-PD-L1 exhibited a higher degree of airway dysfunction as measured by pulse oximetry (64) (Table 1). PD-1-deficient mice infected with IAV exhibited similar airway dysfunction and recover weight more slowly when compared to wildtype mice (64). Congruent with these data, histological sections of tissue collected from the lower airways of pediatric patients with severe H1N1 influenza A virus reveal increased expression of PD-1 and PD-L1, thereby supporting the role that the expression of PD-1 and its ligand are important in the context of inflammation in human subjects (64).
Acute hepatitis B virus infection can lead to liver cirrhosis, dangerous organ failure and necessary liver transplantation. Studies in human patients examining acute hepatitis B (AHB) virus infection reveal a similar pattern to that observed in the lung tissue of patient's infected with influenza. In a clinical cohort of patients diagnosed with AHB, the authors report that PD-1 expression was significantly upregulated on HBV-specific CD8+ T cells during the early phase of acute HBV, and that viral clearance correlated with a subsequent decrease in PD-1 expression. Importantly, patients with acute liver failure expressed lower levels of PD-1 within 1–2 weeks after the clinical onset of disease (53). Thus, dysfunctional PD-1 expression on HBV-specific CD8+ T cells positively correlates with liver failure (15). Not only do these data support the PD-1 pathway being an important regulator of tissue damage, but they also suggest that the expression of PD-1 may act as a potential indicator for the clinical outcome of AHB infection. Additionally, these studies also suggest that PD-1-mediated signaling not only attenuates HBV-specific CD8+ T cell effector function during the acute phase of infection but also that PD-1 expression early during infection positively correlates with the development of HBV-specific memory CD8+ T cells following disease resolution (53).
Given the known role that PD-1 signaling has on tempering T cell responses, therapeutic blockade of the pathway would theoretically augment T cell effector function during acute infection (Table 1). Indeed, therapeutic blockade of the PD-1 pathway during acute infection with the rabies virus enhanced both the immune response and survival rate in a murine model (65). Additional studies exploring murine models of influenza, reveal that checkpoint blockade improves CD8+ T cell function by increasing IFN-γ and granzyme B production (64, 66). Murine models of acute infection also support a role for the PD-1 pathway in regulating the development of T memory cells. In the absence of PD-1 signaling, antigen-experienced CD8+ T cells taken from mice infected with vaccinia virus preferentially differentiate to central memory CD8+ T cells, whereas wild type CD8+ T cells preferentially differentiate into the effector memory cells. Additionally, antigen-specific CD8+ T cells taken from PD-1-deficient mice produced higher levels of cytokines when compared to antigen-specific CD8+ T cells taken from wildtype mice (100).
Interestingly, data derived from patients infected with the Ebola virus suggests that the PD-1 pathway plays an important role in facilitating Ebola-virus-induced immune subversion. CD4+ and CD8+ T cells taken from patients infected with Ebola virus exhibited markedly increased levels of PD-1 expression, impaired IFN-γ production, and inversion of the CD4/CD8 T cell ratio. Importantly, surviving patients showed significantly lower expression of the PD-1 and the coinhibitory receptor CTLA-4 as well as lower inflammation as measured by pro-inflammatory cytokines (e.g., TNFα, IL-8, MIP-1β, etc.) (101, 102). These observations suggest that PD-1 blockade may help enhance survival of patients infected with Ebola virus.
PD-1 Pathway in Bacterial and Parasitic Infections
The PD-1 pathway has also been examined in the context of numerous bacterial infections. Persistent infections with bacteria have also been associated with suboptimal T cell responses. For example, the gastrointestinal pathogen, Helicobactor pylori establishes chronic infections in humans leading to gastroduodenal ulcers and gastric cancer (103–105). T cell dysfunction and PD-1 expression have been observed in patients infected with H. pylori (67, 106, 107). Human gastric epithelial cells taken from patients infected with H. pylori express higher levels of PD-L1 when compared to controls (67, 107). PD-L1 upregulation induced by H. pylori is mediated by the Sonic Hedgehog signaling pathway (103). This infection-associated PD-L1 expression may help explain the increased frequency of gastric cancer among people infected with H. pylori, whereby PD-L1 expression on premalignant lesions permits immune checkpoint evasion thereby facilitating progression toward malignancy (103). Furthermore, in vitro co-cultures of human gastric epithelial cell lines infected with H. pylori and CD4+ T cells demonstrate that treatment with anti-PD-L1 antibodies results in enhanced CD4+ T cell proliferation and IL-2 production (67). These data not only support the role that H. pylori-induced PD-L1 expression plays in promoting persistent infection, but also implicate H. pylori-induced PD-L1 expression on gastric epithelial cells as an important player in immune evasion and cancer progression.
In contrast to these data, PD-1-deficient mice result in decreased survivability of mice infected with Mycobacterium tuberculosis (Mbt) (108, 109). PD-1-deficient mice infected with Mbt exhibited uncontrolled bacterial proliferation, focal necrosis, and lower numbers of infiltrating T and B cells. Additionally, the pro-inflammatory cytokines TNFα, IL-1, IL-6, and IL-17A were markedly increased in the lung and sera of PD-1-deficient mice infected with Mbt (109). Interestingly, CD4+ T cells exhibit increased and sustained PD-1 expression during Mbt infection in both mice and humans (108–110). However, Mbt-specific CD8+ T cell responses in PD-1-deficient mice were normal (108). These data suggest that Mbt acquires a fitness advantage in the absence of PD-1 and that PD-1 expression on CD4+ T cells specifically exerts a protective effect against Mbt infection. Indeed, there are reports of Mbt reactivation among patients with cancer being treated with PD-1 blockade (111). Depleting CD4+ T cells in PD-1-deficient mice during infection with Mbt rescued the animal's ability to survive infection (108). These data highlight the important role that inhibition through the PD-1 pathway plays in preventing T cell-driven immunopathology and exacerbation of infection with Mbt. These results again support the role of the PD-1 pathway in both shaping the immune response and protecting against severe immunopathology.
Why does PD-1 blockade in H. pylori infection enhance the immune response whereas lack of PD-1 in Mbt infection worsens the extent of infection? These seemingly conflicting responses may be partially explained by the type and number of CTLs, Teff, and Tex cells present at the site of infection, their ratios to Treg cells, and their degree of PD-1 and costimulatory molecule expression. In the context of infection with H. pylori, gastric biopsy samples from infected patients reveal that specimens with higher frequency of both gastric infiltrating Treg cells and PD-1+ CD4+ T cells correlates directly with the density of H. pylori, suggesting that these cell types permit bacterial colonization rather than control it (112). This is in contrast to the Th1 effector cells that are required to control intracellular Mbt infection (113). Despite being an intracellular pathogen, it has also been demonstrated that CD8+ T cells do not recognize Mbt-infected macrophages (114). It follows that targeting the PD-1 pathway in the context of either infection would likely have differential effects owing to the fact that PD-1 blockade would effectively be targeting different cell types. At present, PD-1 blockade as a treatment in bacterial infection is poorly studied and only a limited amount of papers has been published. One possible explanation for the difficulty in defining T cell responses against bacteria is that a single species of bacteria can be commensal, opportunistic, or even pathogenic depending on the host's immune system, location, and route of inoculation. As it follows, T cells must respond to the same organism differently. This inherent complexity may present itself with challenges when attempting assess T cell responses to bacterial infection. Even the bacterial route of entry has been shown to elicit different types of Teff cells, where intravenous inoculation of Listeria monocytogenes favors the differentiation of long-lived Th1 memory cells whereas intra-nasal inoculation favors shorter-lived Th17 cells (115).
While the role of the PD-1 pathway in the context of T cells has namely been studied among a limited number of bacteria, there is a growing body of evidence that supports a role for the PD-1 pathway in directly regulating the response of B lymphocytes and innate immune cells, such as macrophages and dendritic cells. PD-1 expression on B cells has been shown to suppress protective humoral immune responses to Streptococcus pneumoniae by inhibiting clonal expansion and IgG production by S. pneumoniae-specific B cells (116). On dendritic cells, PD-1 suppresses the immune system's protective responses against infection with Listeria monocytogenes (117) and on macrophages, PD-1 expression is associated with macrophage dysfunction and promotes lethality secondary to bacteremia and sepsis (118). These data illustrate the need for more extensive research on the PD-1 pathway in the context of bacterial infection.
T cell exhaustion has been described during infection with a number of parasitic protozoans as well. Chronic infection in context of parasitic infection are common because of the ability of parasites to escape the immune system by increasing anti-inflammatory molecules such as TGF-β and IL-10. In addition, parasitic infections are associated with increased expression of coinhibitory receptors and their ligands (29). For example, PD-1 expression in persistent infections with T. gondii, L. major, and Plasmodium falciparum (70, 119) have been reported. Mice infected with the intracellular protozoan parasite T. gondii exhibit persistent infections and CD8+ T cell exhaustion (68). Blocking the PD-1 pathway in mice infected with T. gondii reinvigorated CD8+ T cell responses and prevented mortality in chronically infected animals (68). Infection with Leishmania major can present with different clinical manifestations. Cutaneous leishmaniasis causes local lesions were parasite replication is contained, not unlike the focal lesions observed in granulomatous tuberculosis. In contrast, visceral leishmaniasis is characterized by disseminated infection, immunosuppression, and severe disease. In both canine and murine models of chronic infection with leishmaniasis, CD4+ and CD8+ T cells express high levels of PD-1 and reduced levels of pro-inflammatory cytokines (120, 121). PD-1 blockade in animals infected with Leishmania infantum resulted in enhanced production of IFN-γ and nitric oxide (NO), whereas IL-4 and IL-10 were decreased (69). Blocking PD-L1 in infections with Leishmania donovani leads to T cell proliferation and decreased parasite burden (70). Interestingly, there is data that supports a differential role between PD-L1 and PD-L2 in murine parasitic infections. For example, PD-L1-deficient mice were protected from symptoms associated with Leishmania mexicana infection whereas PD-L2-deficient mice were not (29). Similarly, increased PD-L2 expression during malaria infection is associated with reduced parasite load and limited T cell exhaustion (29, 122).
PD-1 Pathway and Sepsis
Of particular interest is the role of the PD-1 pathway during sepsis. Sepsis is a heterogeneous syndrome that generally presents with a hyper-inflammatory state that is then followed by a compensatory anti-inflammatory or immunosuppressive state. There are over 30 million cases and 6 million deaths worldwide per year (123). Of the patients that survive the initial hyper-inflammatory state progress to the immunosuppressive state where they still remain difficult to treat. Indeed, the majority of deaths caused by sepsis occur during this latter phase (124). Impaired immunity during sepsis is now recognized to be caused by systemic levels of T cell exhaustion (125). Early studies of PD-1 expression on macrophages in the context of sepsis were associated with reduced bacterial clearance. This sparked a more thorough investigation of the role that the PD-1 pathway plays in bacteremia. In these experiments, macrophages taken from PD-1-deficient mice were resistant to sepsis-induced cellular dysfunction, as defined by diminished bacteriocidal ability, decreased cytokine production, and suppressed antigen presenting capacity (118). Additionally, PD-1 upregulation on CD4+ and CD8+ T cells has been observed as early as 48 h after the onset of sepsis in both mouse and human subjects (125–127). PD-1-deficient mice show improved survival during sepsis when compared to their wild type counterparts, thereby suggesting a relationship between PD-1 expression, immune dysfunction, and the pathology associated with this highly acute inflammatory response (118). Similarly, administering blocking antibodies against the PD-1 pathway in septic mice increased their long-term survival (118, 128).
While PD-1 expression on T cells during sepsis is well-established, it is not clear whether T cells are the major therapeutic targets of PD-1 blockade during septic immune dysfunction. Understanding the role that the PD-1 pathway plays in sepsis is important for the proper selection of patients for treatment with immunomodulatory drugs. Given these results, a number of clinical trials are underway to investigate PD-1 blockade and a growing body of research is quickly developing around the role of the PD-1 pathway in sepsis.
Conclusion
It is clear that the PD-1 pathway plays an important role during chronic infection with numerous pathogens. As mentioned above, the PD-1 pathway permits pathogens to escape elimination by the host's immune system and thereby establish chronic infection. While inhibiting this pathway in some models of infection clearly reinvigorates Tex cells, not all models of PD-1 blockade elicit this response. More work is needed to understand the role of additional coinhibitory receptors in the context of infection. Involvement multiple coinhibitory pathways may explain why blocking the PD-1 pathway in the context of some infections is sufficient to restore Tex cell function while in others it is not. By better describing Tex cells in the context of acute and chronic infections with different pathogens, we may be able to more carefully employ combination therapies aimed at blocking two or more coinhibitory pathways. Indeed, combination therapy has already been successfully employed to treat mice with chronic malaria (129). Indeed, given the promising results seen in humanized mice infected with HIV and primates infected with SIV, investigating the impact that combining ART with blocking antibodies against the PD-1 pathway is of significance.
While the role of the PD-1 pathway during acute infection is less well-defined, there is evidence to suggest that its expression can act as an important biomarker to predict disease outcomes. Using PD-1 expression as a marker for immune cell activation or immune cell dysfunction during acute (and chronic infection) may have advantages when considering treatment options and the cost of care for patients with difficult to treat microbial infections. Furthermore, PD-1 expression on T cells, B cells, and monocytes has been associated with severe immune dysfunction, an example of which is the second phase of sepsis. A broader understanding of the role that the PD-1 pathway plays on both innate and adaptive immune cells may help us better understand the immune dysregulation observed during sepsis. Interestingly, the immune dysregulation observed during the latter phase of sepsis resembles the acute immune dysfunction observed during infection with the Ebola virus. Drawing parallels between these two inflammatory states may help investigators piece together the pathways circumvented during sepsis or infection with the Ebola virus. While rapid translation of blocking antibodies against the PD-1 pathway from bench to clinic has been observed over the past decade, the role of PD-1 blockade for the treatment of infectious disease is still not clear and it requires further investigation. It is important that we better define the role that the PD-1 pathway has in the context of acute and chronic infection as well as during severe immune dysregulation. Not only might PD-1 expression act as an important biomarker to predict disease outcomes or optimize care for patients with difficult to treat infections, but evaluating the efficacy of blocking the PD-1 pathway in the context of difficult to treat infections may add to our arsenal of drugs to fight drug-resistant pathogens.
Author Contributions
All authors contributed to conception and design of the study. JJ, ZB, and FS researched and wrote the content of the review article. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from the National Multiple Sclerosis Society (RG 1809-32591 to FS) and is part of the doctoral thesis of JJ. Because of space restrictions, we were able to cite only a fraction of the relevant literature and apologize to colleagues whose contributions might not be appropriately acknowledged in this review.
References
1. Bucktrout SL, Bluestone JA, Ramsdell F. Recent advances in immunotherapies: from infection and autoimmunity, to cancer, and back again. Genome Med. (2018) 10:79. doi: 10.1186/s13073-018-0588-4
2. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity. (2016) 44:955–72. doi: 10.1016/j.immuni.2016.05.002
3. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
4. Croft M. Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev. (2003). 14:265–73. doi: 10.1016/S1359-6101(03)00025-X
5. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. (2002) 16: 769–77. doi: 10.1016/S1074-7613(02)00323-0
6. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 Promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. (2001) 15:445–55. doi: 10.1016/S1074-7613(01)00191-1
7. Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho IC, Sharpe AH, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol. (2009) 10:167–75. doi: 10.1038/ni.1690
8. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
9. Bennett CL, Yoshioka R, Kiyosawa H, Barker DF, Fain PR, Shigeoka AO, et al. X-linked syndrome of polyendocrinopathy, immune dysfunction, diarrhea maps to Xp11.23-Xq13.3. Am J Hum Genet. (2000) 66:461–8. doi: 10.1086/302761
10. Bennett F, Luxenberg D, Ling V, Wang IM, Marquette K, Lowe D, et al. Program Death-1 engagement upon TCR activation has distinct effects on costimulation and Cytokine-Driven proliferation: attenuation of ICOS, IL-4, and IL-21, But Not CD28, IL-7, and IL-15 responses. J Immunol. (2003) 170:711–8. doi: 10.4049/jimmunol.170.2.711
11. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. (2018) 18:153–67. doi: 10.1038/nri.2017.108
12. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2:261–68. doi: 10.1038/85330
13. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. expression of programmed death 1 ligands by murine T cells APC. J Immunol. (2002) 169:5538–45. doi: 10.4049/jimmunol.169.10.5538
14. LaFleur MW, Muroyama Y, Drake CG, Sharpe AH. Inhibitors of the PD-1 pathway in tumor therapy. J Immunol. (2018) 200:375–83. doi: 10.4049/jimmunol.1701044
15. Attanasio J, Wherry EJ. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity. (2016) 44:1052–68. doi: 10.1016/j.immuni.2016.04.022
16. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
17. Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, et al. Structural and Functional analysis of the costimulatory receptor programmed death-1. Immunity. (2004) 20:337–47. doi: 10.1016/S1074-7613(04)00051-2
18. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
19. Shinohara T, Taniwaki M, Ishida Y, Kawaichi M, Honjo T. Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics. (1994) 23:704–6. doi: 10.1006/geno.1994.1562
20. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. (2011) 186:2772–9. doi: 10.4049/jimmunol.1003208
21. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. (2006) 439:682–7. doi: 10.1038/nature04444
22. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. (2009) 10:29–37. doi: 10.1038/ni.1679
23. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
24. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. (2015) 212:1125–37. doi: 10.1084/jem.20142237
25. Boenisch O, Sayegh MH, Najafian N. Negative T-cell costimulatory pathways: their role in regulating alloimmune responses. Curr Opin Organ Transplant. (2008) 13:373–8. doi: 10.1097/MOT.0b013e328306117f
26. McGrath MM, Najafian N. The role of coinhibitory signaling pathways in transplantation and tolerance. Front Immunol. (2012) 3:47. doi: 10.3389/fimmu.2012.00047
27. Guleria I, Khosroshahi A, Ansari MJ, Habicht A, Azuma M, Yagita H, et al. A critical role for the programmed death ligand 1 in fetomaternal tolerance. J Exp Med. (2005) 202:231–7. doi: 10.1084/jem.20050019
28. Yang W, Li H, Chen PW, Alizadeh H, He Y, Hogan RN, et al. PD-L1 expression on human ocular cells and its possible role in regulating immune-mediated ocular inflammation. Invest Ophthalmol Vis Sci. (2009) 50:273–80. doi: 10.1167/iovs.08-2397
29. Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. (2017) 47:765–79. doi: 10.1002/eji.201646875
30. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
31. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human t cell stimulation, but only receptor ligation prevents t cell activation. J Immunol. (2004) 173:945–54. doi: 10.4049/jimmunol.173.2.945
32. Tkachev V, Goodell S, Opipari AW, Hao LY, Franchi L, Glick GD, et al. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J Immunol. (2015) 194:5789–800. doi: 10.4049/jimmunol.1402180
33. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. (2015) 6:6692. doi: 10.1038/ncomms7692
34. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med. (2012) 209:2485–99. doi: 10.1084/jem.20121015
35. Sage PT, Schildberg FA, Sobel RA, Kuchroo VK, Freeman GJ, Sharpe AH. Dendritic cell PD-L1 limits autoimmunity and follicular T cell differentiation and function. J Immunol. (2018) 200:2592–602. doi: 10.4049/jimmunol.1701231
36. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. (2001) 291:319–22. doi: 10.1126/science.291.5502.319
37. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. (2007) 8:239–45. doi: 10.1038/ni1443
38. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
39. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
40. Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother. (2005) 54:307–14. doi: 10.1007/s00262-004-0593-x
41. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
42. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. (2015) 6:e1792. doi: 10.1038/cddis.2015.162
43. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res. (2007) 13:4345–54. doi: 10.1158/1078-0432.CCR-07-0472
44. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol. (2016). 34:539–73. doi: 10.1146/annurev-immunol-032414-112049
45. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. (2012) 338:1220–5. doi: 10.1126/science.1229620
46. Bertoletti A, Gehring AJ. The immune response during hepatitis B virus infection. J Gen Virol. (2006) 87:1439–49. doi: 10.1099/vir.0.81920-0
47. Larrubia JR, Benito-Martínez S, Miquel J, Calvino M, Sanz-de-Villalobos E, Parra-Cid T. Costimulatory molecule programmed death-1 in the cytotoxic response during chronic hepatitis C. World J Gastroenterol. (2009) 15:5129. doi: 10.3748/wjg.15.5129
48. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. (2006) 12:1198–202. doi: 10.1038/nm1482
49. Organisation WH. Hepatitis B. (2019) Available online at: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b.
50. Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. (2007) 81:4215–25. doi: 10.1128/JVI.02844-06
51. Peng G, Li S, Wu W, Tan X, Chen Y, Chen Z. PD-1 upregulation is associated with HBV-specific T cell dysfunction in chronic hepatitis B patients. Mol Immunol. (2008) 45:963–70. doi: 10.1016/j.molimm.2007.07.038
52. Ye P, Weng ZH, Zhang SL, Zhang JA, Zhao L, Dong JH, et al. Programmed death-1 expression is associated with the disease status in hepatitis B virus infection. World J Gastroenterol. (2008) 14:4551. doi: 10.3748/wjg.14.4551
53. Zhang Z, Zhang JY, Wherry EJ, Jin B, Xu B, Zou ZS, et al. Dynamic programmed death 1 expression by virus-specific CD8 T cells correlates with the outcome of acute hepatitis B. Gastroenterology. (2008) 134:1938–49. doi: 10.1053/j.gastro.2008.03.037
54. Dong Y, Li X, Zhang L, Zhu Q, Chen C, Bao J, et al. CD4(+) T cell exhaustion revealed by high PD-1 LAG-3 expression the loss of helper T cell function in chronic hepatitis B. BMC Immunol. (2019) 20:27. doi: 10.1186/s12865-019-0309-9
55. Fisicaro P, Valdatta C, Massari M, Loggi E, Biasini E, Sacchelli L, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology. (2010) 138:682–93. doi: 10.1053/j.gastro.2009.09.052
56. Tang ZS, Hao YH, Zhang EJ, Xu CL, Zhou Y, Zheng X, et al. CD28 family of receptors on T cells in chronic HBV infection: expression characteristics, clinical significance and correlations with PD-1 blockade. Mol Med Rep. (2016) 14:1107–16. doi: 10.3892/mmr.2016.5396
57. Nakamoto N, Cho H, Shaked A, Olthoff K, Valiga ME, Kaminski M, et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog. (2009) 5:e1000313. doi: 10.1371/journal.ppat.1000313
58. Nakamoto N, Kaplan DE, Coleclough J, Li Y, Valiga ME, Kaminski M, et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology. (2008) 134:1927–37. doi: 10.1053/j.gastro.2008.02.033
59. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. (2006) 443:350–4. doi: 10.1038/nature05115
60. Fromentin R, DaFonseca S, Costiniuk CT, El-Far M, Procopio FA, Hecht FM, et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4(+) T cells from ART-suppressed individuals. Nat Commun. (2019) 10:814. doi: 10.1038/s41467-019-08798-7
61. Palmer BE, Neff CP, Lecureux J, Ehler A, Dsouza M, Remling-Mulder L, et al. In vivo blockade of the PD-1 receptor suppresses HIV-1 viral loads and improves CD4+ T cell levels in humanized mice. J Immunol. (2013) 190:211–9. doi: 10.4049/jimmunol.1201108
62. Seung E, Dudek TE, Allen TM, Freeman GJ, Luster AD, Tager AM. PD-1 blockade in chronically HIV-1-infected humanized mice suppresses viral loads. PLoS ONE. (2013) 8:e77780. doi: 10.1371/journal.pone.0077780
63. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. (2009) 458:206–10. doi: 10.1038/nature07662
64. Erickson JJ, Gilchuk P, Hastings AK, Tollefson SJ, Johnson M, Downing MB, et al. Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J Clin Invest. (2012) 122:2967–82. doi: 10.1172/JCI62860
65. Lafon M, Mégret F, Meuth SG, Simon O, Velandia Romero ML, Lafage M, et al. Detrimental contribution of the immuno-inhibitor B7-H1 to rabies virus encephalitis. J Immunol. (2008) 180:7506–15. doi: 10.4049/jimmunol.180.11.7506
66. McNally B, Ye F, Willette M, Flaño E. Local blockade of epithelial PDL-1 in the airways enhances T cell function and viral clearance during influenza virus infection. J Virol. (2013) 87:12916–24. doi: 10.1128/JVI.02423-13
67. Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during helicobacter pylori infection. J Immunol. (2006) 176:3000–9. doi: 10.4049/jimmunol.176.5.3000
68. Bhadra R, Gigley JP, Weiss LM, Khan IA. Control of toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1–PDL-1 blockade. Proc Natl Acad Sci USA. (2011) 108:9196–201. doi: 10.1073/pnas.1015298108
69. Oliveira Silva KL, Marin CV, Luvizotto VG, Correa LAA, de Almeida BF, De Rezende EF, et al. PD-1 and PD-L1 regulate cellular immunity in canine visceral leishmaniasis. Comp Immunol Microbiol Infect Dis. (2019) 62:76–87. doi: 10.1016/j.cimid.2018.12.002
70. Habib S, El Andaloussi A, Elmasry K, Handoussa A, Azab M, Elsawey A, et al. PDL-1 blockade prevents T cell exhaustion, inhibits autophagy, and promotes clearance of Leishmania donovani. Infect Immun. (2018) 86:1–14. doi: 10.1128/IAI.00019-18
71. Liu Y, Gao LF, Liang XH, Ma CH. Role of Tim-3 in hepatitis B virus infection: an overview. World J Gastroenterol. (2016) 22:2294–303. doi: 10.3748/wjg.v22.i7.2294
72. Abers MS, Lionakis MS, Kontoyiannis DP. Checkpoint inhibition and infectious diseases: a good thing? Trends Mol Med. (2019) 25:1080–93. doi: 10.1016/j.molmed.2019.08.004
73. Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. (2010) 6:e1000947. doi: 10.1371/journal.ppat.1000947
74. Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, Reddy KR, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS ONE. (2013) 8:e63818. doi: 10.1371/journal.pone.0063818
75. Caraballo Cortés K, Osuch S, Perlejewski K, Pawełczyk A, Kazmierczak J, Janiak M, et al. Expression of programmed cell death protein 1 and T-cell immunoglobulin- and mucin-domain-containing molecule-3 on peripheral blood CD4+CD8+ double positive T cells in patients with chronic hepatitis C virus infection and in subjects who spontaneously cleared the virus. J Viral Hepat. (2019) 26:942–50. doi: 10.1111/jvh.13108
76. Pandey A, Ezemenari S, Liaukovich M, Richard I, Boris A. A rare case of pembrolizumab-induced reactivation of hepatitis B. Case Rep Oncol Med. (2018) 2018:5985131. doi: 10.1155/2018/5985131
77. Carter W, Connelly S, Struble K. Reinventing HCV treatment: past and future perspectives. J Clin Pharmacol. (2016) 57:287–96. doi: 10.1002/jcph.830
78. Kohli A, Shaffer A, Sherman A, Kottilil S. Treatment of hepatitis C: a systematic review. JAMA. (2014) 312:631–40. doi: 10.1001/jama.2014.7085
79. WHO. HIV/ AIDS. (2019). Available online at: https://www.who.int/en/news-room/fact-sheets/detail/hiv-aids/.
80. Klenerman P, Hill A. T cells and viral persistence: lessons from diverse infections. Nat Immunol. (2005) 6:873–9. doi: 10.1038/ni1241
81. D'Souza M, Fontenot AP, Mack DG, Lozupone C, Dillon S, Meditz A, et al. Programmed death 1 expression on HIV-Specific CD4+ T cells is driven by viral replication and associated with t cell dysfunction. J Immunol. (2007) 179:1979–87. doi: 10.4049/jimmunol.179.3.1979
82. Salisch NC, Kaufmann DE, Awad AS, Reeves RK, Tighe DP, Li Y, et al. Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. J Virol. (2007) 81:5819–28. doi: 10.1128/JVI.00024-07
83. Tsunemi S, Iwasaki T, Imado T, Higasa S, Kakishita E, Shirasaka T, et al. Relationship of CD4+CD25+ regulatory T cells to immune status in HIV-infected patients. AIDS. (2005) 19:879–86. doi: 10.1097/01.aids.0000171401.23243.56
84. Aandahl EM, Michaëlsson J, Moretto WJ, Hecht FM, Nixon DF. Human CD4+ CD25+ regulatory T cells control T-cell responses to human immunodeficiency virus and cytomegalovirus antigens. J Virol. (2004) 78:2454–9. doi: 10.1128/JVI.78.5.2454-2459.2004
85. Weiss L, Donkova-Petrini V, Caccavelli L, Balbo M, Carbonneil C, Levy Y. Human immunodeficiency virus-driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood. (2004) 104:3249–56. doi: 10.1182/blood-2004-01-0365
86. Peligero C, Argilaguet J, Güerri-Fernandez R, Torres B, Ligero C, Colomer P, et al. PD-L1 blockade differentially impacts regulatory T cells from HIV-Infected individuals depending on plasma viremia. PLoS Pathog. (2015) 11:e1005270. doi: 10.1371/journal.ppat.1005270
87. Teigler JE, Zelinskyy G, Eller MA, Bolton DL, Marovich M, Gordon AD, et al. Differential inhibitory receptor expression on T cells delineates functional capacities in chronic viral infection. J Virol. (2017) 91:1–15. doi: 10.1128/JVI.01263-17
88. Fromentin R, Bakeman W, Lawani MB, Khoury G, Hartogensis W, DaFonseca S, et al. CD4+ T cells expressing PD-1, TIGIT LAG-3 contribute to HIV persistence during art. PLoS Pathog. (2016) 12:e1005761. doi: 10.1371/journal.ppat.1005761
89. Kalams SA, Buchbinder SP, Rosenberg ES, Billingsley JM, Colbert DS, Jones NG, et al. Association between virus-specific cytotoxic T-lymphocyte and helper responses in human immunodeficiency virus type 1 infection. J of Virol. (1999) 73:6715–20. doi: 10.1128/JVI.73.8.6715-6720.1999
90. Porichis F, Kwon DS, Zupkosky J, Tighe DP, McMullen A, Brockman MA, et al. Responsiveness of HIV-specific CD4 T cells to PD-1 blockade. Blood. (2011) 118:955–74. doi: 10.1182/blood-2010-12-328070
91. Nicholas KJ, Zern EK, Barnett L, Smith RM, Lorey SL, Copeland CA, et al. B cell responses to HIV antigen are a potent correlate of viremia in HIV-1 infection and improve with PD-1 blockade. PLoS ONE. (2013) 8:e84185. doi: 10.1371/journal.pone.0084185
92. Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, Metcalf T, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. (2013) 19:494–9. doi: 10.1038/nm.3109
93. Muema DM, Macharia GN, Olusola BA, Hassan AS, Fegan GW, Berkley JA, et al. Proportions of circulating follicular helper T cells are reduced and correlate with memory B cells in HIV-infected children. PLoS ONE. (2017) 12:e0175570. doi: 10.1371/journal.pone.0175570
94. Cho H, Kang H, Kim CW, Kim HY, Jang JW, Yoon SK, et al. Phenotypic characteristics of PD-1 and CTLA-4 expression in symptomatic acute hepatitis Gut Liver A. (2016) 10:288–94. doi: 10.5009/gnl14368
95. Rutebemberwa A, Ray SC, Astemborski J, Levine J, Liu L, Dowd KA, et al. High-programmed death-1 levels on hepatitis C virus-specific T cells during acute infection are associated with viral persistence and require preservation of cognate antigen during chronic infection. J Immunol. (2008) 181:8215–25. doi: 10.4049/jimmunol.181.12.8215
96. Schönrich G, Raftery MJ. The PD-1/PD-L1 axis and virus infections: a delicate balance. Front Cell Infect Microbiol. (2019) 9:207. doi: 10.3389/fcimb.2019.00207
97. Brown KE, Freeman GJ, Wherry EJ, Sharpe AH. Role of PD-1 in regulating acute infections. Curr Opin Immunol. (2010) 22:397–401. doi: 10.1016/j.coi.2010.03.007
98. Erickson JJ, Rogers MC, Tollefson SJ, Boyd KL, Williams JV. Multiple inhibitory pathways contribute to lung CD8+ T cell impairment and protect against immunopathology during acute viral respiratory infection. J Immunol. (2016) 197:233–43. doi: 10.4049/jimmunol.1502115
99. Zdrenghea MT, Johnston SL. Role of PD-L1/PD-1 in the immune response to respiratory viral infections. Microbes Infect. (2012) 14:495–9. doi: 10.1016/j.micinf.2012.01.002
100. Allie SR, Zhang W, Fuse S, Usherwood EJ. Programmed death 1 regulates development of central memory CD8 T cells after acute viral infection. J Immunol. (2011) 186:6280–6. doi: 10.4049/jimmunol.1003870
101. Agrati C, Castilletti C, Casetti R, Sacchi A, Falasca L, Turchi F, et al. Longitudinal characterization of dysfunctional T cell-activation during human acute Ebola infection. Cell Death Dis. (2016) 7:e2164. doi: 10.1038/cddis.2016.55
102. Ruibal P, Oestereich L, Lüdtke A, Becker-Ziaja B, Wozniak DM, Kerber R, et al. Unique human immune signature of Ebola virus disease in Guinea. Nature. (2016) 533:100–4. doi: 10.1038/nature17949
103. Holokai L, Chakrabarti J, Broda T, Chang J, Hawkins JA, Sundaram N, et al. Increased programmed death-ligand 1 is an early epithelial cell response to Helicobacter pylori infection. PLoS Pathog. (2019) 15:e1007468. doi: 10.1371/journal.ppat.1007468
104. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. (1984) 1:1311–5. doi: 10.1016/S0140-6736(84)91816-6
105. Chen HN, Wang Z, Li X, Zhou ZG. Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: evidence from a meta-analysis. Gastric Cancer. (2016) 19:166–75. doi: 10.1007/s10120-015-0462-7
106. Anderson KM, Czinn SJ, Redline RW, Blanchard TG. Induction of CTLA-4-mediated anergy contributes to persistent colonization in the murine model of gastric helicobacter pylori infection. J Immunol. (2006) 176:5306–13. doi: 10.4049/jimmunol.176.9.5306
107. Wu YY, Lin CW, Cheng KS, Lin C, Wang YM, Lin IT, et al. Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection. Clin Exp Immunol. (2010) 161:551–9. doi: 10.1111/j.1365-2249.2010.04217.x
108. Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol. (2011) 186:1598–607. doi: 10.4049/jimmunol.1003304
109. Lázár-Molnár E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, et al. Programmed death-1 (PD-1)–deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci USA. (2010) 107:13402–7. doi: 10.1073/pnas.1007394107
110. Shen L, Shi H, Gao Y, Ou Q, Liu Q, Liu Y, et al. The characteristic profiles of PD-1 and PD-L1 expressions and dynamic changes during treatment in active tuberculosis. Tuberculosis. (2016) 101:146–50. doi: 10.1016/j.tube.2016.10.001
111. Anastasopoulou A, Ziogas DC, Samarkos M, Kirkwood JM, Gogas H. Reactivation of tuberculosis in cancer patients following administration of immune checkpoint inhibitors: current evidence and clinical practice recommendations. J Immunother Cancer. (2019) 7:239. doi: 10.1186/s40425-019-0717-7
112. Wu YY, Chen JH, Kao JT, Liu KC, Lai CH, Wang YM, et al. Expression of CD25(high) regulatory T cells and PD-1 in gastric infiltrating CD4(+) T lymphocytes in patients with Helicobacter pylori infection. Clin Vaccine Immunol. (2011) 18:1198–201. doi: 10.1128/CVI.00422-10
113. Srivastava S, Ernst JD. Cutting edge: direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J Immunol. (2013) 191:1016–20. doi: 10.4049/jimmunol.1301236
114. Yang JD, Mott D, Sutiwisesak R, Lu YJ, Raso F, Stowell B, et al. Mycobacterium tuberculosis-specific CD4+ and CD8+ T cells differ in their capacity to recognize infected macrophages. PLoS Pathog. (2018) 14:e1007060. doi: 10.1371/journal.ppat.1007060
115. Pepper M, Linehan JL, Pagán AJ, Zell T, Dileepan T, Cleary PP, et al. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat Immunol. (2010) 11:83–9. doi: 10.1038/ni.1826
116. McKay JT, Egan RP, Yammani RD, Chen L, Shin T, Yagita H, et al. PD-1 suppresses protective immunity to streptococcus pneumoniae through a B cell-intrinsic mechanism. J Immunol. (2015) 194:2289–99. doi: 10.4049/jimmunol.1401673
117. Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. (2009) 113:5811–8. doi: 10.1182/blood-2009-02-203141
118. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci USA. (2009) 106:6303–8. doi: 10.1073/pnas.0809422106
119. Fonseca R, Salgado RM, Borges da Silva H, do Nascimento RS, D'Império-Lima MR, Alvarez JM. Programmed cell death protein 1-PDL1 interaction prevents heart damage in chronic trypanosoma cruzi infection. Front Immunol. (2018) 9:997. doi: 10.3389/fimmu.2018.00997
120. Esch KJ, Juelsgaard R, Martinez PA, Jones DE, Petersen CA. Programmed death 1-mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J Immunol. (2013) 191:5542–50. doi: 10.4049/jimmunol.1301810
121. Mou Z, Muleme HM, Liu D, Jia P, Okwor IB, Kuriakose SM, et al. Parasite-derived arginase influences secondary anti-Leishmania immunity by regulating programmed cell death-1-mediated CD4+ T cell exhaustion. J Immunol. (2013) 190:3380–9. doi: 10.4049/jimmunol.1202537
122. Karunarathne DS, Horne-Debets JM, Huang JX, Faleiro R, Leow CY, Amante F, et al. Programmed death-1 ligand 2-mediated regulation of the PD-L1 to PD-1 axis is essential for establishing CD4(+) T cell immunity. Immunity. (2016) 45:333–45. doi: 10.1016/j.immuni.2016.07.017
123. WHO. Sepsis. (2018) Available online at: https://www.who.int/news-room/fact-sheets/detail/sepsis.
124. Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care. (2011) 15:1–8. doi: 10.1186/cc10332
125. Niu B, Zhou F, Su Y, Wang L, Xu Y, Yi Z, et al. Different expression characteristics of LAG3 and PD-1 in sepsis and their synergistic effect on T cell exhaustion: a new strategy for immune checkpoint blockade. Front Immunol. (2019) 10:1888. doi: 10.3389/fimmu.2019.01888
126. Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol. (2010) 88:233–40. doi: 10.1189/jlb.0110037
127. Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care. (2011) 15:R99. doi: 10.1186/cc10112
128. Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care. (2010) 14:R220. doi: 10.1186/cc9354
Keywords: PD-1, PD-L1, PD-L2, T cell exhaustion, acute infection, chronic infection, checkpoint inhibitor, infectious disease
Citation: Jubel JM, Barbati ZR, Burger C, Wirtz DC and Schildberg FA (2020) The Role of PD-1 in Acute and Chronic Infection. Front. Immunol. 11:487. doi: 10.3389/fimmu.2020.00487
Received: 04 January 2020; Accepted: 03 March 2020;
Published: 24 March 2020.
Edited by:
Julia Szekeres-Bartho, University of Pécs, HungaryReviewed by:
Birgit Sawitzki, Charité – Universitätsmedizin Berlin, GermanyRaj Raghupathy, Kuwait University, Kuwait
Copyright © 2020 Jubel, Barbati, Burger, Wirtz and Schildberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank A. Schildberg, frank.schildberg@ukbonn.de
†These authors have contributed equally to this work