 Keiko Taguchi
Keiko Taguchi Masayuki Yamamoto
Masayuki Yamamoto- Department of Medical Biochemistry, Graduate School of Medicine, Tohoku University, Sendai, Japan
Cancer cells first adapt to the microenvironment and then propagate. Mutations in tumor suppressor genes or oncogenes are frequently found in cancer cells. Comprehensive genomic analyses have identified somatic mutations and other alterations in the KEAP1 or NRF2 genes and in well-known tumor suppressor genes or oncogenes, such as TP53, CDKN2A, PTEN, and PIK3CA, in various types of cancer. Aberrant NRF2 activation in cancer cells occurs through somatic mutations in the KEAP1 or NRF2 gene as well as through other mechanisms that disrupt the binding of KEAP1 to NRF2. Unregulated NRF2 confers on cancer cells high-level resistance to anticancer drugs and reactive oxygen species (ROS) and directs cancer cells toward metabolic reprogramming. Therefore, NRF2 has been studied as a therapeutic target molecule in cancer. Two strategies have been used to target NRF2 via therapeutic drugs: inhibition of NRF2 and induction of NRF2. NRF2 inhibitors may be effective against NRF2-addicted cancer cells in which NRF2 is aberrantly activated. These inhibitors have not yet been established as NRF2-targeted anticancer drugs for the treatment of human cancers. Diagnosis of NRF2 activation could facilitate the use of NRF2 inhibitors for the treatment of patients with NRF2-addicted cancers. Conversely, NRF2 inducers have been used or are being developed for non-cancer diseases. In addition, NRF2 inducers may be useful for cancer chemotherapy in combination with conventional anticancer agents or even NRF2 inhibitors.
Introduction
NF-E2-related factor 2 (NRF2) is a master regulator of numerous cytoprotective genes (1, 2). After translation, the NRF2 protein is rapidly degraded by the ubiquitin-proteasome system in the cytoplasm (3). Kelch-like ECH-associated protein 1 (KEAP1) is a component of the Cullin 3 (CUL3)-based E3 ubiquitin ligase complex and controls the stability and accumulation of NRF2. The KEAP1 DC domain directly binds NRF2 through DLG and ETGE motifs within the N-terminal Neh2 domain of NRF2 (4, 5) (Figure 1A). Since KEAP1 molecules form homodimers within cells, the stoichiometry of the KEAP1 homodimer and NRF2 is 1:1, and that of a single KEAP1 molecule and NRF2 is 2:1 (6) (Figure 1B). Thus, two KEAP1 molecules and one NRF2 molecule form a trimer, and the formation of this structure accelerates the ubiquitination of lysine residues in the NRF2 Neh2 domain, leading to proteasomal degradation of NRF2 (7–9). This two-site binding of NRF2 and KEAP1 has been shown to be the molecular basis of electrophile-induced NRF2 accumulation (10). Inactivation of KEAP1 strongly induces NRF2, and this phenomenon is often observed in cancer cells; cancer cells can thus acquire malignancy by perverting NRF2 activity. In this review, we will focus on the regulation of cellular NRF2 levels by KEAP1 and the perturbation of KEAP1 regulation in cancer cells.
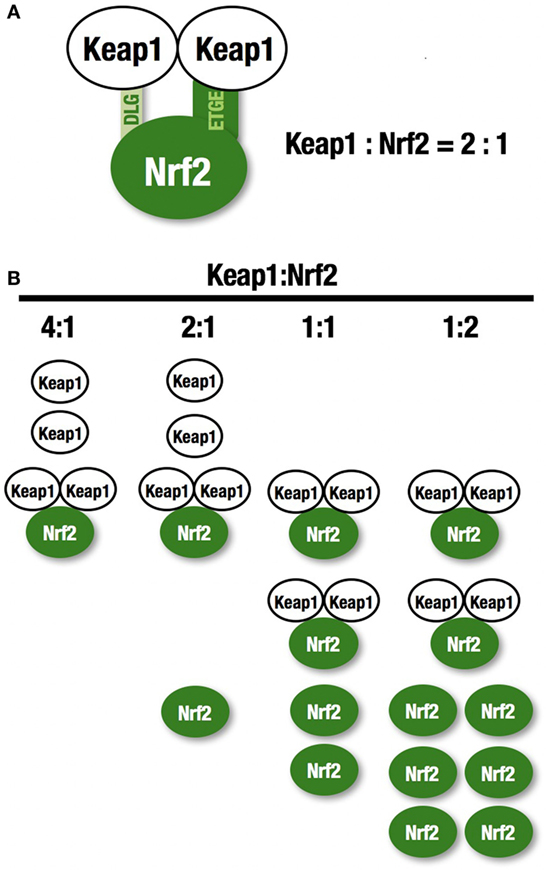
Figure 1. KEAP1 and NRF2. (A) Two KEAP1 molecules and one NRF2 form a trimer through the DC domain in KEAP1 and the DLG and ETGE motifs within the N-terminal Neh2 domain of NRF2. This two-site binding of NRF2 and KEAP1 has been shown to be the molecular basis of electrophile-induced NRF2 accumulation. (B) The ratio of KEAP1:NRF2 binding. KEAP1 is unable to bind a large amount of NRF2.
Mechanisms That Transmit Environmental Stresses to Gene Expression
In normal unstressed conditions, the cellular NRF2 level is very low (11), but it is dramatically increased upon exposure to electrophilic chemicals or reactive oxygen species (ROS) (1). Electrophiles modify reactive cysteine residues in KEAP1 (12). Murine KEAP1 possesses 25 cysteine residues, and these residues are categorized into several classes based on their reactivity to various electrophiles (13–15). For instance, cysteine 151 (C151) and C288 have been shown to sense distinct sets of electrophiles that are produced endogenously or exogenously (15–17). Oxidative modification of KEAP1 has also been shown to attenuate its binding to NRF2 or CUL3 (18, 19), although specific cysteine residues modified by ROS remain to be identified. These electrophilic and oxidative modifications inactivate KEAP1 and thereby stabilize NRF2. Therefore, the increase in NRF2 in response to electrophiles and ROS is not induction in a strict sense but rather is a mechanism referred to as derepression (from the rapid degradation-based repression). In this paper, we use both derepression and induction to describe this phenomenon, i.e., the increase in NRF2.
For example, an electrophile, such as tert-butyl hydroquinone (tBHQ), reacts with reactive cysteine residues in KEAP1 to activate NRF2 (20). Importantly, binding of tBHQ to KEAP1 does not disrupt the binding of NRF2 to KEAP1 (21), demonstrating that simple dissociation of NRF2 from the KEAP1 homodimer cannot explain the electrophile-mediated induction of NRF2 accumulation within cells. Instead, the evidence indicates that since electrophilic modification inactivates KEAP1, newly synthesized NRF2 is able to escape KEAP1 trapping in this situation, so that the newly made NRF2 is stabilized and accumulates. KEAP1 is primarily localized to the perinuclear cytoplasm (22), loosely connected with the actin cytoskeleton (23), and serves to maintain the correct levels of NRF2 (6). KEAP1 forms an E3 ubiquitin ligase complex with CUL3, acting as a substrate recognition/binding subunit. The KEAP1-based ubiquitin ligase complex ubiquitinates NRF2, subjecting NRF2 to rapid proteasomal degradation in the cytoplasm (3) (Figure 2).
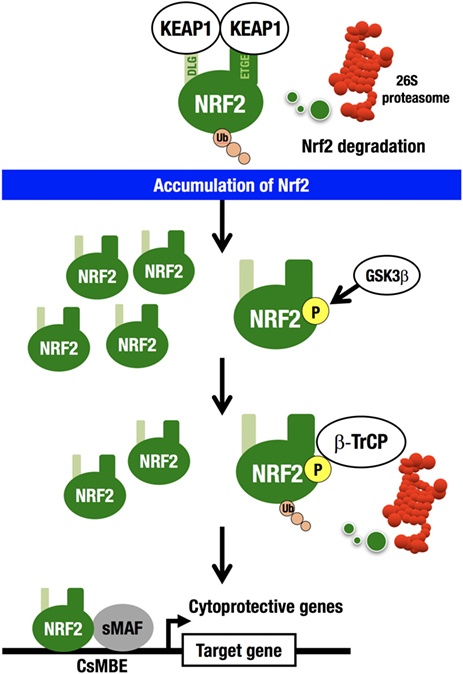
Figure 2. KEAP1–NRF2 system. KEAP1-CUL3 directly binds NRF2, which is then rapidly degraded by the 26S proteasome in the cytoplasm. NRF2 that escapes KEAP1 trapping is stabilized and accumulates in the nucleus. GSK3 phosphorylates the Neh6 domain of NRF2. Phosphorylated NRF2 binds β-TrCP-CUL1 and is then degraded by the 26S proteasome in the nucleus. NRF2 forms a heterodimer with sMAF and binds CNC-sMAF-binding elements (CsMBE), including the consensus ARE/EpRE sequence, TGACNNNGC. NRF2 upregulates cytoprotective genes encoding antioxidant enzymes and detoxifying enzymes.
While this is the major pathway for the cellular response to NRF2 induction (derepression), there exists a second NRF2 degradation pathway within the nucleus. β-TrCP (β-transducin repeat-containing protein) forms an E3 ubiquitin ligase complex with CUL1 and ubiquitinates NRF2. β-TrCP serves as a substrate recognition/binding subunit that recognizes phosphorylated NRF2 (24). In the nucleus, the serine residues in the Neh6 domain of NRF2 are phosphorylated by glycogen synthase kinase 3 (GSK3), which is a downstream effector of the phosphoinositide 3-kinase (PI3K)–AKT pathway (25), and phosphorylated NRF2 is captured by β-TrCP, ubiquitinated, and subjected to proteasomal degradation.
Interestingly, in an experiment using mouse liver, deletion of the PTEN gene and subsequent activation of the PI3K–AKT pathway was insufficient to activate NRF2. Conversely, deletion of the KEAP1 gene significantly activated NRF2. Simultaneous deletion of the PTEN and KEAP1 genes activated NRF2 much more strongly than the KEAP1 single deletion (26). Thus, NRF2 degradation occurs via two pathways (27); the major pathway is localized in the cytoplasm and governed by KEAP1, while the secondary pathway is in the nucleus and governed by β-TrCP. These observations support the notion that cellular NRF2 levels are strictly regulated by two pathways through the protein degradation-repression mechanism: derepression from the KEAP1-based repression causes a rapid increase in NRF2 activity and induction of cellular defense mechanisms against electrophilic and oxidative insults, while β-TrCP-based NRF2 degradation inhibits unnecessary NRF2 over-induction caused by KEAP1 inactivation.
It has been reported that there is a link between NRF2 activity and miRNAs that appears to be relevant to disease (28). For example, in breast cancer, miR-28 regulates NRF2 expression through a KEAP1-independent mechanism (29). miR-200a regulates NRF2 activation by targeting KEAP1 mRNA in breast cancer cells (30). On the other hand, NRF2 regulates miR-1 and miR-206 to direct carbon flux toward the pentose phosphate pathway (PPP) and tricarboxylic acid (TCA) cycle, which reprograms glucose metabolism (31) (see the section on metabolic reprogramming in cancer).
NRF2 Target Genes
Stabilized NRF2 translocates into the nucleus and forms a heterodimer with a small MAF (sMAF) transcription factor (1). The NRF2-sMAF heterodimer binds to antioxidant-responsive element (ARE) (32) or electrophile-responsive element (EpRE) (33) and induces transcription of numerous cytoprotective genes. The consensus ARE/EpRE sequence is TGACNNNGC (34). This sequence is highly similar to the consensus-binding sequence for the erythroid transcription factor NF-E2 (35), which is composed of a p45 subunit and sMAF. Historically, the NRF2-sMAF-mediated regulation of cytoprotective gene expression via ARE/EpRE was identified based on this similarity (1), as both p45 and NRF2 belong to a small transcription factor family named the Cap‘n’collar (CNC) family (36).
Recently, an extensive genome-wide analysis of the NRF2-sMAFF-binding sequence (i.e., the ARE/EpRE) and the MAF homodimer-binding sequence (the MAF responsive element or MARE) was conducted, and the differences between these elements were clarified (37). As a result, it was proposed that ARE, EpRE, and the NF-E2 binding sequence be collectively named CNC-sMAF-binding elements (CsMBE).
Recent chromatin immunoprecipitation-deep sequencing (ChIP-Seq) analyses have revealed target genes of the NRF2-sMAF heterodimer (34, 38–40). NRF2-sMAF appears to globally regulate cytoprotective and metabolic networks. One group of important NRF2-sMAF target genes encodes antioxidative enzymes and detoxifying enzymes. NAD(P)H:quinone oxidoreductase 1 (Nqo1) is representative of this group of genes and is widely used to evaluate NRF2 activity. In addition, NRF2-sMAF regulates genes encoding PPP enzymes, ABC pumps, and some heme-metabolizing enzymes.
Aberrant NRF2 Activation in Cancer
An intriguing finding in human biology and pathology related to the KEAP1–NRF2 regulatory system is that cancer cells occasionally acquire aberrant NRF2 activation (41–43) (Figure 3A). At first, this observation was confusing, as the KEAP1–NRF2 system has been shown to contribute to cancer chemoprevention (44). However, previous observations that cancer cells often acquire strong antioxidative and drug-metabolizing activities explain cancer cell acquirement of malignancy and cytoprotective activity (45, 46).
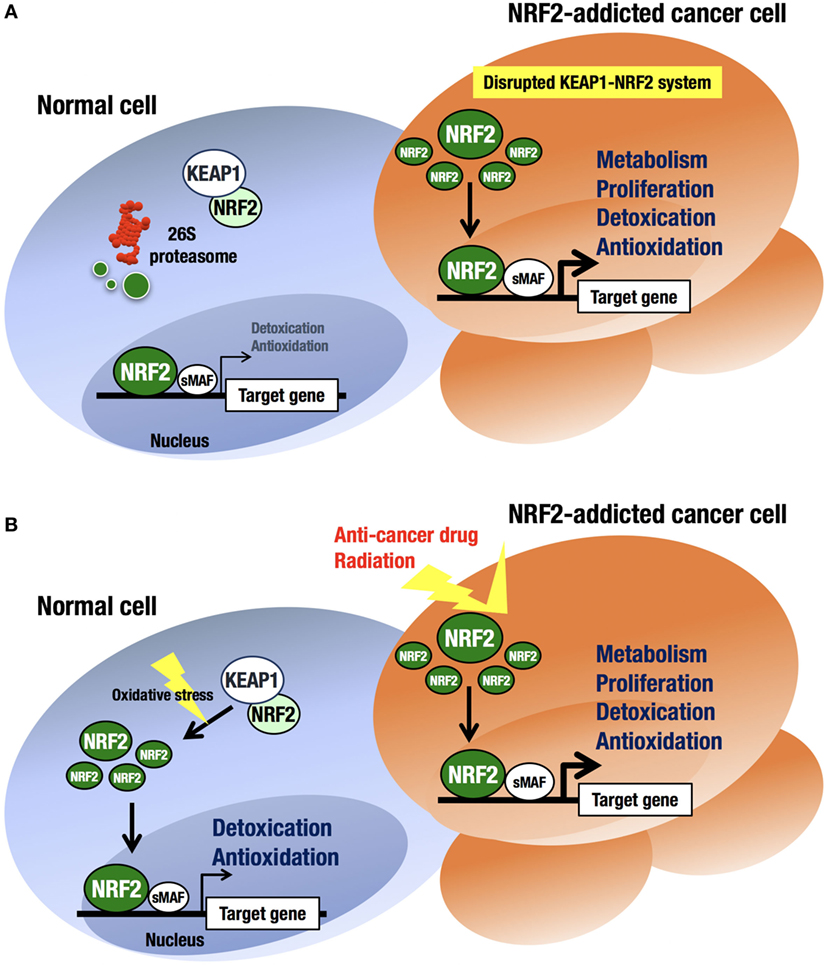
Figure 3. Difference in NRF2 activation between normal cells and NRF2-addicted cancer cells. (A) Unstressed condition. The intracellular NRF2 level is very low in normal cells. In contrast, constitutive NRF2 activation in cancer cells accelerates proliferation and metabolism. (B) Oxidative stress-exposed condition. In normal cells, the cellular NRF2 level is temporarily increased upon exposure to toxic (often electrophilic) chemicals and ROS. In contrast, constitutive NRF2 activation confers resistance on cancer cells to anticancer drugs and radiation.
At least four pathways have been reported to be involved in NRF2 activation in cancer cells (17). First, somatic mutations within the NRF2, KEAP1, or CUL3 genes (41, 47–49) have been shown to cause aberrant NRF2 activation in cancer cells. Second, epigenetic silencing of the KEAP1 gene has also been found to cause KEAP1 downregulation and NRF2 upregulation (50). Third, the accumulation of KEAP1 interacting proteins, such as p62/Sqstm1 (51) and p21 (52), has been found to block NRF2 binding to KEAP1, leading to NRF2 accumulation. Fourth, cysteine modification by oncometabolites such as fumarate affects KEAP1 activity and leads to NRF2 accumulation (53, 54). All these molecular events result in disrupted binding of KEAP1 to NRF2, causing aberrant accumulation of NRF2 in cancer cells. Unregulated NRF2 activates the target genes responsible for cytoprotection, conferring chemo- and radio-resistance on cancer cells (44) (Figure 3B).
The A549 cell line was derived from an adenocarcinoma of the human alveolar basal epithelium and is a typical cancer cell line that exhibits aberrantly active NRF2. There are two mechanisms by which A549 cells acquire constitutive NRF2 activation: one is a somatic mutation of the KEAP1 gene at G333C (47, 55), and the other is epigenetic alteration by methylation in the KEAP1 promoter (50). NRF2 is a key molecule that controls proliferation in NRF2-addicted cancer cells, such as A549 cells (26) (Table 1).
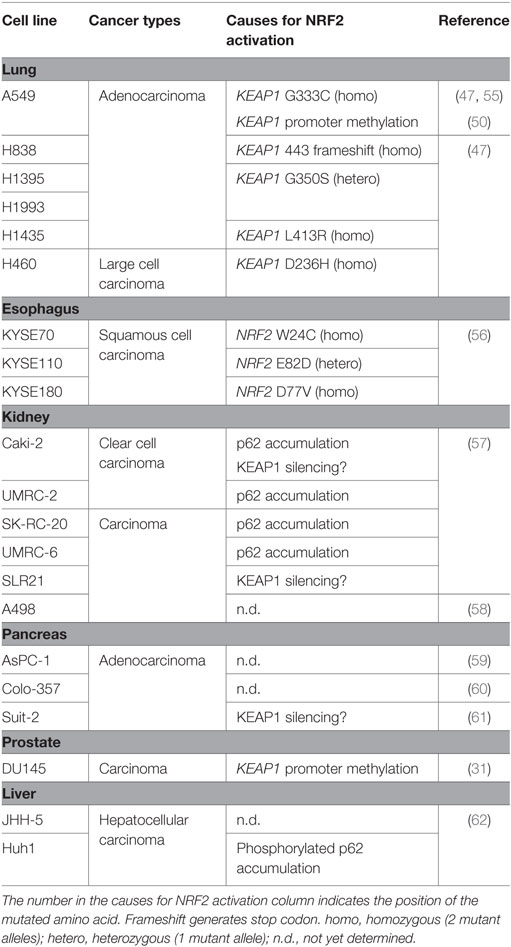
Table 1. NRF2-addicted cancer cell lines.
A catalog of somatic mutations in cancer (COSMIC; http://cancer.sanger.ac.uk/cosmic) reveals 274 coding mutations in the KEAP1 gene and 389 in the NRF2 gene in cancers from various tissues (Figure 4A). Mutations in KEAP1 or NRF2 were found in approximately 0.9% of all cancer samples examined in studies published in COSMIC 2016. Interestingly, the NRF2 mutations are exclusively found in the DLG and ETGE motifs responsible for binding to KEAP1 (Figure 4B). Cancer cells that harbor a somatic mutation in these motifs of the NRF2 gene lose the KEAP1–NRF2 interaction and the subsequent constitutive repression of NRF2 activity during unstressed conditions.
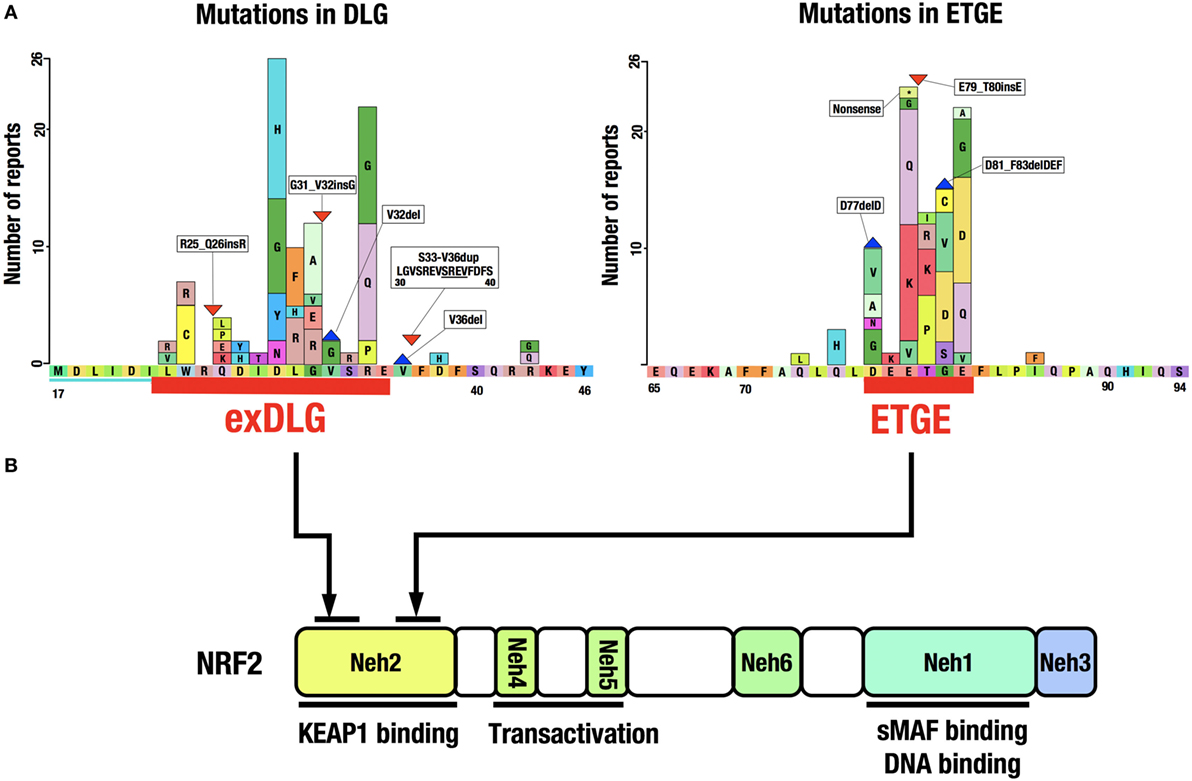
Figure 4. Distribution of somatic mutations in the NRF2 gene in cancers. (A) NRF2 mutations in the exDLG and ETGE motifs. Red lines indicate the DLG motif and the ETGE motif. Please see Ref. (10). (B) The domain structure of the NRF2 protein. NRF2 mutations are exclusively found in the DLG and ETGE motifs responsible for binding to KEAP1.
KEAP1 mutations, however, are widely distributed in the KEAP1 gene and are found in virtually all domains of the protein. Somatic mutations in the KEAP1 gene, similar to those in the NRF2 gene, affect protein–protein interactions, i.e., the binding of NRF2 to KEAP1. Genomic alterations in various cancer-related genes have been comprehensively characterized in squamous cell lung cancers (63). The most frequently mutated gene was TP53, with mutations found in 81% of the analyzed samples. While KEAP1 (12%) and NRF2 (15%) mutations were found in a much lower percentage of samples, gene mutations related to the KEAP1–NRF2 system, including CUL3 (7%) and PTEN (8%), were more frequent than those in the second most frequently mutated gene CDKN2A (15%).
Notably, mutations in KEAP1, NRF2, and PTEN are mutually exclusive and are seldom found in the same cancer cell (63). Therefore, correlations among mutations in KEAP1–NRF2 system genes and other well-known tumor suppressor genes or oncogenes are intriguing but have yet to be precisely studied. It was recently reported that mutations in either KEAP1 or NRF2 were found in approximately 14% of hepatocellular carcinoma (HCC) cases (64). This observation indicates that the frequency of mutations in KEAP1 or NRF2 depends on the cancer type and origin.
Metabolic Reprogramming in Cancer
NRF2 target gene products are involved in cytoprotection, and typical examples include detoxifying enzymes and antioxidant enzymes. A ChIP-Seq analysis of NRF2 and MAFG target genes in A549 cells identified novel NRF2 target genes; for example, those encoding metabolic enzymes (26). In fact, NRF2 regulates the expression of genes encoding PPP enzymes and glutaminolysis-related enzymes. Previous studies have shown that the expression of malic enzyme 1, glucose-6-phosphate dehydrogenase (G6PD), and phosphogluconate dehydrogenase is correlated with NRF2 (65, 66). However, the functional implications of the induction of these enzymes in cancer cells are not clearly understood. Notably, siRNA-mediated NRF2 knockdown in A549 cells that are addicted to NRF2 significantly decreases the proliferative ability of these cells (26). This observation suggests that NRF2 plays an important role in cancer cell proliferation. Based on this observation, we have proposed that NRF2 directs the metabolic reprogramming of cancer cells, a notion that has been supported by recent studies.
Both amplification of the p62 gene and aberrant accumulation of phosphorylated p62 protein have been implicated in the acceleration of tumor development. Phosphorylation of p62 at S349 activates NRF2 and directs glucose metabolism to the glucuronate pathway and glutamine metabolism to glutathione synthesis (67). These changes make HCC cells resistant to anticancer drugs and enhance their proliferation ability. Phosphorylated p62 accumulates in tumor regions positive for hepatitis C virus (HCV). Similarly, hepatitis B virus (HBV) also stimulates NRF2 activation and upregulates G6PD, the first and rate-limiting enzyme of the PPP (68). NRF2 also reportedly regulates miR-1 and miR-206 to direct carbon flux toward the PPP and TCA cycle, and NRF2 also regulates the reprogramming of glucose metabolism in cancer cells (31).
KEAP1–NRF2 System as a Therapeutic Target
NRF2 is an attractive molecule as a therapeutic target in cancer. There are two main strategies used to target NRF2 by therapeutic drug treatment: one is NRF2 inhibition, and the other is NRF2 induction. NRF2 inducers have been shown to accelerate the detoxification of carcinogens (often electrophiles) from the environment and protect the body from chemical carcinogenesis (Figure 5A). Of note, the NRF2 inducer dimethyl fumarate has been approved by the FDA for multiple sclerosis treatment, and bardoxolone methyl (CDDO-Me or RTA 402) is now in phase II clinical trials for pulmonary hypertension and chronic kidney diseases. Some phytochemicals, such as sulforaphane from broccoli sprouts (69), curcumin from turmeric (70), or carnosic acid from rosemary (71), also activate NRF2. These chemicals have been used as dietary supplements (72).
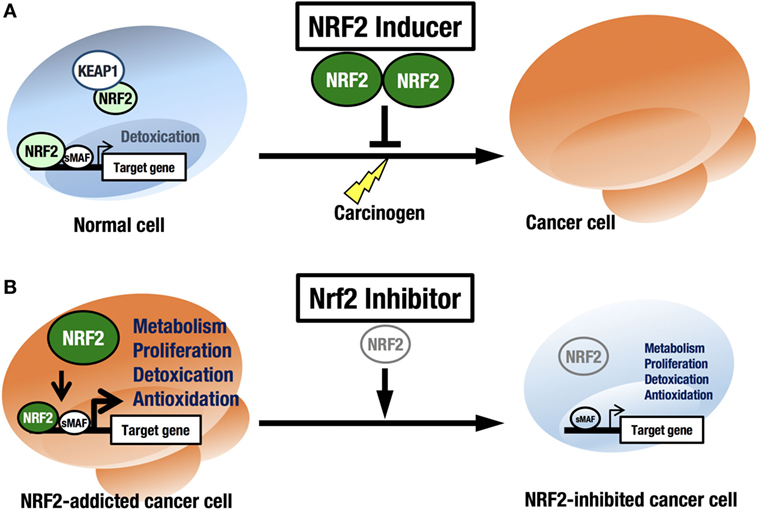
Figure 5. Two strategies for cancer therapy focused on NRF2. (A) Chemoprevention against carcinogens by NRF2 inducers in normal cells. (B) Anticancer therapy against NRF2-addicted cancer cells by NRF2 inhibitors.
As described in the previous section, the chemicals described above act to modify KEAP1 cysteine residues (the cysteine code). Therefore, concerns remain regarding glutathione depletion or redox side effects. An alternative approach for the development of NRF2 inducers is the use of chemicals that disrupt the KEAP1–NRF2 interface, especially chemicals that target a pocket that resides in the bottom of the KEAP1 DC domain (10, 67).
In this regard, it should be noted that there are many cancers in a variety of tissues that show intrinsically high NRF2 activity. We refer to these cancers as NRF2-addicted cancers, as NRF2 provides cytoprotection to these cancer cells by activating detoxifying and antioxidative enzymes and through metabolic reprogramming. For these cancers, NRF2 inhibitors may show therapeutic effects (73) (Figure 5B).
NRF2 Inhibitors
Brusatol is a type of degraded diterpenoid isolated from the Brucea javanica plant (74). Brusatol has been used in traditional Chinese medicine to treat a variety of ailments, including cancer, amoebic dysentery, and malaria. Brusatol inhibits DNA, RNA, and protein synthesis (75, 76), and inhibition of overall protein synthesis by brusatol has been observed in many types of cancer cells (74, 76, 77). Brusatol has recently been reported to act as an NRF2 inhibitor (78), inhibiting NRF2 in all cell lines tested, regardless of whether the KEAP1 or NRF2 genes were mutated. A subsequent paper showed that brusatol induces its potent cytotoxic effects in a manner independent of KEAP1–NRF2 activity and with a profile similar to a protein translation inhibitor. Therefore, brusatol does not specifically inhibit NRF2 but rather functions as a global protein synthesis inhibitor (79).
ML385, a thiazole-indoline compound, is a probe molecule that specifically binds to Neh1, the DNA, and sMAF-binding domain in NRF2, and inhibits the expression of downstream target genes (80). Combined with doxorubicin or Taxol, ML385 substantially enhances cytotoxicity in non-small cell lung cancer (NSCLC) cells compared to single agents. ML385 shows specificity and selectivity for NRF2-addicted NSCLC cells with KEAP1 mutations, such as A549 and H460 cells. In preclinical models of NRF2-addicted NSCLC, ML385 shows significant antitumor activity in combination with carboplatin.
Febrifugine is the bioactive component of the traditional Chinese medical herb Dichroa febrifuga (81). The febrifugine derivative halofuginone has been optimized to be less toxic than febrifugine. Halofuginone is a specific inhibitor of collagen type-I synthesis (82) and of prolyl-tRNA synthetase (83). Febrifugine derivatives have been used as treatments for cancer, malaria, fibrosis, and inflammatory diseases. Halofuginone has been tested in phase 2 clinical trials for cancer (84) and fibrotic diseases (85) including Duchenne muscular dystrophy (86). Though HT-100, an oral analog of halofuginone, was studied in a phase 2 clinical trial for bladder cancer treatment, development of the compound was discontinued. Quite recently, halofuginone was found to inhibit NRF2 (87).
In NRF2-addicted cancer cells, halofuginone decreases NRF2 protein synthesis by inhibiting prolyl-tRNA synthetase. This inhibition is rescued by the addition of proline, supporting the hypothesis that halofuginone inhibits NRF2 by affecting the prolyl-tRNA synthetase. Halofuginone treatment of NRF2-addicted cancer cells, such as lung cancer-derived A549 cells or esophagus cancer-derived KYSE70 cells, attenuates proliferation in vitro. Co-treatment with halofuginone also enhances the effects of conventional anticancer drugs, such as cisplatin or doxorubicin, in a xenograft tumor model. While halofuginone is not a specific NRF2 inhibitor, it exerts a strong inhibitory effect on NRF2. This may be because NRF2 is a rapidly turned-over protein with a half-life of less than 20 min, and NRF2 synthesis is somehow linked to the amino acid starvation machinery. Thus, halofuginone could serve as a chemosensitizer in the treatment of various NRF2-addicted cancers.
Another challenge related to NRF2 inhibition is the inhibition of the protein–protein interaction between phosphorylated p62 and KEAP1. In many cases of HCC, phosphorylated p62 accumulates and inhibits KEAP1 activity by interacting with the NRF2-binding pocket of KEAP1. For example, Huh1 cells express a high level of phosphorylated p62 (64). Therefore, a specific inhibitor of the interaction between phosphorylated p62 and KEAP1 would enable KEAP1 to bind and rapidly degrade NRF2; such a drug would be expected to function as an anticancer drug, particularly for cancers such as HCC, in which phosphorylated p62 accumulates and inhibits KEAP1 activity and thereby increases NRF2 activity. K67 (N-[2-acetonyl-4-(4-ethoxybenzenesulfonylamino)naphthalene-1-yl]-4-ethoxybenzenesulfonamide) is an inhibitor of the phosphorylated p62–KEAP1 interaction that reduces the NRF2 level in HCC (67). K67 suppresses the proliferation of HCC cells and accelerates the effects of anticancer agents. K67 could also make cancer cells less resistant to anticancer drugs, especially in HCV-positive HCC patients.
Other chemicals have also been reported as NRF2 inhibitors, although the molecular mechanisms through which these chemicals inhibit NRF2 have not been well elucidated. The coffee alkaloid trigonelline suppresses proteasomal activity in pancreatic cancer cells following NRF2 inhibition (60). Chrysin (5,7-dihydroxyflavone) and apigenin (4′,5,7-trihydroxyflavone) sensitize doxorubicin-resistant human liver cancer-derived Bel-7402 cells to doxorubicin, which is associated with the downregulation of NRF2 (88). Luteolin (3′,4′,5,7-tetrahydroxyflavone) reduces NRF2 expression at both the mRNA and protein levels in NSCLC A549 cells (89).
NRF2 Inducers
Aflatoxin B1 (AFB1) is a carcinogen contaminant in food. Exposure to AFB1 is one of the causes of HCC in Asia and Africa. While AFB1-driven HCC is experimentally reproducible in rats, it cannot be reproduced in mice. Mice are innately resistant to AFB1, because they express high levels of glutathione S-transferases (GSTs), which play an important role in AFB1 detoxification. CDDO-Im (1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl] imidazole) is a synthetic oleanane triterpenoid and a powerful NRF2 inducer. CDDO-Im suppresses AFB1-induced toxicity and preneoplastic lesion formation (GST-P-positive foci) and completely protects rats against AFB1-induced HCC (90). With CDDO-Im treatment, the integrated level of urinary AFB1-N7-guanine is significantly reduced and aflatoxin-N-acetylcysteine, a detoxification product, is consistently elevated after the first AFB1 dose. In AFB1-treated rats, the hepatic burden of GST-P-positive foci increases, but the foci largely disappear after CDDO-Im intervention. The toxicogenomic RNA expression signature characteristic of AFB1 is absent in rats dosed with AFB1 in combination with CDDO-Im. The remarkable efficacy of CDDO-Im as an anticarcinogen is observed even in the presence of a significant aflatoxin adduct burden.
In this regard, GST-P has also been used as a marker of DEN (diethylnitrosamine)-induced hepatic micronodules in the Solt–Farber-resistant hepatocyte experiments (45). In the hepatic micronodules, GST-P appears to protect cancer cells from anticancer chemotherapy as well as from oxidative stress that originated within the microenvironment. This observation suggests that upon cancer chemotherapy, anticancer drugs strongly attack the healthy cells in the microenvironment rather than the cancer cells, which makes the chemotherapy less effective. To counteract this activity, the combined use of NRF2 inducers with anticancer drugs may be useful to overcome this limitation of conventional cancer chemotherapy.
Oltipraz (4-methyl-5-(2-pyrazinyl)-3H-1,2-dithiole-3-thione) is a schestosomicide that is, and also a well-known NRF2 inducer. Oltipraz inhibits the formation of various cancers in rodent models (44, 91). The clinical cancer chemoprevention trials of this drug have progressed (92, 93). Whereas Oltipraz attenuates the progression of non-alcoholic steatohepatitis (NASH) and is now in phase 3 clinical trial against NASH in South Korea, Oltipraz has not yet been developed as an anticancer drug.
There are some NRF2 inducers in dietary supplements. Sulforaphane from broccoli sprouts (69) induces cytoprotective enzymes through direct binding to C151 of KEAP1 (94). It has been shown that curcumin from turmeric activates NRF2 through the inhibition of KEAP1 (70); however, it remains unclear whether curcumin reacts with cysteine residues of KEAP1. Carnosic acid from rosemary activates NRF2 by directly binding to cysteine residues in KEAP1 (71). To demonstrate the protective effect of broccoli sprouts containing precursors of sulforaphane against carcinogens such as aflatoxin and airborne toxicants, a clinical trial using broccoli sprouts was performed in China (95). Individuals who received a broccoli sprout-based beverage exhibited reduced toxic metabolite levels and increased detoxified metabolite levels.
In addition to these chemo-preventive uses of NRF2 inducers, there may be alternative uses of NRF2 inducers in the chemo- and radio-therapy of cancer patients. NRF2-addicted cancers are resistant to these cancer therapies due to their high intrinsic NRF2 activity. Therefore, anticancer drugs attack micro-environmental cells or immune cells more severely than the cancer cells, which already express a high level of NRF2 and cytoprotective enzymes. To this end, the combined use of an NRF2 inducer with conventional anticancer agents may better protect the host cells and improve the efficacy of anticancer drugs against NRF2-addicted cancers.
Conclusion
Both NRF2 inducers and NRF2 inhibitors are expected to function as anticancer drugs. However, the targets of these drugs are significantly different: NRF2 inducers act to protect normal cells from carcinogens, whereas NRF2 inhibitors act to suppress the proliferation of cancer cells that have acquired aberrant NRF2 activation or NRF2 addiction. However, many questions related to the NRF2 inducers and inhibitors remain and must be resolved before the NRF2 inducers and inhibitors can be judiciously applied in anticancer therapy. For example, methods to assess the NRF2 addiction status of each cancer need to be established. Notably, in human HCC biopsies, G6PD and NQO1 mRNAs can be used as markers that correlate well with metastatic status and poor prognosis (96). These markers could also be used as accurate indicators of NRF2 activity or NRF2 addiction. In addition, an emerging possibility is to use NRF2 inducers for cancer chemotherapy in combination with conventional anticancer agents or even NRF2 inhibitors. In this regard, there is a concern whether long-term application of NRF2 inducers may eventually support the growth of cryptic cancer-initiating cells into real cancer cells. While available lines of evidence from us and other laboratories do not support this concern, further investigations are needed to exclude this possibility.
Author Contributions
Both authors equally contribute to the description.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by funding from MEXT/JSPS KAKENHI (24249015, 26111002, and 15H02507 to MY; 26460384, 15KK0325, 16H01190 to KT), AMED-CREST, AMED (MY), MEXT [a research program of the Project for Cancer Research and Therapeutics Evolution (P-CRETE)], the Mitsubishi Foundation (MY), and the Takeda Science Foundation (MY), the Gushinkai Foundation (KT), Gonryo Medical Foundation (KT), the Waksman Foundation of Japan (KT), Kato Memorial Bioscience Foundation (KT), and the Naito Foundation (KT and MY).
References
1. Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun (1997) 236(2):313–22. doi: 10.1006/bbrc.1997.6943
2. McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, et al. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res (2001) 61(8):3299–307.
3. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol (2004) 24(16):7130–9. doi:10.1128/MCB.24.16.7130-7139.2004
4. Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol (2006) 26(8):2887–900. doi:10.1128/MCB.26.8.2887-2900.2006
5. Kim J, Keum YS. NRF2, a key regulator of antioxidants with two faces towards cancer. Oxid Med Cell Longev (2016) 2016:2746457. doi:10.1155/2016/2746457
6. Iso T, Suzuki T, Baird L, Yamamoto M. Absolute amounts and status of the Nrf2-Keap1-Cul3 complex within cells. Mol Cell Biol (2016) 36(24):3100–12. doi:10.1128/MCB.00389-16
7. Itoh K, Ishii T, Wakabayashi N, Katoh Y, Yamamoto M. Nrf2 activation by electrophiles is mediated by the inhibition of proteasomal degradation of Nrf2 protein in activated macrophages. FASEB J (2001) 15(5):A899–899. doi:10.1128/MCB.26.1.221-229.2006
8. McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem (2003) 278(24):21592–600. doi:10.1074/jbc.M300931200
9. Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol (2004) 24(24):10941–53. doi:10.1128/MCB.24.24.10941-10953.2004
10. Fukutomi T, Takagi K, Mizushima T, Ohuchi N, Yamamoto M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol Cell Biol (2014) 34(5):832–46. doi:10.1128/MCB.01191-13
11. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev (1999) 13(1):76–86. doi:10.1101/gad.13.1.76
12. Dinkova-Kostova AT, Holtzclaw WD, Wakabayashi N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry (2005) 44(18):6889–99. doi:10.1021/bi047434h
13. Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol (2003) 23(22):8137–51. doi:10.1128/MCB.23.22.8137-8151.2003
14. Takaya K, Suzuki T, Motohashi H, Onodera K, Satomi S, Kensler TW, et al. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic Biol Med (2012) 53(4):817–27. doi:10.1016/j.freeradbiomed.2012.06.023
15. Saito R, Suzuki T, Hiramoto K, Asami S, Naganuma E, Suda H, et al. Characterizations of three major cysteine sensors of Keap1 in stress response. Mol Cell Biol (2015) 36(2):271–84. doi:10.1128/MCB.00868-15
16. Eggler AL, Luo Y, van Breemen RB, Mesecar AD. Identification of the highly reactive cysteine 151 in the chemopreventive agent-sensor Keap1 protein is method-dependent. Chem Res Toxicol (2007) 20(12):1878–84. doi:10.1021/tx700217c
17. Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells (2011) 16(2):123–40. doi:10.1111/j.1365-2443.2010.01473.x
18. Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem (2000) 275(21):16023–9. doi:10.1074/jbc.275.21.16023
19. Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol (2008) 21(3):705–10. doi:10.1021/tx700302s
20. Abiko Y, Miura T, Phuc BH, Shinkai Y, Kumagai Y. Participation of covalent modification of Keap1 in the activation of Nrf2 by tert-butylbenzoquinone, an electrophilic metabolite of butylated hydroxyanisole. Toxicol Appl Pharmacol (2011) 255(1):32–9. doi:10.1016/j.taap.2011.05.013
21. Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, et al. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol (2006) 26(1):221–9. doi:10.1128/MCB.26.1.221-229.2006
22. Watai Y, Kobayashi A, Nagase H, Mizukami M, McEvoy J, Singer JD, et al. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells (2007) 12(10):1163–78. doi:10.1111/j.1365-2443.2007.01118.x
23. Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci U S A (2004) 101(7):2046–51. doi:10.1073/pnas.0308347100
24. Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol (2012) 32(17):3486–99. doi:10.1128/MCB.00180-12
25. Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene (2013) 32(32):3765–81. doi:10.1038/onc.2012.388
26. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell (2012) 22(1):66–79. doi:10.1016/j.ccr.2012.05.016
27. Taguchi K, Hirano I, Itoh T, Tanaka M, Miyajima A, Suzuki A, et al. Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol Cell Biol (2014) 34(5):900–13. doi:10.1128/MCB.01384-13
28. Cheng X, Ku CH, Siow RC. Regulation of the Nrf2 antioxidant pathway by microRNAs: new players in micromanaging redox homeostasis. Free Radic Biol Med (2013) 64:4–11. doi:10.1016/j.freeradbiomed.2013.07.025
29. Yang M, Yao Y, Eades G, Zhang Y, Zhou Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res Treat (2011) 129(3):983–91. doi:10.1007/s10549-011-1604-1
30. Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem (2011) 286(47):40725–33. doi:10.1074/jbc.M111.275495
31. Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest (2013) 123(7):2921–34. doi:10.1172/JCI66353
32. Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem (1990) 265(24):14648–53.
33. Friling RS, Bensimon A, Tichauer Y, Daniel V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc Natl Acad Sci U S A (1990) 87(16):6258–62. doi:10.1073/pnas.87.16.6258
34. Hirotsu Y, Katsuoka F, Funayama R, Nagashima T, Nishida Y, Nakayama K, et al. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res (2012) 40(20):10228–39. doi:10.1093/nar/gks827
35. Mignotte V, Navarro S, Eleouet JF, Zon LI, Romeo PH. The extinction of erythroid genes after tetradecanoylphorbol acetate treatment of erythroleukemic cells correlates with down-regulation of the tissue-specific factors NF-E1 and NF-E2. J Biol Chem (1990) 265(36):22090–2.
36. Sykiotis GP, Bohmann D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell (2008) 14(1):76–85. doi:10.1016/j.devcel.2007.12.002
37. Otsuki A, Suzuki M, Katsuoka F, Tsuchida K, Suda H, Morita M, et al. Unique cistrome defined as CsMBE is strictly required for Nrf2-sMaf heterodimer function in cytoprotection. Free Radic Biol Med (2016) 91:45–57. doi:10.1016/j.freeradbiomed.2015.12.005
38. Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res (2010) 38(17):5718–34. doi:10.1093/nar/gkq212
39. Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res (2012) 40(15):7416–29. doi:10.1093/nar/gks409
40. Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci (2013) 34(6):340–6. doi:10.1016/j.tips.2013.04.005
41. Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell (2006) 21(5):689–700. doi:10.1016/j.molcel.2006.01.013
42. Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res (2008) 68(5):1303–9. doi:10.1158/0008-5472.CAN-07-5003
43. Leinonen HM, Kansanen E, Polonen P, Heinaniemi M, Levonen AL. Dysregulation of the Keap1-Nrf2 pathway in cancer. Biochem Soc Trans (2015) 43(4):645–9. doi:10.1042/BST20150048
44. Ramos-Gomez M, Dolan PM, Itoh K, Yamamoto M, Kensler TW. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis (2003) 24(3):461–7. doi:10.1093/carcin/24.3.461
45. Solt DB, Medline A, Farber E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am J Pathol (1977) 88(3):595–618.
46. Kartner N, Shales M, Riordan JR, Ling V. Daunorubicin-resistant Chinese hamster ovary cells expressing multidrug resistance and a cell-surface P-glycoprotein. Cancer Res (1983) 43(9):4413–9.
47. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med (2006) 3(10):e420. doi:10.1371/journal.pmed.0030420
48. Ooi A, Dykema K, Ansari A, Petillo D, Snider J, Kahnoski R, et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res (2013) 73(7):2044–51. doi:10.1158/0008-5472.CAN-12-3227
49. Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med (2016) 22(7):578–93. doi:10.1016/j.molmed.2016.05.002
50. Wang R, An J, Ji F, Jiao H, Sun H, Zhou D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun (2008) 373(1):151–4. doi:10.1016/j.bbrc.2008.06.004
51. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol (2010) 12(3):213–23. doi:10.1038/ncb2021
52. Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell (2009) 34(6):663–73. doi:10.1016/j.molcel.2009.04.029
53. Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell (2011) 20(4):524–37. doi:10.1016/j.ccr.2011.09.006
54. Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell (2011) 20(4):511–23. doi:10.1016/j.ccr.2011.08.024
55. Taguchi K, Shimada M, Fujii S, Sumi D, Pan X, Yamano S, et al. Redox cycling of 9,10-phenanthraquinone to cause oxidative stress is terminated through its monoglucuronide conjugation in human pulmonary epithelial A549 cells. Free Radic Biol Med (2008) 44(8):1645–55. doi:10.1016/j.freeradbiomed.2008.01.024
56. Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia (2011) 13(9):864–73. doi:10.1593/neo.11750
57. Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, et al. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell (2013) 24(6):738–50. doi:10.1016/j.ccr.2013.10.025
58. Probst BL, McCauley L, Trevino I, Wigley WC, Ferguson DA. Cancer cell growth is differentially affected by constitutive activation of NRF2 by KEAP1 deletion and pharmacological activation of NRF2 by the synthetic triterpenoid, RTA 405. PLoS One (2015) 10(8):e0135257. doi:10.1371/journal.pone.0135257
59. Hong YB, Kang HJ, Kwon SY, Kim HJ, Kwon KY, Cho CH, et al. Nuclear factor (erythroid-derived 2)-like 2 regulates drug resistance in pancreatic cancer cells. Pancreas (2010) 39(4):463–72. doi:10.1097/MPA.0b013e3181c31314
60. Arlt A, Sebens S, Krebs S, Geismann C, Grossmann M, Kruse ML, et al. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene (2013) 32(40):4825–35. doi:10.1038/onc.2012.493
61. Lister A, Nedjadi T, Kitteringham NR, Campbell F, Costello E, Lloyd B, et al. Nrf2 is overexpressed in pancreatic cancer: implications for cell proliferation and therapy. Mol Cancer (2011) 10:37. doi:10.1186/1476-4598-10-37
62. Ichimura Y, Waguri S, Sou Y, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell (2013) 51(5):618–31. doi:10.1016/j.molcel.2013.08.003
63. Cancer Genome Atlas Research. Comprehensive genomic characterization of squamous cell lung cancers. Nature (2012) 489(7417):519–25. doi:10.1038/nature11404
64. Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet (2015) 47(5):505–11. doi:10.1038/ng.3252
65. Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem (2003) 278(14):12029–38. doi:10.1074/jbc.M211558200
66. Wu KC, Cui JY, Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci (2011) 123(2):590–600. doi:10.1093/toxsci/kfr183
67. Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun (2016) 7:12030. doi:10.1038/ncomms12030
68. Liu B, Fang M, He Z, Cui D, Jia S, Lin X, et al. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis (2015) 6:e1980. doi:10.1038/cddis.2015.322
69. Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci U S A (1992) 89(6):2399–403. doi:10.1073/pnas.89.6.2399
70. Balogun E, Hoque M, Gong P, Killeen E, Green CJ, Foresti R, et al. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem J (2003) 371(Pt 3):887–95. doi:10.1042/bj20021619
71. Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem (2008) 104(4):1116–31. doi:10.1111/j.1471-4159.2007.05039.x
72. Krajka-Kuzniak V, Paluszczak J, Baer-Dubowska W. The Nrf2-ARE signaling pathway: an update on its regulation and possible role in cancer prevention and treatment. Pharmacol Rep (2016) 69(3):393–402. doi:10.1016/j.pharep.2016.12.011
73. Zhu J, Wang H, Chen F, Fu J, Xu Y, Hou Y, et al. An overview of chemical inhibitors of the Nrf2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic Biol Med (2016) 99:544–56. doi:10.1016/j.freeradbiomed.2016.09.010
74. Lee KH, Okano M, Hall IH, Brent DA, Soltmann B. Antitumor agents XLV: bisbrusatolyl and brusatolyl esters and related compounds as novel potent antileukemic agents. J Pharm Sci (1982) 71(3):338–45. doi:10.1002/jps.2600710320
75. Willingham W Jr, Stafford EA, Reynolds SH, Chaney SG, Lee KH, Okano M, et al. Mechanism of eukaryotic protein synthesis inhibition by brusatol. Biochim Biophys Acta (1981) 654(2):169–74. doi:10.1016/0005-2787(81)90168-4
76. Hall IH, Liou YF, Okano M, Lee KH. Antitumor agents XLVI: in vitro effects of esters of brusatol, bisbrusatol, and related compounds on nucleic acid and protein synthesis of P-388 lymphocytic leukemia cells. J Pharm Sci (1982) 71(3):345–8. doi:10.1002/jps.2600710229
77. Liou YF, Hall IH, Okano M, Lee KH, Chaney SG. Antitumor agents XLVIII: structure-activity relationships of quassinoids as in vitro protein synthesis inhibitors of P-388 lymphocytic leukemia tumor cell metabolism. J Pharm Sci (1982) 71(4):430–5. doi:10.1002/jps.2600711208
78. Ren D, Villeneuve NF, Jiang T, Wu T, Lau A, Toppin HA, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A (2011) 108(4):1433–8. doi:10.1073/pnas.1014275108
79. Vartanian S, Ma TP, Lee J, Haverty PM, Kirkpatrick DS, Yu K, et al. Application of mass spectrometry profiling to establish brusatol as an inhibitor of global protein synthesis. Mol Cell Proteomics (2016) 15(4):1220–31. doi:10.1074/mcp.M115.055509
80. Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem Biol (2016) 11(11):3214–25. doi:10.1021/acschembio.6b00651
81. Coatney GR, Cooper WC, Culwell WB, White WC, Imboden CA Jr. Studies in human malaria. XXV. Trial of febrifugine, an alkaloid obtained from Dichroa febrifuga lour., against the Chesson strain of Plasmodium vivax. J Natl Malar Soc (1950) 9(2):183–6.
82. Pines M, Knopov V, Genina O, Lavelin I, Nagler A. Halofuginone, a specific inhibitor of collagen type I synthesis, prevents dimethylnitrosamine-induced liver cirrhosis. J Hepatol (1997) 27(2):391–8. doi:10.1016/S0168-8278(97)80186-9
83. Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol (2012) 8(3):311–7. doi:10.1038/nchembio.790
84. Elkin M, Ariel I, Miao HQ, Nagler A, Pines M, de-Groot N, et al. Inhibition of bladder carcinoma angiogenesis, stromal support, and tumor growth by halofuginone. Cancer Res (1999) 59(16):4111–8.
85. Pines M, Snyder D, Yarkoni S, Nagler A. Halofuginone to treat fibrosis in chronic graft-versus-host disease and scleroderma. Biol Blood Marrow Transplant (2003) 9(7):417–25. doi:10.1016/S1083-8791(03)00151-4
86. Nevo Y, Halevy O, Genin O, Moshe I, Turgeman T, Harel M, et al. Fibrosis inhibition and muscle histopathology improvement in laminin-alpha2-deficient mice. Muscle Nerve (2010) 42(2):218–29. doi:10.1002/mus.21706
87. Tsuchida K, Tsujita T, Hayashi M, Ojima A, Keleku-Lukwete N, Katsuoka F, et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic Biol Med (2016) 103:236–47. doi:10.1016/j.freeradbiomed.2016.12.041
88. Gao AM, Ke ZP, Shi F, Sun GC, Chen H. Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem Biol Interact (2013) 206(1):100–8. doi:10.1016/j.cbi.2013.08.008
89. Tang X, Wang H, Fan L, Wu X, Xin A, Ren H, et al. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic Biol Med (2011) 50(11):1599–609. doi:10.1016/j.freeradbiomed.2011.03.008
90. Johnson NM, Egner PA, Baxter VK, Sporn MB, Wible RS, Sutter TR, et al. Complete protection against aflatoxin B1-induced liver cancer with a triterpenoid: DNA adduct dosimetry, molecular signature, and genotoxicity threshold. Cancer Prev Res (Phila) (2014) 7(7):658–65. doi:10.1158/1940-6207.CAPR-13-0430
91. Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res (2004) 64(18):6424–31. doi:10.1158/0008-5472.CAN-04-1906
92. Kelloff GJ, Crowell JA, Boone CW, Steele VE, Lubet RA, Greenwald P, et al. Clinical development plan: oltipraz. J Cell Biochem Suppl (1994) 20:205–18.
93. Kensler TW, Helzlsouer KJ. Oltipraz: clinical opportunities for cancer chemoprevention. p. J Cell Biochem Suppl (1995) 22:101–7. doi:10.1002/jcb.240590813
94. Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A (2002) 99(18):11908–13. doi:10.1073/pnas.172398899
95. Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, et al. Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People’s Republic of China. Cancer Epidemiol Biomarkers Prev (2005) 14(11 Pt 1):2605–13. doi:10.1158/1055-9965.EPI-05-0368
Keywords: NRF2, KEAP1, cancer, metabolic reprogramming, cancer therapy
Citation: Taguchi K and Yamamoto M (2017) The KEAP1–NRF2 System in Cancer. Front. Oncol. 7:85. doi: 10.3389/fonc.2017.00085
Received: 13 February 2017; Accepted: 18 April 2017;
Published: 04 May 2017
Edited by:
Amedeo Columbano, University of Cagliari, ItalyReviewed by:
Mina Konigsberg, Universidad Autónoma Metropolitana, MexicoMaria Rosa Ciriolo, University of Rome Tor Vergata, Italy
Copyright: © 2017 Taguchi and Yamamoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masayuki Yamamoto, masiyamamoto@med.tohoku.ac.jp