


Abstract
5-[(2-Chloro-6-fluorophenyl)acetylamino]-3-(4-fluorophenyl)-4-(4-pyrimidinyl)isoxazole (AKP-001) is a potent p38 mitogen-activated protein kinase inhibitor that is being developed to specifically target the intestines for the treatment of inflammatory bowel disease. According to the ante-drug concept, AKP-001 was designed to be metabolized to inactive forms via the first-pass metabolism to avoid undesirable systemic exposure. The purpose of this study is to investigate the pharmacokinetic characteristics of AKP-001 and its metabolites (M1 and M2) in rats, utilizing a simple physiologically based pharmacokinetic (PBPK) model. In vitro metabolic activity of AKP-001 in the S9 fraction of rat liver was examined, and plasma concentration-time profiles were developed following intravenous and/or oral administration of AKP-001 and its metabolites. AKP-001 was primarily metabolized to M1; however, M2 was not detected in liver S9 fractions. In accordance with this observation in vitro, M2 was detected in plasma after oral dosing of AKP-001 with a lag time of 1.5 hours, but not after intravenous dosing. To analyze pharmacokinetics in rats in vivo, a simple PBPK model was developed by simultaneous fitting of the plasma concentrations after treatment with AKP-001 and its metabolites. The observed plasma concentration-time profiles of AKP-001 and metabolites were described by the model adequately. Intestinal and systemic exposures of AKP-001 were simulated using the model to assess the relationship between pharmacokinetics and efficacy/safety. Model analysis suggested that oral bioavailability of intestine-targeting ante-drugs should be low to avoid systemic side effects. The pharmacokinetic properties of AKP-001 meet this criterion owing to extensive first-pass metabolism.
Introduction
Inflammatory bowel disease (IBD) describes a group of chronic and relapsing inflammatory diseases of the gastrointestinal (GI) tract, among which Crohn’s disease and ulcerative colitis are the most common forms. Crohn’s disease and ulcerative colitis are multifactorial diseases caused by the interplay of genetic, environmental, and immunologic factors (Thoreson and Cullen, 2007). Although the cause of IBD is unknown, a key trigger of IBD pathogenesis is the secretion of inflammatory cytokines, such as tumor necrosis factor-α, interleuken-1β, and interleuken-6, from activated leukocytes and macrophages in inflamed intestinal tissue (Papadakis and Targan, 2000).
Mitogen-activated protein kinase (MAPK) p38 is a crucial mediator of inflammation (Branger et al., 2002). p38 MAPK directly controls the activity of several transcription factors that regulate the production of proinflammatory cytokines, which are upregulated in IBD. Furthermore, p38 MAPK can modulate a number of different aspects of inflammatory signaling cascades, including transcriptional regulation of genes encoding the cytokines involved in IBD (tumor necrosis factor-α, interleuken-1, and interferon-γ in monocytes and lymphocytes), as well as the degranulation of neutrophils (Mócsai et al., 2000; Kyriakis and Avruch, 2012). However, p38 MAPK inhibitors have not been approved for clinical use in the management of inflammatory disorders, including IBD and rheumatoid arthritis, because systemic inhibition of p38 MAPK may cause various adverse effects, including abnormal liver function, dizziness, and skin reactions (Schreiber et al., 2006; Cohen et al., 2009; Genovese, 2009).
Recently, much work has focused on developing local drug delivery systems that target the intestines for the treatment of GI tract disorders (Chourasia and Jain, 2003). For example, rifaximin, a rifamycin derivative, remains in the GI tract and has extremely low systemic exposure owing to poor solubility (Descombe et al., 1994; Rizzello et al., 1998). Sulfasalazine, which is used to treat IBD, is delivered to the distal intestine through an efflux mechanism mediated by transporters (MRP2 and BCRP) expressed in the intestinal epithelium (Dahan and Amidon, 2009). Sulfasalazine is extensively metabolized to its pharmacologically active forms, sulfapyridine and 5-aminosalicylate, in the colon, followed by cleavage via intestinal bacteria (Peppercorn and Goldman, 1972). The ante-drug/soft drug concept is an alternative approach for local delivery to the intestine (Lee et al., 2002; Khan et al., 2005). An ante-drug is a drug designed to exert a specific effect at the administration site, and then be rapidly metabolized to inactive compounds upon circulation. One of the successful therapeutic agents for IBD, budesonide, is highly metabolized to inactive forms in the liver (O’Donnell and O’Morain, 2010). This drug design concept offers a significant advantage through minimization of systemic off-target effects of the parent drug.
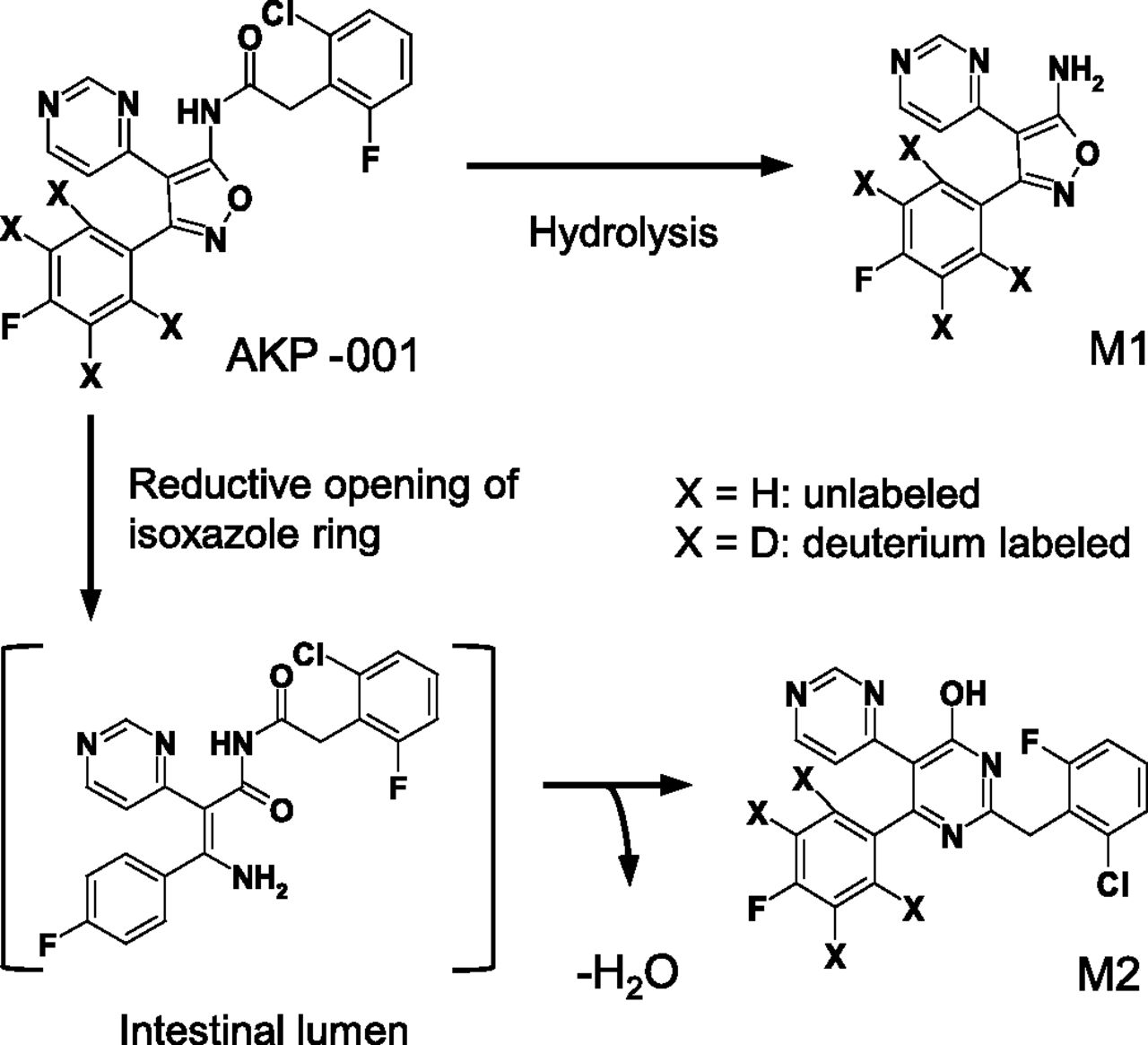
5-[(2-Chloro-6-fluorophenyl)acetylamino]-3-(4-fluorophenyl)-4-(4-pyrimidinyl)isoxazole (AKP-001; Fig. 1) is a novel p38 MAPK inhibitor synthesized by ASKA Pharmaceutical Co. Ltd. that potently inhibits the ability of p38α and p38β to phosphorylate target proteins (IC50 = 10.9 and 326 nM, respectively). AKP-001 ameliorated experimental colitis induced by dextran sulfate sodium in mice and by 2,4,6-trinitrobenzene sulfonic acid in rats (Hasumi et al., 2014). AKP-001 was designed according to the ante-drug concept, and is quickly metabolized to an inactive form, M1 (Fig. 1), through first-pass metabolism in the intestine and liver after oral administration. AKP-001 is partially metabolized in the intestinal lumen, producing a second metabolite, M2 (Fig. 1). Thus, in evaluating pharmacokinetics in target organs, it is necessary to consider the metabolism of AKP-001 to both M1 and M2.
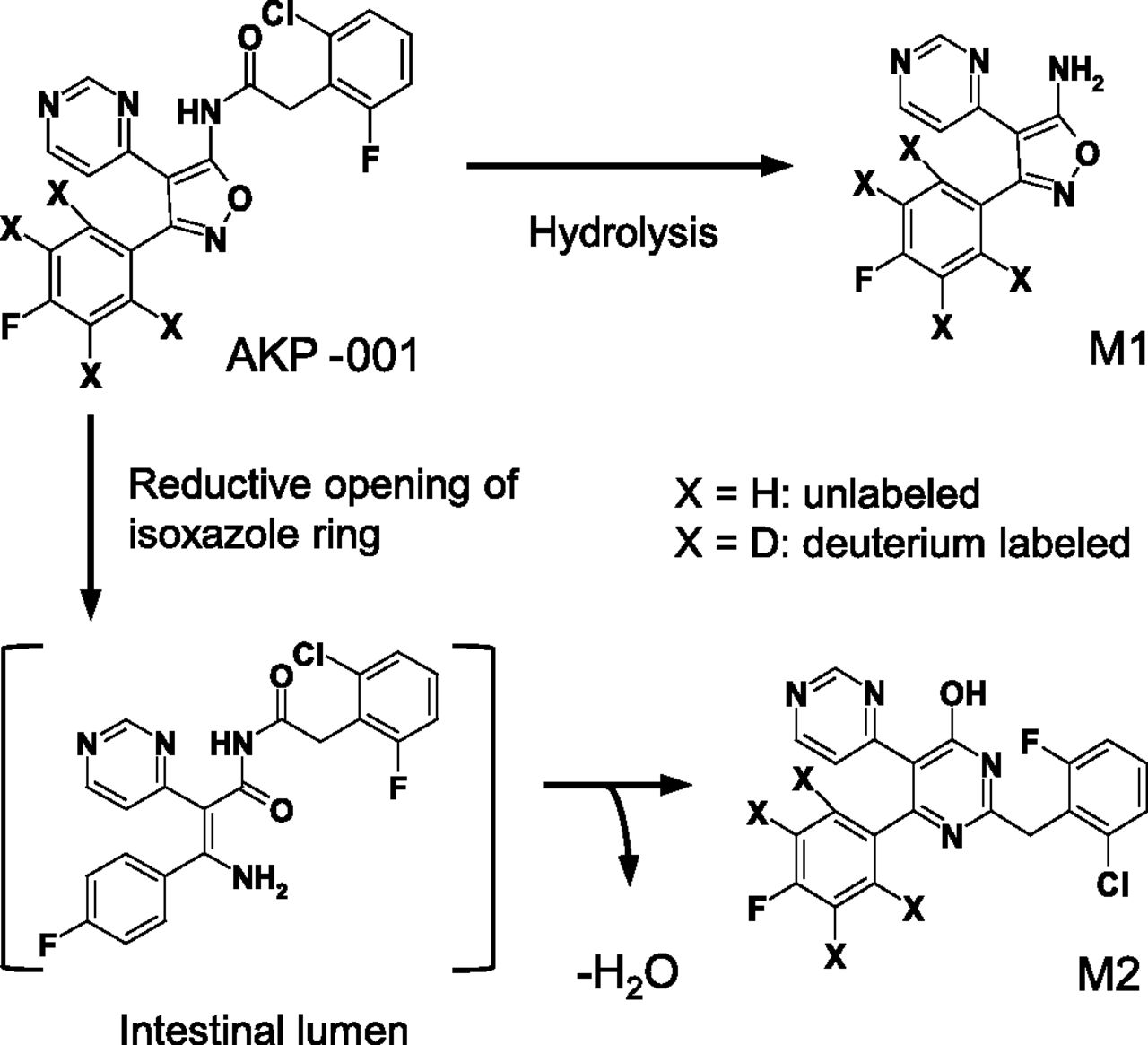
Chemical structures and metabolic pathways of AKP-001 and its metabolites (M1 and M2).
The aim of this study was to assess the factors affecting the pharmacological efficacy/safety of AKP-001 through the characterization of the pharmacokinetic profiles of AKP-001 and its metabolites (M1 and M2) in rats. The plasma concentrations of AKP-001, M1, and M2 after intravenous and oral administration of each compound were determined. Further, we developed a simple physiologically based pharmacokinetic (PBPK) model of AKP-001 and its metabolites in rats on the basis of plasma concentration data. Finally, we simulated plasma and intestinal concentration profiles of AKP-001 to clarify factors affecting intestinal and systemic exposure of AKP-001. A systematic understanding of the pharmacokinetics of AKP-001 would be necessary for the evaluation of the efficacy and safety of APK-001 in humans, and may also facilitate the further development of other ante-drugs targeting the intestine.
Materials and Methods
AKP-001, 5-amino-3-(4-fluorophenyl)-4-(4-pyrimidinyl)isoxazole (M1), and 2′-(2-chloro-6-fluorobenzyl)-6′-(4-fluorophenyl)-[4,5′]bipyrimidinyl-4′-ol (M2) (Fig. 1) were synthesized by the Synthetic Research Department of ASKA Pharmaceutical Co., Ltd. Deuterium-labeled compounds (d4-AKP-001, d4-M1, and d4-M2) used as internal standards were also synthesized by ASKA Pharmaceutical Co., Ltd.
Rat liver S9 was purchased from BD Gentest (Woburn, MA). β-NADP+, glucose 6-phosphate, glucose 6-phosphate dehydrogenase, and bis-p-nitrophenyl phosphate were purchased from Sigma-Aldrich (St. Louis, MO). Tween 80 was purchased from MP Biomedicals (Solon, OH). Polyethylene glycol-400 (mean molecular weight, 400) was purchased from Nacalai Tesque (Kyoto, Japan). Isoflurane was purchased from Mylan (Tokyo, Japan). All other chemicals and reagents were of high-performance liquid chromatography or analytical grade.
Animals.
Male Sprague-Dawley rats (6 weeks old) were purchased from Charles River Laboratories Japan (Yokohama, Japan). The animals were quarantined and acclimated for at least 7 days after receipt. Animals were maintained in a temperature-controlled (23°C) room under a 12-hour light/dark cycle and allowed free access to water and food during the experimental period. All animal study procedures were approved by the Animal Research Committee of ASKA Pharmaceutical Co., Ltd., and were conducted in compliance with the Law Concerning the Protection and Control of Animals (Japanese Law 105, October 1, 1973; revised on June 22, 2005).
Determination of Blood-to-Plasma Concentration Ratio in Rats.
The blood-to-plasma concentration ratios (Rb) of AKP-001 and its metabolites in rats were determined using an in vitro method described previously (Yan et al., 2012). Briefly, AKP-001, M1, and M2 were individually added to prewarmed rat blood and blank plasma (reference plasma), to obtain a final concentration of 0.1 μM. After incubation at 37°C for 30 minutes, rat blood was centrifuged at 2000g for 10 minutes at 4°C to separate plasma. The hematocrit value of the rat blood used in this study was 0.45. Plasma samples were analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS) as described below for the determinations of AKP-001 and its metabolites. The stability of AKP-001 throughout the experimental process was 66% of the prepared concentration. Rb was calculated by dividing the concentrations in the reference plasma (nominal blood concentration) by the concentrations in the compound-treated plasma, under the assumption that the stability of AKP-001 in rat blood was equal to that in rat plasma.
Determination of Unbound Fractions in Rat Plasma.
The unbound fractions of AKP-001, M1, and M2 in rat plasma (fp) were determined by ultracentrifugation (Giuliano et al., 2005). AKP-001, M1, and M2 were individually spiked into rat plasma containing less than 1% acetonitrile to obtain a final concentration of 1 μM. After incubation at 37°C for 1 hour, the sample (200 μl) was transferred to a polycarbonate ultracentrifugation tube, and centrifuged at 356,000g for 3 hours at 4°C in an ultracentrifuge (CS100FNX, Hitachi-Koki, Tokyo, Japan). For AKP-001, an additional sample was prepared to assess hydrolysis in plasma. The concentrations of AKP-001 and its metabolites in plasma and supernatant were analyzed by LC-MS/MS, as described below. The stability of AKP-001 in plasma throughout the experimental process was 48% of the initial concentration. The plasma unbound fraction was determined by dividing the concentration of the test compounds in the supernatant by the concentration in the plasma.
Pharmacokinetic Study.
AKP-001, M1, and M2 were individually suspended in 0.5% Tween 80 (w/v) to prepare oral dosing formulations. The doses were 70.3, 117, and 73.0 μmol/kg, respectively. For intravenous dosing, AKP-001 or M2 was dissolved in a mixed solution of dimethylsulfoxide, polyethylene glycol-400, and saline (5:65:30). The doses were 2.34 and 7.30 μmol/kg, respectively. Male Sprague-Dawley rats (n = 3 per condition) received doses by oral gavage or by intravenous injection into the femoral vein. Blood samples were collected via the tail vein for 24 hours after administration. Plasma was separated by centrifugation (12,000g, 3 minutes, 4°C), and was stored at –20°C until analysis.
Determination of Unbound Fractions in Rat Liver S9 Fractions.
AKP-001, M1, or M2 was added to rat liver S9 (fS9) fractions suspended in 50 mM phosphate buffer (pH 7.4, 0.5 mg protein/ml), to obtain a final concentration of 1 μM. After incubation at 4°C for 1 hour, an aliquot of each mixture was centrifuged at 356,000g, for 3 hours at 4°C. For AKP-001, an additional sample was prepared to assess hydrolysis in the incubation mixture. Aliquots (50 μl) of the mixture and supernatant were collected and stored at –20°C until analysis. The concentration of AKP-001 and its metabolites in the samples was determined by LC-MS/MS, as described below. The stability of AKP-001 in plasma throughout the experimental process was 81% of the initial concentration. The unbound fraction (fS9) of AKP-001 and its metabolites in rat liver S9 fractions was calculated by the same method used for plasma protein binding.
Metabolic Stability of AKP-001 and Its Metabolites in Rat Liver S9.
AKP-001, M1, and M2 were individually spiked at a final concentration of 1 μM to 50 mM phosphate buffer (pH 7.4), containing 3.3 mM MgCl2, 1.0 mM β-NADP+, 2.5 mM glucose 6-phosphate, and 2 IU/ml glucose 6-phosphate dehydrogenase, or to liver S9 fractions (0.5 mg protein/ml). The metabolic reaction was terminated after incubation for 2, 5, 15, or 30 minutes at 37°C by the addition of a 4-fold volume of ice-cold acetonitrile containing 0.1 μM internal standard. The samples were centrifuged at 2000g for 10 minutes, and the supernatant was subjected to LC-MS/MS. Intrinsic clearance (CLint) of the test compounds in rat liver S9 fractions was calculated according to eq. 1: (1)where Ke is the elimination rate constant of the test compound, V is the initial incubation volume, and proteinS9 represents the initial amount of protein in the incubation mixture. The CLint, S9 values of AKP-001, M1, and M2 were extrapolated to the unbound intrinsic hepatic clearance (CLint,u,in vitro), by correcting with the corresponding unbound fraction in rat liver S9 fractions, and the physiologic scaling factors of 96.1 mg S9 protein/g liver (Watanabe et al., 2009) and 40 g liver/kg body weight (Davies and Morris, 1993).
(1)where Ke is the elimination rate constant of the test compound, V is the initial incubation volume, and proteinS9 represents the initial amount of protein in the incubation mixture. The CLint, S9 values of AKP-001, M1, and M2 were extrapolated to the unbound intrinsic hepatic clearance (CLint,u,in vitro), by correcting with the corresponding unbound fraction in rat liver S9 fractions, and the physiologic scaling factors of 96.1 mg S9 protein/g liver (Watanabe et al., 2009) and 40 g liver/kg body weight (Davies and Morris, 1993).
To determine the enzyme involved in the metabolism of AKP-001 to M1 in rat liver S9 fractions, the NADPH dependence and the effect of a typical carboxylesterase inhibitor (Masaki et al., 2007) bis-p-nitrophenyl phosphate (BNPP) on the metabolism of AKP-001 were investigated. For the evaluation of NADPH dependence, AKP-001 (1 μM) was incubated with rat liver S9 fractions in the presence or absence of an NADPH-generating system. The role of carboxylesterases in the metabolism of AKP-001 was evaluated in the presence of an NADPH-generating system by the addition of BNPP (100 μM). The experimental procedure was the same as described above, except that the reaction time was 5 minutes. The intrinsic clearance for the disposition of AKP-001 and the formation of M1 was calculated according to eq. 1, using the rate constant of M1 formation instead of Ke, based on the assumption that the sequential metabolism of M1 was negligible.
LC-MS/MS Analysis.
The LC-MS/MS system consisted of an Alliance 2795 separation module (Waters, Milford, MA) and a Quattro Ultima Pt triple quadrupole mass spectrometer (Micromass, Manchester, UK) with an electron spray ionization interface. Stock solutions of the test compounds (AKP-001, M1, and M2) and internal standards (d4-AKP-001, d4-M1, and d4-M2) were prepared in acetonitrile (0.2 mg/ml and 0.1 mg/ml, respectively). The stock solutions of AKP-001, M1, and M2 were mixed, and the mixtures were further diluted with acetonitrile to yield working solutions (10, 100, and 1000 ng/ml). For the preparation of calibration standards (1–1000 ng/ml) and quality controls (2.5, 50, and 800 ng/ml), standard working solutions were diluted in an appropriate matrix. Aliquots of the samples (50 μl) were added and vortex-mixed, followed by addition of 50 μl of the internal standard working solution (d4-AKP-001, d4-M1, and d4-M2, 100 ng/ml), 0.5 M sodium hydrogen carbonate solution (0.5 ml), and diethyl ether (2 ml). After vigorously shaking for 10 minutes and centrifugation (2000g, 5 minutes), the organic layer was collected and evaporated under a nitrogen gas stream. The samples were reconstituted with 100 μl of acetonitrile, and were transferred to high-performance liquid chromatography vials. Analytes (10 μl) were separated on a Discovery HS F5 column (2.1-mm i.d. × 50 mm, 3 μm; Supelco, Bellefonte, PA) at 40°C with a linear gradient program consisting of 10 mM ammonium formate (pH 3) (A) and acetonitrile (B) at a flow rate of 0.2 ml/min. Mobile phase composition was initially 70% A and 30% B, and was increased from 30% B to 70% B from 1 to 5.5 minutes. Thereafter, the mobile phase composition was held for 1 minute at a flow rate of 0.5 ml/min, and the column was re-equilibrated for 3 minutes at the initial conditions before the next injection. The optimum operating conditions of the mass spectrometer used were as follows: electron spray capillary voltage, 3.5 kV; source temperature, 120°C; and probe desolvation gas temperature, 400°C. Calibration curves for AKP-001, M1, and M2 were linear from 1 to 1000 ng/ml (R2 > 0.99). Intra- and interday precision (coefficient of variance percentage) and accuracy (percentage relative error) of the quality control samples for these compounds were within 15%. The mass spectrometer was operated in positive ion mode using multiple reaction monitoring: AKP-001, m/z 427.0 → 240; M1, m/z 257.0 → 121; M2, m/z 411.0 → 199; d4-AKP-001, m/z 431.0 → 244; d4-M1, m/z 261.0 → 121; and d4-M2, m/z 415.0 → 203.
Model Development.
A simplified PBPK model was constructed to describe the pharmacokinetics of AKP-001 and its two metabolites (M1 and M2) in rats (Fig. 2). In the first step, we individually constructed the pharmacokinetic models of AKP-001, M1, and M2 as parent compounds, on the basis of in vivo plasma concentration-time profiles after intravenous and/or oral administration. A simple PBPK model, which consisted of a conventional two-compartment pharmacokinetic model with an incorporated gut compartment, was applied for the pharmacokinetic modeling of AKP-001.
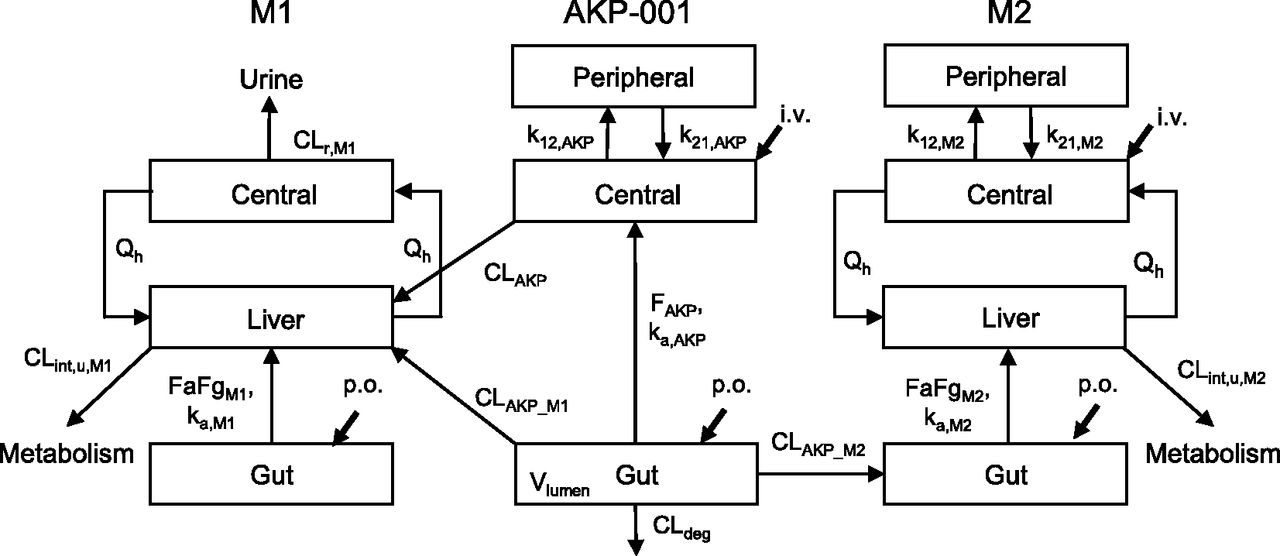
Model scheme of AKP-001 and metabolite (M1 and M2) disposition in rats. Distinct model structures were developed for the parent compound (AKP-001) and the metabolites (M1 and M2). A conventional two-compartment pharmacokinetic model with an incorporated gut compartment was applied for AKP-001 and was connected with that of M1 by first-pass metabolism and liver metabolism, assuming that hepatic metabolism mainly contributed to systemic formation of M1. For the production of M2, the pharmacokinetic model was linked to gut metabolism. Qh represents the hepatic blood flow. ka represents the absorption rate constant. k12 and k21 are the first-order rate constants for transfer from the central to peripheral and from the peripheral to central compartment, respectively. FaFg represents the intestinal availability. F represents the availability. CLint,u represents the hepatic unbound metabolic clearance. CLAKP represents the systemic clearance of AKP-001. CLAKP_M1 represents the clearance for metabolic conversion of AKP-001 to M1 by first-pass metabolism. CLAKP_M2 represents the metabolic clearance of AKP-001 to M2 in the gut compartment. CLdeg represents the clearance for metabolic conversion from AKP-001 to other metabolites. CLr represents the renal clearance. Subscripts: AKP, AKP-001; M1, metabolite M1; M2, metabolite M2.
The mass balance equations for AKP-001 were expressed as eqs. 2–4: (2)
(2) (3)
(3) (4)where X2,AKP and Xlumen,AKP represent the amounts of AKP-001 in the peripheral and intestinal lumen compartments, respectively; C1,AKP is the blood concentration (Cp × Rb) of AKP-001 in the central compartment; V1,AKP is the volume in the central compartment; ka,AKP is the absorption rate constant of AKP-001; k12,AKP and k21,AKP are the first-order rate constants for transfer from the central-to-peripheral and from the peripheral-to-central compartments, respectively; FAKP is the availability of AKP-001 and was calculated by dividing the dose-normalized area under the plasma concentration-time curve (AUC) after oral administration by the AUC after intravenous administration; CLAKP is the systemic clearance of AKP-001.
(4)where X2,AKP and Xlumen,AKP represent the amounts of AKP-001 in the peripheral and intestinal lumen compartments, respectively; C1,AKP is the blood concentration (Cp × Rb) of AKP-001 in the central compartment; V1,AKP is the volume in the central compartment; ka,AKP is the absorption rate constant of AKP-001; k12,AKP and k21,AKP are the first-order rate constants for transfer from the central-to-peripheral and from the peripheral-to-central compartments, respectively; FAKP is the availability of AKP-001 and was calculated by dividing the dose-normalized area under the plasma concentration-time curve (AUC) after oral administration by the AUC after intravenous administration; CLAKP is the systemic clearance of AKP-001.
For the pharmacokinetic modeling of M1, a one-compartment model incorporated with the liver and intestinal lumen was applied. The mass balance equations for M1 were expressed as eqs. 5–7: (5)
(5) (6)
(6) (7)where Xlumen,M1 represents the amount of M1 in the intestinal lumen compartment; Qh is the liver blood flow; C1,M1 and Ch,M1 are the concentrations of M1 in the central and liver compartments, respectively; V1,M1 and Vh are the volumes in the central and liver compartments, respectively; ka,M1 is the absorption rate constant of M1; FaFgM1 is the intestinal availability of M1; CLr,M1 is the renal clearance of M1, which is estimated by dividing the total amount of M1 excreted in urine by the AUC after oral administration of AKP-001; CLint,u,M1 is the unbound intrinsic clearance of M1 in the liver compartment; fp,M1 is the unbound fraction of M1 in plasma; Rb,M1 is the blood/plasma concentration ratio of M1; and Kp,M1 is the liver-plasma partition coefficient of M1.
(7)where Xlumen,M1 represents the amount of M1 in the intestinal lumen compartment; Qh is the liver blood flow; C1,M1 and Ch,M1 are the concentrations of M1 in the central and liver compartments, respectively; V1,M1 and Vh are the volumes in the central and liver compartments, respectively; ka,M1 is the absorption rate constant of M1; FaFgM1 is the intestinal availability of M1; CLr,M1 is the renal clearance of M1, which is estimated by dividing the total amount of M1 excreted in urine by the AUC after oral administration of AKP-001; CLint,u,M1 is the unbound intrinsic clearance of M1 in the liver compartment; fp,M1 is the unbound fraction of M1 in plasma; Rb,M1 is the blood/plasma concentration ratio of M1; and Kp,M1 is the liver-plasma partition coefficient of M1.
For the pharmacokinetic modeling of M2, a two-compartment model incorporated with the liver and intestinal lumen compartments was applied. The mass balance equations for M2 were expressed as eqs. 8–11: (8)
(8) (9)
(9) (10)
(10) (11)where Xlumen,M2 represents the amount of M2 in the intestinal lumen compartment; C1,M2 and Ch,M2 are the concentrations of M2 in the central and liver compartments, respectively; V1,M2 is the volume in the central compartment; ka,M2 is the absorption rate constant of M2; CLint,u,M2 is the unbound intrinsic clearance of M2 in the liver compartment; fp,M2 is the unbound fraction of M2 in plasma; Rb,M2 is the blood/plasma concentration ratio of M2; and Kp,M2 is the liver-plasma partition coefficient of M2. FaFgM2 is the intestinal availability of M2. FaFgM2 was calculated using eqs. 12 and 13:
(11)where Xlumen,M2 represents the amount of M2 in the intestinal lumen compartment; C1,M2 and Ch,M2 are the concentrations of M2 in the central and liver compartments, respectively; V1,M2 is the volume in the central compartment; ka,M2 is the absorption rate constant of M2; CLint,u,M2 is the unbound intrinsic clearance of M2 in the liver compartment; fp,M2 is the unbound fraction of M2 in plasma; Rb,M2 is the blood/plasma concentration ratio of M2; and Kp,M2 is the liver-plasma partition coefficient of M2. FaFgM2 is the intestinal availability of M2. FaFgM2 was calculated using eqs. 12 and 13: (12)
(12) (13)where FM2 represents oral bioavailability of M2, which was calculated by dividing the dose-normalized AUC after oral administration by the AUC after intravenous administration; Fh,M2 represents the hepatic availability; and CLh,M2 represents the hepatic clearance of M2. In case of M2, the CLh,M2 value was assumed to be equal to the total body clearance, since urinary excretion of M2 after intravenous administration was negligible in preliminary experiments.
(13)where FM2 represents oral bioavailability of M2, which was calculated by dividing the dose-normalized AUC after oral administration by the AUC after intravenous administration; Fh,M2 represents the hepatic availability; and CLh,M2 represents the hepatic clearance of M2. In case of M2, the CLh,M2 value was assumed to be equal to the total body clearance, since urinary excretion of M2 after intravenous administration was negligible in preliminary experiments.
To construct the pharmacokinetic model describing the disposition of AKP-001 and its metabolites after oral administration of AKP-001, the pharmacokinetic models of AKP-001, M1, and M2 were combined according to the respective metabolic pathway of AKP-001. The differences of mass balance equations for AKP-001 and metabolites were expressed as eqs. 14–16:
For AKP-001,
 (14)
(14)For M1,
 (15)
(15)For M2, (16)where Vlumen is the volume in the intestinal lumen compartment; CLAKP_M1, CLAKP_M2, and CLdeg are the intrinsic clearances for the metabolic pathways corresponding to the formation of M1 and M2, and for degradation in the intestinal lumen compartment, respectively.
(16)where Vlumen is the volume in the intestinal lumen compartment; CLAKP_M1, CLAKP_M2, and CLdeg are the intrinsic clearances for the metabolic pathways corresponding to the formation of M1 and M2, and for degradation in the intestinal lumen compartment, respectively.
Noncompartmental analysis for the calculation of AKP-001 and metabolites pharmacokinetic parameters was performed using Phoenix WinNonlin software (version 6.1; Pharsight, Mountain View, CA). Calculation of the differential equations constructed in this study was performed by Napp (ver. 2.31; available from http://plaza.umin.ac.jp/~todaiyak/download.php) with the Runge-Kutta-Fehlberg method using a weighting factor of 1/observation (Hisaka and Sugiyama, 1998). The predicted pharmacokinetic parameters (Cmax, tmax, and t1/2) of AKP-001 and its metabolites after oral administration of AKP-001 were numerically calculated using the Napp program. The predicted AUC was determined by trapezoidal integration. The goodness-of-fit to the model was assessed by visual comparison of the observed data to the predicted concentration-time profiles for the test compounds and by comparison of the observed pharmacokinetic parameters by noncompartmental analysis to the predicted parameters.
Results
Blood-to-Plasma Concentration Ratio and Plasma Protein Binding of AKP-001 and Its Metabolites.
The blood-to-plasma concentration ratio (Rb) and plasma unbound fractions of AKP-001, M1, and M2 are summarized in Table 1. The unbound fraction of M2 in rat plasma was extremely low (<0.01) compared with AKP-001 and M1. The Rb values of AKP-001 and its metabolites were relatively low (<1) in rat blood. The Rb value of M2 was almost equal to the plasma abundance in rat blood, suggesting that M2 is minimally distributed to blood cells.
Blood-to-plasma concentration ratio and plasma unbound fraction of AKP-001 and metabolites in rats
Plasma Concentration-Time Profiles of AKP-001 and Its Metabolites in Rats.
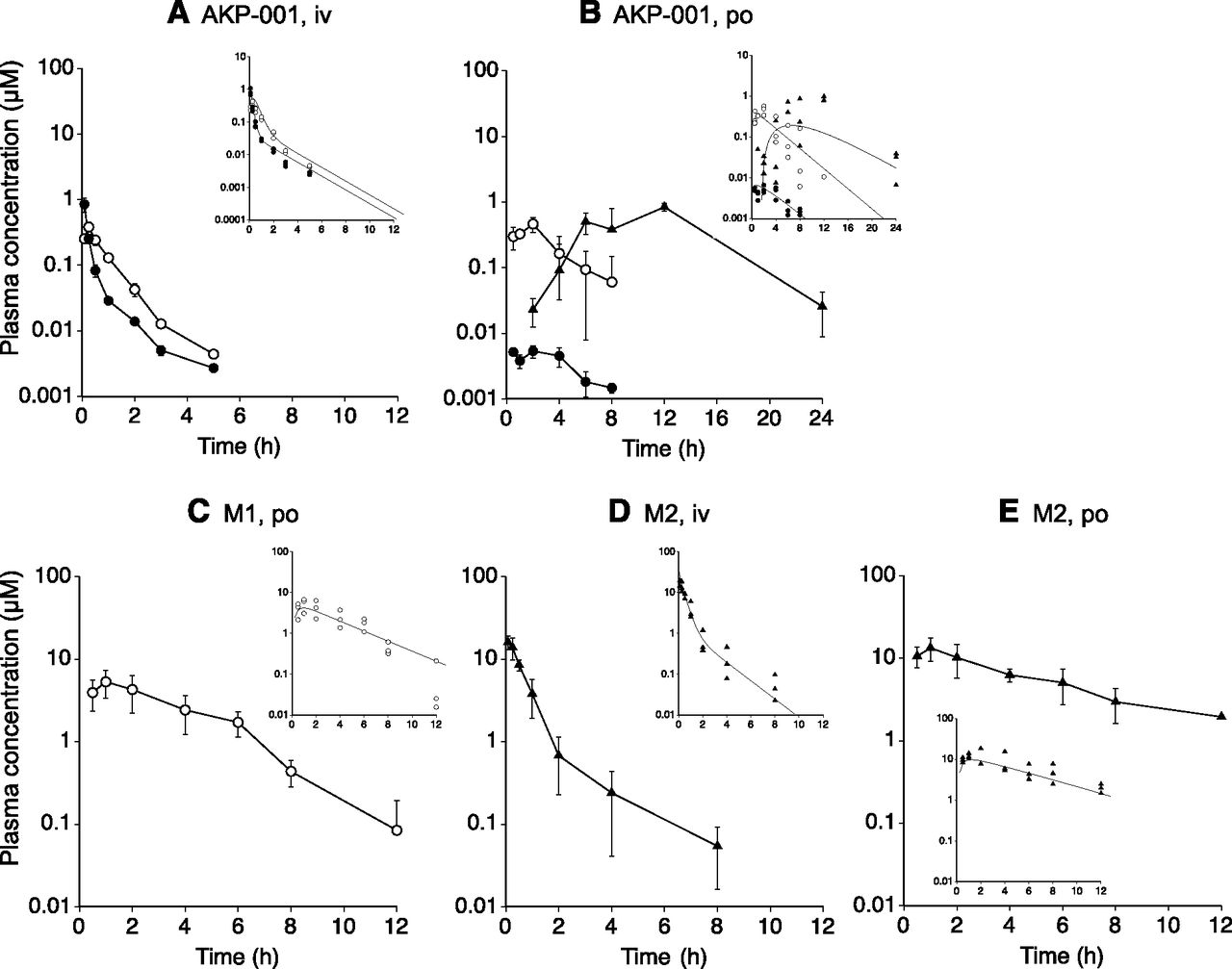
The plasma concentration-time profiles of AKP-001 and its metabolites in rats are shown in Fig. 3. After intravenous administration of AKP-001 (2.34 μmol/kg), the plasma concentration of AKP-001 biphasically decreased and was less than the limits of quantification (1 ng/ml) 8 hours after dosing (Fig. 3A). M1 appeared in plasma immediately after administration, with the tmax being 0.5 hours after dosing. M1 levels declined in parallel with AKP-001 for up to 8 hours. M2 was not detected in plasma following intravenous administration of AKP-001. After oral administration of AKP-001 (70.3 μmol/kg), plasma concentrations of AKP-001 were low (< 0.01 μM) and could only be detected for 8 hours after administration (Fig. 3B). Plasma concentrations of M1 reached a maximum concentration (Cmax) of approximately 0.5 μM at 2 hours, and the concentration declined thereafter. M2 appeared in plasma 1–2 hours after administration, and increased gradually, reaching a Cmax of approximately 1 μM at 8–12 hours.

Plasma concentration-time profiles of AKP-001 (closed circle), M1 (open circle), and M2 (closed triangle) after intravenous administration of AKP-001 (2.34 μmol/kg) (A), oral administration of AKP-001 (70.3 μmol/kg) (B), oral administration of M1 (117 μmol/kg) (C), intravenous administration of M2 (7.30 μmol/kg) (D), and oral administration of M2 (73.0 μmol/kg) (E). Data represent the mean and standard deviation of the observed plasma AKP-001 or metabolite concentration from three rats. Insets: individual plasma concentrations and fitted lines generated by simultaneous fitting.
After oral administration of M1 (117 μmol/kg), plasma concentrations of M1 reached Cmax at 1–2 hours after dosing, and monophasically declined (Fig. 3C). The plasma concentration-time profile of M1 was similar to the profile observed after oral administration of AKP-001. Intravenous administration of M2 (7.3 μmol/kg) resulted in plasma M2 concentrations that biphasically declined, and were less than the limits of quantification (1 ng/ml) 8 hours after dosing (Fig. 3D). After oral administration of M2 (73.0 μmol/kg), plasma concentrations reached Cmax at 2 hours, and decreased thereafter (Fig. 3E). The plasma concentration-time profile of M2 after dosing of the parent compound was different from the profile observed after oral administration of AKP-001, suggesting that the formation and absorption of M2 in the gut might account for these differences.
Pharmacokinetic Modeling of AKP-001 and Its Metabolites in Rats.
Based on the results of the in vivo plasma concentration-time profiles, a simple PBPK model describing the disposition of AKP-001, M1, and M2 was constructed (Fig. 2). The major metabolic pathways of AKP-001 were the hydrolysis of the amide bond (M1) and the opening of the isoxazole ring, followed by cyclization with dehydration (M2) (Fig. 1). AKP-001 was metabolized to M1 in various tissues of the body, including the liver, intestinal epithelium, and plasma. To include the production of M1 in the absorption process in this model, presystemic metabolic clearance from AKP-001 to M1 (CLAKP_M1) was parameterized. The contribution of systemic formation of M1 from AKP-001 was assessed by CLAKP under the assumption that AKP-001 entering into systemic circulation was primarily metabolized to M1 in the liver. The systemic production of M2 was negligible, and the metabolism of AKP-001 to M2 occurred predominantly in the intestinal lumen (Fig. 3, A and B). The metabolism of AKP-001 to M2 in the intestinal lumen was parameterized as CLAKP_M2, and a lag time was set on the basis of the plasma concentration-time profile of M2 after oral administration of AKP-001.
To characterize the pharmacokinetic properties of AKP-001 and its metabolites as parent compounds, plasma concentrations of AKP-001, M1, and M2 after oral and intravenous administration were individually fitted to the corresponding model. The physiologic and compound-specific parameters, and the estimated parameters determined by individual fitting are shown in Tables 2 and 3, respectively. The plasma concentrations of M2 after intravenous dosing were well fit to a two-compartment model. However, plasma M2 concentrations after oral administration showed monophasic elimination, suggesting that the observed profile of M2 after oral administration reflects the absorption of M2 following administration. Therefore, the coefficient of variation (CV) values of the estimated parameters of k12,M2 and k21,M2 were relatively high (CV, 65–122%; calculated from S.D. values in Table 3), compared with other pharmacokinetic parameters.
Physiologic and compound-specific fixed model parameters associated with the disposition of AKP-001 and metabolites in rats
Pharmacokinetic model parameters estimated by plasma concentration-time profiles of AKP-001 and metabolites after dosing as a parent compound
The pharmacokinetic model parameters of AKP-001, M1, and M2 after oral administration, estimated by simultaneous fitting of all plasma concentration data, are listed in Table 4. The predicted plasma concentration-time profiles of AKP-001 and its metabolites (Fig. 3, insets) adequately described the observed concentrations of the compounds, although some deviations were found between the observed plasma concentration and the predicted plasma concentration of M2 around tmax after oral administration of AKP-001 (Fig. 3B). The differences between the observed pharmacokinetic parameters and the predicted pharmacokinetic parameters after oral administration of AKP-001 were within 30% for AKP-001 and M1 (Table 5). The plasma concentrations of M2 following oral dosing of AKP-001 was underestimated in the model analysis, and the predicted Cmax and AUC of M2 were approximately 22–25% of the observed values. Most of the pharmacokinetic model parameters of AKP-001 and its metabolites obtained by simultaneous fitting were estimated with adequate precision (CV, ∼50%; Table 4), with the exception of ka,AKP and CLdeg. The total clearance of AKP-001 (CLAKP) was estimated as 10.9 l/h per kilogram, which was approximately 3-fold higher than Qh (3.3 l/h per kilogram), suggesting that elimination pathways of AKP-001 other than hepatic metabolism exist. The intestinal clearance of AKP-001 was the highest in CLdeg, followed by CLAKP_M1 and CLAKP_M2 (Table 4).
Pharmacokinetic model parameters estimated by simultaneous fitting of plasma concentration-time profiles of AKP-001 and its metabolites after dosing of AKP-001 or metabolites in rats
The observed and predicted pharmacokinetic parameters of AKP-001 and its metabolites after oral administration of AKP-001 to rats
The observed pharmacokinetic parameters of AKP-001 and metabolites were calculated by noncompartmental analysis using Phoenix WinNonlin software (version 6.1). Predicted pharmacokinetic parameters were assessed using a fitted plasma concentration-time curve generated by simultaneous fitting. Predicted Cmax, tmax, and t1/2 were numerically calculated by using Napp (version 2.31) and the predicted AUC0–inf was determined by trapezoidal integration.
Metabolism of AKP-001 and Its Metabolites in Biologic Samples.
Metabolism of AKP-001 in rat liver S9 fractions revealed that M1 was the major metabolite (Fig. 4). M2 was not detected in the samples. The unbound hepatic intrinsic clearance (CLint,u,in vitro) was the highest for AKP-001, followed by M2 and M1 (Table 6). The CLint,u,in vitro of M1 and M2 were approximately 5-fold lower than those estimated by model fitting (Tables 4 and 6). To determine the enzyme involved in the formation of M1 in rat liver, the NADPH dependence and the effect of the carboxylesterase inhibitor BNPP on disposition of AKP-001 and formation of M1 were investigated. Metabolic activity of AKP-001 by rat liver S9 fractions was the highest in the presence of NADPH (control), and decreased to 75% of control in the absence of NADPH (Fig. 4). Metabolic activity of AKP-001 was inhibited by the addition of 100 μM BNPP, and decreased to 30% of control. M1 formation from AKP-001 was independent of the presence of NADPH in the reaction mixture, and was almost completely inhibited by BNPP (Fig. 4).

The metabolic stability of AKP-001 in rat liver S9 fractions. For the evaluation of NADPH dependency, AKP-001 (1 μM) was incubated with rat liver S9 fractions (0.5 mg protein/ml) in the presence or absence of an NADPH generating system. The inhibitory effect of BNPP (100 μM) on AKP-001 metabolism was assessed in the presence of NADPH. The open column represents the intrinsic clearance for the disposition of AKP-001, and the closed column represents the intrinsic clearance for the metabolism of AKP-001 to M1. Data represent the mean and S.D. of triplicate determinations.
In vitro hepatic intrinsic clearance of AKP-001 and its metabolites
Simulation of Intestinal and Systemic Exposure of AKP-001 After Oral Administration With Varying Intestinal Clearance And Bioavailability.
To assess the dose efficacy and safety of AKP-001, the pharmacokinetic model was used to simulate the intestinal and systemic exposure to AKP-001 in conditions of varying intestinal clearance and/or the availability. For the evaluation of efficacy, the AKP-001 concentration in the gut compartment was simulated by changing the CLgut (defined as a sum of CLdeg, CLAKP_M1, and CLAKP_M2) at fixed FAKP (0.01; Fig. 5A). The simulated intestinal AKP-001 concentrations were greatly affected by the changes in intestinal clearance, and were under the hypothetical efficacy index (2 μM) at 12 hours after dosing when the CLgut was set at values greater than 3-fold of the estimate. For the assessment of dose safety, the plasma concentration-time profiles of AKP-001 after oral administration were simulated by varying CLgut and/or FAKP (Fig. 5B). The simulated unbound plasma concentration curves indicated that changes to CLgut caused the elimination rate of AKP-001 to increase. In contrast, though the Cmax of AKP-001 increased slightly, it was not in proportion with changes to CLgut. The simulated unbound plasma concentrations of AKP-001 were under the presumed safety indices for any value of CLgut investigated in this simulation when FAKP was set at less than 0.1 (Fig. 5B). When FAKP was set at 0.3, the predicted Cmax of AKP-001 reached the safety index, independent of the CLgut values. These results suggest that the persistence of the pharmacological effects of AKP-001 is affected by the CLgut, and that the safety margin associated with the systemic exposure of AKP-001 is governed by the FAKP.
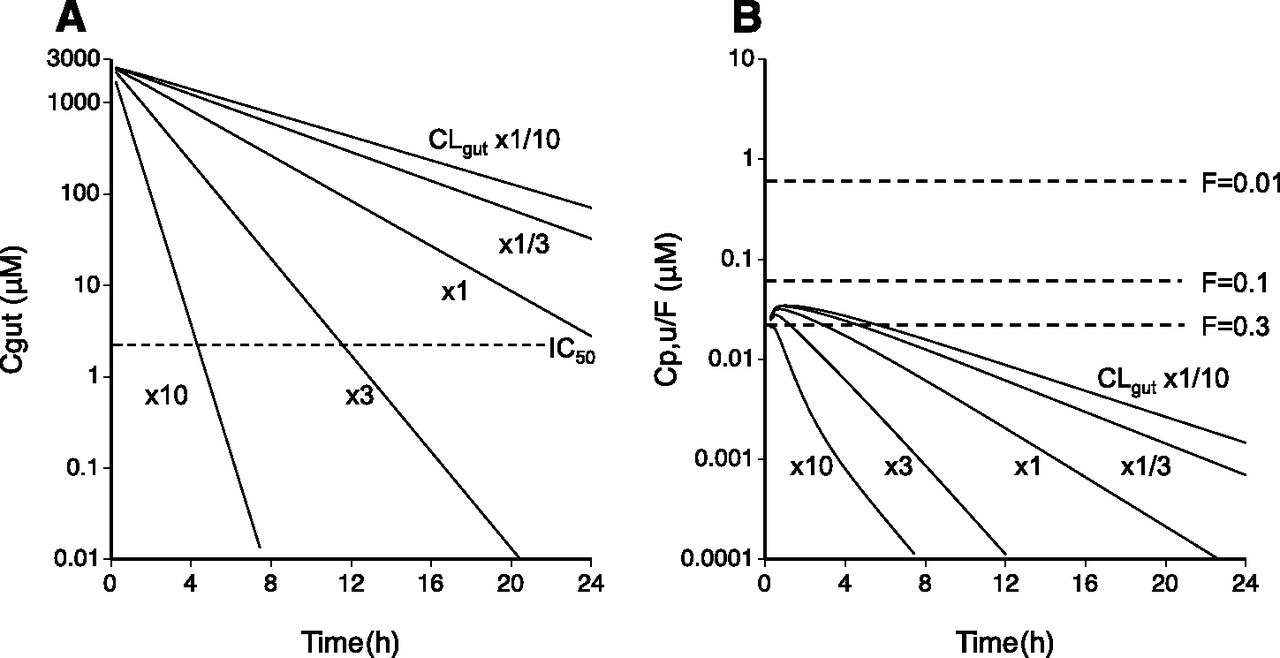
Model predictions of in vivo efficacy (A) and safety (B) of AKP-001 after oral administration with varying intestinal clearance (CLgut) and availability (FAKP). (A) Intestinal concentration-time profiles of AKP-001 after oral dosing (70.3 μmol/kg), using a pharmacologically effective dose for experimental ulcerative colitis in rat, were simulated with varying CLgut (defined as the sum of CLdeg, CLAKP_M1, and CLAKP_M2) at one-tenth, one-third, equal, 3-fold, and 10-fold the estimated CLgut value in the model analysis at fixed FAKP (0.01). The dotted line represents the efficacy index (2 μM) derived from a hypothetical IC50 of AKP-001 in rats, which was determined on the basis of the estimated in vitro IC50 value of AKP-001 in rats by a comparison with the standard p38 MAPK inhibitor SB203580 (Wadsworth et al., 1999). (B) Unbound plasma AKP-001 concentration-time profiles were simulated with varying CLgut when the FAKP was set at 0.01, 0.1, and 0.3. The y-axis was expressed as a FAKP-normalized unbound plasma concentration of AKP-001, and dotted lines represent the in vivo safety indices of AKP-001 at the corresponding FAKP. The safety indices of AKP-001 were assumed on a basis of the in vivo effective dose (25 mg/kg; Wadsworth et al., 1999) of SB203580 for the inhibition of systemic production of inflammatory cytokines following lipopolysaccharide treatment, since no systemic effect related to p38 MAPK inhibition was observed after oral administration of AKP-001. The safety index was determined according to following equation: Safety index = Cp,u,SB × RAKP/SB, where Cp,u,SB represents the unbound plasma concentration of SB203580 in rats after oral administration of SB203580 (25 mg/kg), extrapolated from the plasma concentration at 0.5 hours (approximately 20 ng/ml) at dose of 4.1 mg/kg, and unbound fraction (0.085) of SB203580 in the literature (Ward et al., 2001), assuming the linearity of pharmacokinetics of SB203580. RAKP/SB represents the ratio of IC50 values of AKP-001 (11 nM, unpublished data) to that of SB203580 (45 nM; Wadsworth et al., 1999) for tumor necrosis factor-α production from lipopolysaccharide-stimulated human peripheral blood mononuclear cells.
Discussion
AKP-001, a novel p38 MAPK inhibitor, was synthesized following the principles of ante-drug/soft drug design for IBD. In the current study, we characterized the pharmacokinetic properties of AKP-001 in vitro and in vivo, and constructed a pharmacokinetic model describing the disposition of AKP-001 and its metabolites (M1 and M2) to systematically illuminate the factors affecting the pharmacokinetic characteristics of the parent compound.
Two major metabolites of AKP-001, M1 and M2, were found in plasma after oral administration (Fig. 3B). AKP-001 was rapidly metabolized in rat liver S9 fractions and its hepatic clearance was extrapolated from in vitro data (fb × CLint,u,in vitro, Table 6) as 2.47 l/h per kilogram, which accounted for 75% of the hepatic blood flow rate (Qh). This result indicated that the AKP-001 absorbed from gut was extensively metabolized by hepatic first-pass metabolism. Furthermore, the metabolism of AKP-001 to M1 occurred in various tissues and biologic fluids, including the small intestine and plasma. A preliminary study using rat intestinal S9 fractions revealed that approximately 16% of AKP-001 was metabolized to M1, accounting for 80% of metabolites, after 15 minutes incubation (unpublished data). AKP-001 was also hydrolyzed to M1 in rat plasma, and the total clearance of AKP-001 was compared with the hydrolytic clearance in rat plasma extrapolated from the in vitro elimination rate constant (Supplemental Table 1) to assess the contribution of plasma hydrolysis to the elimination of AKP-001 in rat. The hydrolytic clearance of AKP-001 in rat extracellular fluid was quite low compared with the total clearance, and the contribution of plasma hydrolysis was minimal in rats. The estimated CLAKP (10.9 l/h per kilogram, Table 4) was 3-fold higher than Qh. AKP-001 was not excreted in urine after oral or intravenous administration, suggesting that the metabolic clearance of AKP-001 entering into the systemic circulation occurs via both hepatic metabolism and metabolism occurring in other tissues. In vitro inhibition studies revealed that the formation of M1 in rat liver S9 fractions was primarily mediated by carboxylesterase (Fig. 4). Carboxylesterase activity was observed in various rat tissues (Satoh and Hosokawa, 1998). Therefore, carboxylesterase is one candidate that may contribute to the disposition of the parent compound in rats.
The second metabolite, M2, was found in plasma after oral administration of AKP-001, but not after intravenous administration (Fig. 3, A and B), indicating that M2 metabolism occurs specifically in the gut. The chemical structure of M2 revealed that the metabolism of AKP-001 to M2 involves a reductive opening of the isoxazole ring (Fig. 1). A similar metabolic reaction is known to occur with zonisamide, which has an isoxazole moiety in its chemical structure. It is hypothesized that intestinal bacteria contribute to the reduction of the isoxazole ring (Kitamura et al., 1997). The cecal content prepared from vehicle-treated rats has significant AKP-001 reductase activity, whereas little reduction activity was observed in the cecal content from antibiotic-treated rats (in house data). In the pharmacokinetic study conducted after oral administration of AKP-001 to rats, plasma M2 appeared with a lag time (1–2 hours after dosing; Fig. 3B), in accordance with the rat intestinal transit time (88 minutes; Davies and Morris, 1993). Thus, the formation of M2 from AKP-001 in the GI tract is considered to be mediated by the intestinal microflora.
Based on the pharmacokinetic characteristics of AKP-001 and its metabolites, we constructed a simple PBPK model describing the disposition of the parent compound and metabolites (Fig. 2), and the pharmacokinetic parameters were estimated by simultaneous fitting using plasma concentrations after oral and intravenous administrations. The predicted plasma concentration curves (Fig. 3, insets) fit reasonably with the observed concentrations of AKP-001 and its metabolites, and the pharmacokinetic parameters of AKP-001 and its metabolites were estimated with adequate precision (Table 4). These results suggested that the constructed PBPK model adequately described the plasma concentration-time profiles of AKP-001 and its metabolites in rats after oral administration of AKP-001 (Fig. 3B, inset), although some deviations were found between the observed and the predicted plasma concentration of M2 (Table 5). As described above, the formation of M2 in the GI tract was mediated by gut microflora. The number of microorganisms increased depending on the location in the gut, indicating that the rate of AKP-001 metabolism to M2 may change based on the location in the intestines (Kararli, 1995). The PBPK model constructed in this study assumed that the intestinal compartment was one compartment, and that the intestinal clearance of AKP-001 was constant in the gut (Fig. 2). This may explain the deviation between the observed and predicted values of M2, especially during absorption. Large deviations in the absorption constant of AKP-001 (Table 4) might be explained similarly. The hepatic metabolic clearance of M1 and M2 extrapolated by in vitro data were underestimated, compared with the in vivo estimate by simultaneous fitting (Tables 4 and 6). There are some plausible reasons for the discrepancy in the in vitro-in vivo extrapolations of M1 and M2 hepatic metabolism, such as an inaccurate estimation of the Kp values of the metabolites and the contribution of phase II metabolism in the liver. A preliminary study indicated the existence of conjugated metabolites of M1 and M2 in rats; therefore, the discrepancy between the in vitro and in vivo hepatic metabolism may be improved by using hepatocytes to estimate the hepatic metabolic clearance of AKP-001 metabolites.
A PBPK modeling approach was used in the current study to evaluate factors affecting the efficacy and safety of AKP-001. Simulations revealed that varying the CLgut value dramatically affected the AKP-001 concentration in the gut compartment (Fig. 5A), which was directly involved in the pharmacological activity of AKP-001. To evaluate the dose-response of the AKP-001 safety profile, the unbound plasma concentrations of AKP-001 were simulated with varying CLgut and/or FAKP (Fig. 5B). The model predicted that changes in the Cmax of AKP-001 were sensitive to FAKP, compared with changes in CLgut. The simulated Cmax/F of AKP-001 at FAKP = 0.3 was over the hypothetical safety index (22.4 nM) at all values of CLgut investigated (Fig. 5B). Thus the systemic inhibitory effect of AKP-001 might not be negligible. From the simulation, intestine-targeting ante-drugs should satisfy two key parameters. First, the drug concentration should be appropriately retained in the target tissue. Specifically, the drug should be stable in the intestine, particularly if it is an intestine-targeting agent. Second, the availability should be low to avoid undesirable adverse effects. For AKP-001, the FAKP was approximately 0.01 because of extensive first-pass metabolism in rats. Therefore, AKP-001 met the above criteria as an intestine-targeting ante-drug. The safety of metabolites should be determined for a risk assessment of ante-drug. Metabolite M1 had a weak inhibitory potential for p38 MAPK isozymes and the IC50 values of M1 for p38α and p38β were 0.792 μM and 9.71 μM, respectively (Hasumi et al., 2014). M2 had no inhibitory effect on any of the p38 MAPK isoforms, and IC50 values were greater than 10 μM for both isozymes (unpublished data). The inhibitory activity of the metabolites was 30-fold higher than that of AKP-001 (IC50: 10.9 and 326 nM for p38α and p38β) (Hasumi et al., 2014). The observed unbound Cmax of M1 and M2 after oral dosing of AKP-001 at 70 μmol/kg was 56.1 nM and 8.47 nM (Tables 1 and 5), which was sufficiently low compared with the IC50 values. From these results, there is a low possibility that systemic exposures to AKP-001 metabolites will cause adverse effects owing to the inhibition of p38 MAPK in rats.
Extrapolation of the pharmacokinetic data for AKP-001 and its metabolite from rats to humans is currently a challenge, because there is limited information regarding species differences in AKP-001 metabolism. However, a preliminary study using human liver S9 fractions suggested that the metabolic activity of AKP-001 in human liver S9 fractions was approximately 3-fold higher than observed rats, and was almost completely inhibited by BNPP. These results indicate that the intrinsic hepatic clearance of AKP-001 in humans was higher than in rats. Human carboxylesterase is also expressed in various tissues, such as liver and intestine (Imai, 2006). Therefore, it is expected that systemic exposure to AKP-001 after oral administration in humans would be quite low owing to extensive first-pass metabolism, thus avoiding the undesired side effects arising from systematic inhibition of p38 MAPK.
In conclusion, a simple PBPK model, which adequately described the pharmacokinetics of AKP-001 and its metabolites M1 and M2, was constructed by simultaneously fitting to the plasma concentration of AKP-001 and its metabolites in rats. The simulation indicated that low oral bioavailability was the predominant factor influencing the wide safety margin, and that AKP-001 satisfies pharmacokinetic criteria for an intestine-targeting ante-drug. This PBPK modeling approach could be applied for the prediction of safety margins and efficacy in clinical situations, and it could guide the systematic development of intestine-targeting ante-drugs in the future.
Authorship Contributions
Participated in research design: Shirota, Kaneko, Sasaki, Minato, Fujikata, Hisaka, Suzuki.
Conducted experiments: Shirota, Kaneko, Sasaki, Minato, Fujikata, Ohta.
Contributed new reagents or analytic tools: Ohta, Hisaka.
Performed data analysis: Shirota, Hisaka, Suzuki.
Wrote or contributed to the writing of the manuscript: Shirota, Hisaka, Suzuki.
Footnotes
- Received July 15, 2014.
- Accepted November 20, 2014.
↵1 Current affiliation: Geriatric Pharmacology and Therapeutics, Graduate School of Pharmaceutical Sciences, Chiba University, Chiba, Japan.
Abbreviations
- AKP-001
- 5-[(2-Chloro-6-fluorophenyl)acetylamino]-3-(4-fluorophenyl)-4-(4-pyrimidinyl)isoxazole
- AUC
- area under the plasma concentration-time curve
- BNPP
- bis-p-nitrophenyl phosphate
- CLint
- intrinsic clearance
- CV
- coefficient of variation
- GI
- gastrointestinal
- IBD
- inflammatory bowel disease
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MAPK
- mitogen-activated protein kinase
- PBPK
- physiologically based pharmacokinetic
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}