Article Text

Abstract
Background Immunotherapy with chimeric antigen receptor (CAR)-engineered T-cells is effective in some hematologic tumors. In solid tumors, however, sustained antitumor responses after CAR T-cell therapy remain to be demonstrated both in the pre-clinical and clinical setting. A perceived barrier to the efficacy of CAR T-cell therapy in solid tumors is the hostile tumor microenvironment where immunosuppressive soluble factors like transforming growth factor (TGF)-β are thought to inhibit the cellular immune response. Here, we analyzed whether CAR T-cells specific for the receptor tyrosine kinase-like orphan receptor 1 (ROR1) antigen, that is frequently expressed in triple-negative breast cancer (TNBC), are susceptible to inhibition by TGF-β and evaluated TGF-β-receptor signaling blockade as a way of neutralizing the inhibitory effect of this cytokine.
Methods CD8+ and CD4+ ROR1-CAR T-cells were prepared from healthy donors and their antitumor function analyzed using the TNBC cell line MDA-MB-231 in vitro and in a microphysiologic 3D tumor model. Analyses were performed in co-culture assays of ROR1-CAR T-cells and MDA-MB-231 cells with addition of exogenous TGF-β.
Results The data show that exposure to TGF-β engages TGF-β-receptor signaling in CD8+ and CD4+ ROR1-CAR T-cells as evidenced by phosphorylation of small mothers against decapentaplegic homolog 2. In the presence of TGF-β, the cytolytic activity, cytokine production and proliferation of ROR1-CAR T-cells in co-culture with MDA-MB-231 TNBC cells were markedly impaired, and the viability of ROR1-CAR T-cells reduced. Blockade of TGF-β-receptor signaling with the specific kinase inhibitor SD-208 was able to protect CD8+ and CD4+ ROR1-CAR T-cells from the inhibitory effect of TGF-β, and sustained their antitumor function in vitro and in the microphysiologic 3D tumor model. Combination treatment with SD-208 also led to increased viability and lower expression of PD-1 on ROR1-CAR T-cells at the end of the antitumor response.
Conclusion We demonstrate the TGF-β suppresses the antitumor function of ROR1-CAR T-cells against TNBC in preclinical models. Our study supports the continued preclinical development and the clinical evaluation of combination treatments that shield CAR T-cells from TGF-β, as exemplified by the TGF-β-receptor kinase inhibitor SD-208 in this study.
- immunotherapy
- receptors, chimeric antigen
- breast neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Triple-negative breast cancer (TNBC) accounts for approximately 15% of breast cancer cases and is insensitive to established endocrine and antibody-based therapies because of the lack of progesterone receptor, estrogen receptor and human epidermal growth factor receptor 2 expression. Recently, the use of anti-programmed death ligand 1 checkpoint blockade in combination with chemotherapy has led to improved outcomes in patients with metastasized TNBC, and highlighted the potential susceptibility of TNBC to immunotherapy.1 Adoptive immunotherapy with gene-engineered T-cells expressing a synthetic chimeric antigen receptor (CAR) has obtained clinical proof-of-concept in B-cell leukemia and lymphoma with CD19 as the exemplary target antigen. We have been pursuing the receptor tyrosine kinase-like orphan receptor 1 (ROR1) as a target for CAR T-cells in hematology (eg, chronic lymphocytic leukemia, mantle cell lymphoma) and oncology indications. Several studies have demonstrated ROR1 expression in TNBC, with high-level expression by immunohistochemistry in 22.4%2 and 57%3 of cases. Some studies even pointed to a role of ROR1 in disease pathophysiology with high ROR1 expression being associated with shorter disease-free and overall survival. We have previously reported on the development of an optimized ROR1-specific CAR with a binding domain derived from the R12 anti-human ROR1 monoclonal antibody (mAb), that confers specific recognition of ROR1+ lymphoma and TNBC cell lines in vitro.4 But a perceived challenge for CAR T-cell therapy in TNBC and other solid tumors is the immunosuppressive and nutrient-deprived tumor microenvironment. Transforming growth factor (TGF)-β is an immunosuppressive cytokine commonly found in the microenvironment of TNBC. Apart from TNBC cells, stromal fibroblasts, macrophages, neutrophils, regulatory T-cells and platelets produce TGF-β in the tumor tissue.5 In human T-cells, binding of TGF-β to TGF-β-receptor I leads to phosphorylation of the intracellular signal transducers small mothers against decapentaplegic homolog (SMAD) 2 and 3, which subsequently associate with SMAD 4 to enter the nucleus, where they promiscuously bind to available transcription factors. The resulting transcription complexes regulate the transcription of genes involved in apoptosis, immunosuppression, and neogenesis of extracellular matrix in a cell-type and context-dependent manner.6 TGF-β has been shown to be a potent inhibitor of tumor-infiltrating T-cells, and exerts the strongest effect on resting (compared with activated) T-cells.7
Here, we studied the effect of TGF-β on the antitumor function of ROR1-CAR T-cells and used the TNBC cell line MDA-MB-231 as a pre-clinical model for analyses in vitro and in a microphysiologic 3D tumor model. To interfere with TGF-β-induced signaling, we used SD-208, a highly selective, competitive and orally bioavailable TGF-β-receptor I kinase inhibitor that has previously been evaluated for safety and efficacy in several tumor entities.8
Methods
Primary cells and cell lines
Peripheral blood was obtained from healthy donors after written informed consent to participate in research protocols approved by the Institutional Review Board of the University of Würzburg. The MDA-MB-231 cell line was purchased from the American Type Culture Collection and transduced with a fusion protein of firefly luciferase and green fluorescent protein (ffluc_GFP).
CAR T-cell manufacture
CD8+ and CD4+ T-cells were isolated from peripheral blood mononuclear cells (PBMCs) using immunomagnetic beads (Miltenyi Biotec). T-cells were activated with anti-CD3/CD28 beads (Dynabeads Human T-Activator, ThermoFisher Scientific) and transfected with minicircle DNA encoding SB100X transposase and a pT2 transposon encoding the CAR expression cassette.9 In brief, the CAR cassette comprised an scFv derived from the anti-human ROR1 mAb R12 (VH-linker-VL), a short IgG4-Fc hinge spacer, 4-1BB and CD3ζ signaling domains in cis with a T2A element and a truncated epidermal growth factor receptor (EGFRt) as detection and selection marker.4 CAR-modified T-cells were enriched by immunomagnetic selection using biotin-conjugated anti-EGFR mAb and streptavidin microbeads (Miltenyi Biotec) and expanded using a rapid expansion protocol prior to functional testing.
Phenotyping
T-cells were stained with specific conjugated mAbs to detect CD3, CD4, CD8 and PD-1 (clone REA1165) (all from Miltenyi Biotec). 7-Aminoactinomycin (7-AAD, Biolegend) was used as a viability marker to exclude dead cells from analysis. Flow cytometric analyses were done on a FACSCanto II (BD Biosciences) and data analyzed with FlowJo software (Tree Star).
Analysis of TGF-β signaling
Western blot: to evaluate the phosphorylation of SMAD2, approximately 5×106 CD8+ or CD4+ CAR T-cells were treated with 10 ng/mL TGF-β (Isoform 1, R&D Systems, cat. no. 240-B-010/CF) for 30 min. Then, total cellular protein was extracted for Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting using antibodies specific for SMAD2 and phospho-SMAD2 (Cell Signaling Technology). Western blots were analyzed on a ChemiDoc instrument using Image Lab V.5.2.1 software (Bio-Rad). Intracellular staining: CD4+ and CD8+ CAR T-cells were cultured in serum-free medium overnight and then treated with 10 ng/mL TGF-β for 20 min. T-cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions. Staining for pSMAD was done with 5 µl of anti-Smad2 (pS465/pS467)/Smad3 (pS423/pS425) antibody (clone O72-670) (BD Biosciences).
Analysis of CAR T-cell function
Prior to assay setup, T-cells were treated with 1 µM SD-208 (Tocris) for 20 min, and then with 10 ng/mL TGF-β for 24 hours unless otherwise stated. In control conditions, T-cells were treated with either SD-208 or TGF-β alone. The cytolytic activity was assessed in a luminescence-based assay. 5×103 ffluc-transduced MDA-MB-231 target cells were co-cultured with CAR T-cells at defined effector-to-target (E:T) ratios for 4 hours. Just prior to analysis, D-luciferin firefly (Biosynth) was added to the co-culture and luminescence measured on an Infinite 200 Pro instrument (Tecan). Cytokine secretion was analyzed in supernatant obtained from 24 hours co-cultures of T-cells with MDA-MB-231 target cells at an E:T ratio of 4:1 (5×104 T-cells) using multiplexed cytokine analysis (ProcartaPlex Kit, ThermoFisher Scientific) and measured on a Luminex Magpix instrument. T-cell proliferation was analyzed by CFSE (Sigma-Aldrich) dye dilution from 72 hours co-cultures with MDA-MB-231 target cells (triplicate wells; E:T ratio 4:1). The proliferation index was calculated using FlowJo software.
Microphysiologic 3D tumor model
The development of microphysiologic 3D tumor models and their use with CAR T-cells has been described.10 In brief, collagen scaffolds derived from decellularized porcine gut were fixed in cell crowns and placed in 12-well plates, each well with 2.5 mL medium. 1×105 MDA-MB-231_ffluc cells were seeded on the luminal side of the scaffold and cultured for 11 days with complete medium exchanges every 2–3 days and immediately prior to T-cell treatment. On day 0, 5×104 CD8+ ROR1-CAR T-cells or mock T-cells were administered and 1 µM SD-208, 10 ng/mL TGF-β or both added directly to the medium. To measure IFN-γ and IL-2 by ELISA (Biolegend), samples were obtained every 24 hours, and the volume replaced by fresh medium in each well (containing SD-208, TGF-β or both). The amount of tumor cells was quantified by bioluminescence imaging (BLI) on day 0 (prior to treatment) and on day 5 (end of treatment). BLI was done after adding D-luciferin to a final concentration of 20 µg/mL to each well and data acquired on an In-vivo Extreme II System and analyzed with MI SE V.7.5.2 software (Bruker).
Statistical analysis
Prism Software (GraphPad, V.7) was used for statistical analyses. Two-sided paired and unpaired t-tests or one-way analysis of variance were used with a CI of 95%. p-values <0.05 were considered significant.
Results
TGF-β induces SMAD phosphorylation in ROR1-CAR T-cells
We prepared polyclonal CD8+ and CD4+ T-cells that were gene-modified to stably express the R12 ROR1-CAR. We confirmed by flow cytometry that >95% of the T-cells were transgene-positive as evidenced by expression of the EGFRt marker (figure 1A). Then, we treated T-cells with exogenous TGF-β and analyzed the phosphorylation of SMADs. First, we performed intracellular staining for pSMAD2 and pSMAD3 and detected a uniform signal in both CD8+ and CD4+ T-cells flow cytometry (figure 1B). Second, we perform Western blots and detected a strong signal for pSMAD2 in T-cells that had been exposed to TGF-β, whereas in non-TGF-β treated T-cells, no (CD8+) or only a minor signal (CD4+) was detectable (figure 1C). Together, these data show that exposure to TGF-β leads to engagement of the TGF-β signaling cascade in CD8+ and CD4+ ROR1-CAR T-cells.

Transforming growth factor (TGF)-β induces small mothers against decapentaplegic homolog (SMAD) 2 phosphorylation in CD8+ and CD4+ receptor tyrosine kinase-like orphan receptor 1 (ROR1) chimeric antigen receptor (CAR) T-cells. (A) Phenotype of CD8+ (upper panel) and CD4+ (lower panel) CAR T-cells. Truncated epidermal growth factor receptor (EGFRt) is a detection marker encoded in cis with the CAR-transgene. (B, C) Analysis of SMAD2 phosphorylation in CD8+ (upper panel) and CD4+ (lower panel) ROR1-CAR T-cells assessed by intracellular flow cytometry staining (B) and Western blotting (C) under untreated control conditions and after incubation with TGF-β (10 ng/mL). (B) The histograms show data from a representative experiment. The right diagram shows fold change in SMAD2 phosphorylation as compared with untreated cells. Data shown are mean values±SD from n=3 independent experiments with **p<0.01 by Student’s t-test.
Blockade of TGF-β-receptor signaling protects the function of ROR1-CAR T-cells
We sought to investigate whether TGF-β affects the antitumor function of ROR1-CAR T-cells. We found that in the presence of TGF-β, the cytolytic activity of CD8+ ROR1-CAR T-cells against MDA-MB-231 TNBC cells was markedly reduced. The reduction of cytolytic activity was consistent over multiple E:T ratios and with ROR1-CAR T-cells that had been prepared from multiple donors (n=3) (figure 2A). We hypothesized that the TGF-β-receptor kinase inhibitor SD-208 might be able to protect ROR1-CAR T-cells from the inhibitory effect of TGF-β. Indeed, ROR1-CAR T-cells that had been treated with SD-208 prior to TGF-β exposure conferred similarly high levels of cytolysis as T-cells that had not been exposed.

Effects of transforming growth factor (TGF)-β and SD-208 on cytolytic activity and cytokine production of receptor tyrosine kinase-like orphan receptor 1 (ROR1)-chimeric antigen receptor (CAR) T-cells. (A–C) ROR1-CAR T-cells were pretreated either with SD-208 (1 µM) or/and TGF-β (10 ng/mL) or were left untreated. (A) Specific lysis of MDA-MB-231 by ROR1-CAR CD8+ T-cells at different E:T ratios after a 4-hour co-culture. The left diagram shows specific killing of one representative experiment. Lysis percentage values are normalized to that of the mock control T-cell. The right diagram shows fold change of cytotoxicity as compared with untreated cells from three independent experiments at E:T=5:1. (B) CD8+ ROR1-CAR T-cells were fixed, permeabilized, and stained for intracellular perforin. The histogram shows data from one representative experiment. The right diagram shows fold change of perforin mean fluorescence intensity (MFI) as compared with untreated cells (n=3). (C,D) Luminex multiplex bead assays were performed on supernatants obtained after a 24-hour co-culture with MDA-MB-231 at an E:T ratio of 4:1. Data shown are mean values±SD of fold change concentration for each cytokine compared with its counterpart cytokine secreted by untreated cells. Cytokine production of CD8+ (C) and CD4+ (D) T-cells were obtained from n=10 (except TNF-α n=4) and n=9 different donors, respectively. Statistical analysis was performed using Student’s t-test (A,B) or one-way analysis of variance (C,D). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns=not significant.
We performed intracellular staining and detected lower levels of perforin in ROR1-CAR T-cells that had been exposed to TGF-β (figure 2B). However, treatment with SD-208 prior to TGF-β exposure was able to prevent this effect (figure 2B). Of note, treatment with SD-208 led to increased intracellular perforin also in T-cells that had not been exposed to exogenous TGF-β, suggesting the potential for autocrine production and signaling (figure 2B).
Next, we investigated the effect of TGF-β (and SD-208) on cytokine production in CD8+ and CD4+ ROR1-CAR T-cells in co-culture with MDA-MB-231 TNBC cells. By multiplexed cytokine analysis we found significantly lower levels of IFN-γ, IL-2, TNF-α, GM-CSF, IL-4 and IL-10 (figure 2C,D). Again, treatment with SD-208 protected T-cells from the inhibitory effect of TGF-β. Even without addition of exogenous TGF-β, we observed a marked increase in cytokine production in both T cell subsets, supporting our earlier notion of autocrine TGF-β production and signaling.
Blockade of TGF-β-receptor signaling augments proliferation and viability of ROR1-CAR T-cells
We investigated the effect of TGF-β and SD-208 on ROR1-CAR T-cell proliferation and viability in co-culture with MDA-MB-231 TNBC cells. TGF-β impaired the proliferation of CD8+ and CD4+ ROR1-CAR T-cells with both—the percentage of T-cells that was induced to proliferate and the number of cell divisions that the proliferating T-cells underwent—being reduced (figure 3A and B). Treatment with SD-208 restored the proliferation of ROR1-CAR T-cells to the level of non-TGF-β treated T-cells. Notably, exposure to TGF-β was associated with a significant loss of viable ROR1-CAR T-cells. However, this negative effect was completely neutralized by SD-208 (figure 3C and D).
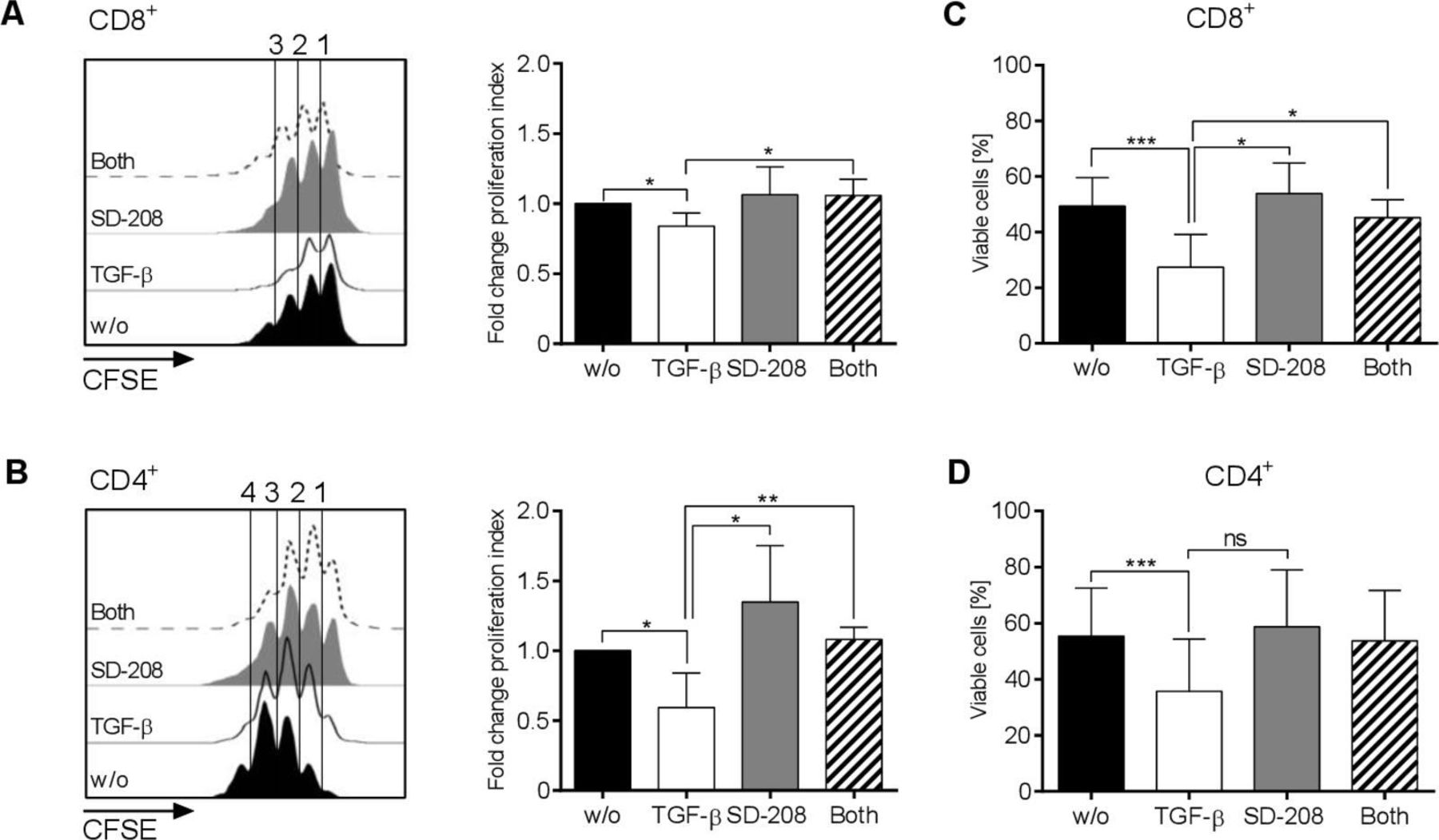
Effects of transforming growth factor (TGF-β) and SD-208 on proliferation and viability of receptor tyrosine kinase-like orphan receptor 1 (ROR1)-chimeric antigen receptor (CAR) T-cells. (A–D) CD8+ and CD4+ ROR1-CAR T-cells were co-cultured for 72 hours with MDA-MB-231 cells at E:T=4:1 in the presence of 1 µM SD-208 or/and 10 ng/mL TGF-β or were left untreated. Proliferation of CAR T-cells was measured by carboxyfluorescein succinimidyl ester (CFSE)-dilution in CD8+ (A) and CD4+ (B) T-cells. The diagrams on the right show fold change of the proliferation index compared with untreated cells. Percentages of viable CD8+ (C) and CD4+ (D) T-cells were assessed by 7-Aminoactinomycin staining. Data shown are mean values±SD from three independent experiments with *p<0.05, **p<0.01, ***p<0.001 by Student’s t-test, ns=not significant.
Taken together, these data show that in the presence of TGF-β, the antitumor function of ROR1-CAR T-cells against TNBC cells is markedly reduced. However, a short treatment course with SD-208 is able to protect ROR1-CAR T-cells from the inhibitory effect of TGF-β and enables sustained antitumor function.
Enhanced potency of ROR1-CAR T-cells in combination with TGF-β-receptor blockade
We sought to evaluate the combination treatment of ROR1-CAR T-cells with SD-208 in a microphysiologic 3D TNBC model (10). In this model, T-cells have to combat a large tumor cell mass and tumor regression can be monitored by BLI over a several-day period.
The data show that in the presence of TGF-β, the antitumor effect of ROR1-CAR T-cell therapy was substantially impaired. By BLI, ROR1-CAR T-cells were less than half as effective as in the absence of TGF-β (figure 4A and B). By ELISA, we found significantly lower levels of IFN-γ and IL-2, with almost undetectable levels of IL-2 toward the end of the analysis period (figure 4C). Also, in this advanced tumor model, SD-208-mediated blockade of TGF-β-receptor signaling was able to protect ROR1-CAR T-cells and to sustain their antitumor function. Similar to our prior in vitro analysis, the cytokine production in the presence of SD-208 was increased even when no exogenous TGF-β had been provided. Phenotypic analyses at the end of the 5-day assay period showed that despite their superior antitumor reactivity, there were lower levels of PD-1 detectable on ROR1-CAR T-cells that had been shielded from TGF-β by SD-208 (figure 4D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of transforming growth factor (TGF)-β and SD-208 on specific lysis and cytokine secretion by CD8+ chimeric antigen receptor (CAR) T-cells in a static 3D tumor model. Over 5-day assay period, 5×104 mock or receptor tyrosine kinase-like orphan receptor 1 (ROR1)-CAR T-cells were allowed to invade in microphysiologic 3D MDA-MB-231 models under control conditions or in the presence of 10 ng/mL TGF-β or/and 1 µM SD-208. (A) Images depicting the bioluminescence signal from viable tumor cells on day 0 and day 5 for one representative T-cell donor. (B) Reduction in tumor burden based on the bioluminescence signal encompassing the entire surface of the cell crowns normalized to the signal of mock cells. (C) Fold change of interferon (IFN-γ) and interleukin (IL-2)concentrations in supernatants at different time points as compared with untreated cells. (D) Proportion of PD-1+ cells from living (7-Aminoactinomycin−) and CD45+ cells collected from the supernatant on day 5. Data shown are mean values±SD from three independent experiments with *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by one-way analysis of variance, ns=not significant.
Collectively, these data show that blockade of TGF-β-receptor signaling leads to a sustained improvement of antitumor function in ROR1-CAR T-cells and suggest that protection from TGF-β may prevent CAR T-cell exhaustion.
Discussion
Solid tumors possess an immunosuppressive microenvironment that can reduce or abrogate the antitumor function of CAR T-cells. TGF-β is well-known as an immunosuppressive cytokine in the tumor microenvironment and has been associated with poor prognosis, short overall survival, and increased likelihood of relapse in TNBC patients.5 Interfering with TGF-β-signaling is therefore a rational strategy for improving CAR T-cell therapy against TNBC. Here, we show that TGF-β negatively affects the effector and helper functions of CD8+ and CD4+ ROR1-CAR T-cells against MDA-MB-231 TNBC in vitro. These insights are consistent with prior data obtained with non-gene modified tumor-reactive T-cells by other investigators.11 To protect ROR1-CAR T-cells from TGF-β, we used the kinase inhibitor SD-208. Our data show that SD-208 preserves (and even enhances) the function of ROR1-CAR T-cells against TNBC cells in 2D in vitro culture and in a microphysiologic 3D tumor model. Of particular interest, we found that exposure to TGF-β led to reduced viability of ROR1-CAR T-cells and the acquisition of an exhausted phenotype. The notion that shielding from TGF-β may prevent CAR T-cell exhaustion is congruent with recent clinical data showing that TGF-β was associated with the exclusion of tumor-reactive T-cells from the tumor microenvironment in metastatic ovarian, urothelial and colon cancer.12–15
Several strategies attempting to modulate TGF-β signaling in immune cells are being explored in pre-clinical and clinical studies including interfering with TGF-β synthesis, preventing activation of latent TGF-β, inhibiting ligand-receptor interaction and intracellular signal transduction.16 A strategy that has been evaluated in the context of T-cell engineering is expression of a dominant-negative TGF-β receptor that led to improved efficacy of prostate-specific membrane antigen-specific CAR T-cells in murine xenograft models.17 Another exemplary strategy is TGF-β blockade with mAbs like fresolimumab that in patients with advanced melanoma provided preliminary evidence of antitumor activity without dose-limiting toxicity,18 and in patients with metastatic breast cancer conferred longer median overall survival in combination with focal irradiation.19 Further, a bifunctional fusion protein M7824 (MSB0011359C), composed of a mAb against programmed death ligand 1 fused to the extracellular domain of TGF-β receptor II to act as a “TGF-β trap,” may enhance the efficacy of immune checkpoint blockade.20 Preliminary data from the phase 1 trial using M7824 demonstrated an objective response rate of 9,1% in TNBC patients.21 A well advanced strategy is the use of small molecule-based TGF-β-receptor kinase inhibitors, with Galunisertib and Vactosertib currently being tested in clinical trials. Notably, the ability of SD-208 to protect immune cell effector functions in the presence of TGF-β has previously been demonstrated in an in vivo model of glioblastoma.8
It is known that TGF-β plays a pivotal role in repair mechanisms of the heart and in pre-clinical development, small-molecule ALK5 inhibitors like SD-208 were found to be cardiotoxic on prolonged application in rats,22 while in clinical trials in humans cardiac toxicity was found to be acceptable.23 A preferred strategy for combining TGF-β-receptor kinase inhibition with ROR1-CAR T-cell therapy may therefore be to use SD-208 shortly after CAR T-cell infusion to create a time window of protection from TGF-β. Ideally, a single (or short term) treatment with SD-208 may render CAR T-cells resistant to TGF-β for a sufficient amount of time to augment the antitumor effect. A phase I/IIa clinical trial to determine the safety and potential efficacy of ROR1-CAR T-cells in patients with TNBC is ongoing (NCT02706392). In pre-clinical models, ROR1-CAR T-cells have proven to be effective against ROR1+ hematologic and solid tumors,4 and did not induce clinical toxicity in non-human primates.24
The use of SD-208 to block TGF-β-receptor signaling is also compatible with CAR T-cell products that target alternative antigens in TNBC, for example, mesothelin, B7-H3 and GD2, and may be combined with other strategies such as immune checkpoint blockade that are currently being investigated to augment the potency of CAR T-cell therapy.25 Further, TGF-β-receptor signaling blockade may be applied to augment CAR T-cell potency in other cancer entities that have been demonstrated to produce TGF-β, eg pancreatic, esophageal, stomach, colon and prostate cancers.26
Conclusion
This study shows that ROR1-CAR T-cells are susceptible to the immunosuppressive effects of TGF-β. The study also shows that blockade of TGF-β-receptor signaling with the small-molecule inhibitor SD-208 protects the antitumor function of ROR1-CAR T-cells and may prevent exhaustion. The investigation of combinatorial treatment regimens of CAR T-cell therapy and TGF-β-receptor blockade is clinically feasible and is anticipated to improve outcome with a favorable therapeutic index in oncology indications.
Acknowledgments
The authors thank Dr. Andreas Mades for providing medical writing support in accordance with good publication practice guidelines.
References
Footnotes
TS and RM contributed equally.
Contributors TS, RM, LW and JK performed experiments, and analyzed data. All authors were involved in the design of experiments, interpretation and discussion of data. SLN and GD supervised experiments in the 3D model. All authors read and approved the final manuscript. JW and MH conceived the project and supervised the study.
Funding TS has been supported by a grant from the Interdisciplinary Center for Clinical Research (Interdisziplinäres Zentrum für klinische Forschung, IZKF) at the University of Würzburg and is a fellow of the Else-Kröner Research Program “Translational Immunology” (Else-Kröner-Forschungs-Kolleg) funded by the Else Kröner-Fresenius-Stiftung. MH is a member of the Young Scholar Program (Junges Kolleg) and Extraordinary Member of the Bavarian Academy of Sciences (Bayerische Akademie der Wissenschaften). This work was supported by a grant from German Cancer Aid (Deutsche Krebshilfe e. V., Max Eder Program, grant no. 70110313 to MH), the m4 Award in Personalized Medicine (Free State of Bavaria, grant no. BIO-1601-0002 to MH), and an intramural grant from the IZKF Würzburg (grant no. B-354 to SLN, GD and MH).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study protocol has been approved by the Institutional Review Board of the University of Würzburg.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.