Article Text

Abstract
Background Peritoneal carcinomatosis (PC) is a common and devastating manifestation of colon cancer and refractory to conventional anticancer therapeutics. During the peritoneal dissemination of colon cancer, peritoneal immunity is nullified by various mechanisms of immune evasion. Here, we employed the armed oncolytic vaccinia virus mJX-594 (JX) to rejuvenate the peritoneal antitumor immune responses in the treatment of PC.
Methods PC model of MC38 colon cancer was generated and intraperitoneally treated with JX and/or anti-programmed cell death protein 1 (PD-1) antibody. The peritoneal tumor burden, vascular leakage, and malignant ascites formation were then assessed. Tumors and peritoneal lavage cells were analyzed by flow cytometry, multiplex tissue imaging, and a NanoString assay.
Results JX treatment effectively suppressed peritoneal cancer progression and malignant ascites formation. It also restored the peritoneal anticancer immunity by activating peritoneal dendritic cells (DCs) and CD8+ T cells. Moreover, JX selectively infected and killed peritoneal colon cancer cells and promoted the intratumoral infiltration of DCs and CD8+ T cells into peritoneal tumor nodules. JX reinvigorates anticancer immunity by reprogramming immune-related transcriptional signatures within the tumor microenvironment. Notably, JX cooperates with immune checkpoint inhibitors (ICIs), anti-programmed death-1, anti-programmed death-ligand 1, and anti-lymphocyte-activation gene-3 to elicit a stronger anticancer immunity that eliminates peritoneal metastases and malignant ascites of colon cancer compared with JX or ICI alone.
Conclusions Intraperitoneal immunotherapy with JX restores peritoneal anticancer immunity and potentiates immune checkpoint blockade to suppress PC and malignant ascites in colon cancer.
- translational medical research
- tumor microenvironment
- immunotherapy
- oncolytic virotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Background
The peritoneal cavity is an immunologically unique compartment.1–5 It has distinct immunological features, in comparison to systemic immunity, with relatively abundant dendritic cells (DCs), predominant CD8+ T cells over CD4+ T cells, and abundant soluble factors in peritoneal fluids, which establishes a robust immune competency.4 6 However, cancer cells can nullify this peritoneal immunity through various immune evasive mechanisms, and rapidly metastasize into the peritoneal cavity, making it the second most common site of metastasis in patients with colon cancer.1 3 6–9
During peritoneal dissemination of cancer cells, they are known to inactivate DCs and induce T cell exhaustion in the peritoneal cavity.4 6 10 Specifically, tumor cells overexpress PD-L1 on their surface and attenuate cell lysis induced by CD8+ cytotoxic T cells.9 Moreover, tumor cell-derived vascular endothelial growth factor (VEGF) generates malformed and leaky neovessels along the surface of the peritoneal cavity, hinders the effective delivery of anticancer drugs and cells, and promotes the accumulation of malignant ascites, in excess of immunosuppressive cytokines and immune cells.4 11 12 This unique tumor microenvironment (TME) of the peritoneal cavity severely disturbs the efficacy of conventional cytotoxic chemotherapeutics and targeted agents, leading to a poor prognosis in patients with peritoneal carcinomatosis (PC).10 13 14 Recently, to infiltrate this peritoneal TME, hyperthermic intraperitoneal chemotherapy was attempted in patients with colon cancer PC after complete cytoreductive surgery. However, the results did not show a meaningful survival benefit, and PC of colon cancer remains a significant clinical challenge.15 16
Over the past decade, cancer immunotherapy has emerged as a potent and effective therapeutic strategy for advanced cancers.17–20 Indeed, antibodies targeting immune checkpoints, including cytotoxic T-lymphocyte associated protein (CTLA)-4, programmed cell death protein 1 (PD-1), and programmed death-ligand 1 (PD-L1), elicit durable antitumor effects in patients with various malignancies.21–24 However, these immune checkpoint inhibitors (ICIs) showed limited efficacy as a monotherapy against colon cancer, especially peritoneal metastases.19 23 25 Therefore, a novel immunotherapeutic agent is urgently needed to overcome this limitation through the activation of peritoneal immunity.
Oncolytic virotherapy (OV) has been classified as a novel type of immunotherapy.26–28 It preferentially induces direct destruction of tumors through the selective infection and lysis of tumor cells.27 29 In this process, immunogenic cell death occurs with the widespread release of tumor-associated antigens, which are presented by DCs to activate antitumor immunity.28 Therefore, it acts as an in situ cancer vaccine within TME.29 In this regard, OV is an attractive therapeutic modality that can induce antigen-specific T cell response with the expansion of cytotoxic effector cells.30
Vaccinia virus, belonging to the poxvirus family, is highly immunogenic and has optimal characteristics that make it feasible for clinical development.31 32 JX-594 (pexastimogene devacirepvec, Pexa-vec) is an oncolytic vaccinia virus armed with GM-CSF, and it showed promising antitumor efficacies through its oncolytic, antiangiogenic, and immune-stimulating mechanisms, in both preclinical and clinical studies.33–36 Recently, we have reported that murine version of JX-594 (mJX-594, hereafter referred to as JX) robustly elicits the innate and adaptive immune responses within TME, thereby augmenting the efficacy of ICIs in immunotherapy-resistant kidney and breast cancers.36
Here, we demonstrate that intraperitoneal JX immunotherapy can rejuvenate peritoneal anticancer immunity, and enhance ICIs to suppress PC and malignant ascites in colon cancer.
Methods
Mice and cell lines
Male C57BL/6N mice between 8 and 12 weeks of age were purchased from Orient Bio Inc (Seongnam, Korea). Mice were housed in a specific pathogen-free animal facility at CHA University (Seongnam, Korea), and all experiments were approved by the Institutional Animal Care and Use Committee (IACUC, #180081) of CHA University.
The MC38 murine colon cancer cell line and ID8 ovarian cancer cell lines were obtained from the National Cancer Center (Goyang, Korea). MC38 and ID8 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C, and was harvested at 80% confluence for the relevant experiments. The cell morphology and growth characteristics were conformant, and they were tested regularly for Mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza, New Jersey, USA).
Construction and production of virus
JX, provided by SillaJen Inc (Seoul, Korea), is a Western Reserve strain of the vaccinia virus encoding murine GM-CSF in the vaccinia thymidine kinase gene locus under the control of the p7.5 promoter.37 38 The generation and quantification of the virus were previously described.36 The virus titer was determined using a plaque assay of U-2 OS cells.
PC model and treatment regimens
To generate peritoneal tumors, we intraperitoneally injected either 5 × 105 MC38 colon cancer cells or 1.5 × 107 ID8 ovarian cancer cells into the peritoneal cavity of wild-type C57BL/6 mice. Tumor-implanted mice were randomized to each experimental group 7 days after implantation. Mice were treated with an intraperitoneal injection of 1 × 107 plaque-forming units (pfu) of JX. For combination immunotherapy, we also administered anti-PD-1 (10 mg/kg, clone J43, BioXCell), anti-VEGFR2 (25 mg/kg, clone DC101, BioXCell), anti-PD-L1 (10 mg/kg, clone 10F.9G2, BioXCell), and anti-LAG-3 (10 mg/kg, clone C9B7W, BioXCell) intraperitoneally at given time points. The optimal doses for checkpoint blockade were determined from previous studies.36 39 Mice in the control group were treated with an intraperitoneal injection of the same volume of phosphate-buffered saline (PBS). Tumor-bearing mice were weighed twice weekly and monitored daily for the clinical sign of swollen bellies indicative of ascites formation. During the sacrifice, ascitic fluid was aspirated entirely directly from the peritoneal cavity of all mice using a 26-gauge needle. The tumor nodules in the peritoneal cavity and peritoneum were harvested and weighed, and peritoneal cells were prepared performing a peritoneal lavage by washing the peritoneum with 3 mL of 3% FBS in PBS, containing 2 mmol/L EDTA. The survival of each mouse was monitored, and the overall survival was calculated.
Flow cytometry analysis of tumor-associated immune cells
For flow cytometry analysis, harvested tumors were minced into small pieces with scissors and incubated in digestion buffer, comprised of 2 mg/mL collagenase D (COLLD-RO, Roche) and 40 µg/mL DNase I (10104159001, Roche), for 1 hour at 37°C. The cell suspensions were filtered through a 70 µm cell strainer (352350, Falcon) and incubated for 3 min at room temperature in ammonium chloride-potassium lysis buffer (A1049201, Gibco) to remove cell clumps and red blood cells. After washing with PBS, the cells were filtered through a 40 µm nylon mesh and resuspended in FACS buffer (1% FBS in PBS). Peritoneal cells, collected from the peritoneal cavity using lavage, were lysed with ACK buffer as described above. In the same way, the cells were filtered and resuspended in FACS buffer. Next, single-cell suspension isolated from tumor tissues and peritoneal cavity were incubated on ice for 30 min in Fixable Viability Dye eFluorTM 450 (65-0863-18, eBioscience) to exclude dead cells before antibody staining. Then the cells were washed with FACS buffer and incubated with mouse Fc receptor binding inhibitor (CD16/32, clone 2.4G2, BD Pharmingen) for 15 min at room temperature before staining with surface antibodies against CD45 (clone 30-F11, BD Pharmingen), CD3 (clone 17A2, eBioscience), CD4 (clone RM4-5, eBioscience) and CD8 (clone 53-6.7, eBioscience) for 30 min on ice. Cells were further permeabilized using a FoxP3 fixation and permeabilization kit (eBioscience), and stained for FoxP3 (clone FJK-16s, eBioscience) or Granzyme B (clone NGZB, eBioscience). For intracellular cytokine staining, cells from peritoneal cavity were stimulated for 4 hours with 20 ng/mL PMA (Sigma) and 1 μM Ionomycin (Sigma) in the presence of 3 µg/mL Brefeldin A (eBioscience). After stimulation, cells were fixed, permeabilized, and stained for interferon (IFN)-γ (clone XMG1.2, eBioscience) and TNF-α (clone MP6-XT22, BD Pharmingen). Tumor cells (CD45−CD31−), CD4+ T cell (CD45+CD4+), CD8+ T cell (CD45+CD8+), DCs (CD45+CD11c+), myeloid cell (CD45+CD11b+) and Tregs (CD4+CD25+) were sorted from tumors using MoFlo XDP cell sorter (Beckman Coulter). Flow cytometry was performed using a CytoFLEX flow cytometer (Beckman Coulter) and analyzed using FlowJo software (Tree Star Inc, Ashland, Oregon, USA).
Immune-related gene expression profiling using NanoString
Total RNA was extracted from fresh tumor tissues using TRIzol reagent (Invitrogen) and purified with ethanol. RNA concentration and quality were confirmed with a Fragment Analyzer instrument (Advanced Analytical Technologies, Iowa, USA). We used 100 ng of total RNA from each tumor sample for the digital multiplexed profiling with NanoString nCounter PanCancer Immune Profiling mouse panel (NanoString Technologies), as per our previously established protocol.36
Gene expression analysis using RT2 Profiler PCR array
Total RNA was extracted from sorted cells using the TRIzol reagent (Invitrogen) and purified with ethanol. RNA was reverse-transcribed using the GoScript Reverse Transcription kit (Promega) for cDNA synthesis and then loaded on to RT2 Profiler PCR Array Mouse Cancer Inflammation and Immunity Crosstalk according to manufacturer’s instructions (Qiagen). Reactions were performed in the LightCycler96 (Roche) for 10 min at 95°C followed by 45 cycles of 15 s at 95°C and 1 min at 60°C.
Enzyme-linked immunosorbent assay (ELISA)
To quantify damage-associated molecular patterns (DAMPs), 4 × 105 MC38 cells seeded into six-well plates overnight were infected with JX at MOIs of 0 and 10 in 10% FBS-containing DMEM for 24 hours. Culture supernatants were harvested to measure calreticulin and HMGB1, and cell pellets were lysed to measure annexin A1. Calreticulin (Biomatik, Ontario, California, USA), HMGB1 (Biomatik, Ontario, CA), and annexin A1 (Abcam, Burlingame, California, USA) were quantified using the ELISA kit following the manufacturer’s instructions.
IFN-γ enzyme-linked immunospot (ELISPOT) assay
For the quantification of tumor-specific cytotoxic T cells, splenocytes, tumor-infiltrating lymphocytes (TILs), and peritoneal cells from each group were isolated 5 days after the last treatment. Harvested cells were labeled using the Dead Cell Removal Kit (Miltenyi Biotec, Auburn, 130090101) according to the manufacturer’s instructions, and purified using MACS (Miltenyi Biotec, Auburn, California, USA). Purified live cells were stimulated with MC38 tumor cells at a 10:1 ratio in mouse IFN-γ-precoated 96-well plates (MABTECH AB, Nacka Strand, Sweden), which were then incubated for 36 hours at 37°C in a CO2 incubator. After being washed, plates were stained with 1 µg/mL of the biotinylated antimouse IFN-γ antibody, R4-6A2-biotin, for 2 hours, followed by incubation with a streptavidin-ALP solution for 1 hour at room temperature. After the addition of BCIP/NBT-plus substrate solution, the number of spots was counted using ImageJ software (http://rsb.info.nih.gov/ij).
Histologic analysis via immunofluorescence
For immunofluorescence staining, tumor samples were fixed in 1% PFA at room temperature and were washed several times with PBS, dehydrated overnight with 20% sucrose, and embedded in tissue-freezing medium (Leica). Frozen tissues were sectioned into 50 µm thick blocks, which were permeabilized with 0.3% PBS-T (Triton X-100 in PBS), and then blocked with 5% goat serum in 0.1% PBS-T for 1 hour. Next, the samples were incubated overnight with the following primary antibodies: Rabbit anti-vaccinia virus (Abcam), hamster (clone 2H8, Millipore) and rabbit anti-CD31 (Abcam), rat anti-CD8 (clone 53–6.7, BD Pharmingen), rat anti-CD4 (clone RM4-5, Invitrogen), hamster anti-CD11c (clone HL3, BD Pharmingen), rat anti-Granzyme B (clone NGZB, eBioscience), rat anti-IFN-γ (clone XMG1.2, eBioscience), rat anti-TNF-α (clone MP6-XT22, BD Pharmingen) or rat anti-FoxP3 (clone FJK-16s, eBioscience). After several washes, the samples were incubated for 2 hours at room temperature with the following secondary antibodies: FITC-conjugated, Cy3-conjugated, or Cy5-conjugated anti-rabbit IgG (Jackson ImmunoResearch), FITC-conjugated anti-rat IgG (Jackson ImmunoResearch), FITC-conjugated or Cy3-conjugated anti-hamster IgG (Jackson ImmunoResearch). The cell nuclei were counterstained with 4′6-diamidino-2-phenylindole (Invitrogen). Finally, samples were mounted with fluorescent mounting medium (DAKO), and images were acquired using an LSM 880 confocal microscope (Carl Zeiss).
Morphometric analysis
Density measurements of the JX, blood vessels, T lymphocytes, and DCs in the same area were performed using ImageJ software. The JX+ density per random 0.49 mm2 area was calculated in tumor sections to determine the level of JX infection. Blood vessel density was measured by calculating the CD31+ density per random 0.49 mm2 area in tumor sections. The degree of cytotoxic T lymphocyte infiltration was calculated as the percentage CD8+ and CD4+ per random 0.49 mm2 area in intratumoral regions. The density of DCs was determined by calculating the percentage CD11c+ in random 0.49 mm2 area. All measurements were performed in at least five areas per mouse.
Statistical analysis
Statistical analyses were performed using GraphPad Prism V.7.0 software (GraphPad Software, La Jolla, California, USA) and PASW statistics V.18 (SPSS). Values are represented as mean±SE unless otherwise indicated. The Shapiro-Wilk normality test was performed for all datasets to analyze whether each dataset follows a normal distribution pattern. If the dataset followed a normal distribution, we applied parametric tests such as the Student’s t-test and one-way analysis of variance. If the dataset did not follow normal distribution owing to small sample size, we used non-parametric tests such as the Mann-Whitney U test and Kruskal-Wallis test. Adjusted p values were used to analyze Nanostring data according to the Benjamini-Hochberg method. Gene set enrichment analysis was used to test the enrichment of a specific gene set, and core enrichment genes were determined. Survival curves were plotted using the Kaplan-Meier method, and statistical differences between curves were analyzed using the log-rank test. The level of statistical significance was set at p<0.05.
Results
JX treatment restores the peritoneal immunity
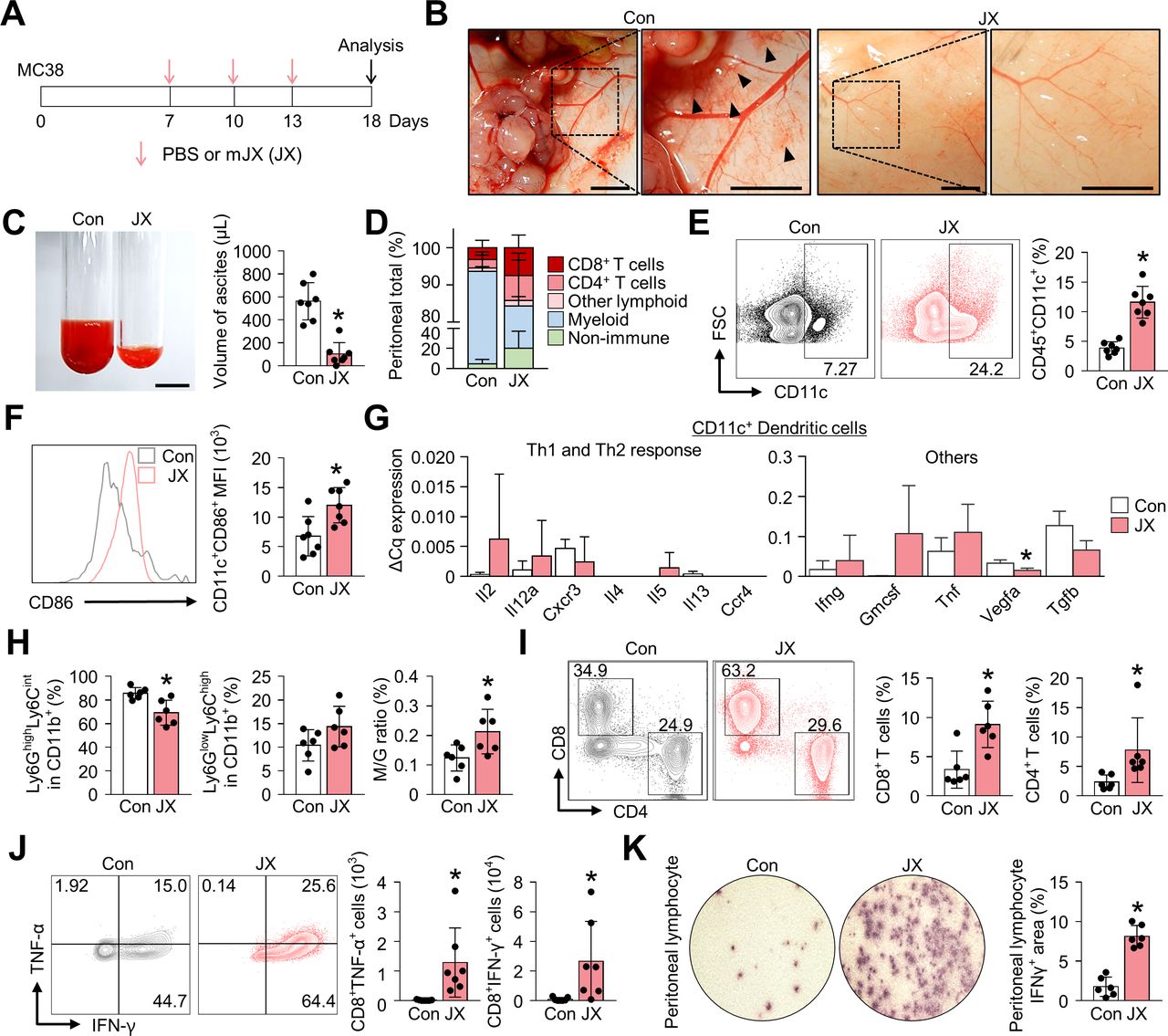
The PC model was established via the intraperitoneal injection of MC38 colon cancer cells into C57BL/6 mice. One week after tumor implantation, mice were treated with intraperitoneal injections of either JX or PBS (figure 1A). At day 18 of tumor implantation, all PBS-treated control mice developed multiple peritoneal seeding metastases on the surface of the visceral and parietal peritoneum and accumulated malignant ascites in the peritoneal cavity (figure 1B). In PBS-treated mice, malformed neovessels were observed on the peritoneal surface that is close to the tumor mass, and these vessels were accompanied by the foci of small peritoneal hemorrhages. In contrast, JX-treated mice had a remarkably fewer number of tumor nodules which were smaller compared with that of the control mice in the peritoneal cavity, and the volume of malignant ascites was 82% less compared with that of the control mice (figure 1B,C). Moreover, there were fewer neovessels on the peritoneal surface, and less peritoneal hemorrhage was observed, indicating suppressed angiogenesis by JX in the peritoneal cavity (figure 1B). Intraperitoneal JX treatment was generally well-tolerated, except for the transient and slight weight loss (~1 g) within 3 days of the first injection, which was self-limited and completely resolved within a week. There were no treatment-related mortalities.
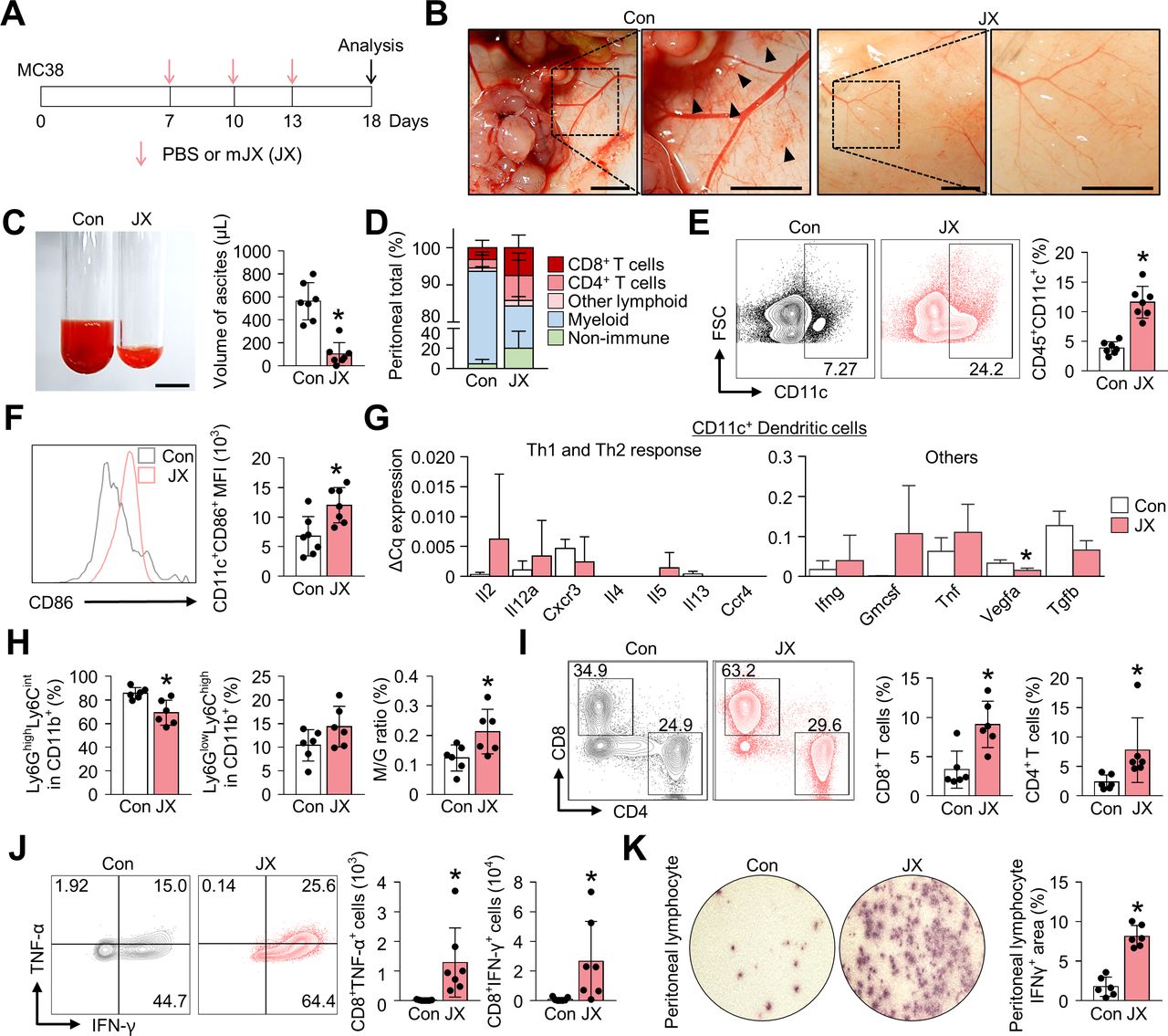
JX treatment activates the peritoneal immunity. MC38 colon tumor cells were implanted intraperitoneally into mice, which were treated intraperitoneally with PBS or mJX-594 (JX, 1 × 107 pfu) three times. (A) Diagram of the treatment schedule. Red arrows indicate treatment with intratumoral delivery of JX, and black arrows indicate sacrifice. (B) Representative images of the parietal peritoneum and its blood vessels. Black arrowheads indicate peritoneal hemorrhages. (C) Representative images and comparisons of malignant ascites in PBS-treated or JX-treated mice. (D) Diagram depicting the comparison of the peritoneal cell population. (E and F) Representative flow cytometric plot and comparisons of CD11c+ DCs (E) and their CD86 expression (F). (G) Comparisons of cytokine and chemokine expressions in CD11c+ DCs in PBS-treated and JX-treated tumors. Intratumoral DCs were isolated using fluorescence-activated cell-sorting. (H) Comparisons of fractions of Ly6GhighLy6Cint granulocytic myeloid cells and Ly6GlowLy6Chigh monocytic myeloid cells. (I) Representative flow cytometric plot and comparisons of CD8+ and CD4+ T cell fractions. (J) Representative flow cytometric plot and comparisons of TNF-α and IFN-γ-expressing fraction in CD8+ T cells. (K) Images and comparisons of IFN-γ ELISPOT in peritoneal CD8+ T lymphocytes from PBS or JX-treated mice. Pooled data from two experiments with n=6 to 7 per group. Values are mean±SD. *p<0.05 versus control. Two-tailed Student’s t-test and Mann-Whitney U test were used (C and E–K). Scale bars, 5 mm (B), 10 mm (C). DCs, dendritic cells; PBS, phosphate-buffered saline; pfu, plaque-forming units.
In parallel, flow cytometry analyses revealed the alteration of innate and adaptive immunity in the peritoneal cavity (figure 1D). At first, JX treatment increased the number of intraperitoneal CD11c+ DCs by 3.0-fold (figure 1E). Notably, the expression of CD86 was enhanced in CD11c+ DCs, suggesting the activation of peritoneal DCs after JX treatment (figure 1F). Intriguingly, DCs within JX-treated tumors induced Th1-predominant responses compared with those within PBS-treated tumors. Moreover, JX-activated DCs upregulated Gmcsf, Tnf, Ifng, while downregulating Vegfa and Tgfb (figure 1G). In addition, the analysis of CD11b+ myeloid cells showed a significant decrease in the number of Ly6GhighLy6Cint granulocytic cells with a slight increase in the number of Ly6GlowLy6Chigh monocytic cells, resulting in an increased ratio of the monocytic myeloid cells to granulocytic myeloid cells (figure 1H).
Finally, regarding the adaptive immunity in the peritoneal cavity, JX treatment increased the number of peritoneal CD8+ cytotoxic T cells by 2.7-fold and that of CD4+ T cells by 3.3-fold compared with the control group (figure 1I). Indeed, the number of TNF-α- or IFN-γ-secreting CD8+ T cells were also dramatically increased after the JX treatment (figure 1J). IFN-γ ELISPOT assay was also performed using peritoneal lymphocytes in the presence of MC38 tumor cells to confirm the tumor-specific effector function of the intraperitoneal T cells after JX treatment, and IFN-γ spots were observed 4.62-fold more frequently in the JX-treated mice compared with the control (figure 1K). Collectively, these findings indicate that the JX treatment effectively suppressed the peritoneal progression of colon cancer and malignant ascites formation, via enhanced innate and adaptive immunity in the peritoneal cavity.
JX treatment suppresses tumor angiogenesis and facilitates immune cell infiltration into the tumor
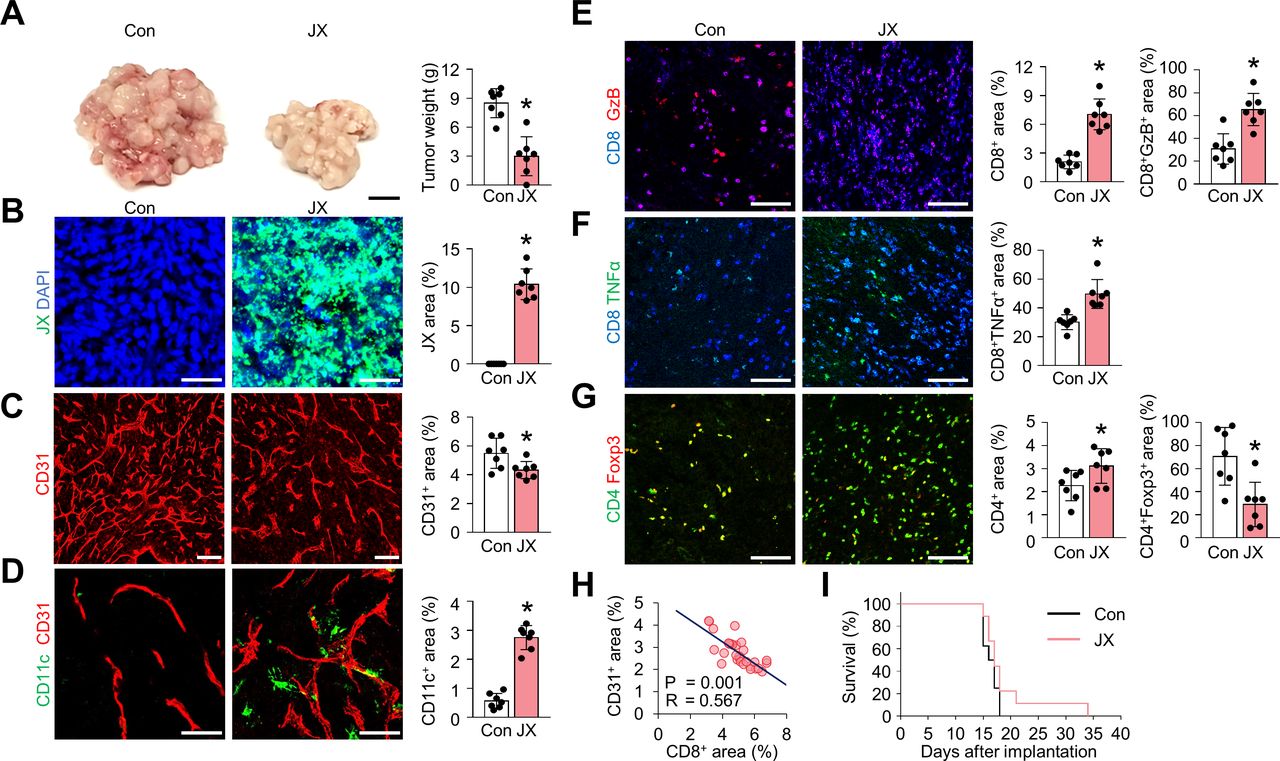
To confirm the effect of JX on TME, we analyzed tumor-infiltrating immune cells within the peritoneal tumors. After consecutive injections of JX, intraperitoneal tumor burden was reduced by 65% when compared with the control group (figure 2A). Consistent with previous reports,36 37 40 JX selectively infected and destroyed colon cancer cells but also disrupted tumor blood vessels within TME (figure 2B,C). These JX-infected, dying tumor cells released DAMPs, such as calreticulin and annexin A1, which are involved in the activation of DCs (online supplemental figure S1).41 42 Additionally, JX itself further induced the activation and maturation of DCs within TME because it was genetically engineered to secrete mGM-CSF. Accordingly, the number of intratumoral DCs were markedly increased by 3.9-fold compared with that of the PBS-treated tumors (figure 2D). Furthermore, the intratumoral infiltration of CD8+ T cells increased 3.4-fold, and most cells upregulated the expression of granzyme B (GzB) and TNF-α after JX treatment (figure 2E,F). FoxP3-expressing CD4+ T cells decreased by 59% with intraperitoneal JX treatment (figure 2G). The JX-induced anti-angiogenic effect also correlated with increased CD8+ T cells within peritoneal tumors (figure 2H). The degree of aberrant tumor vasculatures suppressed with JX treatment correlated with the number of CD8+ cytotoxic T cells infiltrated into TME. Although JX monotherapy-induced dramatic remodeling of TME, it did not show a remarkable survival benefit as a monotherapy, suggesting the need for optimal combination partners for JX (figure 2I).
Supplemental material

JX treatment reduces peritoneal metastases by suppressing angiogenesis and promoting immune cell infiltration into peritoneal tumors. MC38 tumor-bearing mice were intraperitoneally treated with PBS or JX. (A) Representative images and comparisons of tumor burden in the PBS-treated or JX-treated mice. (B–D) Representative images and comparisons of JX infection (B), CD31+ tumor blood vessels (C), and CD11c+ DCs (D). (E–G) Representative images and comparisons of CD8+GzB+ T cells (E), CD8+TNF-α+ T cells (F), and CD4+Foxp3+ T cells (G) within tumors. (H) Correlation between intratumoral CD8+ T cells and CD31+ blood vessels in JX-treated tumors (n=30). (I) Kaplan-Meier survival curves for overall survival in PBS-treated or JX-treated mice. Unless otherwise denoted, pooled data from two experiments with n=7 to 8 per group. Values are mean±SD. p<0.05 versus control. Two-tailed Student’s t test and Mann-Whitney U test were used (A–G). Scale bars, 10 mm (A), 50 µm (B and D), 100 µm (C and E–G). DCs, dendritic cells; GzB, granzyme B; PBS, phosphate-buffered saline.
Overall, these results suggested that JX could suppress tumor angiogenesis and promote the infiltration of CD11c+ DCs and CD8+ T cells into tumor nodules, eliciting an effective antitumor immune response in TME.
JX treatment reinvigorate effector functions of intratumoral T cells
In order to evaluate the role of JX on TME further, we compared the phenotype of tumor-infiltrating T cells in control or JX-treated tumors. First, JX treatment notably increased the number of intratumoral lymphocytes by 6.6-fold compared with control mice, while the number of myeloid cells was decreased by 22% (figure 3A). In particular, the number of CD8+ and CD4+ T cells were increased by 10.6-fold and 2.9-fold, respectively, compared with that of the control group (figure 3B). To evaluate the activation status of intratumoral CD8+ T cells, we assessed the expression of GzB and inducible T-cell costimulator (ICOS), which are known as the T cell activation and costimulatory markers.36 In CD8+ T cells of JX-treated tumors, the expression of GzB was increased 35.3-fold, and the expression of ICOS was also upregulated by 16.0-fold compared with that of control tumors (figure 3C).
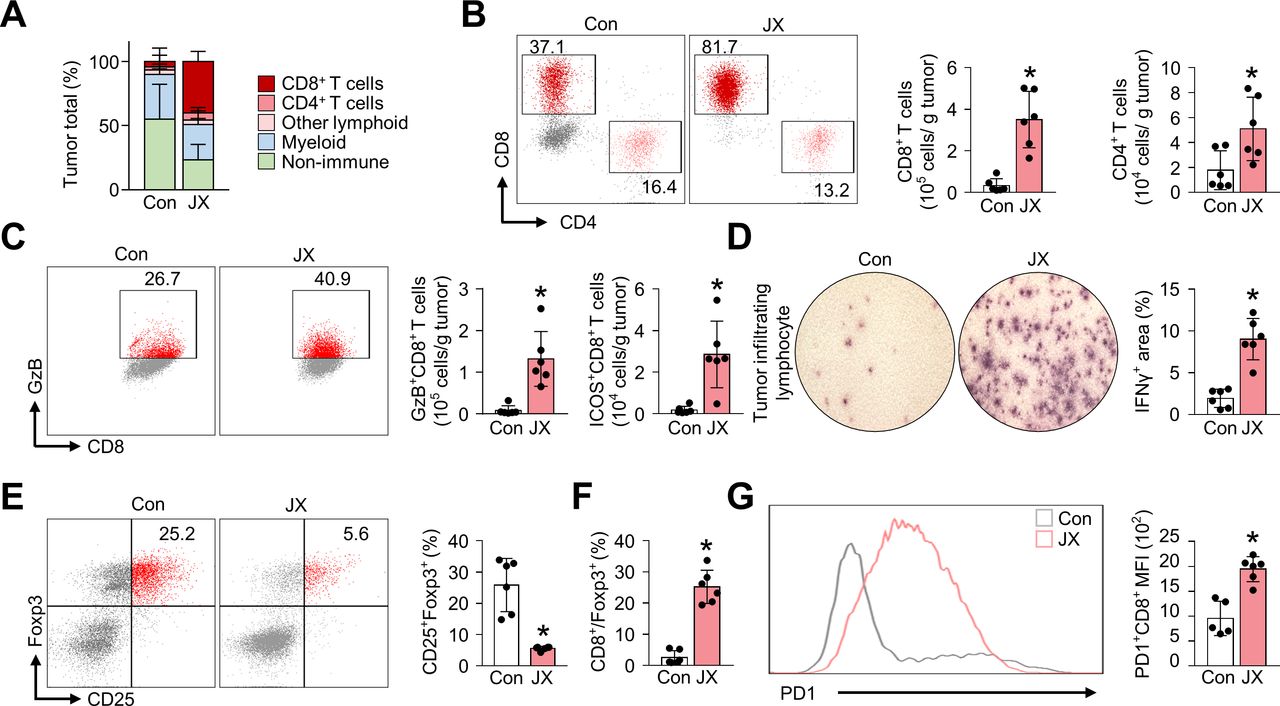
JX reinvigorate the effector function of tumor-infiltrating T cells. Mice were implanted with MC38 tumor cells and treated with intraperitoneal injections of PBS or JX. (A) Diagram depicting the comparison of the tumor-infiltrating cell population. (B) Representative flow cytometric images and comparisons of CD8+ and CD4+ T cell fractions. (C) Comparisons of fractions of CD8+GzB+ and CD8+ICOS+ cells. (D) Images and comparisons of IFN-γ ELISPOT in CD8+ tumor-infiltrating lymphocytes from PBS-treated or JX-treated mice. (E) Representative flow cytometric plot showing CD25+Foxp3+ (Treg) fraction in CD4+ T cells. (F) Comparisons of CD8/Treg ratio in the tumor. (G) Representative histogram showing PD-1 expression in CD8+ T cells. Pooled data from two experiments with n=5 to 7 per group. Values are mean±SD. p<0.05 versus control. Two-tailed Student’s t-test and Mann-Whitney U test were used (B–G). ELISPOT, enzyme-linked immunospot; GzB, granzyme B; PBS, phosphate-buffered saline; PD-1, programmed cell death protein 1.
Additionally, the TILs in JX-treated tumors secreted increased levels of IFN-γ (4.7-fold) compared with the control tumors (figure 3D). Also, the number of CD4+CD25+Foxp3+ regulatory T cells (Treg) was reduced by 78.3% in JX-treated tumors compared with that of the control tumors (figure 3E). Moreover, the ratio of cytotoxic T cells to Tregs was increased by 9.7-fold after JX treatment, indicating the increase in the overall effector function of the intratumoral T cells (figure 3F). On the other hand, although JX activated anticancer immunity, it also significantly increased the number of inhibitory checkpoint molecules, such as PD-1 in CD8+ T cells, suggesting the induction of negative feedback mechanism to counter-balance immune response in the TME (figure 3G). Collectively, these findings demonstrated that the JX treatment rejuvenated the effector functions of CD8+ T cells in TME, thereby generating a robust antitumor immunity.
JX reprograms the immune-related transcriptional signature in peritoneal metastasis
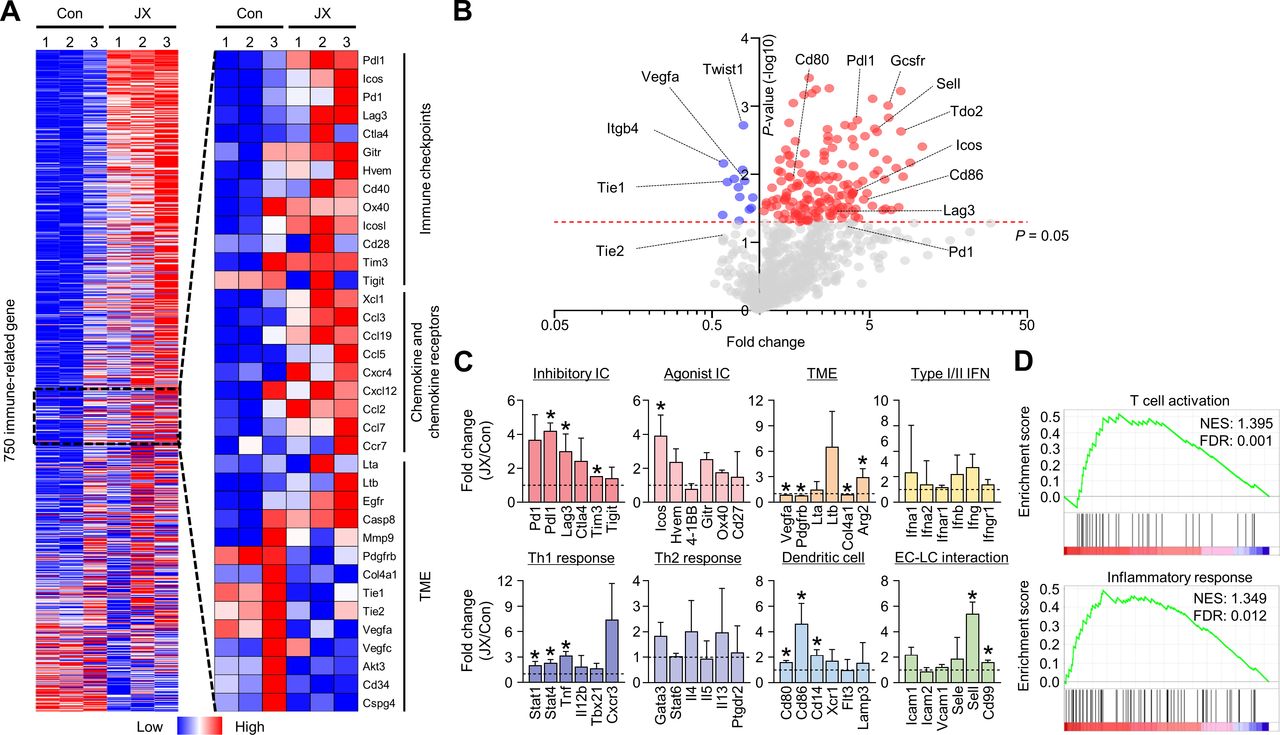
To comprehensively analyze the JX-induced immune changes in the peritoneal metastases of colon cancer, we assessed the transcriptional changes of immune-related genes using the NanoString PanCancer Immune profiling panel. The results revealed distinct immunologic reprograming in response to JX treatment, especially in immune checkpoint molecules, chemokines, and chemokine receptors (figure 4A, online supplemental table 1). Overall, 102 genes were upregulated more than twofold after JX monotherapy (figure 4B). Notably, the genes related to immune checkpoint (Pdl1, Lag3, Tim3), TME (Arg2), Th1 (Stat1, Stat4, Tnf), DCs (Cd80, Cd86, Cd14), and endothelial-lymphocyte interaction (Sell, Cd99) were significantly upregulated in JX-treated tumors compared with the control tumors (figure 4C). Intriguingly, intratumoral Vegfa expression was significantly suppressed after JX treatment, which is consistent with our observation of reduced neovessels in the peritoneal cavity. Furthermore, gene sets related T cell activation and inflammatory response were more enriched in JX-treated tumors compared with control tumors (figure 4D). JX treatment induced differential changes in various cell populations. JX upregulated Ifng, Tnf, and Gmcsf in most of immune cells, but not in tumor cells. Myeloid cells and DCs upregulated Th1 cytokines. Pdl1 expression was increased in most of immune cells, whereas Pd1 upregulation was observed in CD8+ and CD4+ T cells (online supplemental figure S2).
Supplemental material
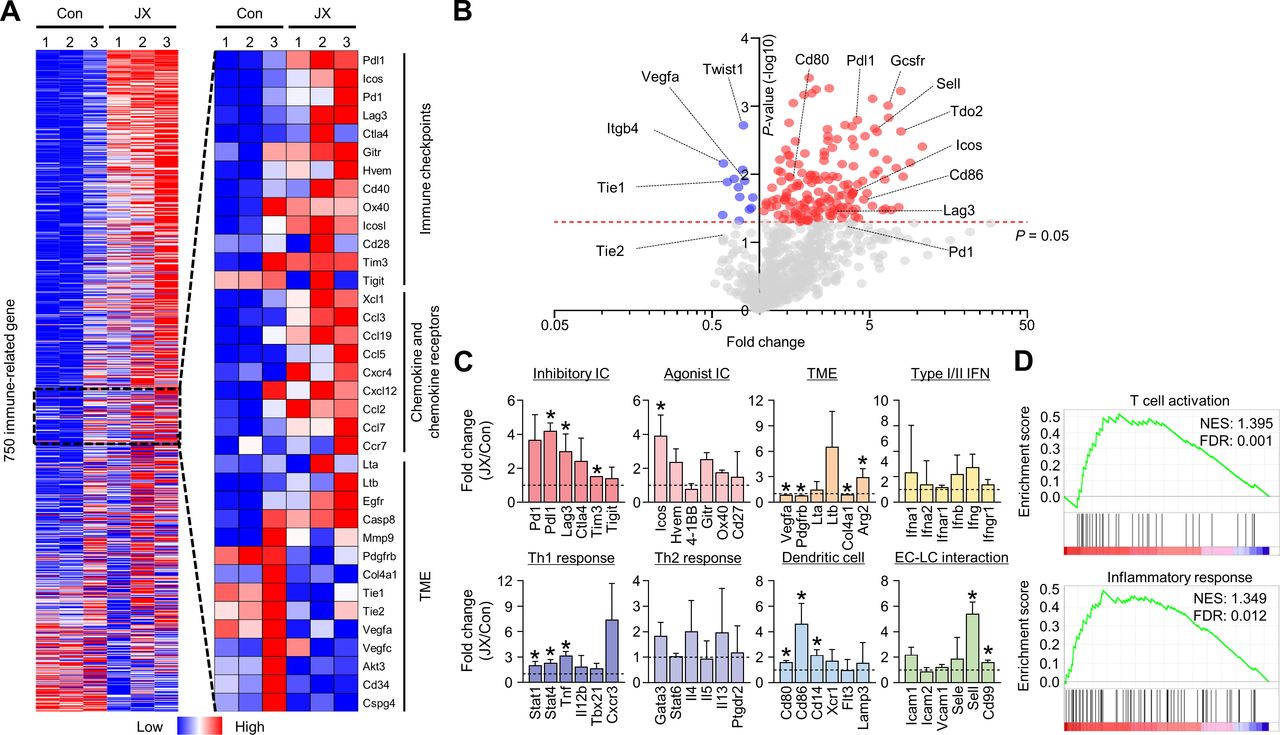
JX reprograms the peritoneal TME to T cell-inflamed tumors. MC38 tumor cells were implanted intraperitoneally into mice and treated with intraperitoneal injections of PBS or JX. (A) NanoString immune-related gene expression heat map. Red and blue color represent upregulated and downregulated genes, respectively. (B) Volcano plot showing the change of gene expression profile in JX-treated tumors of mice. Red line indicates p<0.05. (C) Comparisons of gene expressions related to inhibitory and agonist immune checkpoints, TME, type I/II IFNs, Th1 response, Th2 response, DC, and endothelial cell-lymphocyte interaction. (D) GSEA of gene sets involved in T cell activation and an inflammatory response. Pooled data from two experiments with n=3 per group. Values are mean±SD. p<0.05 versus control. Two-tailed Student’s t-test was used (C). DC, dendritic cell; GSEA, gene set enrichment analysis; PBS, phosphate-buffered saline; TME, tumor microenvironment.
Consequentially, these results demonstrated that immune checkpoints, Th1 response, and type I/II IFN-related genes in the intraperitoneal seeding nodules were extensively reprogrammed in response to JX treatment, thereby converting non-inflamed peritoneal tumors to T cell-inflamed tumors.
JX cooperates with PD-1 blockade to elicit potent anticancer immunity that eliminates peritoneal metastases of colon cancer
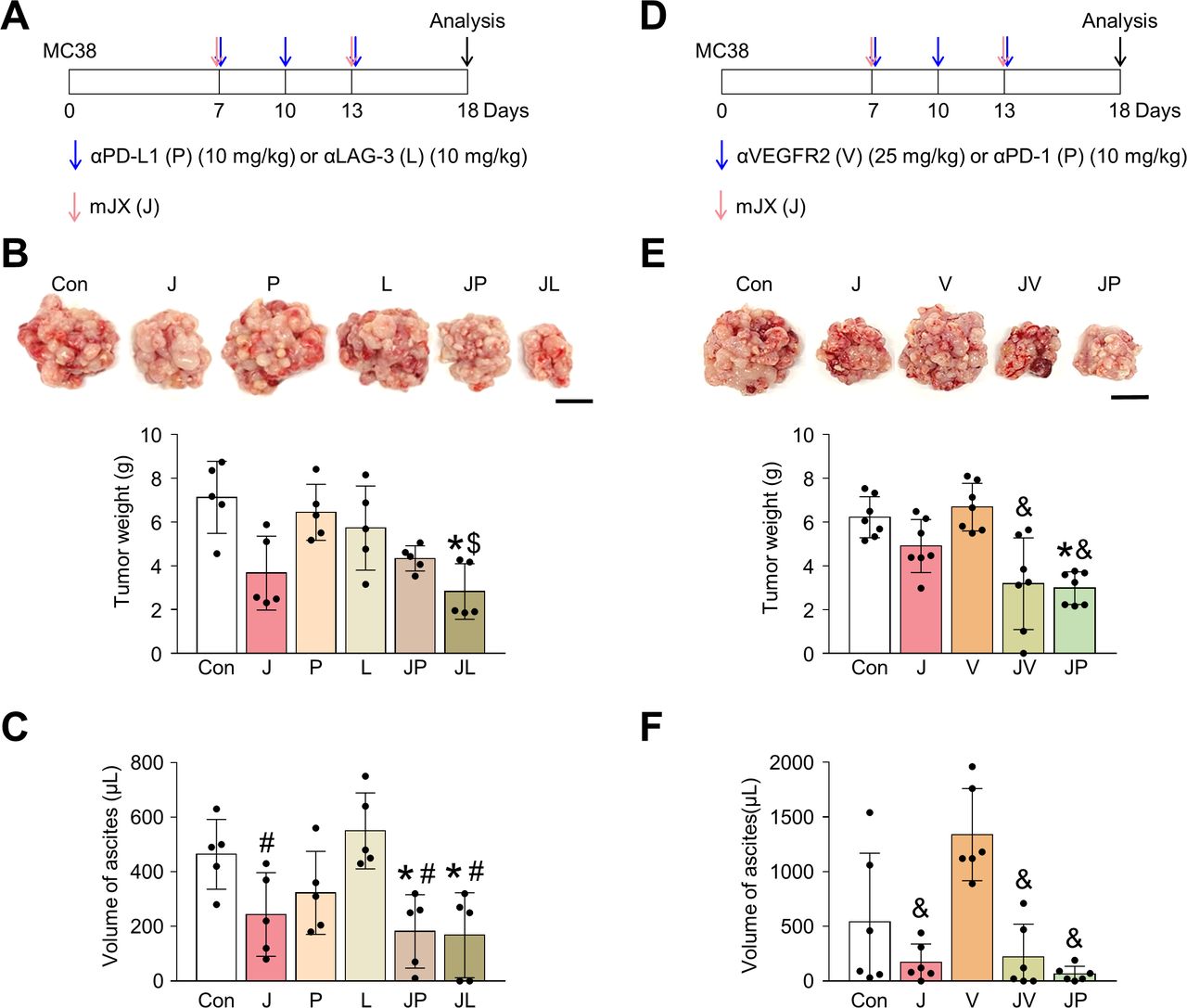
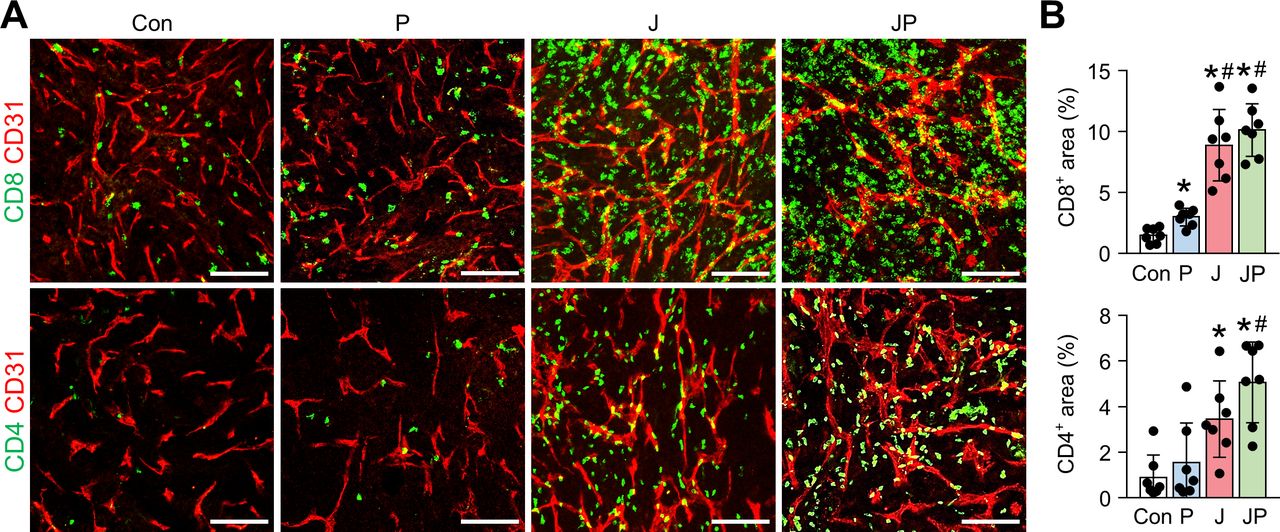
As shown earlier, we confirmed the robust activation of peritoneal and intratumoral immunity against peritoneal metastases of colon cancer after JX treatment. However, since its therapeutic efficacy was modest as a monotherapy and could induce PD-1 upregulation in CD8+ T cells, we combined anti-PD-1 antibody with JX therapy to overcome the limitations. Peritoneal tumor-bearing mice were treated with either JX and/or anti-PD-1 on indicated days (figure 5A). As a result, anti-PD-1 monotherapy delayed tumor growth by 15.9%, JX monotherapy reduced tumor growth by 63.2%, and the combination therapy inhibited tumor growth by 86.3% (figure 5B,D). Mice treated with a combination immunotherapy of JX and anti-PD-1 showed a better overall survival compared with those in other groups (figure 5C). Notably, two mice in combination group experienced complete regression of peritoneal tumors and remained tumor free. Moreover, the combination therapy attenuated hemorrhage of the peritoneal tumor vessels (figure 5D,E). Besides, when the volume of malignant hemorrhagic ascites was compared, combination therapy decreased the volume of ascites by 97%, and four mice remained ascites free after the treatment (figure 5). When the TME of peritoneal tumors was analyzed, the combination therapy increased the number of tumor-infiltrating CD8+ T cells by 6.77-fold, whereas anti-PD-1 and JX monotherapy increased infiltrating CD8+ T cells by 2-fold and 5.94-fold, respectively, when compared with the controls. Moreover, the combination therapy also increased the number of tumor-infiltrating CD4+ T cells by 5.76-fold, whereas anti-PD-1 and JX monotherapy increased infiltrating CD4+ T cells by 1.71-fold and 3.37-fold, respectively when compared with the controls (figure 6A,B).
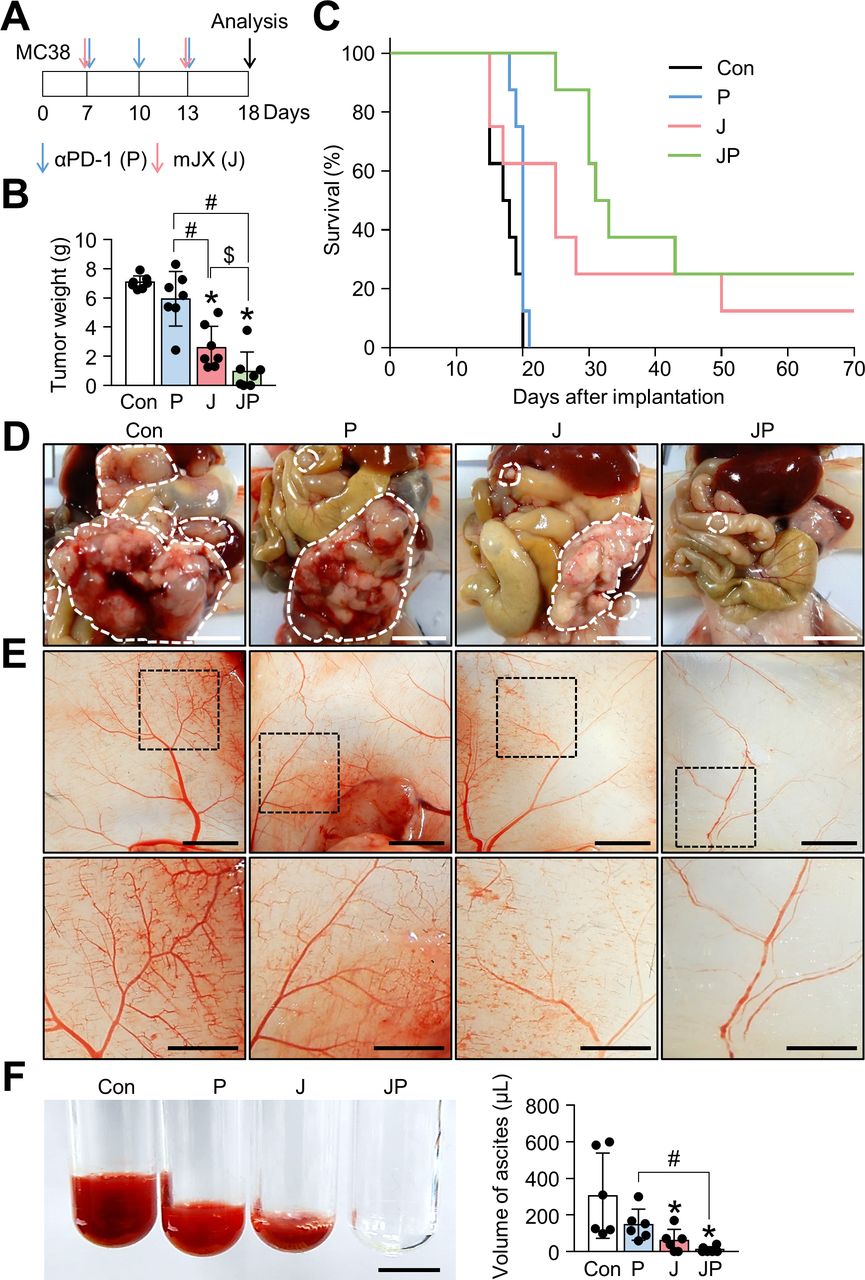
JX cooperates with PD-1 blockade to suppress PC and malignant ascites formation in colon cancer. Mice were intraperitoneally implanted with MC38 tumor cells and treated with JX and/or anti-PD-1 on indicated days (arrows). Black arrow indicates mice sacrifice. (A) Diagram of the treatment schedule. (B) Comparisons of tumor weight in mice. (C) Kaplan-Meier survival curves for overall survival. (D) Representative images and comparisons of peritoneal tumors. (E) Representative images and comparisons of the parietal peritoneum and its blood vessels. (F) Representative images and comparisons of malignant ascites. Pooled data from two experiments with n=6 to 8 per group. Values are mean±SD *p<0.05 versus control; #p<0.05 versus anti-PD-1; $p<0.05 versus JX. Kruskal-Wallis test was used (B and F). Scale bar, 5 mm (D and E) and 2.5 mm (F). PC, peritoneal carcinomatosis; PD-1, programmed cell death protein 1.
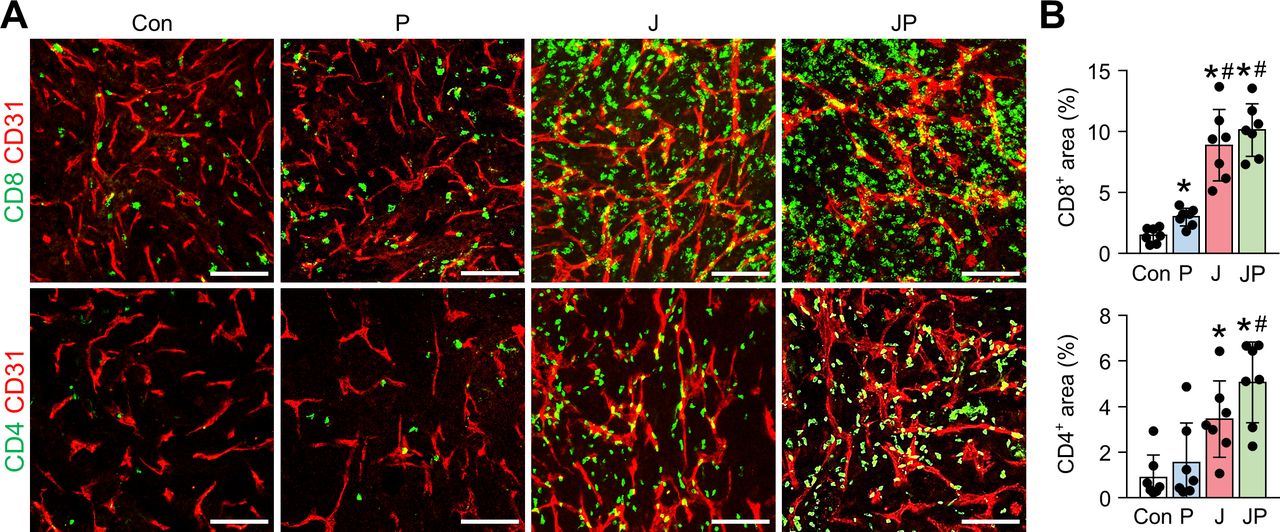
The combination immunotherapy of OV with anti-PD-1 augments the intratumoral infiltration of CD8+ and CD4+ T cells. MC38 tumor-bearing mice were intraperitoneally treated with JX and/or anti-PD-1. Representative images (A) and comparisons (B) of CD8+ T cells and CD4+ T cells in tumor. Pooled data from two experiments with n=7 per group. Values are mean±SD. *p<0.05 versus control; #p<0.05 versus anti-PD-1. One-way ANOVA and Kruskal-Wallis test were used. Scale bar, 100 µm. ANOVA, analysis of variance; OV, oncolytic virotherapy; PD-1, programmed cell death protein 1.
To validate the potential of combining JX and anti-PD-1 to treat PC, we examined ID8 ovarian cancer model. Mice were intraperitoneally injected with syngeneic ID8 ovarian cancer cells and treated with JX and/or anti-PD-1-antibody on indicated days (online supplemental figure S3A). When mice were sacrificed 28 days after tumor implantation, those treated with PBS showed diffuse peritoneal metastases and malignant ascites, while those treated with the combination of JX and anti-PD-1 showed suppressed peritoneal tumor dissemination by 98% and, more importantly, eradication of malignant ascites (online supplemental figure S3B–D). Overall survival was prolonged in both the JX monotherapy and JX + anti-PD-1 combination therapy groups compared with that in the control and anti-PD-1 monotherapy groups (online supplemental figure S3E). Thus, the combined immunotherapy of JX and anti-PD-1 has therapeutic potential in both ovarian cancer and colon cancer PC.
Collectively, the combination of JX with anti-PD-1 further enhanced the adaptive immune response and, therefore, resulted in the better control of peritoneal metastases and malignant ascites in advanced colon cancer.
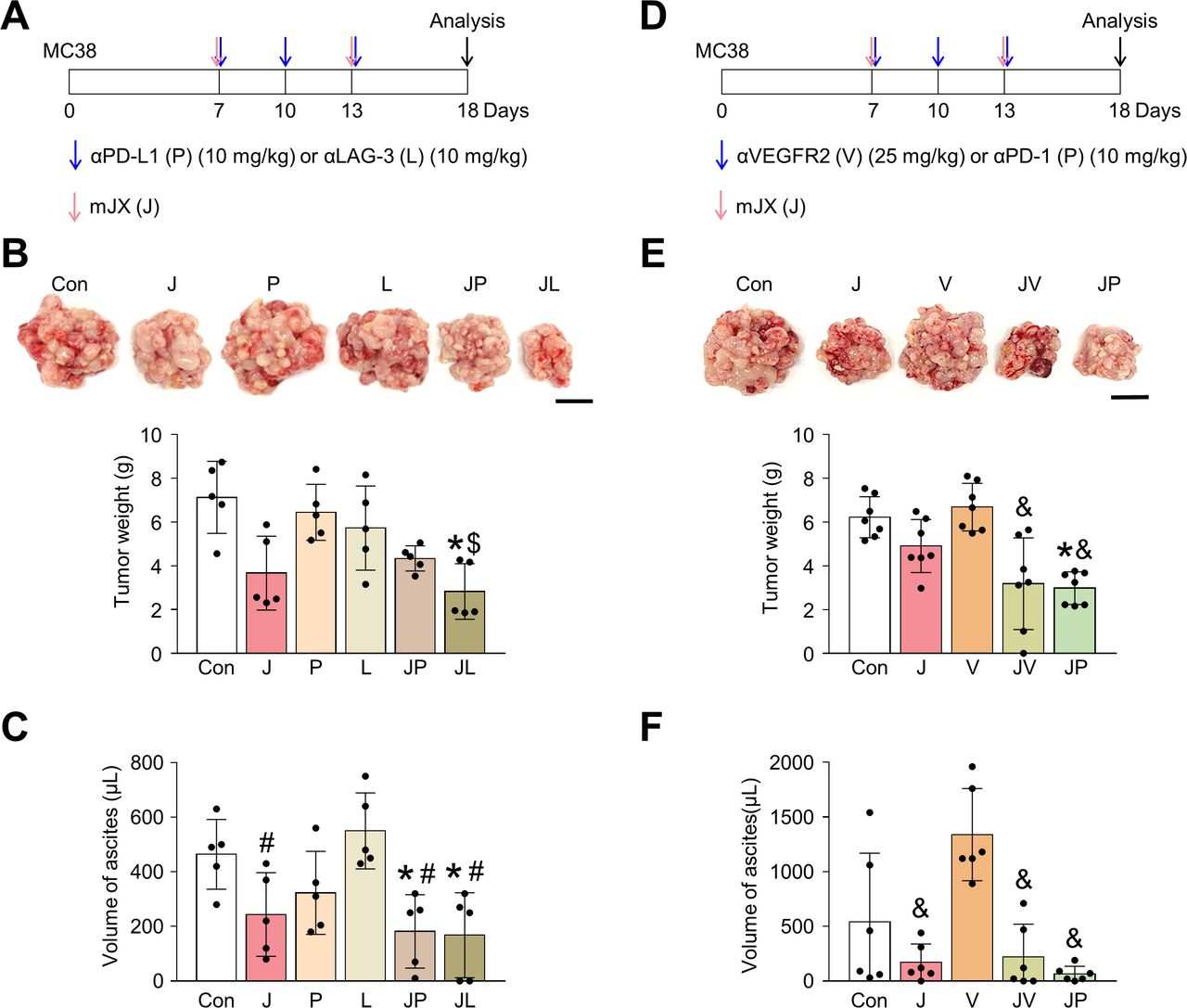
JX collaborates with other ICIs or antiangiogenic therapy to suppress peritoneal metastases of colon cancer
Because JX upregulated PD-L1 or LAG-3 within TME, these immune checkpoint molecules could limit the immunotherapeutic efficacy of JX monotherapy. Therefore, to further strengthen JX-based immunotherapy, we treated peritoneal MC38 tumor-bearing mice with anti-PD-L1 or anti-LAG-3 (figure 7A–C). While monotherapy with anti-PD-L1 or anti-LAG-3 showed only marginal efficacies, the combined therapy of JX to anti-PD-L1 or anti-LAG-3 revealed stronger anti-tumor efficacies and further suppressed the formation of malignant ascites within the peritoneal cavity compared with monotherapies. Therefore, the concurrent blockade of these immune checkpoints is a valid strategy to overcome the limitations of JX monotherapy.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The combination treatment of JX with other ICIs or anti-VEGFR2 antibody elicits an improved antitumor effect. Mice were intraperitoneally implanted with MC38 tumor cells and treated with JX and/or ICI (anti-PD-L1, anti-LAG-3, or anti-PD-1) or anti-VEGFR2 antibody on indicated days (arrows). (A) Diagram of the treatment schedule. Red arrows indicate JX treatment. Blue arrows indicate anti-PD-L1 or anti-LAG-3 treatment. (B) Representative images and comparisons of tumor burden. (C) Representative images and comparisons of malignant ascites. (D) Diagram of the treatment schedule. Red arrows indicate JX treatment. Blue arrows indicate anti-VEGFR2 or anti-PD-1 treatment. (E) Representative images and comparisons of tumor burden. (F) Representative images and comparisons of malignant ascites. Pooled data from two experiments with n=5 to 7 per group. Values are mean±SD. *p<0.05 versus control; #p<0.05 versus anti-LAG-3; $p<0.05 versus anti-PD-L1; &p<0.05 versus anti-VEGFR2. One-way ANOVA and Kruskal-Wallis test were used (B, C, E and F). Scale bar, 10 mm (B and E). ANOVA, analysis of variance; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1.
Next, because the antiangiogenic therapy is a critical part of the current targeted therapy against advanced cancers and JX also has antiangiogenic effects, we examined the possible cooperation between these two agents for treating PC of colon cancer.43 44 We treated MC38 peritoneal tumor-bearing mice with anti-VEGFR2 (DC101) or anti-PD-1 (figure 7D–F). While the anti-VEGFR2 monotherapy did not show any significant efficacy in suppressing peritoneal tumor growth and malignant ascites formation, it induced stronger antitumor effects when combined with intraperitoneal JX treatment. Some mice treated with the combination of JX and anti-VEGFR2 showed complete resolution of malignant ascites within the peritoneal cavity. Moreover, the efficacy of this combination was almost comparable to that of JX and anti-PD-1 combination therapy. Thus, the combination therapy of JX and antiangiogenic agents is also effective when treating PC of colon cancer.
Discussion
PC is a common and devastating manifestation of colon cancer which is usually accompanied by the accumulation of malignant ascites within the peritoneal cavity.1 2 6 In PC, a significant amount of tumor cells are free-floating within the malignant ascites or seeded widespread along the surface of the peritoneum. Since these tumor cells are poorly vascularized or not vascularized at all, the systemic delivery of conventional chemotherapeutics to peritoneal tumor cells is severely hampered, making PC as a formidable challenge when treating patients with colon cancer.6 14 Therefore, intraperitoneal therapy could be an attractive route of intervention, which would guarantee the direct exposure of a higher concentration of the drug for a longer duration.6
Here, we took advantage of intraperitoneal injection to maximize the efficacy of JX against peritoneal tumor cells. JX selectively infected and lysed peritoneal tumor cells, because it lacks thymidine kinase. It also successfully activated peritoneal DCs because it was engineered to express GM-CSF. Moreover, it reduced immunosuppressive myeloid-derived cells, restored the effector functions of CD8+ T cells within the peritoneal cavity and facilitated their infiltration into peritoneal tumor nodules. This JX-induced anticancer immunity effectively suppressed peritoneal tumors.
The accumulation of malignant ascites is mediated by VEGF, a pivotal proangiogenic factor, which is known to be mainly secreted from tumor cells.12 45 46 Excessive VEGF in the peritoneal cavity promotes robust tumor angiogenesis both on the peritoneal surface and within the peritoneal tumor nodules, generating a myriad of hyperpermeable tumor neovessels.4 12 In the present study, intraperitoneal JX treatment effectively suppressed the malignant ascites formation within the peritoneal cavity through multiple mechanisms. First, in our results, it destroyed peritoneal tumor cells and downregulated Vegfa within the tumor. Next, it promoted T cells to secrete IFN-γ, which is a potent antiangiogenic factor that impairs the proliferation and survival of endothelial cells.39 47 Moreover, IFN-γ could reduce the ascites formation because it is known to recruit pericytes to tumor endothelial cells, enhancing the integrity of tumor vasculatures.47 48
Though ICIs made a massive breakthrough in the treatment of advanced cancers, their therapeutic efficacy is limited in the treatment of PC, especially in patients with colon cancer.19 Consistently, in our study, ICI monotherapy revealed minimal efficacy in peritoneal tumors and malignant ascites, indicating our model is indeed an immunotherapy-resistant model. However, intraperitoneal JX treatment induced the intense infiltration of CD8+ T cells into the peritoneal tumors reactivated their effector functions, thus reprogramming immunotherapy-resistant peritoneal tumors into T cell-inflamed tumors that can respond well to ICI therapy. Accordingly, the combination therapy of JX and ICI (anti-PD-1, anti-PD-L1, or anti-LAG-3) showed dramatic control of peritoneal metastases and malignant ascites, even leading to complete regression in some mice.
Intriguingly, the JX-based combination immunotherapy remarkably repressed malformed vasculatures on the surface of the peritoneum. Recently, the mutual regulation of tumor vasculatures and anticancer immunity is increasingly recognized as an essential determinant of tumor progression.20 48–50 Tumor angiogenesis disturbs anticancer immunity, whereas anticancer immunity suppresses tumor angiogenesis.48 Therefore, a robust anticancer immunity elicited by combination immunotherapy would contribute to the suppressed tumor angiogenesis in the peritoneum.
Conclusions
In conclusion, our study demonstrated that intraperitoneal immunotherapy with JX activates peritoneal anticancer immunity and potentiate immune checkpoint blockade to suppress PC and malignant ascites in colon cancer.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
YSL, WSL and CWK contributed equally.
Contributors YSL designed and performed the experiments, analyzed the data, and drafted the manuscript. WSL performed and guided the experiments, interpreted the data, and drafted the manuscript. SJL, HY, and SJK performed the experiments and provided technical support. JN, K-MY, BK, WRK, and CWK discussed the project. HJC and CK conceived and designed the study, oversaw research activities, guided the experiments, analyzed the data, and drafted the manuscript. All authors read and approved the final manuscript.
Funding This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2020R1A2C2004530 to CK, NRF-2020R1C1C1010722 to HJC, and NRF-2020R1C1C1008212 to CWK).
Competing interests K-MY is an employee of SillaJen, Inc and a member of a Board. JN is an employee of SillaJen Biotherapeutics, Inc. CK and HJC are named inventors on a patent application 'Oncolytic vaccinia virus and checkpoint inhibitor combination therapy'. The other authors declare that they have no competing interests.
Patient consent for publication Not required.
Ethics approval Ethics approval and consent to participate: all animal experiments in our study were approved by the Institutional Animal Care and Use Committee of CHA University (IACUC, #180081) and were conducted in accordance with the approved protocols.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.