Article Text

Abstract
Immunotherapeutic strategies targeting B-cell acute lymphoblastic leukemia (B-ALL) effectively induce remission; however, disease recurrence remains a challenge. Due to the potential for antigen loss, antigen diminution, lineage switch or development of a secondary or treatment-related malignancy, the phenotype and manifestation of subsequent leukemia may be elusive. We report on two patients with multiply relapsed/refractory B-ALL who, following chimeric antigen receptor T-cell therapy, developed myeloid malignancies. In the first case, a myeloid sarcoma developed in a patient with a history of myelodysplastic syndrome. In the second case, two distinct events occurred. The first event represented a donor-derived myelodysplastic syndrome with monosomy 7 in a patient with a prior hematopoietic stem cell transplantation. This patient went on to present with lineage switch of her original B-ALL to ambiguous lineage T/myeloid acute leukemia. With the rapidly evolving field of novel immunotherapeutic strategies, evaluation of relapse and/or subsequent neoplasms is becoming increasingly more complex. By virtue of these uniquely complex cases, we provide a framework for the evaluation of relapse or evolution of a subsequent malignancy following antigen-targeted immunotherapy.
- receptors
- chimeric antigen
- immunotherapy
- adoptive
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Single antigen-targeted immunotherapies, including antibody-drug conjugates, bispecific T-cell engagers, and chimeric antigen receptor (CAR) T cells, have been highly successful in treating B-cell acute lymphoblastic leukemia (B-ALL). Despite their efficacy, disease relapse is not uncommon. Approximately 30%–60% of patients relapse after receiving anti-CD19 CAR T cells: the majority with CD19 negative disease, and less commonly with lineage switch to a myeloid leukemia.1–4 Furthermore, development of myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) as a de novo treatment-related malignancy is a well-established mechanism for emergence of myeloid malignancies.5 6 The development of subsequent neoplasms following novel immunotherapies is not well described but remains of concern, particularly in patients who have received extensive prior therapy or those receiving CAR T cells, given a theoretical risk of insertional mutagenesis with utilization of retroviral vectors.7
We highlight the unique presentations of two patients with multiply relapsed/refractory ALL who were effectively treated with CAR T cells and had subsequent development of myeloid malignancies. By virtue of these complex cases, we describe our diagnostic approach and provide insights into optimal evaluation of patients who develop myeloid malignancies post-CAR T-cell therapy.
Case 1
Secondary myeloid sarcoma in an 18-year-old with concurrent B-ALL relapse (figure 1A–D).
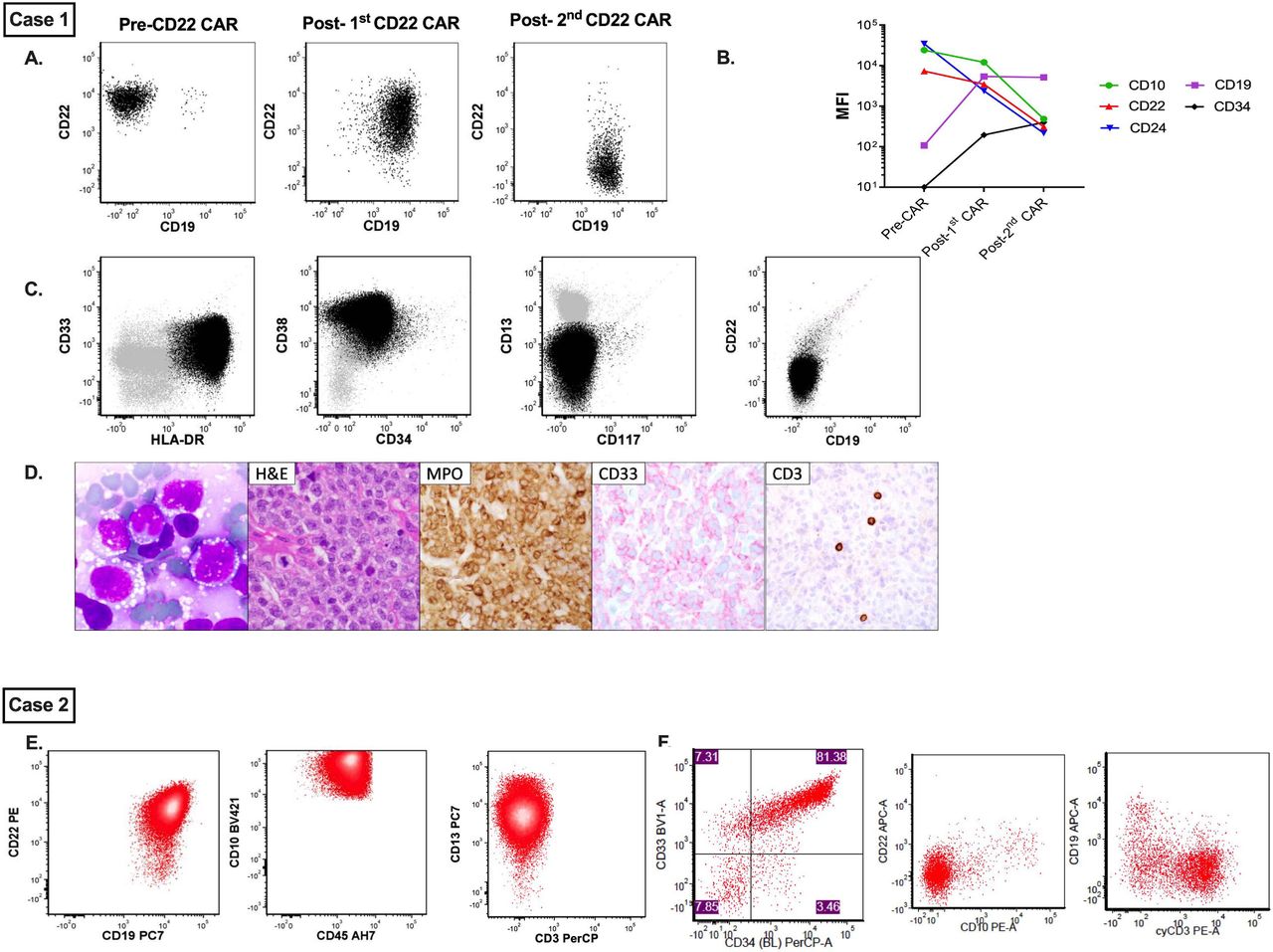
{kind=link}
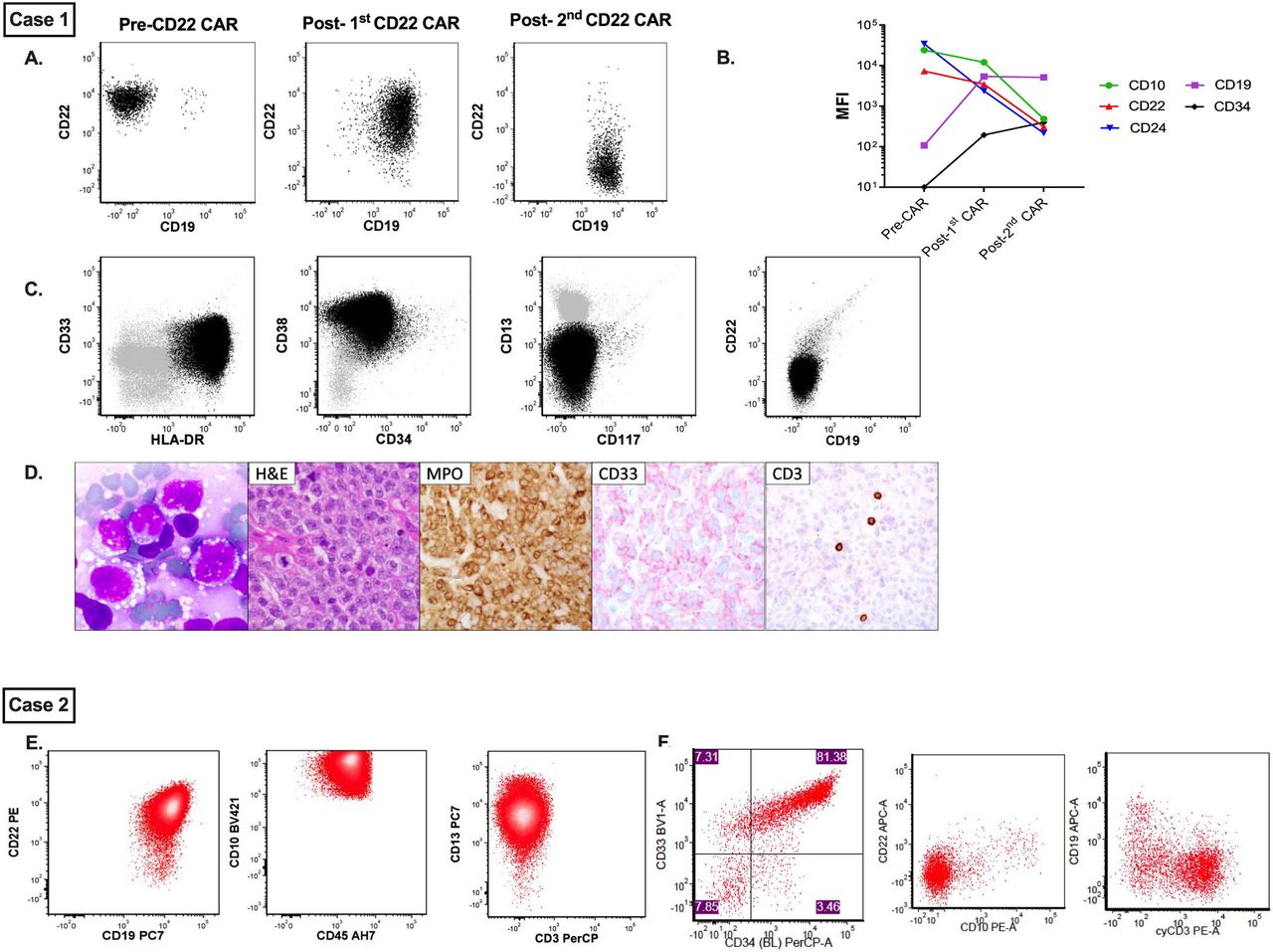
Immunophenotypic evolution of disease in cases 1 and 2. Case 1. (A) Represents sequential flow cytometric evolution of CD19 and CD22 expression following CD22 CAR T-cell immunotherapy. (B) Concurrent changes in MFI of several B cell antigens, including CD10, CD19, CD22, CD24 and CD34. (C) Demonstrates immunophenotypic evaluation of concurrent myeloid sarcoma alongside morphological appearance of myeloid blasts from the aspirate. (D) H&E, MPO and CD33 immunohistochemistry from the myeloid lesion. Case 2. (E) Select flow plots from B-ALL sample prior to treatment with CD22 CAR T cells, demonstrating both CD19 and CD22 positivity. (F) Select flow plots from T/myeloid ALL at relapse demonstrating loss/diminution of CD19 and CD22 with new expression of CD3 and CD33. B-ALL, B-cell acute lymphoblastic leukemia; CAR, chimeric antigen receptor; MFI, mean fluorescence intensity; MPO, myeloperoxidase.
An 18-year-old man with Down syndrome (DS) presented with multiply relapsed CD22+/CD19 negative B-ALL with non-central nervous system (CNS) extramedullary disease (EMD) following multiple cycles of chemotherapy, hematopoietic stem cell transplantation (HSCT), and blinatumomab. His medical history was notable for a diagnosis of MDS, at age 11 (years), and CD19+ B-ALL (online supplemental appendix). He was referred for a phase I study of CD22 CAR T cells (NCT02315612). His CD22 CAR T-cell course was complicated by grade 2 cytokine release syndrome (CRS), following which he achieved minimal residual disease negative complete remission (MRD-CR) with clearance of EMD.
Supplemental material
At 1 year post-CAR, cerebrospinal fluid (CSF) analysis revealed CD19+/CD22+ blasts, a phenotypic change from his prior CD19 negative expression. Although bone marrow was negative for residual leukemia, fluorodeoxyglucose-positron emission tomography (FDG-PET) scan and MRI demonstrated spinal canal involvement. He received a re-infusion of CD22 CAR T cells and achieved MRD-CR in the CNS at 1 month and full eradication of PET avidity 3 months post-CAR.
Eight months post-CAR re-infusion, he developed an extraorbital soft tissue mass in the inferior aspect of his left eye. Biopsy revealed a recipient-derived myeloid sarcoma with no immunophenotypic evidence of B-ALL. RNA sequencing analysis revealed a potentially novel PEX1-CDK6 fusion confirmed by PCR and Sanger sequencing (online supplemental appendix).8 Efforts at analyzing the original biopsy from his MDS for cytogenetic analysis were unsuccessful and could not be used for comparison.
Full disease restaging revealed spinal leptomeningeal disease with evidence for CD19+/CD22−negative ALL on CSF sampling, thus demonstrating both ALL (now CD22−negative) and concurrent AML. He had ongoing B-cell aplasia with no evidence for bone marrow disease. Residual CD22 CAR T cells were detected in both blood and bone marrow, and replication competent lentivirus (RCL) testing was negative. Digital drop PCR revealed essentially no CAR T-cell DNA in the AML sample (two copies of CAR T cells/10,780 cells), making CAR integration-associated leukemia unlikely. He died 4 months later from infectious complications.
Case 2
T/myeloid lineage switch in a 19-year-old with B-ALL and monosomy 7 donor-derived MDS (figure 1E,F).
A 19-year-old woman with post-HSCT relapsed CD19+/CD22+ B-ALL with t(12; 21) ETV6-RUNX1 gene rearrangement, who was initially diagnosed at age 14, was referred for CD22 CAR T cells (online supplemental appendix). She had grade 1 CRS and achieved MRD-CR by day 28. She had persistent cytopenias, and a bone marrow aspirate and biopsy at day 50 post-CAR showed <10% marrow cellularity with trilineage hypoplasia and ongoing remission.
At approximately 6 months post-CAR, she presented with abdominal pain and was found to have a pancreatic head mass consistent with B-ALL (biopsy-confirmed). Marrow aspirate showed disease recurrence with 0.2% CD19+/CD22+ ALL. FDG-PET scan demonstrated uptake in the spleen, mesentery, and medial left breast. Residual CAR T cells were detected in the blood and bone marrow, and all RCL testing was negative. Bone marrow had improved cellularity (average 30%) with no evidence of marrow dysplasia.
Following palliative radiation therapy for symptom management, she was referred for CD19 CAR T cells (NCT02028455). Pre-CAR evaluations revealed recipient-derived B-ALL with persistent ETV6-RUNX1 rearrangement, marrow dysplasia and a new donor-derived XY clone positive for monosomy 7, consistent with concurrent MDS. She attained MRD-CR of her ALL following CD19 CAR T cells and was referred for a second HSCT, both for ALL remission consolidation and for definitive treatment of her persistent monosomy 7, which had additionally acquired a trisomy 8 clone.
She underwent a haploidentical HSCT with pre-emptive post-HSCT blinatumomab to prevent ALL relapse (NCT02790515). At 1 year post-HSCT, bone marrow revealed 40% blasts. Flow cytometry revealed a single homogeneous population positive for T cell and myeloid makers, and cytogenetic testing demonstrated ETV6-RUNX1 gene rearrangement. PET-CT showed EMD in the bilateral axilla/breasts. Biopsy results revealed an identical phenotype. Ultimately, this was consistent with a recipient-derived acute leukemia of ambiguous lineage, representing a switch from B-ALL to a T/myeloid CD19 negative immunophenotype. She died from complications of refractory disease.
Discussion
The etiology of myeloid malignancies following B cell-directed immunotherapy is multifactorial, and mechanisms by which these occur are not fully understood. Here, we report on two cases that illustrate the complexity inherent in describing and identifying the origin of a new myeloid malignancy following B-ALL antigen-directed CAR T-cell therapy. In context of these cases, we have developed a diagnostic framework for evaluation of such malignancies (table 1).
Diagnostic Approach to Evaluation of Leukemia Detection Following B-cell Targeted Immunotherapy
An important first step in post-immunotherapy disease evaluation is to maintain a broad differential diagnosis and specifically include the possibility of finding a myeloid malignancy. The work-up should aim to investigate all possible mechanisms of recurrence. Because of potential for antigen modulation, immunophenotypic evaluation cannot solely rely on the initial leukemia-associated immunophenotype. Given the concern for antigen loss, knowledge of prior immunotherapies received is essential, as is incorporating flow cytometric methods that expand on traditional gating strategies and include antigens such as CD22, CD24 and intracellular CD79a to more clearly identify occult disease.9 A ‘different from normal’ analysis should be applied to identify all abnormal immunophenotypes. Importantly, the evaluation should also incorporate myeloid markers (eg, CD13, CD33, CD117, CD34) in order to detect lineage-switched or newly developed myeloid neoplasms. Furthermore, antigen modulation may not represent a permanent state. As illustrated by case 1 and in our collective experience,10 CD19 negativity following blinatumomab may potentially be transient, and monitoring for antigen evolution is important in surveillance for disease recurrence.
Beyond phenotypic changes, genomic monitoring of the recurrent malignancy will help inform whether disease is clonally related to the prior disease (eg, lineage switch) or if there is a new neoplasm. Detection of unique cytogenetic abnormalities may provide insight into the possibility of treatment-related events (eg, monosomy 7), as seen in case 2. Chimerism studies in post-HSCT settings will also provide insight into the disease origin. Accordingly, we report on two potentially novel findings. To our knowledge, case 1 is the first report of PEX1-CDK6 fusion implicated in AML, highlighting the importance of a comprehensive genomic evaluation to identify potentially targetable lesions, particularly in patients with limited options. Our second case demonstrates a lineage switch (T/myeloid) in a patient with multiply relapsed CD19+ ALL with ETV6-RUNX1 fusion. ETV6-RUNX1 has not historically been associated with lineage switch, and we believe that this is the first case seen in the context of CAR T cells. Recent literature focused on the genomics of mixed phenotypic acute leukemia report on ETV6 and RUNX1 mutations, particularly in those with T/myeloid phenotypes, suggesting that ETV6-RUNX1 could potentially predispose to phenotypic switching.11
Our cases also suggest that it is important to consider the development of a myeloid malignancy in patients with genetic predisposition to lineage switch (eg, KMT2Ar)12 13 or in those with a history of a myeloid malignancy. Although lineage switch specific to DS following immunotherapy has not been well described, based on the history of MDS in case 1, an AML evaluation was warranted. Further monitoring of patients with DS with B-ALL who receive B cell-targeted therapies is needed to determine if this population is at higher risk of lineage switch. Additional evaluations for cancer predisposition syndromes should also be undertaken in those with a family history.
Cytopenias are increasingly recognized as an effect of CAR T-cell therapy,14 15 the etiology of which is multifactorial and may be due in part to an ongoing inflammatory milieu, confounded by the impact of prior therapy, among other potential factors. However, for those who are heavily pretreated and have ongoing cytopenias, diagnostic evaluation for MDS should be considered, despite the expectedness of CAR T cell-mediated effects. This is well illustrated in case 2, whose initial cytopenias were attributed to ongoing CAR T-cell persistence but ongoing findings led to the identification of an MDS.
Another important consideration, in particular given the relative infancy of the field, is the unknown long-term impact of CAR T cells on risk of secondary neoplasms. Due to the long latency for development of subsequent neoplasms, this may be particularly hard to monitor for; however, ongoing surveillance is warranted and required by most regulatory agencies governing gene therapy. In case 1, our patient had residual CAR T cells at the time of diagnosis with myeloid sarcoma, raising the potential for CAR T-cell-mediated leukemogenesis. Although integration site analysis was not necessary for case 1 given the low presence of CAR T cells, it nonetheless remains an important component of the diagnostic evaluation, raising concern for potential clonal expansion of CAR T cells if present at higher frequency.7
In both cases, it is important to note that the patients were very heavily pretreated, which in and of itself increases the risk for secondary malignancies. Although the concern for CAR T cell-mediated neoplasm remains of concern, it is conceivable that as CAR T cells are used earlier in a patient’s course (before they have received extensive therapy) that this may spare patients additional chemo and/or radiation therapy and potentially diminish the risk of secondary neoplasms. Ongoing monitoring will be imperative in elucidating the risk of secondary neoplasms as the treatment paradigm of CAR T-cell therapy shifts.
Lastly, these cases highlight the essential role for biopsy of EMD. EMD in B-ALL is most frequently noted in the CNS or in the testes but is likely underappreciated in other sites.16 Relapse with EMD frequently occurs in those who have undergone prior HSCT,17–19 and further study to evaluate the incidence of EMD relapse following immunotherapy is warranted.3 20 Based on our cases and the potential for leukemic evolution following sequential immunotherapies, we strongly recommend imaging for EMD evaluation and consideration for biopsy of a new EMD site following immunotherapy, particularly for patients with a history of HSCT who may be predisposed to EMD relapse.
In conclusion, we highlight two complex cases of relapse following B cell lineage-directed immunotherapies, through which we demonstrate antigen modulation, evolution of myeloid sarcoma, donor-derived treatment-related MDS and lineage switch. While the pathogenesis of myeloid malignancies in the context of B cell-targeted immunotherapies is not yet fully understood, given the rapidly evolving field of immunotherapy and increased utilization of CAR T cells, such cases may become more frequent. We provide our framework as a practical guide and systematic approach to the evaluation of a new myeloid malignancy following immunotherapy in B-ALL.
Acknowledgments
We gratefully acknowledge the study participants and their families, referring medical care teams, the faculty and staff of the NIH Clinical Center who provide their expertise in the management of the study participants, and the data managers involved with this work. We would also like to acknowledge Dr Jayashree Motwani for her assistance in providing relevant details regarding medical history and Dr John Choi for his assistance with generating flow cytometric plots for case 2. This work was supported in part by the Intramural Research Program, National Cancer Institute and NIH Clinical Center, National Institutes of Health. This research was made possible through the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, Genentech, the American Association for Dental Research, the Colgate-Palmolive Company, and other private donors. Both patients and/or families provided consent for publication of this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors GM and NNS wrote the first version of the manuscript. SAS, ACT, BY, HS, MGD, JFS, SC, KP, TJF, BMT, RG, and NNS, all contributed to patient care and analyzed patient data and outcomes. XW performed CAR T cell detection studies by PCR. H-WW and KRC provided expertise in analysis of disease immunophenotype and disease detections. YS and JK performed genomic analysis and interpretation of the data. H-WW, BMT, XW, JK, KRC, and RG, all contributed to writing. All authors reviewed and approved the final submission.
Funding This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research and the Warren Grant Magnuson Clinical Center.
Competing interests BMT received travel support from Miltenyi Biotec to the EBMT annual meeting in 2018 to present published data. Additionally, MGD receives funding from Pfizer for an investigator-initiated clinical trial and has no other disclosures.
Patient consent for publication Parental/guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.