Article Text

Abstract
Background T follicular helper cells (Tfh) are essential to shape B cell response during germinal center formation. Tfh accumulation has been reported in various human cancers, with positive or negative prognostic roles. However, the mechanisms explaining the accumulation of Tfh and their role in cancer remain obscure.
Methods In vitro differentiated and mouse cell sorted Tfh phenotype was evaluated by flow cytometry and quantitative PCR (qPCR). Antitumor effect of Tfh was evaluated by adoptive transfer in different tumor-bearing mice models. The involvement of immune cells, cytokines and chemokines was evaluated, using depleting antibodies. Chemokines and cytokines expression and production were evaluated by qPCR and ELISA. In human, the impact of immune cells and chemokines on survival was evaluated by analyzing transcriptomic data from public databases and from our own patient cohorts.
Results In this study, we show that Tfh exert an antitumor immune effect in a CD8+-dependent manner. Tfh produce interleukin-21, which sustains proliferation, viability, cytokine production and cytotoxic functions of exhausted T cells. The presence of Tfh is required for efficacy of antiprogrammed cell death ligand-1 therapy. Tfh accumulate in the tumor bed and draining lymph nodes in different mouse cancer models. This recruitment is due to the capacity of transforming growth factor β to drive Chemokine (C-X-C motif) Ligand 13 expression, a chemoattractant of Tfh, by intratumor CD8+ T cells. Accumulation of Tfh and exhausted CD8+ T cells predicts cancer outcome in various cancer types. In patients treated with anti-programmed cell death-1 mAb, accumulation of Tfh and CD8+ at the tumor site is associated with outcome.
Conclusion This study provides evidence that CD8+/Tfh crosstalk is important in shaping antitumor immune response generated by immunotherapy.
- CD8-positive T-lymphocytes
- immunotherapy
- programmed cell death 1 receptor
Data availability statement
Data are available on reasonable request. Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
CD4 T cell support is essential for B cell activation, somatic hypermutation, isotype switching and production.1 2 After CD40 engagement, B cells proliferate and reach the germinal center where they undergo somatic hypermutation, enabling antibody diversification and affinity maturation. T cells can also migrate into the germinal center to prolong their interaction with B cells and participate in the generation of plasma cells and memory B cells.3 A specific CD4 T cell subset called T follicular helper cells (Tfh) has been described and is dedicated to this role.1 2 Tfh cells are characterized by expression of the C-X-C motif chemokine Receptor 5 (CXCR5) membrane marker, the receptor of Chemokine (C-X-C motif) ligand 13 (CXCL13), required for their migration to the germinal center, as well as by the expression of B-cell lymphoma 6 (BCL6), Achaete-Scute family bHLH transcription factor 2 (ASCL2), interleukin (IL)-21, programmed cell death-1 (PD-1), and inducible T cell costimulator (ICOS).4 These molecules have been shown to be essential for the regulation of Tfh development, migration, and function. Importantly, strong expression of PD-1 in combination with CXCR5 is a reliable way to identify Tfh cells,5 and is a new independent subset of Th cells that is different from Th1, Th2, Treg and Th17.
The accumulation of Tfh cells has been described in different cancer types in humans, such as colorectal, non-small-cell lung cancer (NSCLC), and breast cancer, and has been associated with better prognosis. In breast and lung cancer, high Tfh cell levels are associated with an increase in interferon γ (IFNγ)-producing Th1 cells, CD8+ T cells and B cells within neoplastic lesions.6–8 However, a contrasting report in melanoma proposed that intratumor Tfh-like cells exert an immunosuppressive function and inhibit the functions of CD8+ and Th1 cells.9 In all cancer types, the presence of Tfh is associated with the presence of tertiary lymphoid structures (TLS), which are commonly associated with better outcome.10 11 Although Tfh accumulation in human solid tumors appears to be associated with good prognosis in most tumor types, the mechanism that dictates their prognostic role is unexplored.
PD-1, and its main ligand (PD-L1), is an important immune checkpoint that maintains tolerance to self-antigens and fights against excessive immune response due to antigen exposure persistance.12 This mechanism is used by cancer cells to promote immune escape.13 Accordingly, anti-PD-1 and PD-L1 antibodies have shown substantial clinical response14 and have been shown to be of major interest, either when used alone or in combination with chemotherapy in many cancer types. However, the role of Tfh in the efficacy of checkpoint inhibitors remains unknown.
To address this question, we investigated the effects of accumulation of Tfh cells on CD8+ T cell immune response and tumor growth in different murine tumor models. We observed that Tfh cells accumulate in the tumor bed. This accumulation was dictated by the ability of intratumor CD8+ T cells to produce CXCL13 in a transforming growth factor β (TGF-β)-dependent manner. The Tfh antitumor effect depended on the expression of IL-21, which sustained effector function of exhausted T cells. Tfh cells were also required for the efficacy of anti-PD-1 therapy. Importantly, we validated these murine data in human patients with lung cancer.
Methods
Study design
The aim of this study was to investigate the importance of the crosstalk between Tfh and CD8+ T cells in cancer progression and immunotherapy response, namely through CXCL13 and IL-21 production. To this end, we performed tumor growth experiments in several murine models and with many blocking mAbs, immunophenotyping, in vitro culture of cancer cell lines and cell sorted immune cells. Experiments were conducted in replicate as indicated in the figure legends. We also analyzed transcriptomic data from public databases and from our own patient cohorts.
Cell culture
Cell lines were cultured at 37°C in 5% CO2 in Dulbecco’s Modified Eagle’s Medium (B16F-10, Lewis Lung Carcinoma, LLC1 and MC38) or in RPMI-1640 Medium (CT26) with 10% (vol/vol) Fetal Calf Serum supplemented with Penicillin-Streptomycin/Amphotericin B (PSA), and 4 mM of 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid.
Animals and in vivo models
OTI and OTII mice were bred at the Cryopreservation, Distribution, Typage et Archivage animal; Orléans, France. Mice with transgenic expression of a TCR on CD4 T cells that recognizes the antigen TRP-1 were provide by Lionel Apetoh and were from JAX. J.A. Harker (Imperial College London) kindly provided Bcl6fl/fllCD4Cre and CD4Cre mice. Balb/cJ and C57BL/6J mice were purchased from Charles River Laboratories (Saint Germain sur l’Arbresle, France). Female animals between 6 and 10 weeks of age were used in all experiments.
To induce tumor lung invasion, retro-orbital intravenous injection of 2.105 B16F-10 murine melanoma cells and B16OVA (OVA-transfected B16F-10 cells) was performed in C57BL/6 syngeneic mice.
For the model of spontaneous lung cancer, mice received weekly intraperitoneal injections of urethane (ten injections of 1 g per kg body weight). All mice were sacrificed after 4 months.
To induce subcutaneous tumor formation, 5.105 MC38 murine colon adenocarcinoma cells, 2.105 B16F-10 murine melanoma cells, 2.105 murine LLC1 cells (syngeneic from C57BL/6 mice), 2.105 CT26 murine colon carcinoma cells, or 2.105 4T1 murine mammary cancer cells (syngeneic from Balb/cJ mice) were injected s.c. When tumors reached 50–70 mm2 (between day 9 and 11), (intraperitoneal (i.p.) antimouse PDL-1 antibody (10F.9G2; BioXcell) injections were initiated at 200 µg per mouse three times a week. For CD8, CD19, IFNγ, CXCL13, and IL-21 depletion, antimouse CD8 (2.43; BioXcell), CD19 (1D3; BioXcell), IFNγ (XMG1.2; BioXcell), CXCL13 (143608; R&D) IL-21 (FFA21; Thermofisher) antibodies were used. BioXcell antibodies were administered i.p. 3 times a week at 200 µg per mice, IL-21 antibody was injected i.p. 3 times a week at 40 µg per mouse and CXCL13 antibody was administered i.t. 3 times a week at 1 µg per mouse.
For T cell transfer, 2.106 in vitro generated Th1 or Tfh cells or 0.5.106 in vitro generated Trp1 Th1 or Tfh cells were intravenously injected three times. All experiments were carried out in accordance with the guidelines laid down by the Ethics Committee at the University of Burgundy.
T cell purification and in vitro differentiation
Naive CD4+T cells (CD4+CD62Lhi) and naive CD8+ T cells were obtained from spleens and lymph nodes of C57BL/6 wild-type. CD4+ T cells were then purified using the MACS Cell Separation system (CD4+ CD62L+ T Cell isolation kit, Miltenyi Biotec) or coupled with flow cytometry (CD4 (L3T4) MicroBead, Miltenyi Biotec and BD ARIA III with FACSDiva Software). CD8+ T cells were then purified using the MACS Cell Separation system (Naive CD8a+ T Cell Isolation Kit, Miltenyi Biotec) or coupled with flow cytometry (CD8a (Ly-2), Miltenyi Biotec and BD ARIA III with FACSDiva Software). The purity of isolated T cell populations routinely exceeded 90%–95%.
Naive CD4+ T cells were stimulated with plate-bound antibodies against CD3 (145–2 C11, 2 µg/mL) and CD28 (PV-1, 2 µg/mL) in the presence of either no cytokine and anti-IL-4 and anti-IL-12 antibodies (Th0); 10 ng/mL of IL-12, anti-IL-4 antibody (Th1); 10 ng/mL of IL-4, anti-IFNγ antibody (Th2); 4 ng/mL of TGF-β, anti-IL-4 and anti-IFNγ antibodies (Treg); 2 ng/mL of TGF-β and 20 ng/mL of IL-6, anti-IL-4 and anti-IFNγ antibodies (Th17); and 50 ng/mL of IL-6 andIL-21, anti-IL-4, anti-IFNγ and anti-TGF-β antibodies (Tfh). Naive CD8+ T cells were stimulated with plate-bound antibodies against CD3 (145–2 C11, 2 µg/mL) and CD28 (PV-1, 2 µg/mL) in the presence of IL-21 (30 ng/mL). Mouse recombinant IL-4, IL-6, IL-12, IL-21 and TGF-β were all purchased from R&D Systems. Anti-IL-4 (clone 1B11), IFNγ (XMG1.2) (10 mg/mL) and TGF-β (1D11.16.8) (20 mg/mL) blocking antibodies were obtained from BioXcell (West Lebanon, New Hampshire, USA).
Measurement of cytokines
Secreted cytokines were measured after 72 hours of cultured by ELISA for CXCL13 (LSBio) according to the manufacturers’ instructions. For intracellular cytokine staining, cells were cultured as described above and then stimulated for 3 hours at 37°C in culture medium containing PMA (50 ng/mL; Sigma), ionomycin (1 mg/mL; Sigma), and protein transport inhibitor cocktail (500×; Thermofisher). After staining for surface markers (see Flow Cytometry section), cells were fixed and permeabilized according to the manufacturer’s instructions (BD Biosciences), then stained for intracellular products (see the Flow cytometry section).
Flow cytometry
Antibodies used for flow cytometry experiments are in the key resources table (online supplemental table S1). In order to phenotype and sort distinct immune cell populations by flow cytometry, lungs, lymph nodes, spleens and tumors were harvested, dissociated with specific organ or tissue dissociation kits (Miltenyi) and cells were stained in Flow Cytometry Staining Buffer (eBioscience) and Brilliant Stain Buffer (BD Bioscience). Intracellular staining was carried out according to the manufacturer’s protocol using the fixation/permeabilization solution (eBioscience). All events were acquired by a BD LSRII or a LSRFortessa cell analyser or sorted by a FACSAria Cell Sorter (BD Bioscience) equipped with BD FACSDiva software (BD Biosciences), and data were analyzed using FlowJo software (Tree Star). The antibodies used are described in online supplemental table S1.
Supplemental material
Quantitative PCR analysis
Total RNA from T cells was extracted with Trizol (Invitrogen). A 300 ng of RNA was reverse transcribed into cDNA by M-MLV reverse transcriptase, Random Primers, and RNaseOUT inhibitor (Invitrogen). cDNA were quantified by real-time PCR with a SYBR Green Real-time PCR kit (Applied Biosystems) on a Fast7500 detection system (Applied Biosystems, France). Relative mRNA levels were determined with the ΔCt method. Values were expressed relative to Actb. The sequences of the oligonucleotides used are described in the Key Resources Table (online supplemental table S2).
Transwell experiments
Th0 or Tfh cells were cultured in a 8.0 µm Transwell insert (Greiner bio-one) loaded in 24 well plates. Cells were placed on the top of a Matrigel matrix (Corning) loaded above the Transwell grid. The other conditions anti-CXCL13 (Clone 143 608 from R&D system) and/or CD8+ cells were placed on the bottom of the underneath wells. After 24 hours of incubation at 37°C at 5% CO2, the Transwell grid was released from the insert system, loaded on microscopy slides (Superfrost Ultra Plus, ThermoFisher Scientific), mounted with a drop of Mounting Medium containing DAPI (Molecular Probes) and covered with a cover slip (Knittelglass). Slides were imaged with a charge-coupled device-equipped upright microscope (Zeiss) and 40× objective with a numerical aperture of 1.4. Images were analyzed with ImageJ software.
Immunohistochemistry
FFPE slides with a 4 µm thickness were cut. Multiplex approach using 3-amino-9-ethylcarbazole (AEC) bleaching was performed as previously described.15 Briefly, a first CD4 staining (HistoSure-HS-360 108, 1/900) with AEC was applied. Slides were digitalized and saved. Next slides were bleached with ethanol and toluene. Then, another antigen retrieval was done and CD8 (HistoSure-HS-361 0003, 1/100) staining was performed. Slides were digitalized, saved and bleached once again. A final antigen retrieval was done before staining with Bcl6 (EuromabNet, GI191E, 1/100) with 3,3' Diaminobenzene (DAB) chromogen. Slides were then permanently mounted and digitalized.
Slides alignment and further analyses were done using Qupath (V.0.2.3) designed by Peter Bankehead and colleagues from Edimburgh.16
Patient material and analyses
RNAseqV2 data with RSEM normalization of 31 cancer types from The Cancer Genome Atlas (TCGA) and corresponding clinical data were downloaded from the TCGA data portal. In addition, three validation cohorts comprizing patients treated with anti-PD1 therapy were collected. The first is a private cohort including 94 patients with NSCLC (51 patients from Québec and 43 patients from Dijon who gave informed consent). The second cohort included 65 patients with melanoma, lung and head and neck cancers. The expression data profiling by array was downloaded from Gene Expression Omnibus (GEO) with GEO accession GSE93157. The third cohort included data from 21 Chinese patients treated for NSCLC with anti-PD-1 as monotherapy. The expression data profiling by array was downloaded from GEO with GEO accession GSE136961.
We used the signatures of Exhausted CD8+ and CD8+ T cell metagenes defined by Danaher et al17 and a Tfh metagene. Then, we defined metagene expression as the average of their gene expressions (online supplemental table S3).
To analyze the association between CXCL13 expression and metagenes, the Pearson correlation coefficient was calculated between CXCL13 and three genes of TGFB (namely TGFB1, TGFB2 and TGFB3) and Tfh.
To analyze the association between the abundance of cells in single cell data, the nine clusters of CD4 cells and seven clusters of CD8+ cells from the single cell analysis provided by Guo et al were downloaded for each of the 11 NSCLC adenocarcinoma patients. Abundances were computed for each cluster. The abundance of cells per group and per patient were calculated and scaled per patient. The Pearson correlation coefficient was calculated between each cluster based on these abundances.
For survival analyses, only patients with available overall survival (OS) were included. OS was limited to 10 years for TCGA cohorts and 1 year for the validation cohorts; progression-free survival (PFS) was limited to 6 months. For each cancer type and each metagene, patients were classified into two groups based on high or low level of metagene expression. The function maxstat.test from the R package was used to determine the optimal cut-off between high and low levels.18 Each group was composed of at least 30% and at most 70% of total observations. The association between the level of metagene expression and OS was evaluated by univariate Cox proportional hazards analysis. P values were based on the log rank test with a significance level of <0.05.
Then, to compare the prognosis between patients with high or low level of expression from a combination of two metagenes among CXCL13, Tfh and CD8+, we grouped patients with a high level of expression for both metagenes, the other patients formed the group ‘others’. For each cancer type, OS or PFS curves were generated using the Kaplan-Meier method.
Flow cytometry analyses on cancer patients’ samples
Study participants
Patients over 18 years old from Georges François Leclerc anticancer center with glioblastoma (n=2), ovarian (O, n=2) and breast (B, n=7) tumors provided written informed consent before enrolment. Whole blood and tumor samples were recovered at the day of surgery between February 2021 and March 2021. Blood and tumor samples were collected as part of the Centre Georges Francois Leclerc (CGFL) biological collection (authorization AC-2014–2260).
Tumor dissociation
Fresh tumor tissues were collected on the day of surgery for each patient. Tumors were mechanically and enzymatically dissociated using a human tumor dissociation kit (130-095-929, Miltenyi Biotec), according to the manufacturer’s instructions. In brief, tumors were cut into small pieces and transferred into the gentlMACS C tubes containing the enzyme mix. The dissociation was done using the gentleMACS Octo Dissociator with heaters and with human tumor dissociation 37C_h_TDK_1 program. Samples were homogenized before being applied to a MACS SmartStrainer 70 µM (130-110-916, Miltenyi Biotec) placed on a 15 mL tube. Filters were washed with 20 mL serum-free RPMI (L0500-500, Dutscher) and then centrifuged at 300 g for 7 min. After complete aspiration of supernatant, tumor cell suspensions were resuspended in RMPI and viable cells were counted with trypan blue. Tumor cell suspensions were then put at a concentration of 10.106 cells/mL.
Flow cytometry analyses
The 1.106 cells of tumor cell suspension in 100 µL of RPMI1640 were stained with anti-CD45-BV785 mAb (clone HI30, BioLegend) in the dark for 15 min at room temperature (RT). The cells were then washed twice and the pellet was resuspended with 50 µL of total heparinized blood. This solution was filed in a DURactive 1 tube (C11101, Beckman Coulter) for 3 hours at 37°C in the dark. After activation, membrane labeling was performed and anti-CXCR5-BV650 (clone RF8B2, BioLegend) and anti-PD-1-PECy5.5 (clone HA5.2B7) were added to DURactive 1 tube and incubated for 15 min at RT in the dark. Then, 100 µL of lysing and fixing OptiLyse B solution (IM1400, Beckman Coulter) were added and incubated for 15 min at RT in the dark. After washing with PBS 1X (702592, Dutscher) and a centrifugation at 300 g for 10 min, the pellet was resuspended in 25 µL of pure fetal bovine serum (FBS, Dutscher) and 300 µL of PerFix-NC R2 buffer was added (PerFix-NC kit, B31168, Beckman Coulter). The antibodies for intracellular labeling are then added to the tube, vortexed and incubated for 1 hour at RT in the dark: anti-IFNγ-FITC (clone 45.15, Beckman Coulter), anti-IL-21-PE (clone 3A3-N2.1, BD Biosciences), anti-IL-4-PECy7 (clone MP4-25D2, Beckman Coulter), anti-CXCL13-AF647 (clone 53610, R&D Systems), anti-IL-17A-AF700 (clone BL168, Beckman Coulter), anti-CD3-AA750 (clone UCHT1, Beckman Coulter), anti-CD4-PacBlue (clone 13B8.2, Beckman Coulter) and anti-CD8-KromeOrange (clone B9.11, Beckman Coulter). At the end of incubation, PBS 1 × (3 mL) was added to the tubes, incubated for 5 min at RT in the dark before centrifugation for 5 min at 500 g. After supernatant removal, the cells were resuspended in 3 mL of 1X PerFix-NC R3 buffer before another 5 min centrifugation at 500 g. The pellet was dried and resuspended in 150 µL of 1X R3 buffer. Acquisition was done on CytoFLEX cytometer and cytometry analyses were done with Kaluza V.1.3 software (Beckman Coulter). The gating strategies were described in online supplemental figure 11.
Quantification and statistical analysis
Results were shown as mean±SD or SEM, and data sets were compared using the unpaired Student’s t-test (test group vs control group) or two-way analysis of variance where appropriate. Differences in tumor foci numbers were assessed using Student’s t-test or the Kruskall-Wallis when appropriated. We performed statistical calculations with GraphPad Prism V.6. All test were two sided. A p<0.05 was considered statistically significant for all experiments.
Results
Adoptive transfer of Tfh cells exerts an antitumor effect, dependent on CD8+ T cells
We generated in vitro ovalbumin-specific Tfh-like cells by stimulating naive CD4 T cells from OTII mice with IL-6 and IL-21. Tfh-like cells shared similar features with Tfh, such as high expression of Bcl6, Pdcd1, Il21, Cxcr5 as assessed by PCR analysis (online supplemental figure 1). Contrary to Th1 cells, Tfh-like cells did not expressed Tbet or IFNγ protein. Concerning their cytotoxic activity, Tfh-like cells and Th1 only weakly express perforin (Prf) and granzyme B (Gzmb) genes (online supplemental figure 1). To address the role of Tfh cells in the antitumor immune response, we carried out adoptive transfer of these ovalbumin-specific Tfh-like cells, or Th1 cells as a control, into mice-bearing subcutaneous tumors or pulmonary foci of B16OVA tumor cells. We observed comparable antitumor efficacy of adoptively transferred Th1 and Tfh cells (figure 1A‒C), while Th2 cells had a protumor role (data not shown). Similar results were observed in the subcutaneous and pulmonary foci of B16–F10 models with adoptive transfer of tyrosinase-specific (Trp1) Tfh or Th1 cells (figure 1D‒F). To determine whether the transferred Tfh cells migrate to the site of the tumor and retain the Tfh phenotype, we injected CD45.2 OTII Tfh cells into CD45.1 B16-F10 tumor-bearing mice. CD45.2 Tfh cells were found in the tumor and in the draining lymph nodes, but not in the non-draining lymph nodes (online supplemental figure 2A). By PCR analysis, the sorted CD45.2 cells from the tumor retained a Tfh phenotype with conserved expression of Cxcr5, Bcl6, Il21 and no expression of transcription factors related to other CD4 subtypes, indicating that the cells had not undergone transdifferentiation into another T helper subtype (online supplemental figure 2B).
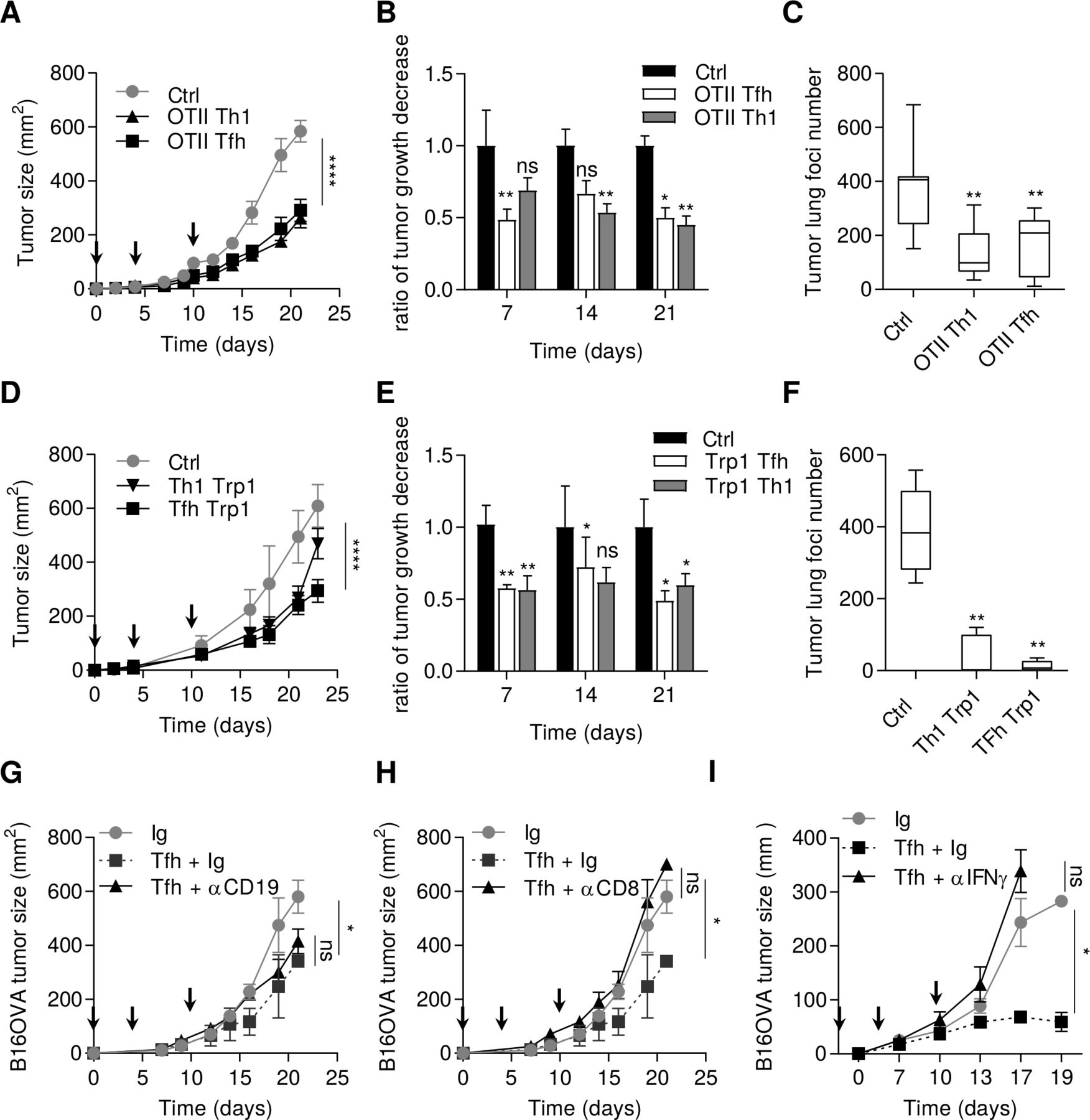
Tfh cells exert an antitumor effect dependent on IFNγ and CD8+ T cells. (A) Wild-type C57BL/6 mice were injected S.C. with B16OVA cancer cells. Mice were injected intravenously or not (Ctrl) with 2.106 in vitro generated OTII Th1 or Tfh cells at day 0, 4 and 10 after tumor cells injection (arrows). Tumor growth was monitored three times a week. (B) The response to adoptive transfer in terms of tumor volume in graph (A) is assessed using the rate of tumor growth, which refers to the ratio of tumor growth to the control mice volume. (C) Wild-type C57BL/6 mice were injected intravenously with B16OVA cancer cells alone (Ctrl) or at day 2 with 2.106 in vitro generated OTII Th1 or Tfh cells. at day 13, the lungs of Ctrl, OTII Th1 and Tfh injected mice were harvested and tumor foci were counted. (D) Wild-type C57BL/6 mice were injected S.C. with B16-F10 cancer cells alone (Ctrl) or at day 4 and 10 postinjection intravenously with 0.5.106 in vitro generated TRP1 Th1 or TRP1 Tfh cells. tumor growth was monitored three times a week. (E) The response to adoptive transfer in terms of tumor volume in graph (D) is assessed using the rate of tumor growth, which refers to the ratio of tumor growth to the control mice volume. (F) Wild-type C57BL/6 mice were injected intravenously with B16-F10 cancer cells alone (Ctrl) or at day 2 with 0.5.106 in vitro generated TRP1 Th1 or Tfh cells. At day 13, the lungs of Ctrl, TRP1 Th1 and Tfh injected mice were harvested and tumor foci were counted. (G–I) Wild-type C57BL/6 mice were injected S.C. with B16OVA cancer cells and intravenously with in vitro generated OTII Tfh cells associated with control Ig or CD19 depletion in (G) CD8 depletion in (H) or IFNγ depletion in (I) tumor growth was monitored three times a week and depletion using 200 µg antibody injection was achieved three times a week. Results are shown as mean±SEM of at least three representative and independent experiments; *p<0.05, **p<0.01, ****p<0.0001, versus Ctrl. IFNγ, interferon-γ; NS, not significant; S.C., subcutaneous; Tfh, T follicular helper cells.
Tfh cells cooperate with B cells to control adaptive immune response.19 To determine whether B cells can cooperate with Tfh cells to induce an antitumor immune response, tumor-bearing mice were depleted of B cells, using an anti-CD19 antibody. This treatment had no effect on the antitumor effect of the adoptive transfer of Tfh cells (figure 1G). In contrast, depletion of CD8+ T cells or IFNγ completely blocked the antitumor effect of adoptive transfer of Tfh cells (figure 1H,I).
Collectively, these data support the hypothesis that Tfh cells mediate their antitumor effect in a CD8+ and IFNγ-dependent manner.
Spontaneous accumulation of Tfh cells in murine spontaneous and transplantable cancer models
In a more physiological model, we examined the spontaneous accumulation of CD4+ PD-1+ CXCR5+ T cells in the spleen, tumor and draining or non-draining lymph nodes (dLN or ndLN) (online supplemental figure 3A). We used mice bearing various subcutaneous models of transplantable tumors in Balb/c (CT26 colorectal cancer and 4T1 triple negative breast cancer) or C57BL/6 mouse strains (B16F-10 melanoma, LLC1 lung cancer, MC38 colorectal cancer, EL4 lymphoma and Tc1 NSCLC). In tumor-free mice, CD4+ PD-1+ CXCR5+ T cells represented less than 1% of CD4+ T cells (figure 2A,B and online supplemental figure 3B). Ten days after tumor cell inoculation, we observed a more or less significant accumulation of CD4+ PD-1+ CXCR5+ Tfh cells in either dLN or the tumor bed in all models, while the detection of these cells varied in the spleen and ndLN, according to models (figure 2A,B). Flow cytometry analysis of 4T1 tumor-bearing mice revealed that CD4+ PD-1+ CXCR5+ cells expressed BCL6, ICOS and did not express CD25 at the protein level (figure 2C). We confirmed by PCR analysis that these cells expressed more Il21, Ascl2, Bcl6, Pdcd1 and Cxcr5 than CD4+ CXCR5- PD-1- cells (figure 2D). These results demonstrate that these cells have a classical Tfh phenotype.

Tfh cells accumulate in murine spontaneous and transplantable cancer models. (A) Representative flow cytometry analysis of PD-1 and CXCR5 expression on CD4+ T cells from 4T1 tumor-bearing mice. (B), Wild-type mice were inoculated with EL4 lymphoma, B16F-10 melanoma, LLC1 lung adenocarcinoma, Tc1 non-small cell lung cancer or MC38 colorectal carcinoma (C57BL/6 mice) or CT26 colorectal carcinoma, 4T1 triple negative breast cancer (BALB/c mice). When tumors reached 100–150 mm2, the spleen, non-draining lymph node (ndLN), tumor-dLN and tumor were excised. The percentage of Tfh among CD4+ T cells was analyzed by flow cytometry. (C) Representative flow cytometry of CD25, ICOS and BCL6 expression on Tfh cells (CD4+ T cells expressing PD-1 and CXCR5) and on CD4+ T cells negative for CXCR5 and PD-1 markers in draining lymph nodes from 4T1 tumor bearing mice. (D) Tfh cells (CD4+CXCR5+PD-1+) and other CD4+ T cells (CD4+CXCR5-PD-1-) were sorted by flow cytometry from spleen. BCL6, ASCL2, Pdcd1, CXCR5 and IL21 expression was determined by real-time PCR. The relative gene expression was normalized to Actb. (E) Wild-type C57BL/6 mice were treated by intraperitoneal injection of urethane once a week for 10 weeks. Then, control mice and treated mice were sacrificed 4 months after the first injection. lungs, spleen and draining mediastinal lymph nodes were harvested and the Tfh cell proportion was assessed by flow cytometry. (F) B16 tumor serial sections were stained with anti-CD4, anti-BCL6 and anti-CD8 antibodies and analyzed by immunohistochemistry. Arrow: enlarge image; dotted red line: TLS. Results are shown as mean±SEM of at least three representative and independent experiments; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, versus ‘naïve’ in (B) versus ‘CXCR5-PD-1‘ in (D) and versus ‘tumor free’ in (E). BCL6, B-cell lymphoma 6; CXCR5, C-X-C motif chemokine Receptor 5; ICOS, Inducible T cell costimulator; IL21, interleukin 21; ns, not significant; PD-1, programmed cell death-1; Tfh, T follicular helper cells.
To generalize these observations, we induced spontaneous lung adenocarcinoma in mice using intraperitoneal injections of urethane carcinogen. In this model, we also observed an accumulation of Tfh cells in lung tumors and dLNs, as well as systemic accumulation in the spleen of tumor-bearing mice (figure 2E). Finally, we investigated the presence of Tfh within the tumor and their colocalization with CD8 T cells by immunohistochemistry. We found Tfh cells (CD4+, BCL6+) in B16, CT26 and MC38 tumors which resided in CD8 T cell clusters (figure 2F and online supplemental figure 4).
Together, our results highlight the accumulation of Tfh in various transplantable models and a carcinoma-induced lung cancer model at the tumor site and in the tumor draining lymph nodes.
CXCL13 producing CD8+ T cells induce Tfh recruitment
Tfh cells express the CXCL13 receptor CXCR5. Using CXCL13 blocking mAb, Tfh cell accumulation was reduced in tumors (figure 3A) and promoted tumor growth (figure 3B), thus suggesting that CXCL13 mediates antitumor Tfh recruitment. Cxcl13 mRNA accumulated in the tumor and dLN in comparison to spleen and ndLN, suggesting that tumors play a role in triggering CXCL13 expression (online supplemental figure 5A). We detected significant expression of CXCL13 at the mRNA and protein levels, in all tumor dissociated cells tested (figure 3C and online supplemental figure 5B) but not in cancer cell lines cultured in vitro, thus suggesting a host and not tumor origin of CXCL13. In addition, we observed a correlation between the level of CXCL13 and the percentage of Tfh in tumor beds (figure 3D). Similarly, mRNA and protein expression of CXCL13 was significantly higher in dLN than in the ndLN, in the tumor models analyzed (4T1, MC38, CT26 and LLC1) (online supplemental figure 5C). To determine the cell types responsible for CXCL13 production within the tumors, we first sorted tumor cells (CD45− CD90−), fibroblasts (CD45− CD90+) and immune cells (CD45+) in the MC38 model and showed that immune cells were the main producers of CXCL13 in these tumors (online supplemental figure 5D). To go further, we next sorted CD19+, CD4+, CD8+ and CD11c+ CD11b+ cells among immune cells. Interestingly, CD8+ T cells from the tumor bed was the cell type with the highest expression of Cxcl13 (figure 3E). As a control, CD8+ T cells from the spleen did not produce CXCL13 (data not shown). In vitro, we validated the ability of CD8+ tumor infiltrated lymphocytes (TILs), but not splenic CD8+ T cells to attract Tfh in a CXCL13-dependent manner, using transwell assays (online supplemental figure 6). In vivo, CD8 depleting mAb limited the accumulation of Tfh in tumors, while it did not affect Th1, Th17 or Treg numbers (figure 3F).

CD8+ T cells recruit Tfh cells via CXCL13 release. Wild-type C57BL/6 mice were injected S.C. with B16-F10 cancer cells and treated three times a week by intratumoral injection of 1 µg anti-CXCL13 antibody or control Ig. (A) The proportion of Tfh cells in tumors was analyzed by flow cytometry at day 10. (B) Tumor growth was monitored three times a week. (C) The production of CXCL13 was analyzed by ELISA in supernatant of cancer cells lines CT26, 4T1, B16-F10, MC38, LLC1 and EL4 and in tumors from tumor bearing mice injected with CT26, 4T1, B16-F10, MC38, LLC1 or EL4 cells. (D) Correlation between CXCL13 production analyzed by ELISA and percentage of Tfh cells in tumors of tumor bearing mice for each cancer model at day 10 analyzed by flow cytometry. (E) CXCL13 expression was determined by real-time PCR in indicated immune cells sorted by flow cytometry from MC38 tumors. The relative gene expression was normalized to Actb. (F) At day 10, proportion of CD4+ T cell subtypes (Th1 (CD4+ CXCR3+ CCR6-), Th2 (CD4+ CXCR3- CCR6- ST2+), Th17 (CD4+CXCR3-CCR6+, Tfh (CD4+ CXCR5+ PD-1+), Treg (CD4+CD25+FoxP3+) in tumors from MC38 tumor-bearing mice treated or not treated with 200 µg of anti-CD8 antibody i.p. injections three times a week. (G) CXCL13 expression was determined by real-time PCR in CD8+ T cells sorted by flow cytometry from tumors of MC38 tumor-bearing mice treated or not treated with 200 µg of anti-TGF-β i.p. injections three times a week. The relative gene expression was normalized to Actb. (H) The proportion of Tfh in spleen, draining lymph nodes (dLN) and tumors was analyzed by flow cytometry in tumor bearing mice treated or not treated with anti-TGF-β i.p. injections. Results are shown as mean±SEM of at least three representative and independent experiments; *p<0.05, **p<0.01 versus Ctrl in (A, B, F, G, H) versus ‘cell lines’ in (C) and versus ‘CD11b+CD11c+CD8−’ in (E). CXCR3, C-X-C motif chemokine Receptor 3; ns, not significant; PD-1, programmed cell death-1; S.C., subcutaneous; Tfh, T follicular helper cells; TGF-β, transforming growth factor-β.
TGF-β was previously described as a potent inducer of CXCL13 in human cancer.20 In vitro, stimulation of naive splenic CD8+ T cells with increasing doses of TGF-β drove their CXCL13 production (online supplemental figure 7A). The supernatant from MC38 tumor cells contained more TGF-β than control normal 3T3 fibroblast supernatant (online supplemental figure 7B) and induced expression of CXCL13 in a TGF-β dependent manner, while 3T3 supernatant could not induce CXCL13 expression (online supplemental figure 7C). In vivo, TGF-β blocking mAb reduced mRNA expression of Cxcl13 in CD8+ T cells from dLN (figure 3G) and decreased Tfh cell accumulation (figure 3H).
Taken together, these data show that TGF-β production induces CXCL13 expression by CD8+ T cells, which is important to promote further Tfh cell recruitment.
IL-21 produced by Tfh cells participates in CD8+ T cell antitumor immune response
Tfh cells are characterized by their capacity to produce IL-21, which is known to affect CD8+ effector functions and to sustain T cell function during chronic viral infection.21 These data raise the hypothesis that IL-21 produced by Tfh can be responsible for intratumor CD8+ T cell activation. At the tumor site, we observed that only Tfh expressed significant amounts of Il21 mRNA (online supplemental figure 8A).To address the role of IL-21 produced by Tfh cells, we first stimulated naive CD8+ T cells through TCR triggering with or without IL-21. IL-21 increased the expression of Tim3 (T cell immunoglobulin and mucin domain-containing protein 3) and Lag3 (lymphocyte-activation gene 3) and decreased the expression of SLAMF6 (signaling lymphocyte activation molecule family member 6) but did not affect PD-1 or T cell immunoreceptor with Ig and ITIM domains, TIGIT) expression (figure 4A). In addition, to these phenotypic changes, we observed that IL-21 enhanced TNFα (tumor necrosis factor α) and granzyme B expression, whereas it did not affect IFNγ expression or proliferation (figure 4B). Then, we sorted CD8+ PD-1+ TILs from MC38 tumors and stimulated them through TCR triggering with or without IL-21. IL-21 enhanced the expression of granzyme B, modestly enhanced IFNγ but did not affect proliferation (figure 4C). At the transcription level, IL-21 increased the expression of Blimp1, Tbx21, interferon regulatory factor 4 (Irf4), Rora, Cd101, Tigit, Lag3, Havcr2, and reduced the expression of Eomes, Tcf1, Bcl6, Slamf6, Cx3cr1, Rorc and Il17 (figure 4D and online supplemental figure 8B). Similarly, the supernatant of Tfh enhanced TNFα and granzyme B expression in naive CD8+ T cells stimulated through TCR triggering and did not affect proliferation or their IFNγ production. This effect was blunted by anti-IL-21 antibody (figure 4E). Together, these data suggest that IL-21 can promote differentiation of CD8+ T cells into exhausted T cells with conserved cytokine production and cytotoxic functions.

IL-21 activates CD8+ T cell antitumor immune response. (A, B) Naive CD8+ T cells activated by anti-CD3/CD28 stimulation and treated or not (Ctrl) with recombinant IL-21 (20 ng/mL) for 72 hours. Analysis of TIGIT, Lag3, Tim3, SLAMF6 and PD-1 (A) Ki67, IFNγ, TNFα and granzyme B (B) expression by flow cytometry. (C) Representative flow cytometry analysis of granzyme B, Ki67 and IFNγ expression in tumor-infiltrating CD8+ T cells from MC38 tumor-bearing mice. (D) Expression assessed by RT-PCR of Tbx21, tox1, rorA, IRF4, Blimp1, TCF1, Eomes and BCL6 in naive CD8+ T cells activated by anti-CD3/CD28 stimulation and treated or not (Ctrl) with recombinant IL-21 (20 ng/mL) for 48 hours. The relative gene expression was normalized to Actb. (E) Analysis of Ki67, IFNγ, TNFα and granzyme B expression by flow cytometry in naive CD8+ T cells treated or not (Ctrl) with Tfh-derived supernatant (sp Tfh) with or without anti-IL-21 antibody (20 µg/mL) for 72 hours. (F) Wild-type (WT) C57BL/6 mice were injected S.C. With B16OVA cancer cells and intravenously injected with 2.106 in vitro generated OTII Tfh cells (arrows) and treated or not with anti-IL-21 antibody or control Ig i.p. tumor growth was monitored three times a week. (G) WT C57BL/6 mice were injected intravenously with B16-F10 cancer cells alone (Ctrl) or with 0.5.106 in vitro generated WT Tfh or TRP1 Tfh cells. At day 13, the lungs of injected mice were harvested and tumor foci were counted. (H, I) MC38 tumor-bearing mice were treated with anti-IL-21 antibody i.p. tumors were harvested and granzyme B, IFNγ, TNFα (H) and Tim3, SLAMF6 (I) expression in CD8+ T cells was analyzed by flow cytometry. results are shown as mean±SEM of at least three representative and independent experiments; NS: not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, versus ‘Ig’ in (F) and versus Ctrl in other panels. IFNγ, interferon-γ; IL-21, interleukin-21; Tfh, T follicular helper cells; TNFα, tumor necrosis factor-α.
To evaluate the role of IL-21 in the antitumor effect of Tfh in vivo, we first showed that the effect of adoptive transfer of Tfh on tumor growth was completely blunted by anti-IL-21 antibody in a B16OVA model (figure 4F). Moreover, we also observed in the B16F-10 lung tumor foci model that polyclonal nonspecific Tfh cells also had antitumor activity, but with less effect than tumor antigen specific Tfh cells (figure 4G). These two experiments suggest that Tfh-derived IL-21 is sufficient to mediate antitumor effect.
In a context of MC38 subcutaneous transplantable tumor, mice were either treated or untreated with anti-IL-21. We observed that IL-21 inhibition reduced PD-1+ CD8+ IFNγ, TNFα and Gzmb proportions (figure 4H), and reduced the ratio of terminal/precursor exhausted T cells, as analyzed either with CD8+ Tim3+/CD8+ SlamF6+ (figure 4I) or CD8+CD101+/CD8+ CX3CR1+ phenotypes22 (online supplemental figure 8C). Similar results were observed using anti-CXC13 antibody (data not shown).
Together, these data prompt us to propose that IL-21 produced by Tfh is involved in sustained production of cytokines and expression of cytotoxic molecules in exhausted T cells, thus enhancing their capacity to control tumor growth.
In vivo Tfhs are required for the antitumor effect of anti-PD-L1 mAb
As IL-21 secreted by Tfh promotes effector functions of tumor infiltrated exhausted T cells, we raised the hypothesis that the presence of Tfh would improve the efficacy of checkpoint inhibitors. To address this question, we used mice deficient in Tfh using Bcl6-/-CD4Cre animals. No Tfh cells were observed in these mice (online supplemental figure 9A). B16OVA tumor-bearing Bcl6-/-CD4Cre or CD4Cre mice were injected with ex vivo activated OTI CD8+ T cells (3 days with TCR triggering to generate precursors of exhausted T cells SLAMF6+ PD-1+ TIM3− (online supplemental figure 9B). In both mouse strains, OTI CD8+ T cells had a significant antitumor immune effect against subcutaneous B16OVA tumors, with a weaker effect in Bcl6-/-CD4Cre mice (figure 5A). Anti-PD-L1 was more effective in CD4Cre mice than in Bcl6-/-CD4Cre mice (figure 5B). Combotherapy using OTI CD8+ T cells and anti-PD-L1 was effective in CD4Cre mice, but not in Bcl6-/-CD4Cre (figure 5C). Similarly, in the MC38 model, anti-PD-L1 therapy lost its antitumor efficacy in Bcl6-/-CD4Cre mice (figure 5D). While anti-PD-L1 generated T cells clusters in CD4Cre mice, this event was not found in Bcl6-/-CD4Cre mice (online supplemental figure 9C).
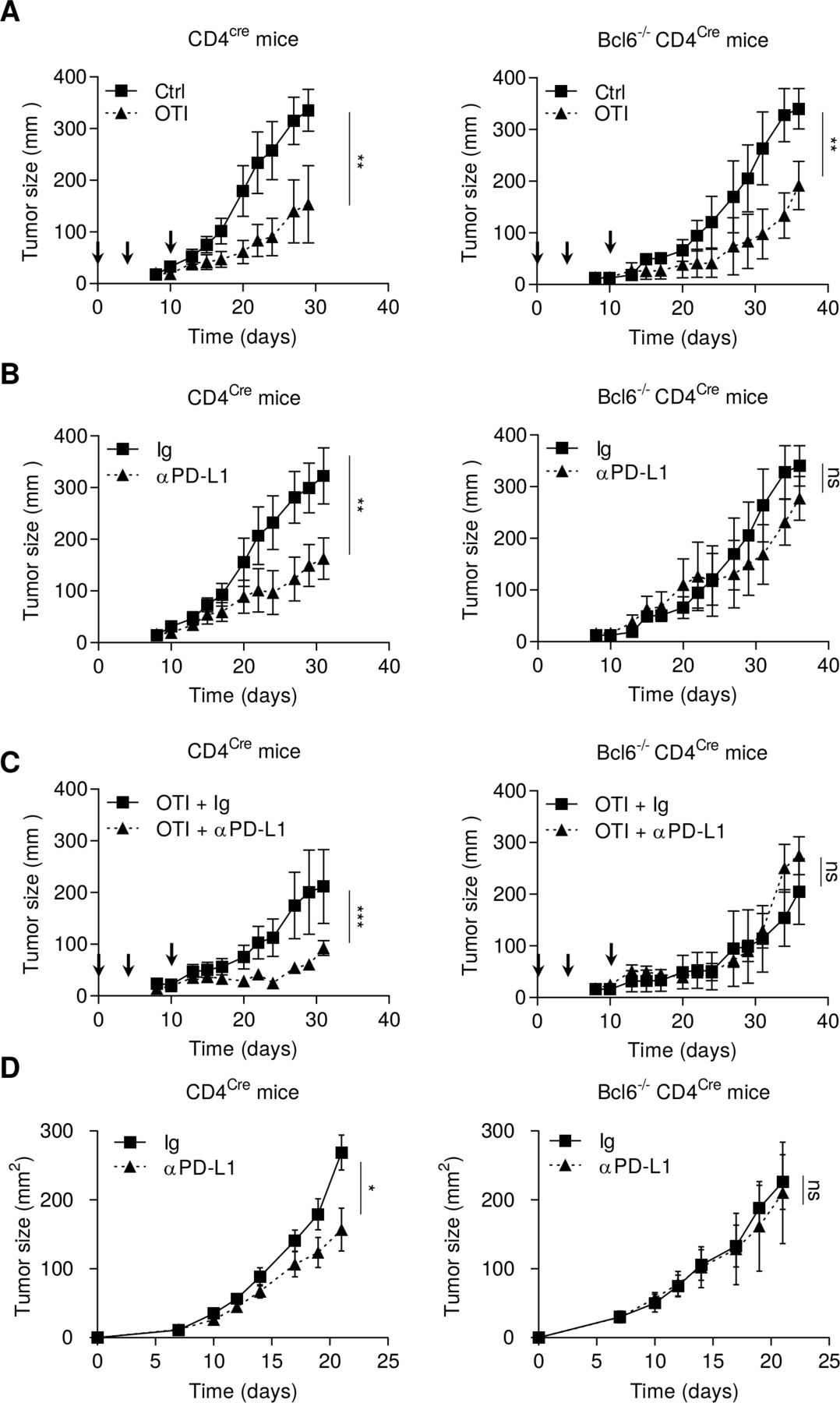
Tfhs are required for the antitumor effect of anti-PD-1 mAb. (A–C) CD4cre and Bcl6-/- CD4cre mice were injected S.C. with B16OVA cancer cells. When tumors reached 50–70 mm2, mice were treated or not (Ctrl) with intravenous injection of 2.106 in vitro generated OTI CD8+ T cells (arrows) and treated or not (Ig) with anti-PD-L1 antibody (200 µg; three times a week). Tumor growth was monitored three times a week. (D) CD4cre and BCl6-/-CD4cre mice were injected S.C. With MC38 cancer cells and treated or not (Ig) three times a week with anti-PD-L1 antibody (200 µg). Tumor growth was monitored three times a week. Results are shown as mean±SEM of at least three representative and independent experiments; *p<0.05, **p<0.01, ***p<0.001, versus control groups. ns, not significant; PD-1, programmed cell death-1; PD-L1, programmed cell death ligand-1; S.C., subcutaneous; Tfh, T follicular helper cells.
Together, these data suggest that Tfhs are required for efficient adoptive CD8+ cellular therapy and anti-PD-L1 therapy.
Human Tfh cell transcriptome is correlated with CXCL13 expression, exhausted CD8+ T cell transcriptome and cancer patients’ prognosis
To validate the clinical relevance of this information, we used transcriptomic data from TCGA. In 12 out of 27 tumor types, we observed that TGFB gene expression is associated with CXCL13 expression (online supplemental figure 10A). We also observed a correlation between the presence of exhausted CD8+ metagene and Tfh metagene in 23 cancer types (figure 6A). Using public data from single cell RNAseq of 11 NSCLC adenocarcinoma patients,23 we also observed a correlation between accumulation of Tfh and exhausted T cells (figure 6B), while Tfhs were not correlated with other CD8+ subsets (online supplemental figure 10B). Interestingly, these cells were the CD8+ subset that expressed the most GZMB,23 like mouse CD8+ T cells stimulated with IL-21 (figure 4A). Using cancer patients’ samples, we observed a correlation between Tfh and CD8 numbers in tumor cores. Moreover, Tfh and CD8 cells expressed CXCL13 and this expression was higher in Tfh cells (figure 6C and online supplemental figure 11). CXCL13 gene, Tfh, CD8+ T cells, and CD8+ exhausted T cell metagenes were associated with better prognosis in, respectively, 12, 13, 12 and 9 cancer types (figure 6D and online supplemental tables S3 and S4). In six tumor types (breast, cervical, colon, head and neck cancers, lung adenocarcinoma and sarcoma), all these parameters shared significant prognostic features. We focused on lung, colon and breast cancers, where Tfhs were previously described as a biomarker of good prognosis.6–8 When we combined exhausted CD8+ T cell metagene with CXCL13 or Tfh metagene, we observed better prognosis in TCGA for patients who had both high exhausted CD8+ T cells and high Tfh metagenes, or high CXCL13 and high exhausted CD8+ T cell metagenes (figure 6E and online supplemental figure 12). In the setting of NSCLC treated with nivolumab or pembrolizumab, analysis of patients included in a pooled cohort from Dijon Cancer center and Quebec Heart and Lung Institute (n=97 patients), showed that patients with both high exhausted CD8+ T cells and Tfh variables or both high exhausted CD8+ T cells and CXCL13 variables had better prognosis than other patients (figure 6F).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Human Tfh cell transcriptome is correlated with CXCL13 expression, exhausted CD8+ T cell transcriptome and cancer patients’ prognosis. (A) Heat MAP showing the Pearson correlation coefficients between exhausted CD8+ and Tfh for each type of cancer provided by the TCGA. Black cells represent significant p-values. (B) Dot plot showing correlations between the cell abundance in two clusters measured from single-cell RNAseq data and characterized by Guo et al: 1-CD8+ exhausted T cells and 2-CD4+ T cells with high expression of CXCL13 and harboring Tfh signature. (C) Flow cytometry analyses on cancer patients’ tumor samples. Correlation (Pearson correlation coefficient and linear regression) between the frequency of Tfh cells in CD4+ TILs and CD8+ T cells in CD3+ TILs (upper panel). The median fluorescence intensity (MFI) of CXCL13 was determined for CD8+ TILs and Tfh cells from tumor samples and compared with the fluorescence minus one (FMO) for CXCL13. *P<0.05. (D) Heat MAP showing, for each type of cancer provided by the TCGA, the HR for CXCL13 gene, Tfh, exhausted CD8+ and CD8+ T cell metagenes. Patients were divided into two groups according to low or high expression for each gene or metagene. The low expression group was taken as a reference. Blue cells represent good prognosis and red cells poor prognosis. *Log rank test p<0.05. (E) Kaplan-Meier curves of overall survival (OS) in breast invasive carcinoma (BRCA), colon and rectum adenocarcinoma (COADREAD) and lung adenocarcinoma (LUAD). Red curves represent patients with high expression of exhausted CD8+ metagene and CXCL13 gene and black curves represent patients with at least one metagene with low expression. (F) Kaplan-Meier curves of progression-free survival (PFS) for non-small cell lung cancer patients treated with nivolumab (n=94 patients). Red curves represent patients with high expression of two metagenes among Tfh, CXCL13 and exhausted CD8+ T cells and black curves represent other patients. CXCL13, chemokine (C-X-C motif) Ligand 13; ns, not significant; Tfh, T follicular helper cells;
Similar results were observed in two other public series of patients treated with anti-PD-1 for NSCLC cancers (GSE136961)24 and for melanoma, lung cancer, and head and neck cancers (GSE93157)25 (online supplemental figure 13).
Discussion
In the present study, we show that accumulation of Tfh cells is a major contributor to CD8+-dependent antitumor immunity and anti-PD-L1 efficacy. The absence of Tfh cells results in CD8+ T cell dysfunction with low cytokine production and cytotoxic functions, thus rendering mice resistant to anti-PD-L1 therapy. On the other hand, Tfh accumulation or adoptive transfer promotes cytokine production and cytotoxic functions in exhausted T cells, thus leading to an increase in their antitumor effect and boosting the anti-PD-L1 antitumor effect. Furthermore, we found that CD8+ T and Tfh cells work through reciprocal crosstalk. On the one hand, exhausted CD8+ T cells stimulated with tumor-derived TGF-β express CXCL13, which drives Tfh recruitment at the tumor site. On the other hand, Tfh cells produce IL-21 at the tumor site, a phenomenon that sustains exhausted T cell function and prevents their terminal exhaustion. The restoration of CD8+ functions and antitumor functions of Tfh is poorly dependent on their antigen specificity, as non-specific Tfh adoptive transfer is also effective in inducing tumor regression. These data provide a potential explanation for some patient outcomes with hot tumors, that is, high tumor antigen burden, high CD8+ infiltration and absence of response to checkpoint inhibitors.26 These observations strongly suggest that CD8+ and Tfh cell infiltration must be studied, to improve prediction of response to anti-PD-L1 therapy. Furthermore, our data support the hypothesis that Tfh cells or IL-21 injection can be a tool to promote efficacy of anti-PD-L1 therapy in tumors lacking Tfh. Conversely, our data should prompt caution regarding the use of TGF-β inhibitors, which prevent Tfh accumulation in the tumor bed. This is an important finding, since many clinical trials combining IL-21 or anti-TGF-β with anti-PD-1 are ongoing.
CXCL13/CXCR5 has a major role during cancer growth. Surprisingly, active CXCL13/CXCR5 signaling has been linked to the development and progression of several advanced human cancers. CXCL13 is produced by some cancer or stromal cells and directly promotes tumor cell survival, or the metastatic process.27 In addition to its direct protumor effect, the CXCL13/CXCR5 signaling axis is important for immune cell trafficking in the tumor microenvironment. CXCL13 is considered as the main organizer of TLS formation in different tumor tissues.28 29 CXCL13 induces homing of Tfh and CXCR5+ B cells in the tumor, to consequently form TLS. These structures enable optimal antigen presentation and T cell activation to mediate the adaptive antitumor immune response. Many reports have underlined the link between accumulation of Tfh or TLS at the tumor site and good outcome.30 31 The mechanism explaining the positive role of CXCL13 and Tfh is not currently clarified. In contrast, some other reports have suggested a negative role of Tfh. Accordingly, the CXCL13/CXCR5 axis may indirectly help malignant cells to escape from effector T cell immunity, through induction of the IL-10 pathway.32 Another recent report suggested that Tfh-like cells can inhibit T cell function in a PD-1/PD-L1-dependent manner in melanoma.9 Despite these controversial roles of CXCL13/Tfh, we observed here an association between CXCL13 or Tfh and good outcome in the TCGA database in 12 and 10 cancer types, respectively, while negative outcomes were only observed in 3 and 2 cancer types respectively. In animal models, we underlined the capacity of tumor Tfh to boost exhausted T cell function in an IL-21-dependent manner.
IL-21 delivers signals through the signal transducer and activator of transcription (STAT), mitogen-activated protein kinase, and phosphoinositide 3-kinase pathways. IL-21 mainly activates STAT3 but can also activate STAT1, STAT5A, and STAT5B.33 This cytokine has broad immunological actions and can regulate natural killer (NK) cells, macrophages, dendritic cells, B cell and CD4 differentiation.34 35 Several reports have shown that IL-21 can affect CD8+ functions in the setting of chronic infection. Some data showed its capacity to limit exhaustion during chronic LCMV infection.36–38 In humans, improved CD8+ T cell response and viral control is associated with higher IL-21 levels in different chronic pathologies, like hepatitis B virus, hepatitis C virus and HIV infection.39–43 IL-21 sustains the expression of IRF4 and (basic leucine zipper transcription factor (BATF), ATF-like) transcriptional factors,21 which can both enhance the effector functions of CD8+ T cells, without reducing expression of checkpoint inhibitors.44 Moreover, in accordance with our results, IL-21 has been described to increase RORα in CD8 T cells. However, an increased expression of RORγt, BATF, IL-17 and TNFα under IL-21 treatment was also reported in CD8 T cells, thus driving their differentiation into Tc17-like cells.45 46 In our hands, IL-21 was not able to induce Il17 and Rorc expression in CD8 T cells, thus arguing for a different role of this cytokine, depending on the cellular context. In the context of cancer, the role of IL-21 on exhausted T cells is poorly addressed and controversial. While a previous report in the context of chronic viral infection suggested that IL-21 is required to prevent the terminal exhaustion phenotype,47 another publication showed that in vitro IL-21 stimulation promotes expression of transcriptomic factors and immune checkpoint molecules associated with a T cell exhaustion phenotype, while maintaining their cytokines and cytotoxic functions.48 Our data show that IL-21 enhanced the exhausted phenotype, with increased expression of Lag3, Tim3, Rora, Irf4 and Blimp1, and decreased expression of Slamf6 and Bcl6. However, IL-21 sustains their cytotoxic and cytokine functions concomitantly with strong induction of Tbx21 a transcriptional factor associated with the effector function of CD8+ T cells. The molecular mechanism that is involved in sustaining CD8+ T cell function remains to be determined.
Antibodies that target PD-1 or PD-L1 can reinvigorate exhausted CD8+ T cells.49–51 However, their efficacy depends on the initial cell activation status of CD8+ T cells. In particular, it was demonstrated that checkpoint inhibitors can reactivate exhausted precursors but cannot resurrect cells with a terminally dysfunctional phenotype. Our data support the rationale that IL-21 and Tfh could be tools to sustain CD8+ T cell functions in the tumor and enable the efficacy of checkpoint inhibitors. While IFNy production by CD8 T cells seems to be important in the antitumor effect of Tfh, we cannot exclude the role of additional molecules, like Gzmb, Prf and TNFα. Checkpoint inhibitors lost their efficacy in Tfh deficient mice, but we cannot exclude that the absence of Tfh may also directly affect follicle formation and TLS generation and thereby influence antitumor effect of immunotherapy or CD8 effector functions. Such data extend previous reports showing that IL-21 was essential for CTL activation and tumor growth inhibition induced by agonist anti-GITR mAb. In this context, GITR costimulation promoted IL21 expression via c-Maf in naïve CD4+ T cells stimulated with IL-4.52
These data support the rationale for using adoptive transfer of Tfh cells or injection of IL-21 to improve the efficacy of checkpoint inhibitors in tumors. While antigen specific and non-specific Tfh share antitumor efficacy, we might suspect that CXCL13 intratumor production is sufficient to drive Tfh recruitment and to increase IL-21 cytokine release at the tumor site, thus providing a signal for CD8+ reactivation. Non-specific Tfh adoptive transfer or CD4+ CAR T cells differentiated into Tfh-like cells could be new tools for enforcing the efficacy of checkpoint inhibitors. Our murine data are confirmed in patients treated with nivolumab for lung cancer, where both CD8+ and Tfh signatures are required to predict better outcome under anti-PD-1 therapy. These data corroborate the rationale for using CD8+ exhausted T cells and Tfh as predictive biomarkers of response to checkpoint inhibitors.
Finally, a particularly interesting observation is that Tfh accumulation at the tumor site is related to CXCL13 production by CD8+ TILs. TGF-β stimulates TILs to produce CXCL13 at the tumor site and anti-TGF-β blunts CXCL13 expression and Tfh recruitment at the tumor site. Our results suggest that TGF-β drives a positive immune loop where CD8+ T cells attract Tfh, which then sustain CD8+ T cell function, induce their proliferation and protect them from apoptosis. TGF-β is a true immunosuppressive cytokine with pleiotropic functions. Its main role is to impede the effector function of T or NK cells and to promote immune exclusion.53 Our results might suggest some ambivalent effects of TGF-β with a capacity to promote Tfh localization in tumor tissue.
This observation is of importance since TGF-β inhibitors are under development for cancer therapy and many ongoing trials associate anti-TGF-β and checkpoint inhibitors.54 55 While TGF-β is a true immunosuppressive cytokine that impedes the effector function of T or NK cells and promotes immune exclusion,56 our data underline a positive effect of TGF-β on antitumor immune response. This should incite caution in the development of anti-PD-1 and anti-TGF-β therapy, notably in tumor types where Tfh cells are essential to drive checkpoint inhibitor efficacy.
Our study underlines a new circle involved in cancer immunity, where CD8+ exhausted T cells and Tfh interact with each other to improve tumor infiltration by immune cells and to promote CD8+ effector functions. These data have several implications. First, Tfh and IL-21 are essential to prevent terminal exhaustion of CD8+ T cells in the tumor and to reverse resistance to anti-PD-1 therapy. This suggests that resistance to anti-PD-1 therapy can be prevented by treatment with intratumor injection of IL-21 or adoptive transfer of Tfh cells. Second, the presence of Tfh and exhausted CD8+ cells could be used to improve prediction of checkpoint inhibitor efficacy in patients treated with anti-PD-1/PD-L1 as monotherapy, and could become an additional predictive biomarker.
Data availability statement
Data are available on reasonable request. Data are available on reasonable request.
Ethics statements
Ethics approval
All animals were bred and maintained according to both the FELASA and the Animal Experimental Ethics Committee Guidelines (University of Burgundy, France). The hospital institutional review board approved the study in accordance with the principles of Good Clinical Practice, the Declaration of Helsinki, and other applicable local regulations.
Acknowledgments
We thank Fiona Ecarnot, PhD (EA3920, University of Franche-Comté, Besançon, France) for careful reading of the manuscript. We thank the Unit of Molecular Biology, Department of Biology and Pathology of Tumors, Georges-François Leclerc for providing RNAseq data. We thank Marc-André Hamel and Andréanne Gagné (Laval University, Quebec, Canada) for providing clinical information and RNAseq data. We thank the animal housing facility at the University of Burgundy (Dijon, France) and the flow cytometry platform of Dijon.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
JN, HB, CR, FV, FG and FC contributed equally.
Contributors FG and FC conceived the study. JN, HB, CR, RM, EB, CT, MT, VD, LH, DR, CH and TA performed data acquisition, analysis, or interpretation. BR and PJ assisted with human samples information. EB and CT performed biostatistical analysis. FG, FC and FV supervised the study. FG, FC and CR wrote and edited the manuscript. JH and FV reviewed the manuscript. All authors approved the final manuscript.
Funding This work was supported by the 'Association pour la Recherche sur le Cancer' (ARC), ISITE-BFC senior Fellowship, LabEx LipSTIC « Lipoprotéines et Santé » ANR-11-LABX0021, Ligue Contre Le Cancer CCIR EST and by the Fondation de France. FG team is 'Equipe labélisée Ligue Nationale Contre le Cancer'. FC is a fellow of ARC and Fondation de France. HB and RM were fellows of the Ministère de la recherche.
Competing interests FG received speaker honoraria from Lilly, Sanofi, BMS, Astra Zeneca and Amgen, received funding for clinical trials from Astra Zeneca, received travel grants from Roche France, Amgen and Servier, and is an advisory board member for Merck Serano, Amgen, Roche France and Sanofi.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.