Article Text

Abstract
Background Modified vaccinia virus Ankara (MVA) are genetically engineered non-replicating viral vectors. Intratumoral administration of MVA induces a cyclic GMP-AMP synthase-mediated type I interferon (IFN) response and the production of high levels of the transgenes engineered into the viral genome such as tumor antigens to construct cancer vaccines. Although type I IFNs are essential for establishing CD8-mediated antitumor responses, this cytokine family may also give rise to immunosuppressive mechanisms.
Methods In vitro assays were performed to evaluate the activity of simvastatin and atorvastatin on type I IFN signaling and on antigen presentation. Surface levels of IFN α/β receptor 1, endocytosis of bovine serum albumin-fluorescein 5 (6)-isothiocyanate, signal transducer and activator of transcription (STAT) phosphorylation, and real-time PCR of IFN-stimulated genes were assessed in the murine fibroblast cell line L929. In vivo experiments were performed to characterize the effect of simvastatin on the MVA-induced innate immune response and on the antitumor effect of MVA-based antitumor vaccines in B16 melanoma expressing ovalbumin (OVA) and Lewis lung carcinoma (LLC)-OVA tumor models. RNAseq analysis, depleting monoclonal antibodies, and flow cytometry were used to evaluate the MVA-mediated immune response.
Results In this work, we identified commonly prescribed statins as potent IFNα pharmacological inhibitors due to their ability to reduce surface expression levels of IFN-α/β receptor 1 and to reduce clathrin-mediated endocytosis. Simvastatin and atorvastatin efficiently abrogated for 8 hours the transcriptomic response to IFNα and enhanced the number of dendritic cells presenting an OVA-derived peptide bound to major histocompatibility complex (MHC) class I. In vivo, intraperitoneal or intramuscular administration of simvastatin reduced the inflammatory response mediated by peritumoral administration of MVA and enhanced the antitumor activity of MVA encoding tumor-associated antigens. The synergistic antitumor effects critically depend on CD8+ cells, whereas they were markedly improved by depletion of CD4+ lymphocytes, T regulatory cells, or NK cells. Either MVA-OVA alone or combined with simvastatin augmented B cells, CD4+ lymphocytes, CD8+ lymphocytes, and tumor-specific CD8+ in the tumor-draining lymph nodes. However, only the treatment combination increased the numbers of these lymphocyte populations in the tumor microenvironment and in the spleen.
Conclusion In conclusion, blockade of IFNα functions by simvastatin markedly enhances lymphocyte infiltration and the antitumor activity of MVA, prompting a feasible drug repurposing.
- cytokines
- drug therapy
- combination
- immunotherapy
- interferon inducers
- vaccination
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Cancer virotherapy is based on the use of viruses for the treatment of tumors. Viruses can be selected or engineered to specifically lyse tumor cells (oncolytic viruses), to enhance antitumor immune response (viral immunotherapy), or to express immunostimulatory (viral gene therapy). Decades of preclinical research in cancer virotherapy have now led to numerous clinical trials and the regulatory approval of an armed oncolytic virus for the treatment of melanoma (talimogene laherparepvec)1 and three ex vivo transduced T lymphocytes with retrovirus or lentivirus to stably express chimeric antigen receptors against CD19.2–4
In viral immunotherapy, viruses are used mainly for their capacity to trigger a potent immune response. Pattern recognition receptors detect several components of viruses. The activation of these receptors leads to the release of proinflammatory cytokines, which activate the innate immune system. This intrinsic immunostimulatory of the virus can be enhanced by expressing immunostimulatory proteins such as cytokines, chemokines, or tumor-associated antigens.5 Recent advances in this field include the use of attenuated viral vaccine strains for yellow fever and rotavirus for intratumoral immunotherapy in mouse tumor models.6 7 However, replication-competent viruses in cancer patients still raise safety concerns. Modified vaccinia virus Ankara (MVA) is an attenuated cytopathic strain that has been recently approved by the Food and Drug Administration as a smallpox vaccine (JYNNEOS). Altogether, MVA is a safe prime candidate for therapeutic vaccine design. The immunogenicity of MVA relies on the activation of both Toll-Like Receptor 9 (TLR9) and the Stimulator of Interferon Genes (STING) pathway. TLR-9 senses extracellular CpG hypomethylated DNA that has entered the endosomes and causes the expression of proinflamatory cytokines and interferons (IFN) through the activation of the transcription factors nuclear factor-κB (NF-κB) and interferon regulatory factor 7.8 In contrast, the DNA-sensing receptor cyclic GMP-AMP synthase (cGAS) detects cytosolic double-stranded DNA and, through the activation of an intracellular signaling cascade that involves STING, triggers the production of type I IFN. Type I IFN is required for the activation of specialized cross-presenting dendritic cells (cDC1s).9
IFN-beta and the various subtypes of IFN-alpha are released as a result of the activation of pathogen-associated or damage-associated molecular pattern receptors. The most potent inductors of type I IFN are the ligands of TLR-3, the ligands of cytosolic RNA receptors such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA-5), or the ligands of cytosolic DNA receptors such as cGAS.10–12 Type I IFNs interact with the IFN-alpha/beta receptor, leading to the activation of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT), MAPK, PI3K, and Akt pathways.13 The activation of these pathways exerts potent antitumor effects. The mechanism involves a direct effect on malignant cell proliferation, the blockade of angiogenesis, and the activation of T lymphocytes and NK cells.14 15 However, type I IFNs also induce the upregulation or downregulation of hundreds of genes that lead to an antiviral program in cells.16 This antiviral cellular reprogramming creates a viral refractory state that involves suppression of viral replication and viral gene transcription. In this sense, the blockade of type I IFN signaling might be beneficial to enhance the viral replication and infection rate of MVA, however might be detrimental for its antitumor efficacy. To prevent this setback, a transient blockade of the type I IFN signaling might allow for efficient viral replication or transgene expression while the IFN receptor (IFNAR) is functionally silenced, but a strong antitumor IFN-mediated activity could be achieved once IFN signaling is reestablished. This transient inhibition concept has been already used to enhance the antitumor activity of oncolytic viruses. Ruxolitinib, a JAK inhibitor, was used to increase the replication of Measles virus,17 herpes simplex virus,18 and vesicular stomatitis virus encoding IFN-β.19 Other pathways acted on to block type I IFN production have been the mammalian target of rapamycin complex 1 (mTORC1)20 and NF-κB.21 Indeed, the drug VSe1-28 has been specifically developed with the purpose of blocking type I IFN activity and enhancing viral replication.22
Here, we evaluated a battery of compounds that could affect type I IFN signaling pathway and increase MVA-dependent antitumor efficacy. Among them, we found that both the simvastatin and atorvastatin exercise a potent and transitory inhibition of type I IFN signaling as a result of suppressing the clathrin-mediated endocytic pathway. Statins are widely used drugs for the treatment of hypercholesterolemia, but they are also potent anti-inflammatory molecules.23 The primary mechanism of action involves the inhibition of the 3-hydroxy-3-methylglutaryl coenzyme A reductase. The blockade of this enzyme decreases cholesterol synthesis and circulating low-density lipoprotein cholesterol levels but also inhibits protein isoprenylation and thereby alters several signaling G-proteins such as RAS, RAC, and Rho.24 Recently, it has been reported that simvastatin blocks the geranylgeranylation of Rab5 in DCs, prolonged antigen presentation and enhance the efficacy of antitumor vaccines.25 In our study, we found that the in vivo treatment of simvastatin combined with MVA virotherapy enhanced MVA antitumor efficacy by increasing the infiltration of effector immune cells into the tumor microenvironment.
Methods
Cell lines and culture media
The L929 murine fibroblast cell line was purchased from American Type Culture Collection and was cultured in medium consisting of DMEM (Gibco, Karlsruhe, Germany) supplemented with 2 mM glutamine (Sigma-Aldrich, Taufkirchen, Germany), 100 IU penicillin, 100 µg/mL streptomycin (Gibco), and 10% (v/v) fetal bovine serum (FBS; Gibco) in a humidified incubator at 37°C and 5% CO2. B16 melanoma expressing ovalbumin (B16-OVA) cells were provided by Dr. Lieping Chen (Yale University, New Haven, Connecticut, USA) and Lewis lung carcinoma (LLC)-OVA were provided by Dr. Daniel Ajona (Cima Universidad de Navarra, Pamplona, Spain). These cells were cultured in medium RPMI 1640 (Gibco) supplemented like DMEM, but with addition of 50 µM β-mercaptoethanol (Sigma-Aldrich) and geneticin G418 400 µg/mL (InvivoGen, San Diego, California, USA).
Reagents
MVA-BN was developed by Bavarian Nordic and is deposited at the European Collection of Cell Cultures (V00083008). All recombinants were generated from a cloned version of MVA-BN in a bacterial artificial chromosome. Infectious viruses were reconstituted from bacterial artificial chromosomes by transfecting bacterial artificial chromosome DNA into BHK-21 cells and superinfecting them with Shope fibroma virus as helper virus. After three additional passages of primary embryo fibroblasts, helper virus-free MVA recombinant viruses were obtained. All viruses used in animal experiments were purified twice through a sucrose cushion. Mouse IFN Alpha 1 protein was obtained from PBL Assay Science (Catalog No. 12 105–1; Piscataway, New Jersey, USA). Simvastatin (PHR1438-1G) and atorvastatin calcium (PHR1422-1G) were purchased from Sigma-Aldrich and were reconstituted in methanol. Bovine serum albumin (BSA)-fluorescein 5 (6)-isothiocyanate (FITC) was acquired from Sigma Chemical Co. (St. Louis, Missouri, USA). Trypan blue was obtained from Sigma-Aldrich.
Flow cytometry analysis
Single-cell suspension of the indicated organs collected at the indicated times were stained as previously described.26 Fluorescence minus one or biological comparison controls were used for cell analysis.27 The following antibodies were used and acquired from BioLegend (San Diego, California, USA): PE anti-mouse IFNAR1 (MAR1-5A3), PE Mouse IgG1 (MOPC-21), Zombie NIR, Brilliant Violet 650 anti-mouse CD19 (6D5), Brilliant Violet 510 anti-mouse CD45 (30-F11), Brilliant Violet 605 anti-mouse TCR β chain (H57-597), PERCPCy5.5 CD44 (IM7), APC CD62L (MEL-14), AF700 Ki67 (16A8), PE-Dazzel 594 Tim3 (B8.2C12), BV785 PD1 (29F.1A12), 7-AAD Viability Staining Solution. These were acquired from BD Biosciences: BUV395 Rat anti-mouse CD8a (53–6.7), BUV496 Rat anti-mouse CD4 (GK1.5), Purified Rat anti-mouse CD16/CD32 antibody (Mouse BD Fc Block), FITC Rat anti-mouse CD4 (RM4-4). These were acquired from eBioscience: PECy7 Tbet (eBio4B10), eFluor 450 Eomes (Dan11mag). PE-conjugated H-2Kb/OVA (257–264 complex was obtained from MBL (Nagoya, Japan). For evaluation of antigen presentation in DCs, CD11c+ cells from spleen were isolated using CD11c-coated magnetic beads (Miltenyi Biotec, Auburn, California, USA) and stained with Zombie NIR, Pacific Blue CD45 (RA3-6B2), BV605 CD11c (HL3) and PE OVA(257-264) (SIINFEKL) peptide bound to H-2Kb (25D1). Flow cytometry analysis was performed using a FACS Canto II flow cytometer (BD Bioscience) and a CytoFLEX LX apparatus (Beckman Coulter, Miami, Florida, USA). Data analysis was performed using FlowJo software (TreeStar).
RNA isolation and quantification of mRNA
RNA was isolated from the studied cell lines and tissues using the Maxwell 16 Total RNA Purification Kit (Promega, Madison, Wisconsin, USA), quantified in a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, Delaware, USA), and retrotranscribed (300 ng) to cDNA with Moloney murine leukemia virus reverse transcriptase from Promega, according to the manufacturer’s instructions.
Quantitative real-time PCR was performed with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) using specific primers for each gene; they were purchased from Invitrogen (Thermo-FisherScientific, USA). As ribosomal protein lateral stalk subunit P0 (PRLP0) mRNA levels remained constant across different experimental conditions, this parameter was used to standardize gene expression. PRLP0 5’-aacatctcccccttctcctt-3’ 5’-gaaggccttgaccttttcag-3’; ubiquitin-specific peptidase 18 (USP18) sense 5’-ccaaaccttgaccattcacc-3’ USP18 as 5’-atgaccaaagtcagcccatcc-3’; interferon-stimulated genes gene 15 (ISG15) sense 5’-gattgcccagaagattggtg-3’ ISG15 as 5’-tctgcgtcagaaagacctca-3’; 2'-5'-oligoadenylate synthetase (2,5 OAS) sense 5’-actgtctgaagcagattgcg-3’ 2,5 OAS as 5’-tggaactgttggaagcagtc-3’; and the open reading frame 082 L of MVA as target for detection of MVA backbone DNA, MVA082L sense 5’-acgtttagccgcctttaatagag-3’ 2 MVA082L as 5’-tggtcagaactatcgtcgttgg-3’ .The amount of each transcript was expressed by the formula 2ΔCt, with Ct being the point at which the fluorescence rises significantly above background levels.
Expression of IFNAR1 and endocytosis evaluation
First, L929 cells (3×104/ well) were seeded in 96-well plates in triplicate in DMEM 10% FBS culture media. After 24 hours, they were washed with phosphate-buffered saline (PBS) and treated for 3 hours with decreasing dilutions of the studied drugs or vehicle on 150 µL of DMEM without FBS. Simvastatin and atorvastatin two-fold dilutions curves were made from 600 µM (atorvastatin) and 50 µM (simvastatin) until 6.5 µM from a 10 mM stock. Then, cells were detached with 50 µL of trypsin-EDTA (Gibco), washed and stained for IFNAR1 detection with 50 µL of a cocktail of PE anti-mouse IFNAR1 (1:200), Zombie NIR (1:1000) and BD Fc Block (1:200) for 20 min at 4°C. The normalized geometric mean was calculated considering as 100% the geometric mean of the cells stained with the anti-IFNAR1 monoclonal antibody (mAb) and as 0% the geometric mean of the cells with isotype control. For the BSA-FITC endocytosis assay, cells were treated with atorvastatin (300 µM), simvastatin (37.5 µM) or vehicle methanol under the same conditions described above, but after 3 hours of incubation, they were washed with PBS and were incubated for 3 hours with DMEM 10% FBS. Then, the culture medium was removed and replaced by 150 µL of DMEM with BSA FITC (100 µg/mL), and cells were incubated for 1 hour for BSA endocytosis. After the last treatment, cells were detached, washed and stained with 7-AAD (1:75) in 50 µL of PBS for 15 min. Finally, they were resuspended in 200 µL of PBS with trypan blue (250 µL/mL) for cytometric analysis.
IFNα signaling evaluation
L929 cells (8×104/ well) were seeded in 24-well plates in triplicate in DMEM 10% FBS culture media. After 24 hours, they were washed with PBS and treated for 3 hours with atorvastatin (300 µM), simvastatin (37.5 µM) or vehicle (methanol) in 250 µL of DMEM without FBS. Afterward, cells were washed with PBS and were kept for 0, 3, 6 and 9 hours with DMEM 10% FBS. Next, cells were stimulated with IFN alpha 1 protein (100 units/mL) for 3 hours in 250 µL of DMEM 10% FBS. Lastly, the culture medium was removed, and the cells collected for RNA extraction.
For Western blot analysis, L929 (1×106/ well) cells were treated with atorvastatin (300 µM), simvastatin (37.5 µM) or vehicle (methanol) in 6-well plates with 2 mL DMEM culture media for 3 hours. Later, the supernatant was eliminated and IFNα alone (3000 units/ml) was added for 30 min and then Western blotting was accomplished using a rabbit polyclonal antibody to β-Actin (A2066, Sigma, Stockholm, Sweden), a rabbit monoclonal antibody to phopho-STAT1 (Tyr701) (58D6, Cell Signaling Technology, Danvers, MA) and a rabbit polyclonal antibody to STAT1 (ref. 9172, Cell Signaling Technology, Danvers, Massachusetts, USA).
For in vitro infection with MVA, L929 cells (6×105) were seeded into 6-well plates. Following overnight incubation, cells were treated with atorvastatin (300 µM), simvastatin (37,5 µM) or vehicle (methanol) in 1 mL DMEM culture media for 3 hours. Supernatants were removed and cells were washed twice with PBS and were infected with MVA OVA at a multiplicity of infection of 5 in DMEM 2% FBS. The viral inoculum was removed 1-hour postinfection, cells were washed twice with PBS and replaced with fresh DMEM 2%. After 6 hours, cells were recollected to RNA analysis.
Animal experimentation
In vivo experiments were performed with 6 to 8 week-old female C57BL/6 mice purchased from Harlan Laboratories (Barcelona, Spain). The mice were kept under specific pathogen-free conditions.
Antitumor activity assay
2.5×105 B16-OVA cells were inoculated subcutaneously on the right hind flank of C57BL/6 mice; the mice were randomized at day 7 and treated at days 7 and 14 after implantation. 20 µg of simvastatin per mouse were injected intraperitoneally or intra-muscularly in 100 µL of PBS. After 2 hours, the MVA vaccine was administered subcutaneously next to the tumor area (peritumorally) at a concentration of 5×107 TCID50 per mouse in 100 µL of PBS. In the case of intramuscular administration, the MVA vaccine was administered simultaneously. Tumor sizes were measured twice weekly using electronic calipers and mice were euthanized when tumors reached 17 mm on the longest axis. Experiments with MVA-gp70 were performed in C57BL/6 inoculated with 2×106 LLC-OVA cells and treated at days 4 and 11.
Lymphocyte population depletions
The tumor model and treatment regimen were as in the antitumor activity assay but mice were depleted of CD4+ T cells using anti-mouse CD4 monoclonal antibody (clone GK1.5, BioXCell, L'Aigle, France), CD8+ T cells using anti-CD8β (clone H35-17.2, in house), NK cells using anti-mouse NK1.1 (clone PK136, BioXCell) or conventional CD4+CD25+Foxp3+ T regulatory cells using anti-CD25 (clone PC-61.5.3, BioXCell). InvivoMab rat IgG2b (clone LTF-2, BioXCell) was used as control. A total of 200 µg of each antibody per mouse were administered intraperitoneally 1 day before first therapeutic treatment administration and on days 2, 6, 9, and 13 after it to maintain immune cell depletion during the course of the experiment. B cells were depleted using 500 µg per mouse of InvivoMab anti-CD19 (clone BE0150, BioXCell) 1 day before first therapeutic treatment administration and on days +2, +7, +12, +17, +22 and +27. The effect of IL10 was neutralized using 250 µg/mice of InvivoMab anti-IL10R (CD210) (clone 1B1.3A, BioXCell) administered after the first therapeutic treatment administration and on days +7, and +14.
Immune cell analysis
The tumor model was performed as in the antitumor activity assay, but, in this case, mice received a single dose of treatment. Tumor tissues, lymph nodes, and spleens were collected on day five after treatment and were processed for cytometric analysis as described in.28 Specific T-cell responses to OVA antigen were assessed ex vivo by a mouse IFN-γ Enzyme-linked Immunosorbent Spot (ELISpot) Assay kit (BD-Biosciences). Ninety-six-well Multiscreen IP Plates (Millipore, Bedford, Massachusetts, USA) were coated with 100 µL of assay diluent containing anti-IFN-γ monoclonal Ab and incubated overnight at 4°C. The plates were washed and then blocked with RPMI-1640 medium containing 10% FBS for 90 min at RT. Splenocytes depleted of erythrocytes were added to wells (4×105) and stimulated with OVA (257-264) peptide (1 µg/mL) or 4×104 irradiated (20 000 rads) B16-OVA, B16.F10 tumor cells in 200 µL/well. Prior to use, tumor cells as a stimulator were treated with 500 IU/mL of IFN-γ for 48 hours to increase MHC-I expression.
Analysis of IFN-stimulated genes in tumor tissues
The tumor model was performed as in the antitumor activity assay, but, in this case, mice received a single dose of the treatment. Tumor tissues were collected in RNA homogenization buffer at 4 hours after treatment and were stored at - 80°C until they were processed.
RNA-Sequencing of UMI-labeled 3’UTR
RNA sequencing was performed by adapting the technology of SCRB-Seq29 to allow for the high cost-efficient multiplexed transcriptome characterization. Briefly, cells were collected in cell lysis buffer, and poly-(A)+RNA were purified using the Dynabeads mRNA DIRECT Purification Kit (ThermoFisher Scientific). poly-(A)+RNA were annealed to a custom primer containing a poly-(T) tract, a Unique Molecule Identifier (UMI), and a sample barcode. Retrotranscription using Template-switching oligonucleotides was then used to synthesize and amplify 3’ untranslated region (3’UTR) enriched cDNA, resulting in barcoded cDNA fragments. Library preparation was performed using the Nextera XT library preparation protocol, which introduces i5-P5 and i7-P7 structures for massive parallel sequencing. Quality control was performed following pre-amplification RT and library preparation to ensure quality and length accuracy and equilibrate sample pooling. Libraries were then circularized and sequenced using a DNBSeq-G400 sequencer (MGI), using the MGIEasy Circularization Kit (MGI). 5–10 million pair-end reads (100 bp) were sequenced for each sample. Raw sequences were called using Zebra caller (MGI) and demultiplexed using Cutadapt. RNAseq was carried out at the Genomics Unit of the CIMA Universidad de Navarra. Quality control was performed using FastQC. Bbduk from BBMap tools (V.38.90) was used to remove the adapter contamination, polyA read-through, and low-quality tails. Nf-core RNAseq pipeline was used to process the reads.30 UMI extraction was done using UMI-tools (V.1.1.1). STAR (V.2.7) was used to align the raw 3’RNA-Seq fastq reads to the mouse reference genome (GRCm38.p6). Read quantification was computed using featureCounts (Subread version 2.0.1). In order to identify viral reads in the 3’RNA-seq data, unmapped reads were mapped to the MVA viral genome (ASM645792v1) using Bowtie2, and read count was performed using featureCounts. Differential analysis was performed with the R package edgeR. False positive discovery <0.05 was set as cut-off for differentially expressed genes (DEGs). GO enrichment analysis was performed for the DEGs using the geneXplain platform (www.genexplain.com). Hierarchical clustering, volcano plots, and barplots were performed using ComplexHeatmap R package, R-base functions, and ggplot2 package, respectively.
Statistical analysis
GraphPad Prism V.8.2.1 software (GraphPad Software, San Diego, California, USA) was used for statistical analysis. Data were analyzed by one-way analysis of variance followed by Sidak’s multiple comparisons test. Longitudinal data were fitted to a third-order polynomial equation and compared with an extra sum-of-squares F test with Bonferroni adjustment for multiple comparisons. Survival analysis was performed in R using the pairwise_sruvdiff function of the package survminer with Benjamini-Hochberg adjustment for multiple comparisons. Values of p<0.05 were considered to be statistically significant.
Results
Screening of potential pharmacological type I IFN inhibitors
We conducted a screening of clinically approved drugs to find novel inhibitors of the type I IFN. The selection was based on two assays. First, we searched for a reduction of the surface expression levels of the IFN-alpha/beta receptor 1. The second assay focused on interference with the endocytosis pathway. These assays were based on the differential intensity of type I IFN signaling depending on the cellular IFNAR density and on the fact that clathrin-dependent endocytosis of activated IFNAR complex is a requirement for type I IFN signaling.31 32 Several compounds such as terbinafine, procainamide, and quinidine successfully reduced both IFNAR1 levels and BSA endocytosis and increased the percentage of GFP positive cells after MVA-GFP infection (online supplemental figure 1). Based on these preliminary data, we focused our attention on statins. Simvastatin, and atorvastatin reduced the membrane levels of IFNAR1 in a dose-dependent manner. The simvastatin EC50 was 26.34 µM, and the atorvastatin EC50 was 68.91 µM. These EC50s are similar to those described for the inhibition of CYP2C8 (2.7 fold-higher in simvastatin and 1.7-fold higher in the case of atorvastatin) and in the same range as the in vivo estimated liver concentrations of both drugs.33 The maximum reduction was approximately 40% of baseline levels. The maximum effect was achieved at 37.5 µM for simvastatin and 300 µM for atorvastatin (figure 1A–D). Simvastatin was not toxic for the L929 cells at the dose range tested, and the level of viable cells analyzed by 7AAD incorporation was higher than 95% in all tested concentrations. Atorvastatin was slightly more toxic in culture, and at the dose of 300 µM, only 69% of cells remained alive. Of note, dead cells were excluded from the IFNAR1 flow cytometry expression analysis. Based on these assays, we selected the effective dose of simvastatin and atorvastatin. Using these doses, the internalization of BSA labeled with FITC was also inhibited (figure 1E,F). We had previously shown that BSA internalization is inhibited at 4°C and by treatment with chlorpromazine, as expected for a clathrin-mediated process.34 Importantly, both statins significantly inhibited fluorescent BSA uptake (figure 1E,F).
Supplemental material
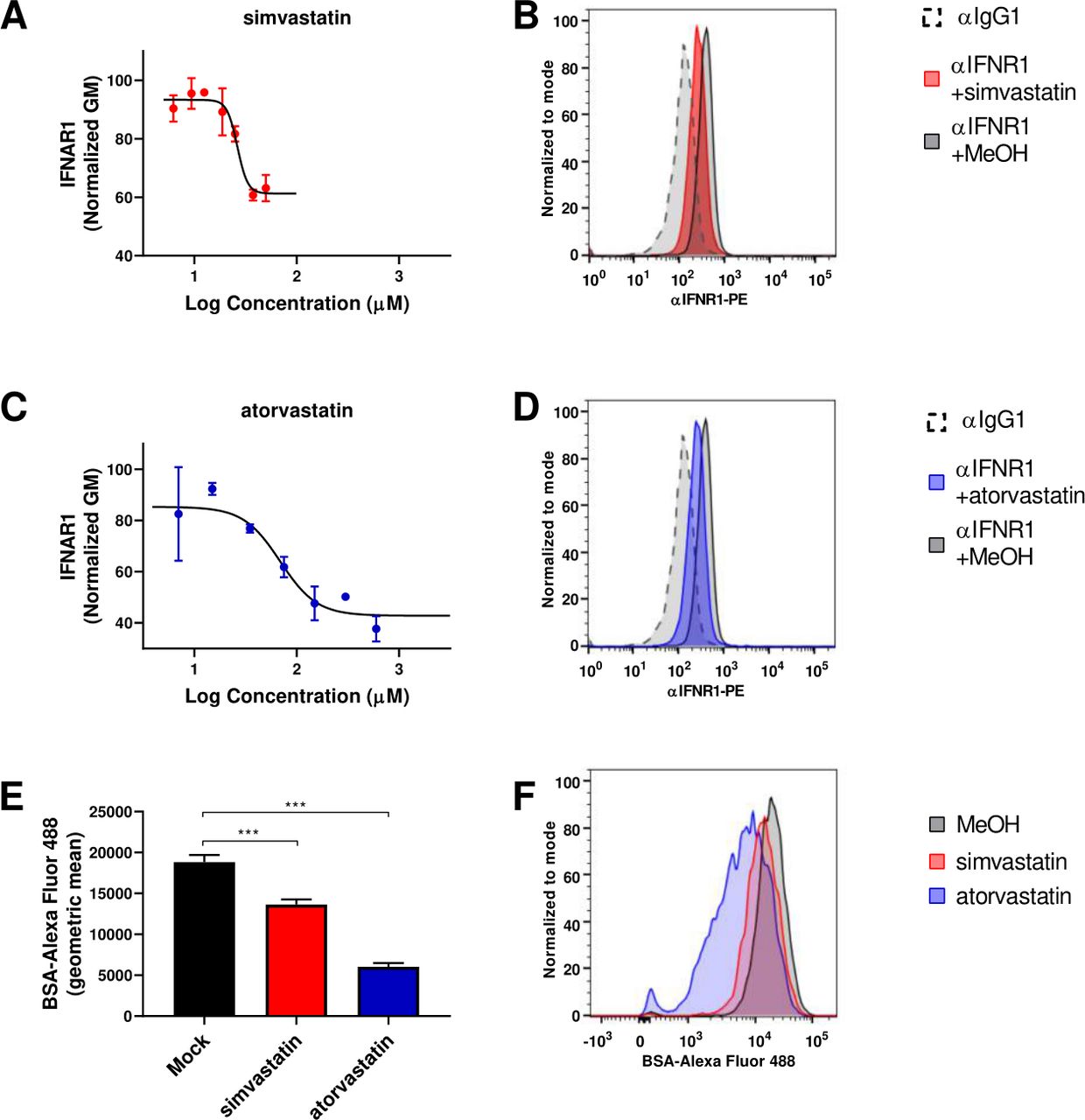
Simvastatin and atorvastatin reduce surface expression of IFNAR1 and protein endocytosis. (A) L929 cells were incubated with different concentrations (from 50 µM until 6.5 µM) of simvastatin for 3 hours, and levels of IFNAR1 on the cell membrane were quantified by flow cytometry. Normalized GM: the normalized geometric mean was calculated considering as 100% the geometric mean of the cells stained with the anti-IFNAR1 mAb and as 0% the geometric mean of the cells with isotype control. N=3. (B) Representative histogram of mock-treated cells without αIFNAR1 staining (black dashed line), simvastatin treated cells stained with αIFNAR1-PE (red line) and mock-treated cells stained with αIFNAR1-PE (black line). (C) As in a but for compound atorvastatin, in this case two fold dilution curve was made from 600 µM. N=3. (D) Representative histogram of mock-treated cells without αIFNAR1 staining (black dashed line), simvastatin treated cells stained with αIFNAR1-PE (blue line) and mock-treated cells stained with αIFNAR1-PE (black line). (E) L929 cells were incubated with 37.5 µM simvastatin or 300 µM atorvastatin for 3 hours, and BSA-FITC internalization was quantified by flow cytometry. N=3. One-way ANOVA followed by Sidak’s post-test. ***P<0.001. (F) Representative histogram of simvastatin treated cells (red line), atorvastatin treated cells (blue line) and mock-treated cells (black line). ANOVA, analysis of variance; BSA-FITC, bovine serum albumin-fluorescein 5 (6)-isothiocyanate; IFNAR1, interferon receptor.
Blockade of IFNα signaling by simvastatin and atorvastatin
Once we verified that the compounds simvastatin and atorvastatin reduced the IFNAR1 membrane expression and endocytosis, we analyzed whether they were able to block recombinant IFNα-elicited signaling effectively. The signaling cascade initiated by the binding of rIFNα to the IFN-α/β receptor leads to the phosphorylation of STATs and the modulation of ISGs. Both simvastatin and atorvastatin reduced the IFNα-mediated STAT1 phosphorylation (figure 2A). To evaluate the timing of the statin effects on the IFNα activity, we analyzed the expression levels of three ISGs commonly used as markers of the activity of rIFNα: 2,5-OAS, ISG 15 (ISG15), and USP18. As expected, the expression of the three genes was markedly upregulated by the rIFNα. Preincubation with simvastatin or atorvastatin 3 hours before exposure to IFNα abrogated ISGs induction (figure 2B). The maximal inhibitory effect was observed when the IFNα exposure immediately followed the withdrawal of statins from the cultures and decreased if the resting period was prolonged. When time between the drug exposure and the IFNα treatment was 9 hours, we could not detect any effect of simvastatin and atorvastatin on the IFNα activity (figure 2C).
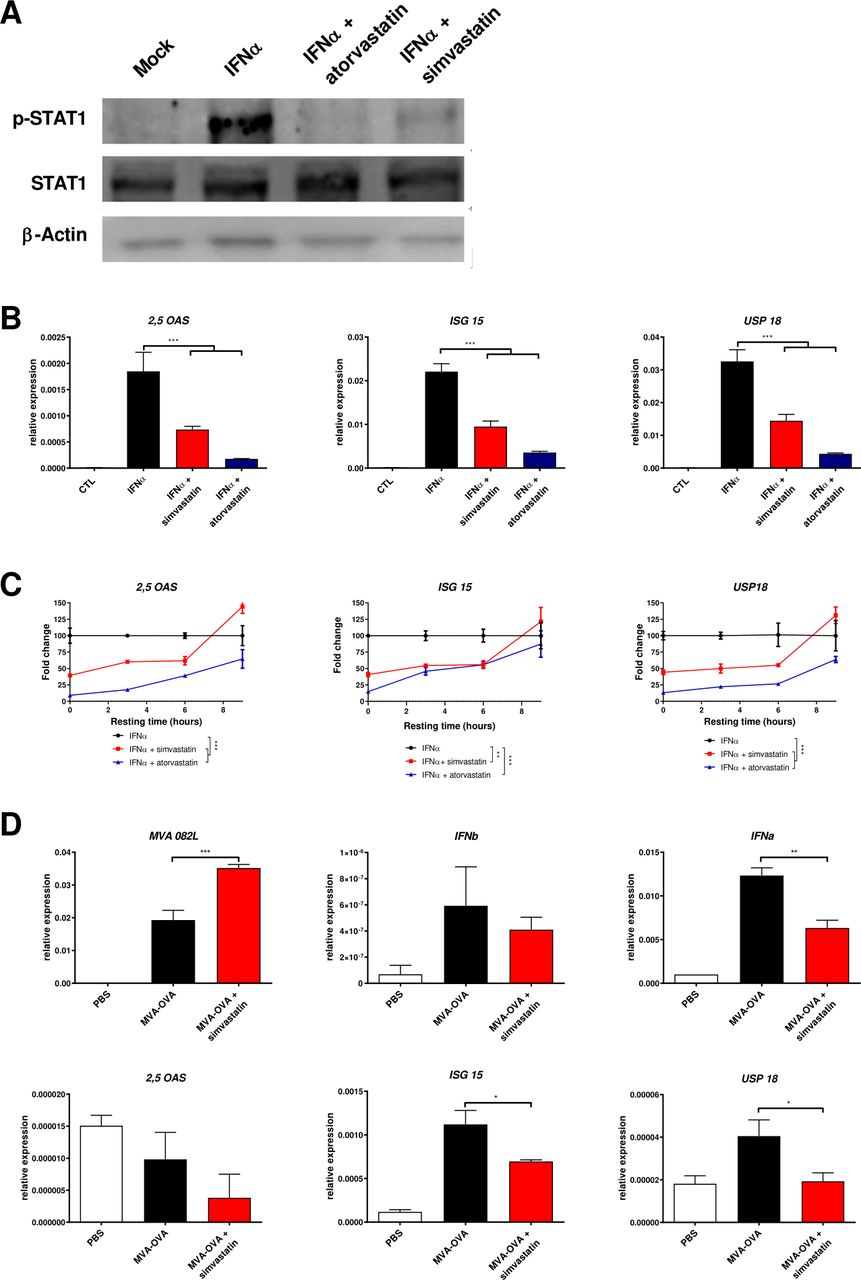
Simvastatin and atorvastatin reduce the expression of interferon α (IFNα) induced genes. (A) L929 cells were treated with vehicle (methanol 7.5 µL) or with atorvastatin (300 µM) or simvastatin (37.5 µM) for 3 hours and then, cell were washed and treated for 30 min with IFNα (3000 units/mL). Then, the phosphorylation of STAT-1 and total STAT-1 were analyzed by Western blot. β-actin was used as a loading control. (B) L929 cells were treated with vehicle, 300 µM atorvastatin, 37.5 µM simvastatin for 3 hours, and then cells were washed. three hours later, IFNα (100 units/mL) was added for 3 hours, and the induction of 2'−5'-oligoadenylate synthetase (2,5-OAS), IFN-stimulated gene 15 (ISG15) and ubiquitin-specific peptidase 18 (USP18) was analyzed by real-time PCR. N=3. One-way ANOVA followed by Sidak’s post-test. ***P<0.001. (C) L929 cells were treated with vehicle, 300 µM atorvastatin, 37.5 µM simvastatin for 3 hours, and then cells were washed. 0, 3, 6, and 9 hours later, IFNα was added for 3 hours, and the induction of 2,5-OAS, ISG15 and USP18 was analyzed by real-time PCR. Data were fitted to a second order polynomial and compared using an extra sum-of-squares F test. N=3. **P<0.001 ***p<0.0001. (D) L929 cells were infected with MVA-OVA at 5 MOI with or without 37.5 µM simvastatin. Six hours later, real-time PCR was used to analyze the expression of MVA 082 L, IFN beta (IFNb), IFNa, 2,5-OAS, ISG15 and USP18. N=3. One-way ANOVA followed by Sidak’s post-test. *P<0.05 **p<0.01. ANOVA, analysis of variance; MOI, multiplicity of infection.
Transient reduction of IFN-α/β signaling by statins was hypothesized to enhance the effects of virotherapy. For experiments with MVA, we selected simvastatin due to the lower doses and reduced toxicity required for the IFNα inhibitory effects in cell cultures. The coincubation of simvastatin and MVA-OVA enhances viral expression as compared with MVA alone. Similar IFN beta expression levels were detected in both experimental conditions reflecting that simvastatin does not interfere with viral detection by of pathogen-associated molecular pattern receptor (figure 2D). However, simvastatin reduced the expression of several ISG such as IFNa, ISG15, or USP18 (figure 2D).
Simvastatin has been shown to exert an adjuvant activity when combined with recombinant proteins. This effect is mediated by prolonging antigen presentation in DCs due to the block of Rab5 lipidation.25 To evaluate whether simvastatin also promotes antigen presentation on MVA infection, mouse DCs were infected for 24 hour with MVA-OVA with or without simvastatin. Flow cytometry analysis revealed that simvastatin increased two-fold the number of DCs presenting the OVA 257–264 (SIINFEKL) bound to MHC class I, indicating that MVA therapeutic vaccination could also benefit of the adjuvant properties of simvastatin (online supplemental figure 2).
Simvastatin enhances the antitumor activity of MVA-OVA
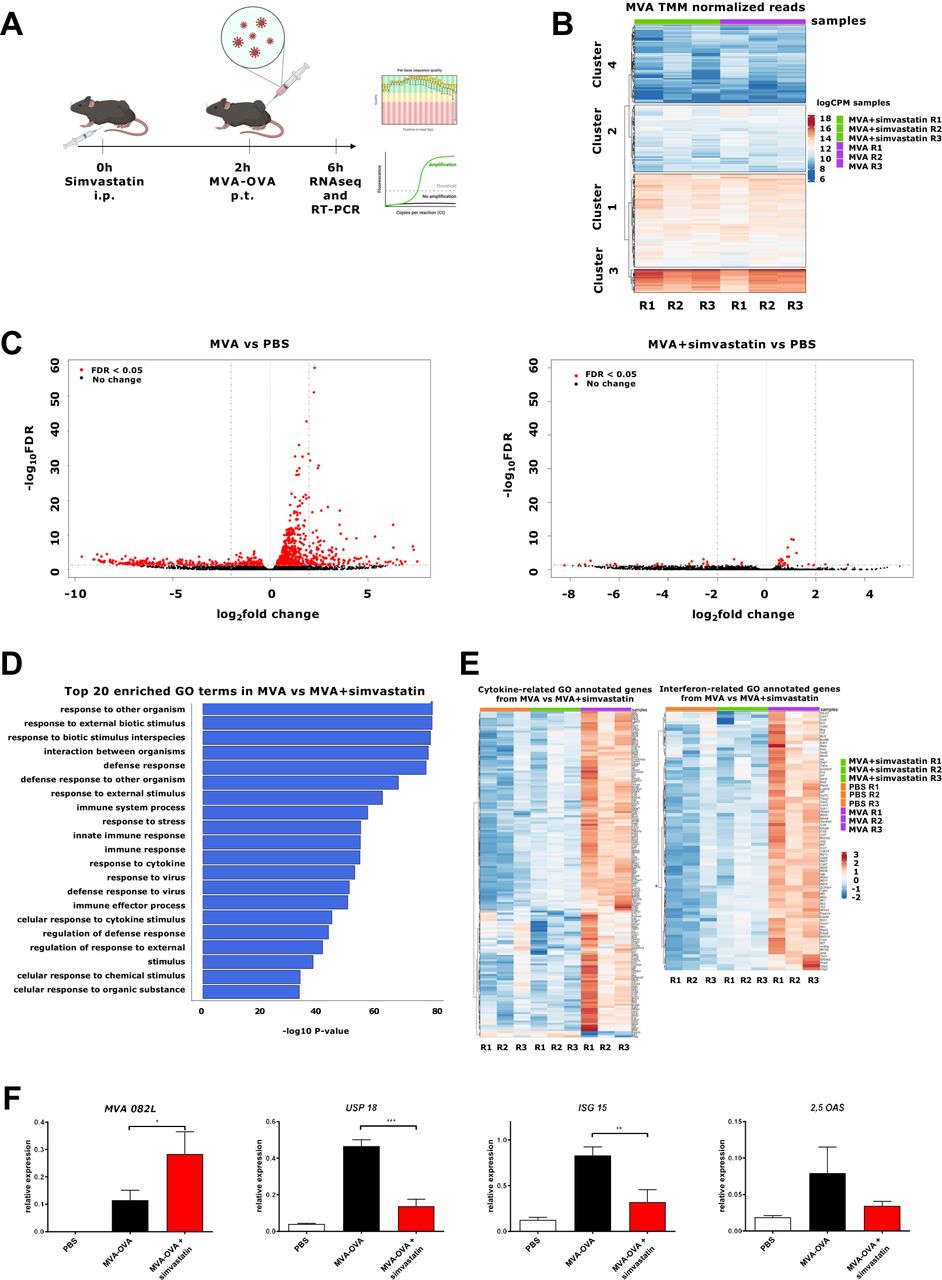
For in vivo experiments, we chose a dose of simvastatin based on the previous literature.25 Peritumoral injection of MVA-OVA in B16-OVA (figure 3A) induced the expression of MVA genes. The viral genes can be grouped in four clusters, depending on the expression level. The administration of simvastatin does not interfere with viral expression, and similar levels of the different clusters were observed (figure 3B and online supplemental figure 3). In contrast, simvastatin dampened the overall MVA-induced genes in the tumor microenvironment (figure 3C). Gene Ontology analysis of the simvastatin-inhibited pathways identified 20 involved in the viral immune response (figure 3D and online supplemental figure 4). The DEGs included a large number of cytokine-related genes and, specifically, several IFN-related genes (figure 3E and online supplemental figures 5 and 6), indicating that additional anti-inflammatory effects of simvastatin are involved in the mechanism of action of this combination in addition to the suppression of the type I IFN signaling. These results were validated by real-time PCR. Intraperitoneal administration of simvastatin slightly enhanced the expression of the viral gene 082 L while the induction of the ISGs was inhibited (figure 3F).

Effect of simvastatin on the MVA-gene expression and innate immune-related genes in tumors. B16-OVA melanoma cells were injected subcutaneously. seven days later, mice were treated intraperitoneally (i.p) with vehicle or simvastatin (20 µg/mice). After 2 hours, MVA-OVA (5×107 TCID50 per mouse) was administered peritumorally (p.t.). Four hours later, mice were sacrificed, and tumor mRNA was analyzed by RNAseq and real-time PCR. (A) Schematic representation of the experimental setting. (B) Heatmap of MVA gene expression in the MVA-OVA treated group (MVA) and the MVA-OVA+ simvastatin group (MVA+ simvastatin). (C) Volcano plots of differentially expressed genes of MVA-OVA-treated mice (MVA) vs PBS-treated tumors (PBS) and MVA-OVA+ simvastatin-treated mice (MVA+ simvastatin) vs PBS-treated tumors (PBS). Red dots indicate false discovery rate (FDR) <0.05. (D) Top significantly differential pathways of MVA vs MVA+ simvastatin. The horizontal bars denote the different pathways based on the p value. (E) Heatmaps of cytokine-related genes and interferon related genes differentially expressed between MVA and MVA+ simvastatin. (F) Real-time PCR analysis of MVA 082 L, ubiquitin-specific peptidase 18 (USP18), 2'−5'-oligoadenylate synthetase (2,5-OAS), and interferon-stimulated gene 15 (ISG15). N=4. One-way ANOVA followed by Sidak’s post-test. *P<0.05 **p<0.01 ***p<0.001. ANOVA, analysis of variance; MVA, modified vaccinia virus Ankara; OVA, ovalbumin.
To evaluate the antitumor effect of the combination, we measured the tumor growth and survival after treatment with MVA-OVA or MVA-OVA combined with simvastatin. The administration of simvastatin alone had no impact on tumor growth (figure 4A). MVA-OVA was injected into the tumor and delayed tumor growth in all mice, showing increased survival but only completely eradicated the tumor in one mouse out of six (figure 4A,B). In contrast, the additional administration of simvastatin either intraperitoneally or intramuscularly markedly enhanced the percentage of cured mice at the end of the experiment. In our hands, the intramuscular administration was slightly more effective and significantly increased survival as compared with the mice treated with MVA-OVA alone (figure 4B). We also evaluated whether the intraperitoneal administration of the MVA synergized with simvastatin. In this case, no differences were observed in the antitumor effect of MVA alone or the MVA combined with simvastatin (online supplemental figure 7). Finally, we evaluated the combination of simvastatin with an MVA encoding an endogenous retroviral tumor-associated antigen such as gp70. The antitumor effect of intraperitoneal administration of simvastatin and the peritumoral administration of an MVA-gp70 exerted a potent antitumor effect in the LLC-OVA model. The antitumor effect of the combination was superior to the effect of MVA-gp70 alone (online supplemental figure 8). Altogether, these results indicate that simvastatin modulates the inflammation triggered by the peritumoral administration of MVA-OVA and promotes the MVA-mediated antitumor effect.
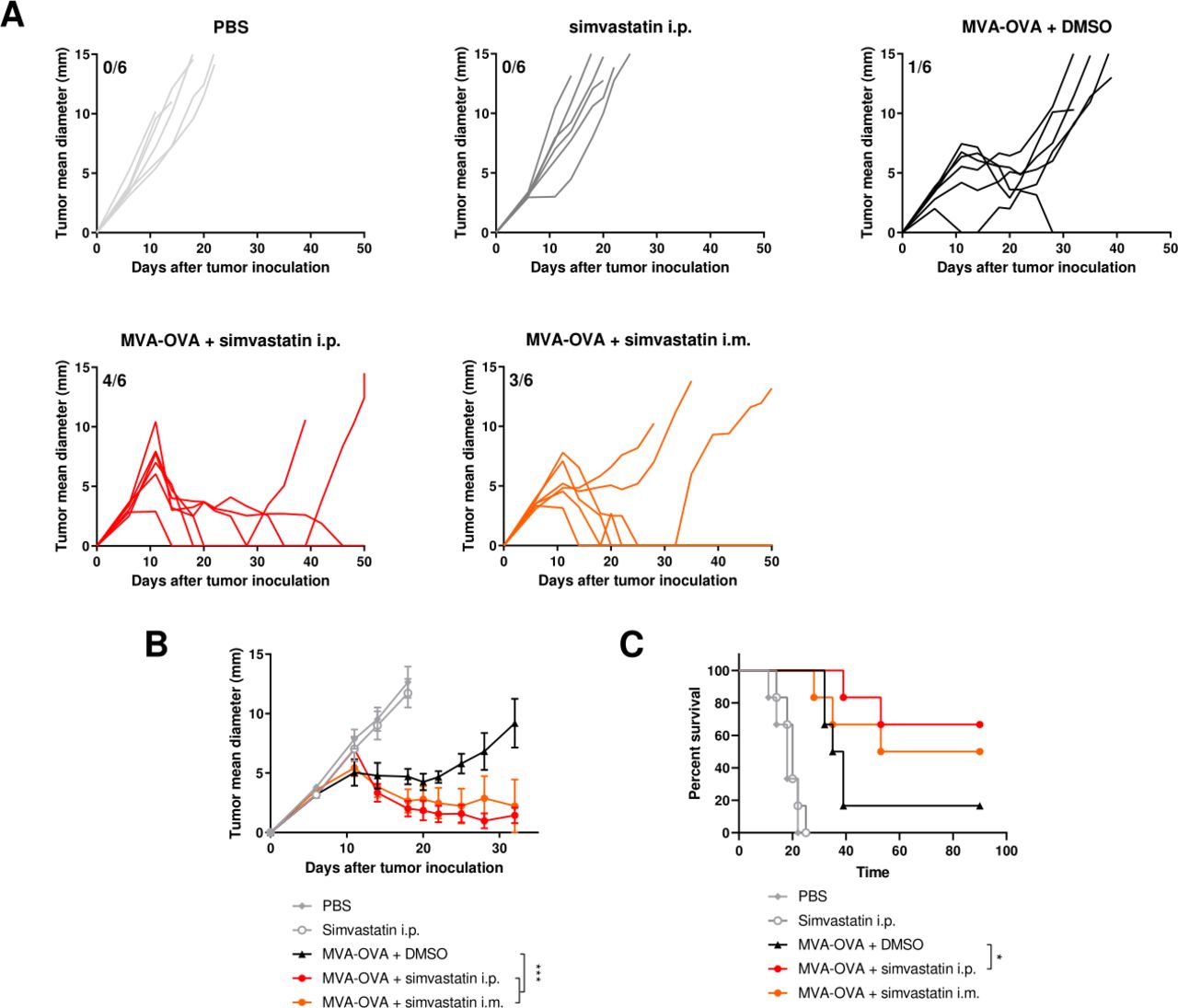
Simvastatin enhances the antitumor activity of MVA-OVA. B16-OVA melanoma cells were injected subcutaneously. Seven and 14 days later, mice were treated intraperitoneally with vehicle or simvastatin (20 µg/mice). After 2 hours, MVA-OVA (5×107 TCID50 per mouse) was administered subcutaneously in the peritumoral area. In the group treated intramuscularly with simvastatin, MVA vaccine was administered simultaneously (A) individual follow-up of mean tumor diameters indicating the fraction of mice completely rejecting established tumors. N=6. (B) Mean+SEM of different experimental groups. Data were fitted to a third order polynomial and compared using extra sum-of-squares F test with Bonferroni correction. ***P<0.0001. (C) Overall survival of the indicated treatment groups. Log-rank test with Benjamini-Hochberg correction. *P<0.05. B16-OVA, B16 melanoma expressing ovalbumin; i.p, intraperitoneally; MVA, modified vaccinia virus Ankara.
MVA-OVA combined with simvastatin enhances immune infiltration in the tumor microenvironment
To further understand the impact of simvastatin on the antitumor effect of MVA-OVA, we used flow cytometry to evaluate the presence of different immune populations in cell suspensions from spleens, tumor-draining lymph nodes, and tumors (figure 5A) following the gating strategy depicted in online supplemental figure 9). B cells, CD4+ lymphocytes, CD8+ lymphocytes, and tumor-specific CD8+ lymphocytes were increased in the tumor-draining lymph node in those animals treated with MVA-OVA alone or with the combination (figure 5C). However, a marked difference between both experimental groups was observed both in the tumor tissue microenvironment and in the spleen (figure 5B,D). While MVA-OVA tended to decrease these immune populations as compared with PBS-treated mice, MVA-OVA combined with simvastatin dramatically increased all these immune populations in the spleen (figure 5B) and also increased the infiltration by B cells, CD8+ T lymphocytes, and, more importantly, tumor-specific CD8+ T lymphocytes into the tumor tissue (figure 5D). To study whether the immune response elicited by the combined treatment was restricted to the MVA-encoded tumor-associated antigens, we evaluated antigen spreading using an IFNγ ELISpot assay. We detected potent immune responses against an immunodominant OVA epitope and the melanoma cells expressing the OVA protein on treatment with MVA or MVA+ simvastatin. However, no response was detected against the parental melanoma cells that do not express the OVA proteins (online supplemental figure 10).
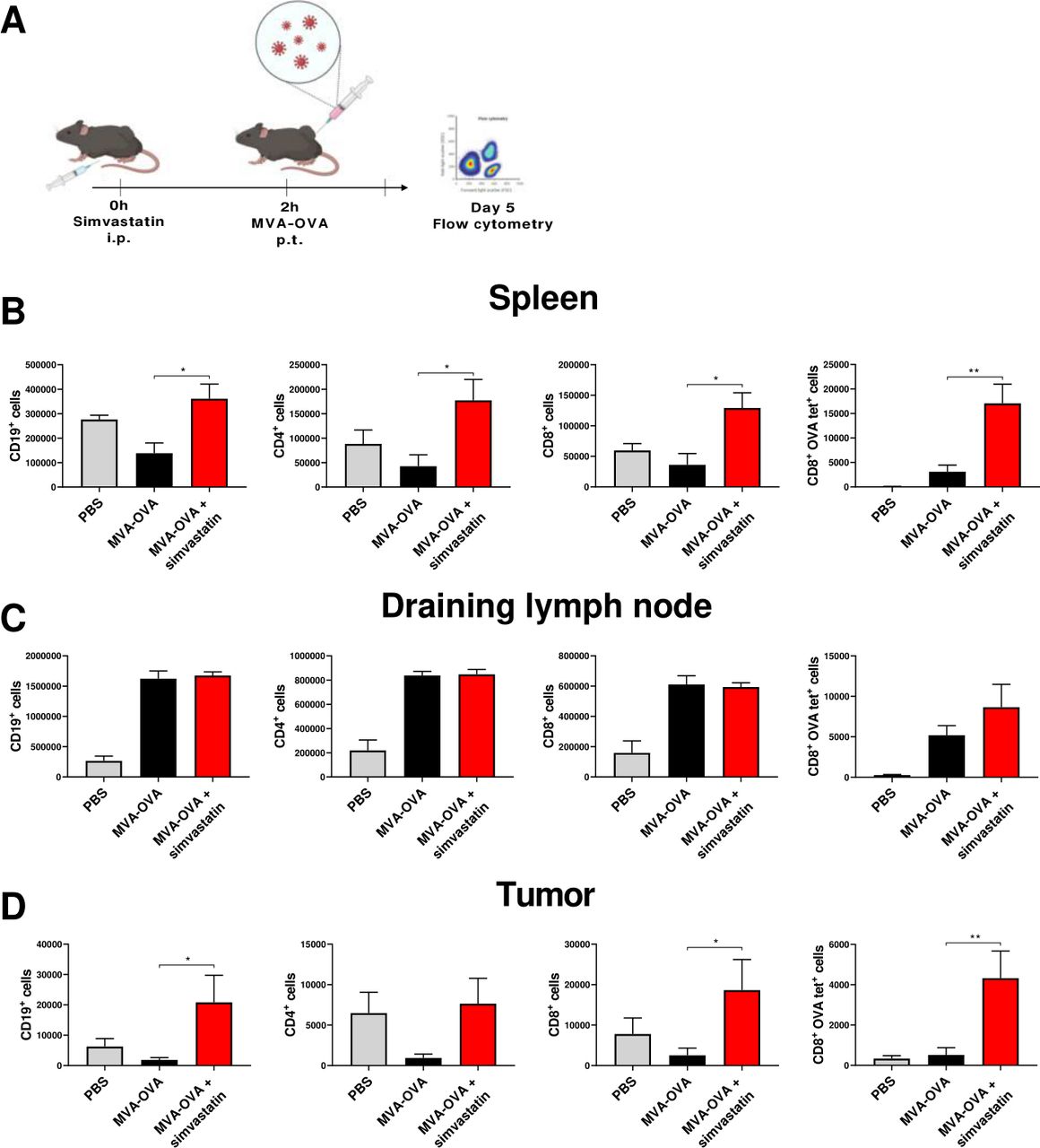
Lymphocyte changes in the tumor-draining lymph nodes and in the tumor microenvironment in response to MVA-OVA combined with simvastatin. B16-OVA melanoma cells were injected subcutaneously. Seven days later, mice were treated intraperitoneally with vehicle or simvastatin (20 µg/mice), after 2 hours, MVA-OVA (5×107 TCID50 per mouse) was administered subcutaneously in the tumor area. Five days later, mice were sacrificed, and immune cells in the spleen, tumor-draining lymph node, and tumors were analyzed by flow cytometry. (A) Schematic representation of the experimental setting. Graphs represent the absolute number of CD19+ cells, CD4+ lymphocytes, CD8+ lymphocytes, and ova tetramer+ CD8+ lymphocytes in spleens (B); in draining lymph nodes (C); and in the tumor microenvironment (D). N=4. One-way ANOVA followed by Sidak’s post-test. *P<0.05, **p<0.01. ANOVA, analysis of variance; B16-OVA, B16 melanoma expressing ovalbumin; MVA, modified vaccinia virus Ankara; OVA, ovalbumin.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
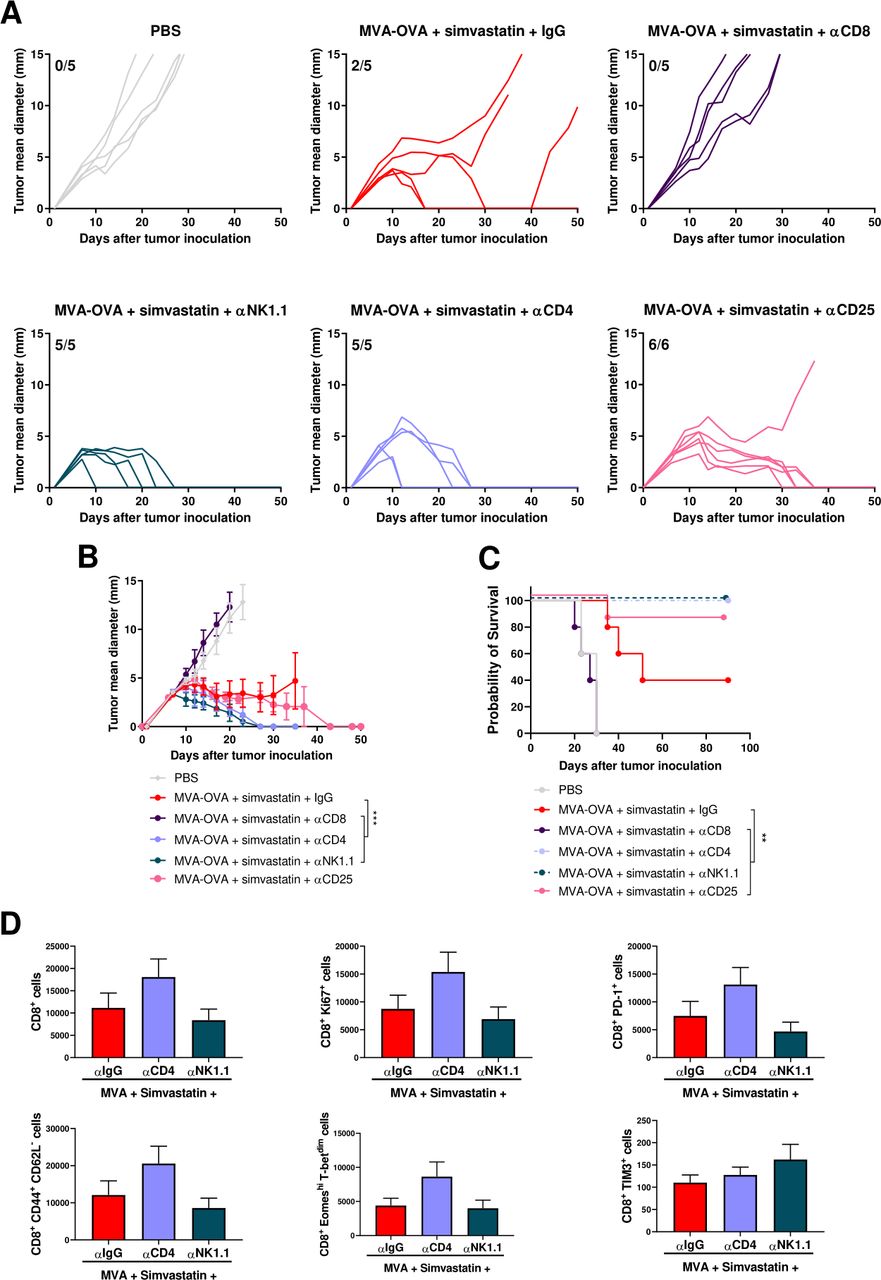
CD8+ T lymphocytes are required for the antitumor activity of MVA-OVA combined with simvastatin, whereas CD4+ T lymphocytes and NK cells are detrimental. B16-OVA melanoma cells were injected subcutaneously. Seven and fourteen days later, mice were treated intraperitoneally with vehicle or simvastatin (20 µg/mice), after 2 hours, MVA-OVA (5×107 TCID50 per mouse) was administered subcutaneously in the tumor area. Anti-CD8 mAb, anti-CD4 mAb or anti-NK1.1 (200 µg/mice) were administered intra-peritoneally 1 day before first therapeutic treatment administration and on days +2, +6, +9 and +13. Anti-CD25 (250 µg/mice) was administered intra-peritoneally 1 day before first therapeutic treatment administration. (A) Individual follow-up of mean tumor diameters indicating the fraction of mice completely rejecting established tumors. N=5 and 6 in the group treated with anti-CD25 (B) Mean+SEM of the different experimental groups. data was fitted to a third order polynomial and compared using extra sum-of-squares F test with Bonferroni correction. ***p<0.0001. (C) Overall survival of the indicated treatment groups. Log-rank test with Benjamini-Hochberg correction. **p<0.01. (D) B16-OVA tumor-bearing mice were treated as described above and 15 days after, immune infiltration in the tumor microenvironment was analyzed by flow cytometry. N=6. One-way ANOVA followed by Sidak’s post-test. No significant differences were detected. ANOVA, analysis of variance; B16-OVA; B16 melanoma expressing ovalbumin; MVA, modified vaccinia virus Ankara.
To evaluate the immune cells involved in the antitumor activity of the combination of MVA-OVA and simvastatin, we selectively depleted CD8+ T lymphocytes, CD4+ T lymphocytes, conventional CD4+CD25+FoxP3+ T regulatory cells or NK cells with monoclonal antibodies following administration of the combined systemic statin and peritumoral MVA-OVA treatment. The antitumor effect was dependent on CD8+ T lymphocytes as depletion of these effector immune cells completely abrogated the antitumor effect (figure 6A–C). Interestingly, CD4+ T lymphocytes and NK cells were detrimental for the antitumor effect and depletion of these immune cells led to the complete eradication of the implanted tumors in all cases (figure 6A–C). Depletion of CD4+ T lymphocytes further enhanced the tumor-infiltration of CD8 T cells and the activation of these intratumoral T lymphocytes. Depletion of NK cells maintained the tumor infiltration promoted by the combination of MVA and simvastatin (figure 6D and online supplemental figure 11). The main CD4+ T lymphocyte subset involved in the mechanism of action of the combined treatment was mainly conventional T regulatory cells as specific depletion of this cell population using anti-CD25 mimicked the effect observed with the anti-CD4 monoclonal antibody (figure 6A–C). IL10 is one of the mediators of the immunosuppressive activity of T regulatory cells.35 Frequent administration of a neutralizing antibody failed to enhance the antitumor activity of combined treatment, indicating that IL10 is not the main mediator of the T regulatory cells in this system (online supplemental figure 12). Finally, a B cell depleting antibody against CD19 was used to interrogate the relevance of the upsurge of B cells in the spleen and tumors treated with the simvastatin +MVA OVA combination. Depletion of this cell population slightly reduced the tumor growth rate but failed to enhance survival, excluding a predominant role of B cells in the mechanism of action of the combination (online supplemental figure 12).
Discussion
Monoclonal antibodies that block the programmed death-1 (PD1)/programmed death-1 ligand 1 (PD-L1) pathway have demonstrated the clinical usefulness of the derepression and normalization of the immune response in the tumor microenvironment.36 Objective clinical responses to these antibodies are characterized in a fraction of patients by long-term tumor growth control and manageable immune-related adverse effects.36 Revolutionary progress based on such approaches calls for clinical development of other immunotherapies designed to enhance effector immune responses against cancer. We have focused our attention on MVA, a viral vector vaccine in clinical development as an anti-cancer drug. The attenuation process through the passage in chicken embryo fibroblasts led to the deletion of several soluble and intracellular factors that block the type I IFN response.37 As a result of these safety reassuring disabilities, MVA is able to induce the release of IFN-beta and alpha. Several genetic strategies have been developed to block MVA-mediated type I IFN expression but the safety of these MVA vectors for in vivo use might be compromised.37 38 Here, we aimed to find a clinically available drug that might increase MVA immunogenicity by transiently blocking the activity of type I IFN. We analyzed several compounds for their ability to reduce the surface levels of IFNAR1 and to block clathrin-mediated endocytosis. Simvastatin and atorvastatin were able to decrease those processes and effectively attenuated type I IFN signaling induced by recombinant IFNα or MVA. Interestingly, simvastatin has been shown to exert a potent adjuvant effect mediated by the blockade of geranylgeranylation of Rab5.25 Here, we demonstrate that simvastatin also enhances antigen presentation in DCs infected by MVA and may underline the early increase of tumor-specific T lymphocytes. Indeed, the antitumor activity of the combination of MVA-OVA and simvastatin relies entirely on CD8+ T lymphocytes, according to depletion experiments. Further experiments should be performed to evaluate the establishment of a memory immune response. Interestingly, MVA-OVA reduced the tumor infiltration of immune cells, perhaps due to the cytotoxic activity of a high local concentration of type I IFN and other proinflammatory mediators.39 40 Thus, transient type I IFN blockade may overcome this early detrimental effect, promoting the infiltration of tumor-specific T cell initiated by the simvastatin-mediated prolonged antigen presentation. In contrast to the beneficial effect of CD8+ T lymphocytes, CD4+ T lymphocytes, conventional T regulatory cells, and NK cells were detrimental for the final outcome of the combined treatment. Although the mechanisms are yet unclear, we have come across similar findings using intratumoral injections of Semliki forest virus vectors expressing sFLT-3L and the vaccine strain of yellow fever.7 41 Both immune NK and CD4+ T cell populations are potent producers of proinflammatory cytokines such as IFNγ and TNF that might induce activation-induced cell death of effector CD8+ T lymphocytes as it has been previously reported for the combination of anti-CTLA-4 and anti-PD-1 monoclonal antibodies.40 42 In the case of anti-CD4 mAb, depletion of Treg cells is also to be considered as a potential explanation,28 and indeed administration of a single dose of anti-CD25 monoclonal antibody also enhanced the antitumor effect of MVA combined with simvastatin. These results might be of clinical relevance since a defucosylated humanized anti-CD4 depleting antibody and several T regulatory depleting monoclonal antibodies are currently being tested in clinical trials.43 44 Regarding the NK1.1 depletion, NK-cell lysis of CD8+ lymphoblast undergoing activation could also be related to the observations.45–47 Indeed, NK cell elimination of CD8+ T cell to regulate exacerbated CD8+ T cell responses has already been reported in several viral infection models,48 49 and could explain the increase in the antitumor efficacy of MVA-OVA in the absence of NK cells. Many mechanisms have been attributed to the control of CD8+ T cells, such as the release of immunosuppressive cytokines like IL-10 and/or the cytolytic protein perforin,48 as well as the recognition of NKG2D ligands and/or Fas on activated CD8+ T cells.47 50
These findings illustrate the complexities of exploiting the antitumor potential of IFNα. Recombinant IFNα was among the first cancer immunotherapies approved for clinical use. This milestone was supported by robust preclinical evidence in preclinical models.15 However, several drawbacks have reduced the clinical use of recombinant IFNα. High, repeated administration of IFNα is associated with frequent adverse effects, including influenza-like symptoms, hematological and neurological toxicities.51 Moreover, although low local concentrations of IFNα are critical for the activation of adaptive antitumor immune responses,52 chronic IFNα unleashes immunoregulatory mechanisms such as upregulation of PD-L1 and indoleamine 2,3-dioxygenase expression in tumor cells, CD8 T cell exhaustion or deletion, and B cell dysfunction.53 To overcome these limitations, new targeted formulations of IFNα or drugs that induce physiological expression of endogenous IFNα such as TLR3 ligand or STING agonists have been evaluated in preclinical models and clinical trials.15 54–56 The regulation of IFNα activity by statins is also a promising strategy to harness the antitumor potential of this cytokine. Previous clinical experience on the combination of recombinant IFNα and statins has led to conflicting reports.57–59 It is likely that timing between the statin and type I administration is critical for the outcome due to the transient effect observed in our in vitro model.
In conclusion, we have identified and preclinically repurposed a widely used drug that is able to potentiate the antitumor effect of an MVA encoding a tumor-associated antigen. The transient and partial inhibition of type I IFN effects of statins holds promise to be generalized to other virotherapy approaches. Indeed, our results propose a feasible and simple clinical intervention that may boost the clinical results of MVA-based vaccines undergoing or about to enter in clinical trials.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. Data are available on reasonable request.
Ethics statements
Ethics approval
The experimental design was approved by the Ethics Committee for Animal Testing of the University of Navarra (097–19).
Acknowledgments
We are grateful to Dr. Paul Miller for English editing, to the Genomics Unit of Cima Universidad de Navarra for RNAseq analysis, and to Gnomics Data Analysis for bioinformatics analysis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors ST, MV and PB designed experiments. ST, MV, MA, IO, MF-S, CADT, NA, CG, AB, and FA performed the experiments and processed samples. JM-E provided MVA-OVA. PB performed all statistical analyses. ST, MV, JM-E, IM and PB analyzed the data. ST, and PB wrote the manuscript. All authors performed a critical revision of the manuscript for important intellectual content and final approval of the manuscript.
Funding This study was supported by Instituto de Salud Carlos III (PI16/00668, PI19/01128, and PI20/00002) cofinanced by Fondos Feder, Gobierno de Navarra Proyecto LINTERNA Ref.: 0011-1411-2020-000075, and Joint Translational Call for Proposals 2015 (JTC 2015) TRANSCAN-2 (code: TRS-2016-00000371). This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 765394. ST received a grant from Gobierno de Navarra (code: 0011-1408-2016-000002). MA was supported by the Spanish Association against cancer research (AECC-2019 Investigator). FA receives a Miguel Servet I (CP19/00114) contract from ISCIII (Instituto de Salud Carlos III) co-financed by FSE (Fondo Social Europeo). AB is recipients of PFIS fellowship from ISCIII (FI20/00058), and MF-S is recipients for a fellowship of the Aid Program Assigned to Projects from the University of Navarra.
Competing interests IM reports advisory roles with Roche-Genentech, Bristol-Myers Squibb, CYTOMX, Incyte, MedImmune, Tusk, F-Star, Genmab, Molecular Partners, Alligator, Bioncotech, MSD, Merck Serono, Boehringer Ingelheim, Astra Zeneca, Numab, Catalym, Bayer and PharmaMar, and research funding from Roche, BMS, Alligator and Bioncotech. PB reports advisory roles with Ferring, Tusk and Moderna, research funding from Sanofi, Ferring, and Bavarian Nordic and speaker honoraria from BMS, MSD, Novartis, Boehringer Ingelheim, and AstraZeneca. JM-E is an employee of Bavarian Nordic. The rest of the authors have no conflict of interest to declare.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Oncolytic and local immunotherapy