Article Text

Abstract
Background Tumor-associated macrophages (TAMs) play a key immunosuppressive role that limits the ability of the immune system to fight cancer and hinder the antitumoral efficacy of most treatments currently applied in the clinic. Previous studies have evaluated the antitumoral immune response triggered by (TLR) agonists, such as poly(I:C), imiquimod (R837) or resiquimod (R848) as monotherapies; however, their combination for the treatment of cancer has not been explored. This study investigates the antitumoral efficacy and the macrophage reprogramming triggered by poly(I:C) combined with R848 or with R837, versus single treatments.
Methods TLR agonist treatments were evaluated in vitro for toxicity and immunostimulatory activity by Alamar Blue, ELISA and flow cytometry using primary human and murine M-CSF-differentiated macrophages. Cytotoxic activity of TLR-treated macrophages toward cancer cells was evaluated with an in vitro functional assay by flow cytometry. For in vivo experiments, the CMT167 lung cancer model and the MN/MCA1 fibrosarcoma model metastasizing to lungs were used; tumor-infiltrating leukocytes were evaluated by flow cytometry, RT-qPCR, multispectral immunophenotyping, quantitative proteomic experiments, and protein–protein interaction analysis.
Results Results demonstrated the higher efficacy of poly(I:C) combined with R848 versus single treatments or combined with R837 to polarize macrophages toward M1-like antitumor effectors in vitro. In vivo, the intratumoral synergistic combination of poly(I:C)+R848 significantly prevented tumor growth and metastasis in lung cancer and fibrosarcoma immunocompetent murine models. Regressing tumors showed increased infiltration of macrophages with a higher M1:M2 ratio, recruitment of CD4+ and CD8+ T cells, accompanied by a reduction of immunosuppressive CD206+ TAMs and FOXP3+/CD4+ T cells. The depletion of both CD4+ and CD8+ T cells resulted in complete loss of treatment efficacy. Treated mice acquired systemic antitumoral response and resistance to tumor rechallenge mediated by boosted macrophage cytotoxic activity and T-cell proliferation. Proteomic experiments validate the superior activation of innate immunity by poly(I:C)+R848 combination versus single treatments or poly(I:C)+R837, and protein–protein-interaction network analysis reveal the key activation of the STAT1 pathway.
Discussion These findings demonstrate the antitumor immune responses mediated by macrophage activation on local administration of poly(I:C)+R848 combination and support the intratumoral application of this therapy to patients with solid tumors in the clinic.
- immunity
- innate
- macrophages
- lung neoplasms
- immunotherapy
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Tumor-associated macrophages (TAMs) accumulate at high density in solid tumors and are key drivers of tumor progression. TAMs sustain immunosuppression in the tumor microenvironment (TME) in a variety of ways1 2 and hinder the antitumoral efficacy of most treatments currently applied in clinical oncology: conventional chemotherapy, antiangiogenic drugs, radiotherapy, and immune checkpoint blockade (ICB).3–6 However, a key feature of macrophages is their phenotypical and functional plasticity, which called attention to TAMs as promising targets for therapeutic intervention based on their repolarization toward an ‘M1-like’ penotype, with antitumor functions, including direct killing of tumor cells and improvement of adaptive immune responses.1 7 8
Toll-like receptors (TLRs) are pattern recognition receptors enriched in innate immune cells that, on engagement by their ligands, activate an immune response.3 9 In macrophages, TLR agonists stimulate an M1-like polarization, making them ideal compounds to reprogram TAMs.10 Despite promising results, their use in the clinic has been limited, as the systemic administration of TLR agonists is burdened with toxicity.11 To avoid adverse effects, intratumoral local immunotherapy has recently gained interest for cancer treatment while limiting systemic toxicity.12 Nowadays, imiquimod (R837, TLR7 agonist) is the only TLR agonist approved by the Food and Drug Administration for topical administration in squamous and basal cell carcinoma.13 However, tumor progression and chemotherapeutic resistance in murine lung adenocarcinoma treated with R837 were reported.14 Early clinical trials are undergoing for poly(I:C) (TLR3 agonist) and resiquimod (R848, imidazoquinoline analog of R837, TLR7/8 agonist).13 Intratumoral injection of poly(I:C) nanocomplexes is being evaluated in clinical trials in combination with anti-PD-1 therapy in patients with aggressive solid tumors,15 while its analog, poly-ICLC (polyinosinic-polycytidylic acid mixed with the stabilizers carboxymethylcellulose and polylysine), has been evaluated as a cancer vaccine.16 R848 has attracted much attention in the past few years for its stronger antitumor activity versus R83717 and has been recently evaluated using R848-loaded cyclodextrin nanoparticles to increase its delivery to TAMs in vivo.18 Despite satisfactory results, these studies have explored only the antitumoral efficacy of single TLR ligands.9 13 In the context of viral and bacterial infections, multi-TLR challenge has led to improved innate immune responses in vitro.19 Thus, the objective of this study was to evaluate the benefit of employing combinations of TLR3 and TLR7/8 agonists over monotherapies and to characterize the induced reprogramming in the TME.
Methods
Leukocyte isolation and differentiation
Human primary monocytes were purified from blood of healthy donors as described,10 differentiated in vitro toward M0 macrophages with recombinant human macrophage colony-stimulating factor (M-CSF) (Peprotech, 25 ng/mL) for 5 days in 5% FBS/RPMI 1640 0.5% penicillin/streptomycin (Gibco) and polarized toward M1 phenotype with lipopolysaccharide (Peprotech, 100 ng/mL) and interferon (IFN)-γ (Peprotech, 50 ng/mL) or M2-phenotype with interleukin (IL)-4 (Peprotech, 20 ng/mL) for 24 hours. Primary murine dendritic cells (DCs) were generated from bone marrow progenitors and were plated at a concentration of 5×106/mL in RPMI 10% FBS with murine granulocyte-macrophage colony-stimulating factor (mGM-CSF) (Peprotech, 50 ng/mL) and 0.5% penicillin/streptomycin (Gibco) for 6 days. At day 3, fresh culture media were added to DCs.
Tumor cell lines
Human pancreatic carcinoma cell line PANC1 (ATCC), murine fibrosarcoma cell line MN/MCA1, and murine lung adenocarcinoma cell line CMT167 (ECACC) were cultured in DMEM (Lonza) with 10% FBS and 0.5% penicillin/streptomycin (Gibco) at 37°C and 5% CO2.
Drugs and reagents
Poly(I:C) HMW (catalog# tlrl-pic), R848 (catalog# tlrl-r848) and R837 (catalog# tlrl-R837) were purchased from InvivoGen and prepared following manufacturer’s indications.
Quantification of cytokine secretion in vitro
Cytokine production was measured by ELISA kits (human CXCL10, CCL5, and IL-10) according to the manufacturer’s instructions (R&D Systems) using the supernatants collected 24 hours after each treatment.
Evaluation of nuclear factor-kappa B (NF-κB) signalling pathway activation in THP-1 cells
THP1-Lucia cells and QUANTI-Luc, luciferase detection reagent, were acquired from InvivoGen. The reporter THP1-Lucia cells have been specifically designed for monitoring the activation of the NF-κB signal transduction pathway in a physiologically relevant monocytic cell line by quantification of secreted luciferase (Lucia). Cells were cultivated in RPMI 1640, 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated fetal bovine serum, 100 µg/mL Normocin, 200 µg/mL Zeocin, and Pen-Strep (100 U/mL–100 µg/mL). The cells were treated with drugs at indicated concentrations for 16 hours, and following manufacturer’s protocols, NF-κB activation was quantified by determining Lucia luminescence and represented as relative luminescence units (RLUs). Fold induction was calculated with the following equation:

Cytotoxic activity of primary human macrophages or murine-derived splenocytes toward cancer cells in vitro
The killing of human cancer cells (PANC1) by pretreated human primary macrophages was evaluated as described.10 For the murine splenocytes, spleens were collected from tumor-rejected or naïve C57BL/6 mice after sacrifice, smashed, filtered (0.7 µm), and treated with ammonium-chloride-potassium (ACK) lysis buffer (Lonza, 3 mL/spleen). Macrophages or splenocytes were cocultured for 2 days with (2.5×104) PANC-1 or CMT167 (1×105) cells, respectively, previously stained with CellTrace Far Red 1 mM (Invitrogen). Then cells were harvested with trypsin (Sigma-Aldrich) and fixed with 1% paraformaldehyde-phosphate-buffered saline (PFA-PBS) for fluorescent activated cell sorter (FACS) analysis using a FACS Canto II (BD Biosciences). For the analysis, high-fluorescent intensity events (corresponding to proliferating cancer cells) were counted for 45 s of acquisition. The values were normalized to non-treated M0 macrophages or splenocytes from healthy mice cocultured with cancer cells, where no cytotoxicity was observed. The following equation was used:

Animals and in vivo experiments
All experiments were performed in compliance with national (D.L.N. 26, G.U. March 4, 2014) and international law and policies (EEC Council Directive 2010/63/EU, OJL 276/33, 22-09-2010; NIH Guide for the Care and Use of Laboratory Animals, US National Research Council, 2011). Authorization was obtained from the Italian Ministry of Health number 453/2020-PR (prot. 6B2B3.103). C57BL/6 mice were purchased from Charles River Laboratories and maintained in specific-pathogen-free (SPF) facility. To generate the subcutaneous lung cancer models, 105 CMT167 cells were implanted in the right flank of C57BL/6 mice or in the left flank for rechallenge experiments, or simultaneously in both flanks for the two-tumor model. To generate the fibrosarcoma model, 105 MN/MCA1 cells were injected into the caudal thigh muscle. Tumor volume was measured with a digital caliper using the following formula: tumor volume≈(width2×length/2). TLR agonists were administered by intratumoral injection (100 µL). Depletion of CD8+, CD4+ T cells and natural killer (NK) cells were achieved by intraperitoneal injection of 200 µg of antimouse CD8α (clone YTS 169.4), antimouse CD4 (clone GK1.5), and antimouse NK1.1 (clone PK136) antibodies, respectively (Bio X Cell). Rat IgG2a isotype (clone 2A3) antibodies were used as control (Bio X Cell). Survival was monitored daily, and tumor volume was measured until maximum allowed size or at the end of the protocol. Surface lung macrometastasis was manually counted on lungs fixed in Bouin’s solution.
Multiplexed tumor immunophenotyping
The murine-specific kit from Akoya (NEL840001KT) was used following the manufacturer’s instructions with additional markers. The kit includes the Alexa Fluor Tyramides Opals 520, 570, and 690, and spectral 4’,6-diamidino-2-phenylindole (DAPI). Opals 540 (FP1494001KT), 620 (FP1495001KT), and 650 (FP1496001KT) were from Akoya. Primary antibodies (Cell Signaling) and experimental details are found in online supplemental table 1. Sample scanning, spectral unmixing, and quantification of signals were conducted with Vectra Polaris Automated Quantitative Pathology Imaging System using the Phenochart and InForm V.2.4 softwares (Akoya). Data were given as number of cells with a specific immunophenotype/total number of cells.
Supplemental material
Flow cytometry
On sacrifice of mice, tumors were prepared for FACS analysis. Cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain (Invitrogen, 1:1000 in PBS −/−) for 30 min at room temperature (RT). Cells were then stained with the antibodies mix, provided in online supplemental table 2, in FACS buffer for 30 min at 4°C. Cells were washed with FACS buffer and fixed with FACS FIX Buffer (1% PFA PBS) for 20 min at 4°C until analysis. Cells were analyzed on FACS Canto II and LSR Fortessa, and data were generated by FACS Diva software (BD Biosciences).
Quantitative real-time PCR
Total RNA was purified from samples in Trizol using the Direct-zol RNA Miniprep Kits (Zymo-Research). cDNA synthesis was performed by random priming from total RNA with the High‐Capacity cDNA Reverse Transcription kit (Applied Biosystems) according to the manufacturer’s instructions. Real‐time PCR was performed with the Fast SYBR Green Master Mix (Applied Biosystems) and QuantStudio V.7 Flex Real Time PCR Systems (Applied Biosystems) in a total volume reaction of 10 µL. The list of primers used is provided in online supplemental table 2. Cycling conditions were 10 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C. For each sample, gene expression is normalized to GAPDH, and results are expressed as fold change to the control using the ΔΔCt method.
CD8+ and CD4+ T-cell proliferation assay
The proliferation capacity of CD8+ and CD4+ T cells was evaluated by a coculture experiment of splenocytes from tumor-rejected or naïve mice with naïve DCs (1:20 ratio, 72 hours) or DCs previously exposed to poly(I:C)+R848 for 16 hours. As control, untreated splenocytes and splenocytes treated with CD3/CD28 beads (Biolegend) were used. Splenocytes were marked with CellTrace Far Red. Cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain (Invitrogen, 1:1000 in PBS −/−) for 30 min at RT. Cells were then stained with anti-CD8 or anti-CD4 antibodies (1 µL each samples) for 30 min at 4°C in FACS buffer (1% FBS PBS–2 mM EDTA). Samples were analyzed on a FACS Fortessa (BD Biosciences). Live CD8+ or CD4+ cells were identified, and the proliferation cycles were defined using the proliferation tool in FlowJo V.10.
Protein extraction and digestion
Frozen murine lung tumor tissue (100 mg) was homogenized in radioimmunoprecipitation assay buffer (RIPA) with antiproteases and antiphosphatases (Sigma-Aldrich, St. Louis, Missouri, USA) in a TissueLyser II (Qiagen, Tokyo, Japan), centrifuged at 14 000 g 4°C for 20 min. Protein concentration was measured using a RC-DC kit (Biorad Lab, Hercules, California, USA). Protein aliquots (100 µg) were concentrated in a single band in a 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), cut and submitted to manual digestion. Finally, the peptides were dissolved in 0.1% formic acid for further analysis.
Generation of the reference spectral library
The spectral library was created using a pool of each treatment group, analyzed by shotgun data-dependent acquisition (DDA) approach by micro-LC-MS/MS (liquid chromatography with tandem mass spectrometry). Samples were separated via micro-LC system Ekspert nLC425 (Eksigen, Dublin, California, USA) using Chrom XP C18 (Eksigen) at a flow rate of 10 µL/min, and a gradient from 5% to 95% of ACN, 0.1% formic acid for a total time of 40 min. The MS coupled was a hybrid quadrupole-TOF (time of flight) mass spectrometer, 6600 (SCIEX, Framingham, Massachusetts, USA) operating with DDA system in positive ion mode. The false discovery rate (FDR) was set to 1 for peptides and proteins with a confidence score above 99%.
Quantification by SWATH and data analysis
The quantitative proteomic analysis was performed by SWATH (sequential window acquisition of all theoretical mass spectra) method in a hybrid quadrupole-TOF mass spectrometer, 6600 (SCIEX, Sciex) as previously described by our group.20 The SWATH–MS acquisition was performed using a DIA (data-independent acquisition mass spectrometry) method. Three biological replicates per group were analyzed. Four micrograms of protein was subjected to chromatographic separation as described.20 The spectral alignment and targeted data extraction were performed by PeakView V.2.2 (SCIEX), matching the reference spectral library with the following settings: 10 peptides/protein and seven fragments/peptide, excluded shared and modified peptides, and FDR below 1%. Protein quantification was performed with Markerview software (SCIEX). Student’s t-test was used to compare the groups in pairs to identify proteins differentially represented using a p value of <0.05 as cut-off.
Protein functional enrichment and network analysis
Differentially regulated proteins were subjected to open access bioinformatic tools. For functional enrichment and interaction network analysis, FunRich was used (http://funrich.org/index.html), implementing hypergeometric test, Benjamini-Hochberg (BH) and Bonferroni.21 For functional and protein–protein interaction network analysis, differentially regulated proteins were subjected to Cytoscape V.3.7 using the STRING app (https://cytoscape.org/).22
Statistical analysis
Statistical analysis were performed using GraphPad Prism V.8 (GraphPad Software). Description of the test is provided in each figure’s caption.
Results
Combination of TLR agonists poly(I:C)+R848 synergistically activates primary macrophages in vitro toward M1 antitumoral effectors
Using primary human macrophages, we first checked the potential toxicity of poly(I:C), R837 and R848, agonists of TLR3, TLR7, and TLR7/8, respectively, alone or in combination at different concentrations (5–50 µg/mL). At 24 hours, TLR agonists did not show significant toxicity, with the exception of a minor reduction in cell viability for R837 alone and in combination with poly(I:C) (poly(I:C)+R837) at the highest concentrations (figure 1A). No toxicity was found also at longer times toward macrophages for concentrations below 10 µg/mL (online supplemental figure 1A), neither toward murine lung cancer cells CMT167, used later for in vivo experiments (online supplemental figure 1B). Subsequent in vitro experiments were performed with 5 µg/mL of each drug for 24 hours.

Toxicological and immunomodulatory evaluation in vitro of intracellular TLR agonists alone or combined using primary human macrophages. Macrophages were in vitro differentiated from purified monocytes stimulated with 25 ng/mL of recombinant human macrophage colony-stimulating factor for 6 days (M0); M1 and M2 polarized macrophages were obtained by stimulation with 100 ng/mL of lipopolysaccharide+50 ng/mL of interferon-γ or 20 ng/mL of IL-4, respectively, for 24 hours. (A) Cell viability (Alamar blue) of M0 macrophages exposed 24 hours to TLR agonists alone or in different combinations. concentrations used were 5, 10, 20 and 50 µg/mL, represented by color gradient from left to right. (B–D) cytokine secretion (ELISA) of (B) CXCL10, (C) CCL5, and (D) IL-10 by macrophages exposed for 24 hours to 5 µg/mL of TLR agonists or untreated M0 macrophages. In each panel, M1 and M2 polarized macrophages from the same individuals are shown as reference populations. Each dot corresponds to macrophages from each blood donor. (E) THP-1-Lucia cells for monitoring the NF-kB signal transduction pathway were exposed for 16 hours to pIC and/or R848 at indicated concentrations. Bars represent mean±SD, n=3. (F,G) Cytotoxic activity of TLR-treated macrophages toward human Panc1 cancer cells stained with CellTrace. Each dot corresponds to macrophages from each blood donor. Bars represent mean±SEM. Statistical comparison was performed using one-way analysis of variance followed by Tukey’s multiple comparison test. Statistically significant differences are represented as follows: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.001. IL, interleukin; NF-kB, nuclear factor-kappa B; ns, non-significant; pIC, poly(I:C); R837, imiquimod; R848, resiquimod; TLR, toll-like receptor.
We next quantified by ELISA the production of proinflammatory chemokines (CXCL10 and CCL5), known to be involved in T-cell recruitment and efficiently produced by M1-polarized macrophages. Notably, the combination of poly(I:C) with R848 (poly(I:C)+R848) showed higher efficacy to trigger CXCL10 and CCL5 secretion than single treatments, and also in comparison with the combination with R837 (poly(I:C)+R837) (figure 1B,C). The anti-inflammatory cytokine IL-10 showed increased production in response to R837 and R848, but not to poly(I:C). The combination of poly(I:C) with either R848 or R837 did not show significantly higher secretion of IL-10 compared with the drugs alone: R848 (p=0.357) or R837 (p=0.955) (figure 1D). The synergistic effect in the production of proinflammatory mediators by poly(I:C)+R848 treatment was confirmed with bone marrow-derived murine macrophages for the secretion of CXCL10, as well as tumor necrosis factor (TNF)-α and nitric oxide (NO) (online supplemental figure 2A–C). The enhanced effect triggered by poly(I:C)+R848 is likely due to a more powerful activation of the NF-kB transduction pathway, compared with single-drug treatments, as observed using the THP-1-Lucia reporter cell system (figure 1E).
To better evaluate the M1 polarization induced by TLR agonists, we set up a functional assay of direct cytotoxic activity of macrophages against human cancer cells (PANC1), which are quantified by FACS at the end of the experiment (figure 1F). As shown in figure 1G, the poly(I:C)+R848 combination showed the highest boosting of macrophage cytotoxicity compared with single-TLR treatments or to the combination including R837 (poly(I:C)+R837). Using murine macrophages, we found that treatment with R848 or poly(I:C)+R848 combination induced a significant cytotoxicity against tumor cells (online supplemental figure 2D). Taken together, these findings demonstrate the advantage of the combination of poly(I:C) with R848, to polarize primary human and murine macrophages in vitro toward M1 antitumoral effectors.
Intratumoral administration of low-dose poly(I:C)+R848 combination induces synergistic antitumor efficacy in immunocompetent murine tumor models
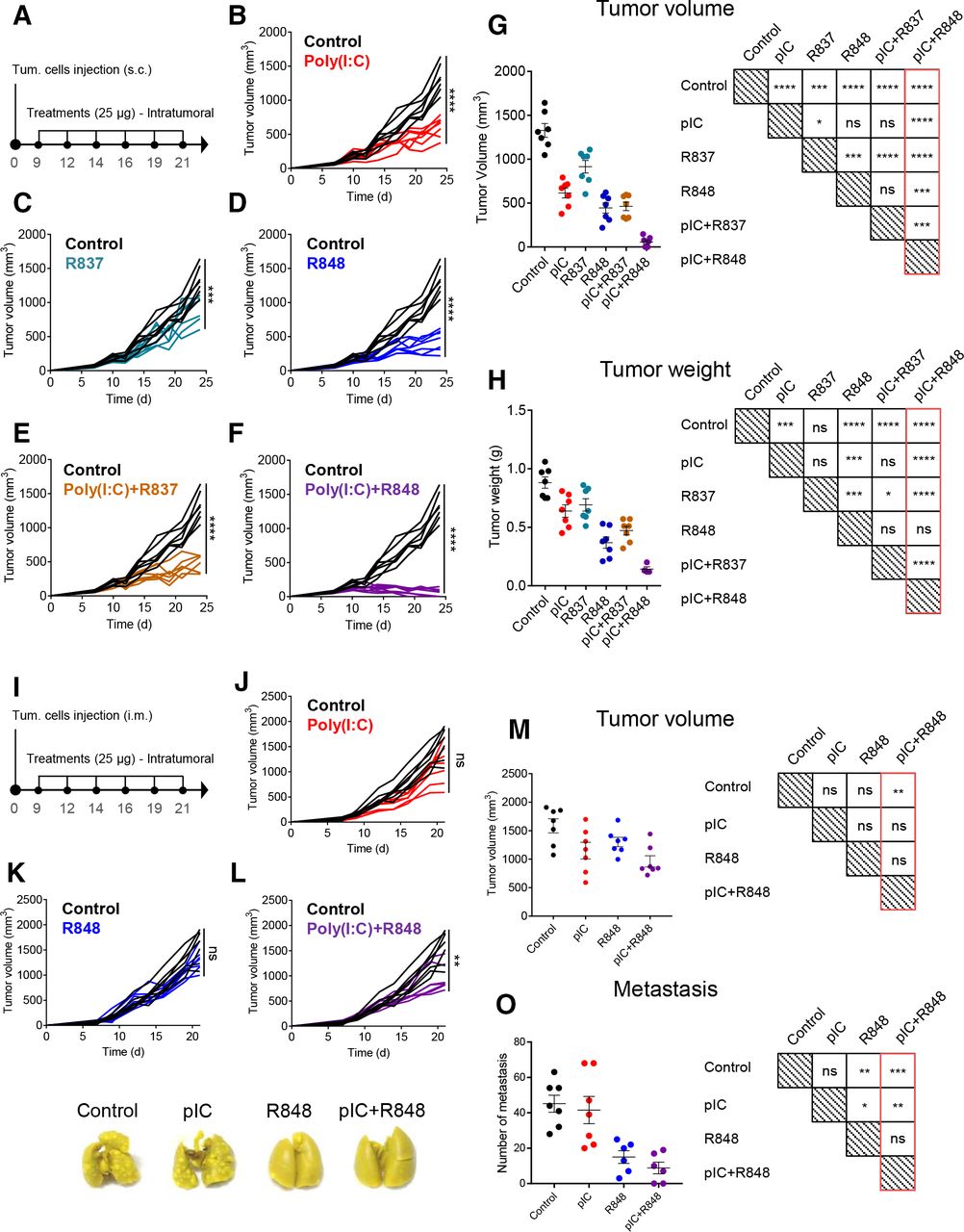
We tested the efficacy of the combination of TLR agonists to prevent tumor progression using an in vivo model of CMT167 murine lung cancer cells subcutaneously implanted in C57BL/6 fully immunocompetent mice (figure 2A). All TLR agonist treatments, intratumorally injected (25 µg, six times), showed significant reduction in tumor volume and weight compared with control mice (figure 2B–H). R837 monotherapy was the least effective (figure 2C,G,H), while poly(I:C) or R848 monotherapies showed a similar partial capacity to reduce tumor growth (figure 2B,D,G,H). No therapeutic improvement was observed for the poly(I:C)+R837 combination versus poly(I:C) alone (figure 2B,E,G,H), while a significantly improved synergistic efficacy of poly(I:C)+R848 was observed, achieving a 96% reduction in tumor volume versus control (figure 2F–H). From this group, three mice were randomly selected and followed up to 3 months, showing complete tumor eradication, while the other four mice were sacrificed, and histological examination at the tumor implantation site revealed a vast majority of fibrotic tissue with few foci of cancer cells (data not shown). Of note, doubling the dose of poly(I:C) from 25 µg to 50 µg did not improve its antitumoral activity (online supplemental figure 3A–E), demonstrating the requirement of TLR3+TLR7/8 agonists for the synergistic antitumoral activity, not achieved with increased drug concentration.

antitumoral and antimetastatic efficacy of intratumoral injections of pIC, R837 and R848, alone or combined, in the immunocompetent lung cancer murine model CMT167 and fibrosarcoma murine model MN/MCA1. (A) Schematic representation of the experimental protocol. CMT167 cells were injected subcutaneously in the flank. From day 9 to day 21, mice received 6 intratumoral injections of TLR agonists (25 µg) as monotherapy or in combination. Control mice received only saline. (B–F) Evolution of tumor growth of mice treated with (B) pIC, (C) R837, (D) R848, (E) combination of pIC+R837, and (F) combination of pIC+R848. (G) Comparison of tumor volume and (H) tumor weight at sacrifice and graphical visualization of the statistical differences (right). Representative experiment of two performed. (I) Schematic representation of the experimental protocol. MN/MCA1 cells were injected intramuscularly in the thigh. From day 9 to day 21, mice received six intratumoral injections of pIC and R848 (25 µg) as monotherapy or in combination. Control mice received only saline. (J–L) Evolution of tumor growth in mice treated with (J) pIC, (K) R848, or (L) pIC+R848. (M) Comparison of tumor volume and (N) the number of surface lung macrometastasis at sacrifice and graphical visualization of the statistical differences (right). (O) Representative pictures of lungs from each treatment group. Bars represent mean±SEM, n=7 per group. Statistical comparison was performed using one-way analysis of variance followed by Tukey’s multiple comparison test. Statistically significant differences are represented as follows: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.001 vs control. ns, non-significant; pIC, poly(I:C); R837, imiquimod; R848, resiquimod; TLR, toll-like receptor.
The murine fibrosarcoma MN/MCA1 is a fast-growing tumor spontaneously metastasizing to the lungs. MN/MCA1 cells were orthotopically (intramuscular) implanted and mice were administered with the same posology of poly(I:C)+R848 as indicated in figure 2I. Although tumor growth inhibition was less marked than in the CMT167 lung cancer model, poly(I:C)+R848 induced a significant antitumoral effect (figure 2J–M). At sacrifice, we evaluated the number of surface lung macrometastasis in the fibrosarcoma-bearing mice, showing strong metastasis reduction for poly(I:C)+R848 and also for R848 monotherapy, but not for poly(I:C) monotherapy (figure 2N,O). As a whole, these results demonstrate the best antitumoral and antimetastatic activity for the low-dose intratumoral poly(I:C)+R848 combination.
Safety of the intratumoral combination of poly(I:C)+R848
To evaluate the safety of the intratumoral TLR treatments, mice bearing CMT167 tumors were monitored until the end of the experiment (24 days). Satisfactorily, the weight of treated mice was not affected (online supplemental figure 4A,B). The circulating systemic levels of acute proinflammatory cytokines (TNF-α and IL-6) showed no significant changes (online supplemental figure 4C,D). The histopathological analysis of relevant tissues (liver, lung, spleen, kidney and heart) showed no alterations (results not shown). The spleens of mice treated with R848 alone or in combination with poly(I:C) showed a minor weight increase (online supplemental figure 4E). However, the flow cytometry evaluation of spleen immune cellular components (ie, macrophages, NK or T cells) showed no significant changes for any treatment (online supplemental figure 4F–K). As a whole, these results demonstrate the safe intratumoral/local administration of the combination treatment (poly(I:C)+R848) with effective antitumoral activity.
Intratumoral combination of poly(I:C)+R848 reprograms TAMs and elicits an adaptive T cell-mediated immune response in the TME
To evaluate the immune infiltration in the TME after different TLR treatments, we performed multiplex immunophenotyping (after six injections, denominated below long-term treatment, as described in figure 2A). This analysis showed a very significant increase in the amount of immune cells versus cancer cells (figure 3A,B) and the high number of macrophages (F4/80+) in animals treated with poly(I:C), R848 and poly(I:C)+R848, but not for R837 alone or in combination poly(I:C)+R837 (figure 3C). CD8+ T-lymphocyte infiltration was significantly higher for poly(I:C) or R848 monotherapies, not showing significant changes for the other TLR treatments (figure 3D). The unexpected low number of CD8+ cells for poly(I:C)+R848 treatment might be explained by the late time point of the evaluation and almost complete tumor eradication. For the most effective treatments, a higher infiltration of CD4+ T lymphocytes was found (figure 3E), and although not significant, the number of regulatory T lymphocytes (FOXP3+/CD4+) was reduced (figure 3F), revealing the dismantling of immunosuppressive cellular mediators in the TME of regressing tumors. This effect was not observed for R837 (figure 3F). For each individual tumor, the correlation between its volume and the immune populations is shown in online supplemental figure 5. The most effective treatments were characterized by a greater immune infiltrate (R2 of 0.52) (online supplemental figure 5A), high percentage of macrophages (R2 of 0.44), and CD4+ T cells (R2 of 0.62) (online supplemental figure 5B,D), but lower amount of immunosuppressive FOXP3 cells among the CD4+ cells (R2 of 0.17) (online supplemental figure 5E). The worst correlation for the FOXP3 population is in part due to the mixed results obtained for tumors treated with R837. No changes were found for the number of endothelial (CD31+) cells for any treatment (results not shown). Tumors treated with poly(I:C), alone or in combination with R848 or R837, presented a higher infiltration of CD11c+ cells (online supplemental figure 6A,B), although in a very low percentage compared with the macrophage populations (figure 3C). Representative pictures, from these multiplex analyses, show clear differences in the TME of tumors treated with the poly(I:C)+R848 combination versus the other treatments or control (figure 3A).

Multiplexed immunofluorescence analysis of immune infiltration in CMT167 tumors treated with TLR agonist monotherapies or combinations. Immunofluorescence analysis of CMT167-derived tumors at sacrifice, on intratumoral treatment with six injections of TLRs agonists, as indicated in figure 2A. (A) Representative images of tumors. (B–F) Quantification of (B) total immune cells, (C) macrophages (F4/80+), (D) CD8+ T cells, (E) CD4+ T cells, and (F) FOXP3+/CD4+ T cells in treated tumors. n=5 per group except pIC+R848, n=3. (G,H) Multiplexed immunofluorescence analysis of TAM polarization in tumors treated with six injections of pIC and R848. (G) Representative images of control and pIC+R848-treated tumors. (H) Ratio of M1 (F4/80+, iNOS+):M2 (F4/80+, Arg1+) macrophages. n=4 per group. Each dot corresponds to a single animal. Bars represent mean±SEM. Statistical comparison was performed using one-way analysis of variance followed by Dunnett’s multiple comparison test. Statistically significant differences are represented as follows: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 vs control. iNOS, inducible nitric oxide synthase; pIC, poly(I:C); R837, imiquimod; R848, resiquimod; TAM, tumor-associated macrophage; TLR, toll-like receptor.
This best combination was further evaluated for inducible nitric oxide synthase (iNOS) and Arg1 expression (as M1 and M2 polarization markers, respectively) (figure 3G,H). Compared with controls, higher expression of iNOS and lower of Arg1 were found in macrophages from treated tumors (figure 3G), showing a higher ratio of M1:M2 macrophages, indicative of the effective reprogramming of TAMs toward M1-like antitumor effector cells (figure 3H).
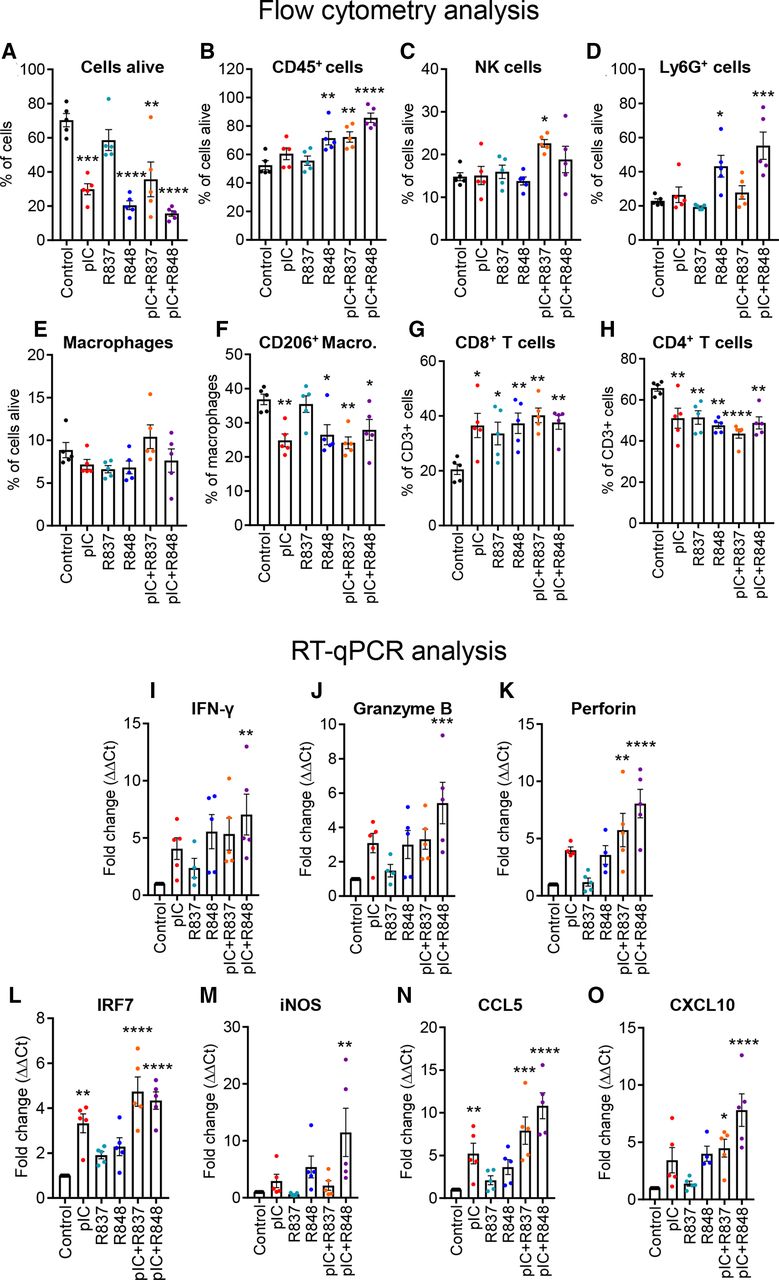
We next investigated the mechanism of action of the different TLR treatments at earlier time points. We performed a similar in vivo experiment and mice were treated with two injections (denominated below as short-term treatment) (online supplemental figure 7A). Even with only two treatments, tumor growth was significantly reduced with poly(I:C) and/or R848 (online supplemental figure 7B–G) but was not significant for R837 and very modest for poly(I:C)+R837 (online supplemental figure 7C,E). The flow cytometry analysis of tumors at sacrifice showed, as expected, a higher number of dead cells for the most effective treatments (figure 4A). Among cells alive, we found a significant increase of leukocyte infiltration (CD45+) in tumors treated with R848, poly(I:C)+R848, and poly(I:C)+R837 (figure 4B). No significant changes were observed for NK cells (figure 4C). In myeloid cells, a high increase in Ly6G+ cells was observed in mice treated with R848 or poly(I:C)+R848 (figure 4D); the percentage of macrophages did not significantly change in response to short-term TLR treatments, but among them, the ratio of M2-like macrophages (F4/80+/CD206+) decreased significantly in all, except for R837-treated tumors (figure 4E,F). The overall proportion of T cells in the tumor did not significantly change (data not shown). However, among T cells, the proportion of CD8+ cytotoxic T cells increased significantly in all TLR-treated tumors (figure 4G), while the proportion of CD4+ helper T cells was decreased (figure 4H). Although not significant, a trend toward an increase in classical DCs was observed after treatment with poly(I:C)+R837 (p=0.07) and poly(I:C)+R848 (p=0.06) (online supplemental figure 6C). The number of preplasmacytoid DCs was increased only in tumors treated with poly(I:C)+R837 (online supplemental figure 6D).

Profiling of immune cell populations and gene expression evaluation in the TME of the CMT167 tumors treated with TLR agonist monotherapies or combinations. CMT167 tumors were intratumorallly treated with two injections of TLR agonists as monotherapy or in combination. Intratumoral leukocytes were analyzed with flow cytometry for the proportion of (A) living cells (LIVE/DEAD Cell Stain) in total cells, (B) leukocytes (CD45+) in living cells, (C) NK cells (NK1.1+), (D) Ly6G+ cells (Ly6G+), (E) macrophages (CD11b+, F4/80+), (F) M2-type macrophages (CD206+) in total macrophages, (G) CD8+ T lymphocytes, and (H) CD4+ T lymphocytes in total T lymphocytes. (I–O) Gene expression analysis of tumors by RT-qPCR of (I) IFN-γ, (J) granzyme B, (K) perforin, (L) IRF7, (M) iNOS, (N) CCL5, and (O) CXCL10 in the TME. Results are expressed as fold change over control. Bars represent mean±SEM. Each dot corresponds to a single animal. n=5 per group. Statistical comparison was performed using one-way analysis of variance followed by Dunnett’s multiple comparison test. Statistically significant differences are represented as follows: *p<0.05), **p<0.01), ***p<0.001, and ****p<0.001 vs control. IFN, interferon; iNOS, inducible nitric oxide synthase; pIC, poly(I:C); R837, imiquimod; R848, resiquimod; TLR, toll-like receptor; TME, tumor microenvironment.
Gene expression of selected markers was evaluated by RT-qPCR in these tumors. The expression of IFN-γ, granzyme B, and perforin, associated with T-cell activation, was increased in tumors treated with poly (I:C)+R848 and was higher than the other treatments (figure 4I–K). Expression of IRF7 and iNOS, associated with M1 macrophage polarization, or CCL5 and CXCL10, implicated in recruitment of T cells, was also the highest in tumor treated with the poly (I:C)+R848 combination (figure 4L–O), reflecting the observation already made in vitro in human macrophages (figure 1B,C).
Thus, T-lymphocyte populations presented significant variations between the early and late stages of the tumor regression, while the infiltration of myeloid cells (F4/80+ or Ly6G+) and a higher macrophage M1:M2 ratio were clearly correlated with the most effective treatments.
Systemic response, activation of adaptive immunity, resistance to tumor rechallenge and antitumoral memory in mice treated with the intratumoral poly(I:C)+R848 combination
To study the systemic antitumoral efficacy of the intratumoral treatment, we implanted the CMT167 syngeneic tumors at two separate sites in the body (figure 5A), and we locally injected the poly(I:C)+R848 combination only in one tumor site. As a result, an abscopal antitumoral response was elicited in the distant, untreated tumor, revealing systemic antitumoral immunity (figure 5B). The poly(I:C)+R848 combination eliminated all treated tumors and delayed the growth of all distant, untreated tumors, even achieving the total eradication of both tumors in two out of five mice. To further understand the contribution of innate and adaptive antitumoral immune responses, we depleted both CD4+ and CD8+ T cells or NK cells in CMT167 tumor-bearing mice treated with intratumoral injections of poly(I:C)+R848, as indicated in figure 5C. While elimination of NK cells did not modify the efficacy of the treatment, depletion of both CD4+ and CD8+ T cells resulted in the complete loss of treatment efficiency (figure 5D).
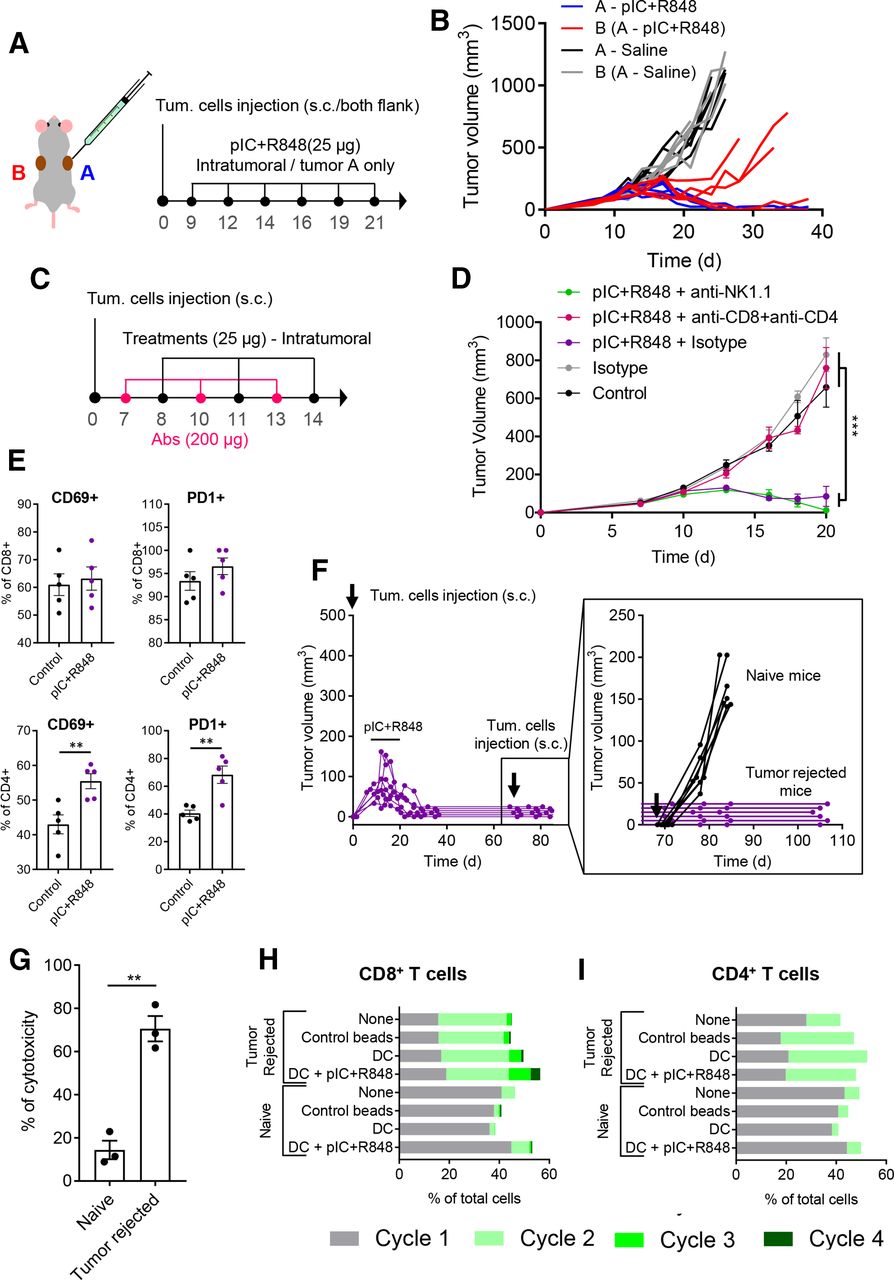
Intratumoral treatment with the pIC+R848 combination activates systemic and memory adaptive antitumor immune responses in mice rejecting tumors. (A,B) pIC+R848 treatment in a two-tumor model. (A) Schematic representation of the experimental protocol. CMT167 cells were subcutaneously injected in the right (A, in blue) and left (B, in red) flanks. From day 9 to day 21, mice received intratumoral injection of pIC+R848 combination (25 µg of each drug) only in one tumor (A). Control mice received only saline. (B) Evolution of each tumor growth (right tumor in blue (A) and left tumor in red (B)) in mice treated with pIC+R848 versus control. n=5 per group. (C,D) Tumor growth in mice depleted for CD4+ and CD8+ T cells or NK cells and treated with pIC+R848. Mean±SEM. n=7 per group. Statistical comparison was performed using one-way analysis of variance followed by Tukey’s multiple comparison test. Statistically significant differences are represented as ***p<0.001. (E) CMT167 tumors intratumorally treated with two injections of pIC+R848 were analyzed with flow cytometry for the proportion of CD69+ and PD1+ cells in CD4+ and CD8+ T cells. n=5 per group. Bars represent mean±SEM. Statistical comparison was performed using a t-test. Statistically significant differences are represented as **p<0.01. (F) Evolution of tumor growth in mice treated with six intratumoral injections of pIC+R848 combination, as indicated in figure 2A. Mice with no sign of tumor growth (tumor rejected mice) were rechallenged with CMT167 tumor cells at day 70 and did not receive any treatments afterward. Tumor growth of individual mice (inset) up to day 105 and comparison with a control group of naïve mice receiving tumor cell injection. (G) In vitro cytotoxicity of spleen macrophages derived from tumor-rejected or tumor-bearing naïve mice in coculture experiments with CMT167 lung cancer cells. Bars represent mean±SEM. Statistical comparison was performed using a t-test. Statistically significant differences are represented as **p<0.01. (H,I) Proliferation of (H) CD8+ or (I) CD4+ T cells from CellTrace stained splenocytes from tumor-bearing naïve or tumor-rejected mice, cocultured for 72 hours with syngeneic healthy naïve mice-derived DCs, which were previously exposed to CMT167 cell lysate with or without pIC+R848. Untreated splenocytes and CD3/CD28 beads were used as controls. DC, dendritic cell; pIC, poly(I:C); R848, resiquimod.
To further investigate T-cell activation, we performed a FACS analysis of tumors treated two times with the combination of poly(I:C)+R848. The results revealed that both CD4+ and CD8+ T cells expressed the activation markers CD69 and PD1. After treatment, the proportion of CD69+ and PD1+ was significantly increased in CD4+ T cells (figure 5E). No changes were observed for other activation markers (ie, CD25 and KLRG1) (online supplemental figure 8).
As complete tumor regression was observed in mice exposed to the intratumoral combination therapy (from two independent experiments figure 2F and online supplemental figure 3F–J), after day 70, cured mice were exposed to a secondary tumor challenge with CMT167 cells. None of the rechallenged mice developed tumors, suggesting that the animals have acquired an effective antitumoral memory (figure 5F). Thus, we evaluated the splenocytes from tumor-rejected mice or naïve mice in in vitro coculture experiments with CMT167 cells. Higher cytotoxic activity against cancer cells was observed from cured mice (figure 5G). T-cell proliferation was enhanced for both CD8+ and CD4+ cells derived from tumor-rejected mice compared with control cells from naïve mice (figure 5H,I); in addition, DCs pulsed with tumor cell lysate and poly(I:C)+R848 further enhanced the proliferation of CD8+ T cells from tumor-rejected mice. As a whole, these data reveal the implication of CD4+ and CD8+ T-cell activation in acute and long-lasting antitumoral responses induced by the poly(I:C)+R848 combination.
Proteomic analysis validates the superior activation of innate and adaptive immunity triggered by the poly(I:C)+R848 combination versus the other TLR agonist treatments
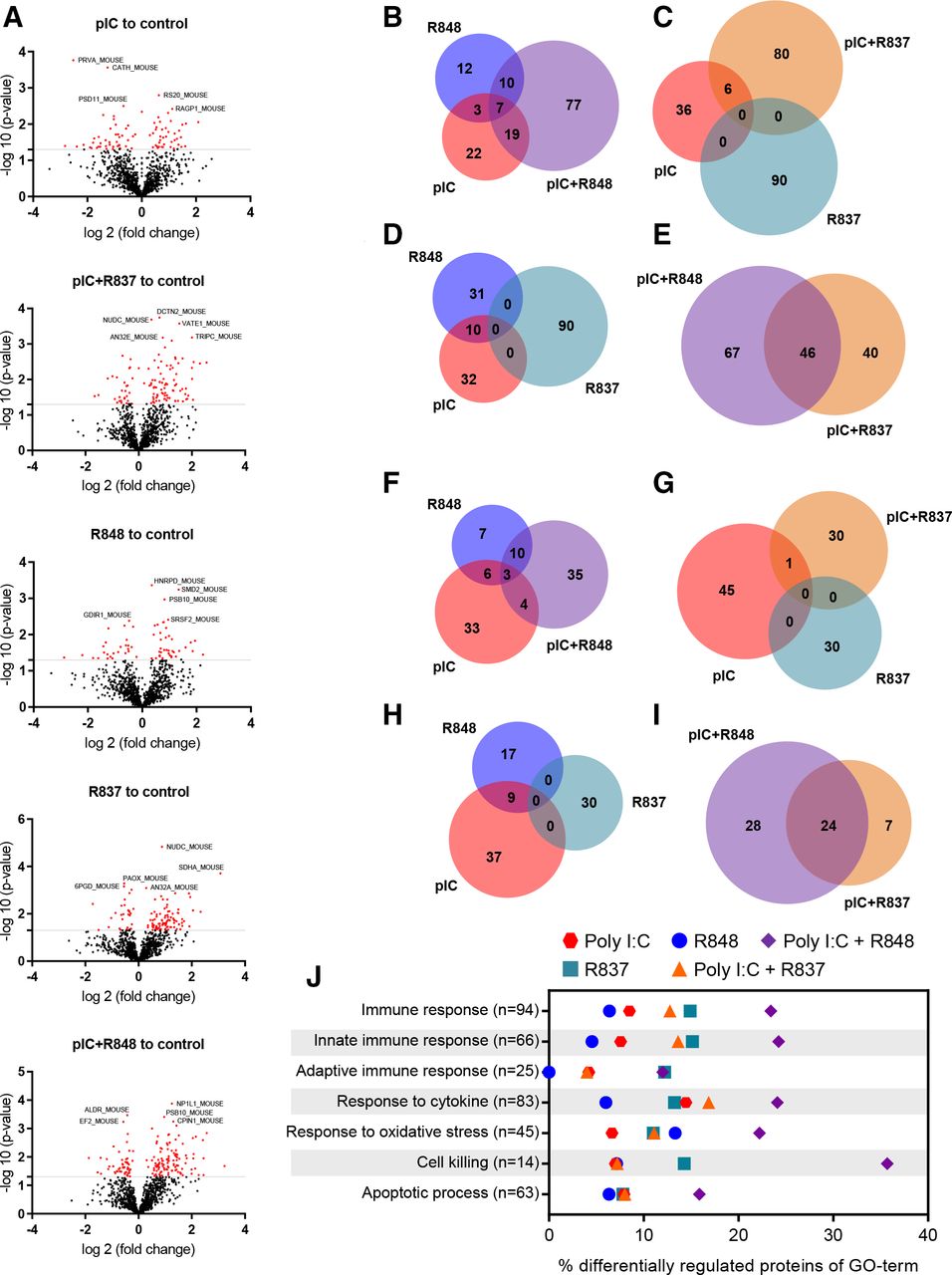
To study the impact on the tumor proteome of each TLR short-term treatment in vivo (online supplemental figure 7A), qualitative and quantitative analyses were performed. Volcano plots revealed the higher alterations in protein expression for poly(I:C)+R848-treated tumors: 16.8% of the identified proteome was affected, with 113 proteins upregulated and 52 downregulated (figure 6A and online supplemental tables 3,7 and 8). However, a significantly lower percentage of proteins was affected by poly(I:C)+R837 (11.9%), or by poly(I:C) or R848 monotherapies (8.9 or 6.83% respectively). Venn diagrams demonstrate the synergistic activity of poly(I:C)+R848 combination, with 78 proteins upregulated (figure 6B–E) and 35 downregulated (figure 6F–I) not affected by the single treatments. Furthermore, seven and three proteins were commonly upregulated and downregulated, respectively, by poly(I:C)+R848 and also by poly(I:C) or R848 monotherapies (figure 6B). A similar number of proteins were synergistically altered by poly(I:C)+R837, but none of them were altered by R837 monotherapy (figure 6C,G). Ten proteins were upregulated by both poly(I:C) or R848 (figure 6D) and nine proteins downregulated (figure 6H), but none of them were affected by the R837 monotherapy. The poly(I:C)+R848 shows some similarities with poly(I:C)+R837 (47 upregulated and 24 downregulated proteins in common), but a higher number of proteins were only affected by the most effective combination (figure 6E,I).

Quantitative proteomic analysis of antitumor immune responses triggered in tumors treated with TLR agonist monotherapies or combinations. Immunocompetent lung cancer murine CMT167 subcutaneous tumors were treated with two intratumoral injections of TLR agonists alone or in combination, as indicated in online supplemental figure 6A. (A) Volcano plots of the SWATH analysis of proteins comparing TLR-treated versus control tumors. X-axis shows log2 (fold change) and Y-axis shows the statistical significance through −log10 (p value). The gray lines represent the cut-off (p≤0.05). Significantly upregulated and downregulated proteins are indicated by red dots. (B–I) Quantitative Venn diagrams showing the number of (B–E) upregulated and (F–I) downregulated proteins versus control, found in the tumor samples of treated mice (p≤0.05). (J) Differential protein expression for GO terms of interest. Each row represents the proportion of significantly changed proteins (p<0.05) involved in the process compared with control. n indicates the number of measured proteins for the process. n=3 tumors per group of treatment. GO, Gene Ontology; pIC, poly(I:C); R837, imiquimod; R848, resiquimod; TLR, toll-like receptor.
The Gene Ontology (GO) analysis of quantitative proteomic data reveals a higher share of differentially regulated proteins related to activation of the immune response, and in particular to innate immunity, triggered by poly(I:C)+R848 (23.40% and 24.24%, respectively, compared with 8.51% and 7.57% for the control), and significantly higher than for any other TLR-treatment (figure 6J and online supplemental table 4). GO terms related to cell killing, response to cytokines, oxidative stress and apoptotic processes showed also the higher number of proteins affected by poly(I:C)+R848 (35.71, 24.09, 22.22%, and 15.87%, respectively) (figure 6J).
Similar enrichment analysis on the qualitative proteomic experiments confirms the higher activation of immune responses, both innate and adaptive, triggered by poly(I:C)+R848 versus any other TLR-treatments (online supplemental figure 9 and supplemental tables 5 and 6). Expression of proteins related to T cell-mediated cytotoxicity, positive regulation of cytokine secretion and T-cell migration was also higher in tumors treated with poly(I:C)+R848, while no significant changes were observed in cellular response to oxidative stress and apoptotic process (online supplemental figure 9 and online supplemental table 6).
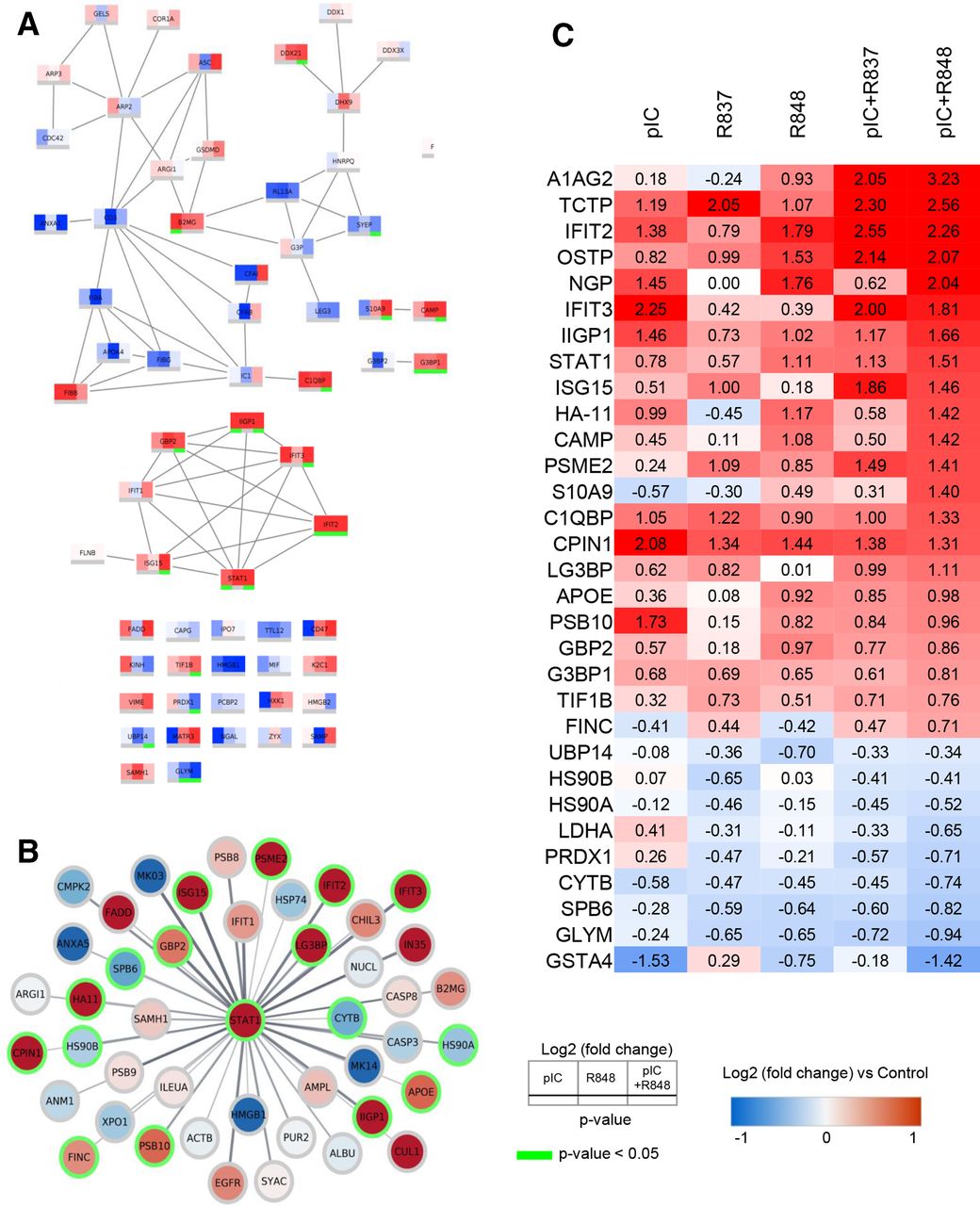
Protein–protein interaction network analysis of the innate immune response activation by poly(I:C)+R848 reveals the upregulation of a STAT1 cluster and other proteins involved in oxidative stress and cell killing processes
The analysis of proteins involved in the innate immune response triggered by poly(I:C)+R848 versus single treatments showed a superior activation of a STAT1 cluster for the combination (figure 7A): IIGP1, IFIT2, IFIT3, ISG15, GBP2, and STAT1 were significantly upregulated. An expanded network of all proteins, in our dataset, connected to STAT1 processes is visualized in figure 7B.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PPI network analysis and heatmap displaying the most significantly altered protein expression. (A) PPI network analysis of innate immune response, with a confidence score of >0.7, showing the differences in protein expression induced by pIC, R848 or pIC+R848 intratumoral treatments. (B) STAT1 centered PPI network, with a confidence score of >0.4, showing the proteins affected by the pIC+R848 combination therapy. (C) Heatmap table showing quantitative protein expression data for selected proteins. Red and blue indicate increased and decreased log2 (fold change) compared with control, respectively. Green color indicates significance (p<0.05). pIC, poly(I:C); PPI, protein–protein interaction; R837, imiquimod; R848, resiquimod.
Using stringApp V.1.5.1 in Cytoscape V.3.8.0 to create a high-confidence (>0.7) protein–protein interaction network for CXCL10 and CCL5, we have found that these proteins are clearly involved in the immune cluster of proteins activated by poly(I:C), R848 and poly(I:C)+R848 (online supplemental figure 10). This validates the correlation between our in vitro and in vivo data on the secretion of chemokines by TLR-treated macrophages. Finally, a heatmap with selected proteins upregulated/downregulated in tumors treated with poly(I:C)+R848, compared with the other TLR-treatments, is presented in figure 7C (full list of quantitative analysis in online supplemental tables 7 and 8).
STAT1 is an essential component of IFN signaling and a major regulator of the antitumor immune response, contributing to the transcription of proinflammatory cytokines,23 but also to apoptosis induction.24 The known ability of TLR agonists to activate STAT1 and related proteins23 was clearly evident in our dataset (figure 7C). R848 monotherapy showed a higher upregulation (log2(FC): 1.11) versus R837 or poly(I:C) (log2(FC): 0.57 and 0.78, respectively), and this activation was further enhanced by the poly(I:C)+R848 combination (log2(FC): 1.51) (figure 7C). Synergistic upregulation by poly(I:C)+R848 versus monotherapies was observed for cell killing and apoptotic-related proteins, such as A1AG2, IFIT2, IFIT3, and IIGP1 (figure 7C)25 26; for proteins with alarming functions: S10A9, CAMP, ISG15, and neutrophilic granule protein (NGP),27–29 and also for proteins involved in phagocytosis and antigen recognition processes: HA11, PSB10, and PSME2.30 31 In accordance with the high infiltration of Ly6G+ cells and macrophages (F4/80+), NGP was clearly upregulated by poly(I:C) or R848, but not altered by R837 (log2(FC): 1.45, 1.76, and 0.00, respectively), and synergistically upregulated by poly(I:C)+R848 (log2(FC): 2.04).
Downregulated proteins in tumors treated with poly(I:C)+R848 were also of interest because some of them may be involved in the process of dismantling the immunosuppression in the TME.3 The glutathione peroxidase GSTA4 and the antioxidant enzyme PRDX1, which can promote cancer proliferation and metastasis,32 were downregulated (figure 7C). Decreases in CYTB and SPB6, involved in preventing cancer progression,33 as well as lactate dehydrogenase A, a glycolytic enzyme that converts pyruvate to lactate and is associated with malignant progression,34 were observed. Similarly, low levels of serine hydroxymethyltransferase 2 (GLYM, log2(FC): −0.94), relevant for the metabolism of folate and the JAK2/STAT3 immunosuppressive pathway,35 were observed (figure 7C). Overall, the proteomic analysis confirms the superior activation of antitumor immune responses by poly(I:C)+R848, and delineates the modulation of proteins which are associated with cell-mediated cytotoxicity, phagocytosis, antigen presentation, cytokine secretion, and T cell-mediated activities.
Discussion
Local intratumoral administration of immunomodulatory drugs (ie, immune receptor agonists, monoclonal antibodies, cytokines, or oncolytic viruses), not only to treat primary tumors but also their metastasis,36 is gaining momentum and some of them have entered clinical trials.37 38 Among this plethora of therapies, the intratumoral administration of TLR agonists might be the simplest and most feasible pharmacological approach for clinical translation, also to treat patients with cancer resistant to current immunotherapeutic regimens (ie, anti-PD1).1 3 9 Despite good preclinical and clinical results using TLR agonists,13 16 comprehensive studies on their combination, synergy, and mechanistic activity in the context of cancer treatment are lacking.
Here, we provide data on the intratumoral administration of poly(I:C) (TLR3 agonist), R837 (TLR7 agonist), or R848 (TLR7/8 agonist), alone or in combination, using fully immunocompetent murine tumor models. The combination of poly(I:C)+R848 was more effective than a combination containing R837 (poly(I:C)+R837) to stimulate macrophage production of cytokines and cytotoxic activity in vitro, and had superior antitumor activity in vivo than single drugs. Mice treated intratumorally with poly(I:C)+R848 showed reduced tumor growth and metastasis in lung cancer and fibrosarcoma models, abscopal antitumor response, and resistance to tumor rechallenge. This systemic and long-lasting antitumoral efficacy was mediated by increased infiltration of macrophages, presenting increased M1:M2 ratio, and CD4+ and CD8+ T-cell activation and proliferation.
As a whole, our findings indicate that the antitumor efficacy elicited by poly(I:C)+R848 is superior over single treatments for myeloid reprogramming and activation of adaptive antitumoral immunity in the TME.
Most of our knowledge on the effects of TLR signaling in innate immunity derives from experiments stimulating single TLRs with their specific ligands; however, some combination studies have been performed. A recent study in vitro, mimicking pathogen infections with multi-TLR agonists on murine macrophages, demonstrated higher secretion of IL-6 on treatment with poly(I:C)+R848 versus other combinations with TLR2/1 or TLR4 agonists.19 In our study, we additionally show that primary human and murine macrophages exposed to poly(I:C)+R848, but not poly(I:C)+R837, produce more T cell-attracting chemokines (CXCL10 and CCL5), proinflammatory TNF-α, and nitric oxide, and show higher ability to kill cancer cells in vitro. These results are well correlated with our in vivo analysis of TLR-treated tumors, showing the increased expression of the same chemokines (CXCL10 and CCL5), M1-polarization markers (IRF7 and iNOS), and other markers of TME reprogramming, which might also be associated with T-cell activation (ie, IFN-γ, granzyme B, and perforin). It can be speculated that this TLR-induced cytotoxic activity by M1-like macrophages will be complemented in vivo by the action of cytotoxic T cells recruited by CXCL10 and CCL5.
Indeed, in response to TLR treatments, tumors presented higher infiltration of immune cells that correlated well with smaller tumors. There was an increase of Ly6G+ myeloid precursor cells and mature macrophages (F4/80+), with a higher M1:M2 ratio in treated tumors, while NK or DC numbers presented minor alterations in the TME. Depletion experiments revealed the key role of CD4+ and CD8+ T cells, while discarding NK cells as partners of M1-like antitumor macrophages in the antitumoral efficacy of poly(I:C)+R848. Notably, in the TME, we found particular differences for the activation of lymphoid cells, at distinct times of the analysis. On short-term treatment (two doses), the TME showed a higher infiltration of CD8+ T cells versus CD4+ T cells, while with long-term treatments (six doses and complete tumor regression), the amount of CD8+ T cells decreased. We attribute these findings to the almost total elimination of the tumor mass, thus non-requiring high numbers of CD8+ T cells at this late stage of remission. Interestingly, CD4+ T cells showed higher expression of the CD69 and PD1 activation markers in the treated samples. Of note, monotherapies with poly(I:C) or R848, but not with R837, induced significant CD8+ T-cell infiltration and partial tumor regression. CD4+ T cells had an opposite trend, being high in the long-term analysis and significantly less, among total T cells, in the short-term treatment due to the relatively higher density of CD8+ lymphocytes. Although the role of CD4+ cells in tumor immunity has been traditionally less appreciated than CD8+ cells, recent studies have demonstrated the importance of macrophage-derived CCL5 for the recruitment of CD4+ T cells, whivh was crucial for effective and sustained antitumoral responses.39 40 An increase in CD4+ and CD8+ T cells was reported in patients treated with BO-112 (poly(I:C) nanocomplexes) monotherapy,41 and an increase in CD8+ accompanied by a decrease in CD4+FOXP3+ T cells was observed in pancreatic murine tumor models treated with R848.17 In these studies, the need for combination therapies was recognized for ‘complete’ antitumoral responses, as also observed in our experiments.
Noteworthy, we also demonstrated the activation of memory adaptive antitumor responses. Mice bearing lung tumors treated with poly(I:C)+R848 had no tumor for up to 70 days post-treatment and further rejected a second inoculum of tumor cells. Spleen CD8+ lymphocytes from these mice showed a significantly enhanced proliferation rate in response to syngeneic DC pulsed with tumor cell lysate, compared with naïve mice, and spleen macrophages were able to kill cancer cells in vitro. Numerous studies have used vaccines loaded with TLR3 or TLR7/8 agonists for DC-mediated T-cell proliferation.42 Interestingly, the stimulation of macrophages with CpG or poly(I:C) to present antigens has been recently reported as a promising strategy to overcome tumor resistance to CAR-T cell therapy43; BO-112 (poly(I:C) nanocomplexes) was able to restore the efficacy of tumor-specific T cells against tumors lacking type I and II IFN sensitivity by induction of NF-kB signaling.44 Along this line, we have observed in the current study that macrophagic cells stimulated with the poly(I:C)+R848 combination presented also a significantly higher activation of the NF-kB pathway.
Our proteomic analysis of tumors treated with the poly(I:C)+R848 combination supports the in vivo findings of tumor growth inhibition and has pointed out pathways of T-cell activation and migration, cytokine secretion, oxidative stress, cell killing, and apoptotic processes. The STAT1 pathway and proteins related to apoptotic and alarmin functions were elucidated as key molecular mediators of poly(I:C)+R848 synergistic antitumoral activity. In an in vitro study, modeling TLR activation by pathogens, Liu et al demonstrated that the JAK-STAT1/2 pathway triggered by poly(I:C)+R848 is required for the synergistic production of cytokines.45 Our findings, using fully immunocompetent in vivo tumor models, confirm the central role of STAT1 on poly(I:C)+R848 treatment, now in the context of cancer (figure 7). This result may be particularly relevant for macrophage reprogramming in patients with lung cancer resistant to ICB, as STAT1 and CXCL10 have been recently identified as key markers of M1hot TAMs signature crucial to support innate and adaptive antitumor responses in these patients.8 Other studies on melanoma and lung cancer found that ICB antitumoral efficacy requires STAT1 activation, which needs to be unleashed by complementary treatments.46 47 As an example, Zemek et al showed great antitumoral efficacy with the combination of IFN-γ+anti-IL-10 mAb+poly(I:C)+ICB therapy but modest results in the absence of ICB.46 Instead, in our study, the TLR combination of poly(I:C)+R848 was sufficient to completely prevent primary tumor growth and metastasis, and treated-mice became resistant to tumor rechallenge (figures 2 and 5).
In conclusion, this study supports the intratumoral application of low doses of poly(I:C)+R848 for the treatment of solid tumors with an immunosuppressive microenvironment dominated by TAMs. Our results reveal the superiority of the poly(I:C)+R848 combination versus each of the single treatments, and it is also more effective than R837, routinely used in the clinic, alone or in combination with poly(I:C). The synergistic activity of poly(I:C)+R848 induces the reprogramming of macrophages into M1hot-antitumor effectors through STAT1 activation, controlling the sustained activation of innate and adaptive antitumor immune responses, while maintaining homeostasis and avoiding an excessive inflammation, to unequivocally fight against the tumor (online supplemental figure 11). In response to treatment, we identified the upregulated production of STAT1/inflammatory/cytotoxic/alarmin-related proteins and downregulation of immunosuppressive molecules in the TME, which eliminate the primary tumor but also its metastasis and even confer antitumor immune memory, demonstrated as resistance to tumor rechallenge in treated mice. These results guarantee future studies to optimize the dose of this combination therapy in tumors refractory to other treatments, such as ICB, antibodies or T-cell therapies, thus providing a valuable tool to improve outcomes in patients with solid tumors.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Procedures involving animals were conducted following standard operating procedures with the appropriate approval of the ethics and scientific committees.
Acknowledgments
We acknowledge Mabel Loza, Rocío Piña Marquez, and Jose Brea for the use of the BioFarma Research Group facilities at CiMUS, Universidade de Santiago de Compostela; Egon Willighagen and Chris Evelo at Department of Bioinformatics, BiGCaT, Maastricht University for their comments on proteomic analysis; and Gabriele De Simone at Humanitas Research Hospital for the help with the FACS analysis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @ftorresandon
Contributors CA and FTA contributed to the design, acquisition, analysis, and interpretation of in vitro and in vivo data, and in the drafting and the revision of the work. FM, ED, AM, and AU contributed to the in vitro and in vivo experiments. AA contributed with FACS analysis. MS and ME provided technical help in the animal facility. SG contributed with in vitro experiments. FE, DS, MR, and AC contributed to the immunohistochemistry and cytokine analysis. MM helped with bioinformatic analysis. SB performed proteomic experiments. AM and PA contributed to the design and the revision of the work.
Funding This work was supported by the 2-INTRATARGET project (PCIN-2017–129/AEI) funded by MINECO-PCIN-2017–129/AEI and the Italian Ministry of Health (IMH), under the frame of EuroNanoMed III (PA, FTA, and CA), by Worldwide Cancer Research UK, and Associazione Italiana Ricerca Cancro (5×1000 grant number 21 147 to AM and PA). AU was supported by a FIRC-AIRC fellowship for Italy. FTA was supported by a Marie Skłodowska-Curie Individual European Fellowship (H2020-MSCA-IF-2014-EF-ST) from the European Commission for the project NANOTAM (number 658592), and he is a recipient of a grant by Fundación de la Asociación Española Contra el Cáncer and an 'Oportunious' program grant by Xunta de Galicia.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.