Article Text

Abstract
Background The prognosis for metastatic Ewing sarcoma family of tumors (EFT) is still poor despite high-dose chemotherapy and radiation treatment. Immunotherapies hold promise, but cancer antigen-targeting immunotherapies have largely failed to induce effective T cell receptor-mediated antitumor response. However, T cell-engaging bispecific antibodies (T-BsAbs) have yet to be adequately explored.
Methods Rehumanized STEAP1-IgG was used to build T-BsAb (named BC261) using the 2+2 IgG-[L]-scFv platform carrying the anti-CD3 huOKT3 scFv as the second specificity. Its binding epitope mapping, species cross-reactivity, tumor cell line staining, and in vitro cytotoxicity were investigated thoroughly. Its potency in driving tumor-infiltrating lymphocytes (TILs) was quantified using bioluminescence, correlated with in vivo antitumor response against cell line-derived or patient-derived xenografts (CDXs or PDXs) and compared with anti-STEAP1 T-BsAbs built on representative antibody platforms.
Results BC261 binding epitope was mapped to its second extracellular domain of STEAP1 shared among canine and primate orthologs. BC261 induced potent cytotoxicity against panels of EFT, prostate cancer, and canine osteosarcoma cell lines despite their low antigen density. BC261 drove significantly more TILs into tumors (30-fold) and exerted superior antitumor effects compared with the other standard BsAb platforms. The antitumor efficacy of BC261 was consistent against EFT and prostate cancer CDXs and PDXs.
Conclusions BC261 was highly efficient in driving T cell infiltration and tumor ablation. Either as stand-alone therapeutics or for ex vivo armed T cells, this novel anti-STEAP1 T-BsAb BC261 has therapeutic potential.
- antibodies
- neoplasm
- antigens
- neoplasm
- immunotherapy
- T-lymphocytes
- sarcoma
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data generated or analyzed during this study are included in this published article.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Ewing sarcoma family of tumors (EFT), which includes Ewing sarcoma of bone, extraosseous Ewing tumor, and peripheral primitive neuroectodermal tumor, is the second most common malignant bone tumor in children and young adults.1 2 EFT is characterized by the oncogenic EWS-ETS translocation including EWS-FLI1, t(11;22)(q24;q12) and EWS-ERG, t(21;22) (q11;q12) and early metastasis.3–5 For localized EFT, cytotoxic chemotherapy has improved the survival from less than 20% to 70%–80%. However, over the last three decades, there has been a modest improvement in survival, and the prognosis for metastatic or relapsed disease has remained poor, with survival rates of less than 20%–30%.6 7 Given the high toxicities associated with intensive chemotherapy and radiotherapy, there is an urgent unmet need for novel therapeutic approaches, and T cell-based immunotherapies are promising alternatives.8 9 However, despite the tumor specificity of EWS-FLI1 fusion sequences, the patients failed to induce strong cytotoxic T cell response because of low MHC expression and defective antigen processing in EFT.10 11 Cancer/testis antigens including membrane-associated phospholipase A1 beta (LIPI),12 X antigen family member 1 (XAGE1),13 and New York esophageal squamous cell carcinoma 1 (NY-ESO-1)14 have also been considered as promising targets for immunotherapy with the advantage of low expression in adult somatic tissues, but the clinical trials have yielded limited efficacy.15 16
The six-transmembrane epithelial antigen of prostate (STEAP) family of proteins which include four members, named STEAP1 to STEAP4, are overexpressed in several human cancers.17 These STEAP proteins, particularly STEAP1 and STEAP2 that control cell proliferation and apoptosis, have potential as therapeutic targets.18 19 However, except for STEAP1, the relationship between overexpression of these other STEAP proteins and tumor aggressiveness has remained largely elusive.20–22 STEAP1 is upregulated in a variety of human cancers, including prostate, bladder, ovarian, rhabdomyosarcoma, and EFT.19 21 The expression of STEAP1 in normal tissues (except secretory tissues of the bladder and prostate) is highly restricted,23 24 suitable for antibody-based immunotherapies.25 ,26 Several therapeutic strategies targeting STEAP1 have been developed including monoclonal antibodies, antibody drug conjugates, DNA vaccines, and small non-coding RNAs for the treatment of prostate cancers.27 Radiolabeled anti-STEAP1 monoclonal antibody, 89Zr-DFO-MSTP2109A (NCT01774071), confirmed its highly restricted tumor tissue distribution,28 29 and antibody-drug (MMAE) conjugate targeting STEAP1 (DSTP3086S) has shown acceptable safety profiles with a modest clinical benefit in patients with metastatic castration-resistant prostate cancer.26 With the advent of T cell-based immunotherapy, T cell engaging bispecific antibodies (T-BsAb) or chimeric antigen receptor (CAR) T cells targeting STEAP1 also are early in human trials.30–34 But most of these therapeutic trials are still in the beginning stage, and the most optimal platforms have yet to emerge.
We previously demonstrated that the interdomain distance and cis-configuration are critical for in vivo antitumor response of T-BsAb.35 We also showed that a 2+2 format (2 tumor binding domains+2 CD3 binding domains) was substantially more potent than a 2+1 format (2 tumor binding domains+1 CD3 binding domain).35 36 Here, we describe the biochemical and cytotoxic properties of the STEAP1 BsAbs built on the IgG-[L]-scFv format, their epitope mapping, species cross reactivity, and their in vivo antitumor activities against a panel of EFT and prostate cancer xenografts.
Methods
Humanization of murine anti-STEAP1 antibody X120
The anti-STEAP1 monoclonal antibody X12037 was rehumanized by grafting complementarity-determining regions (CDRs) onto human IgG1 frameworks based on their homology with human frameworks IGHV4-30-4*01-IGHJ6*01 for variable heavy chain (VH), IGKV4-1*01-IGKJ4*01 for variable light chain (VL), respectively. Six humanized VH (VH1 to VH6) and four humanized VL (VL1 to VL4) sequences were paired to create 24 humanized versions, with identical CDR sequences to the mouse antibody X12037 using CHO cells. Instead of surface plasmon resonance analysis, mean fluorescence intensity (MFI) of antibody binding to EFT cell line TC-32 was used to rank binding affinity of 24 antibodies due to conformational epitope of the STEAP1 (online supplemental figure 1A), and eight IgG clones pairing VH1 or VH2 with VL1 to VL4 and VH5+VL1 were chosen for binding avidity test. After eight cycles of repeated washing using phosphate buffered saline (PBS/EDTA dissociation buffer, four clones (VH1+VL1, VH2+VL1 and VH2+VL2, and VH5+VL1) retained >50% of their baseline MFIs, higher than that of the chimeric antibody (online supplemental figure 1B). Binding affinities of these four clones were compared with that of the chimeric antibody using a cell-based fluorescent method38 (online supplemental table 1). Antibody purity was analyzed by size-exclusion high-performance liquid chromatography (SEC-HPLC), and antibody stability was monitored by SEC-HPLC after freeze–thaw cycles or after heat stress at 40°C for 4 weeks. These four clones with >85% sequence humanness showed high binding affinities to targets and high purity and stability after multiple freeze–thaw cycles or heat stress at 40°C over 4 weeks when assayed by HPLC (online supplemental table 2) and were chosen for developing T-BsAb.
Supplemental material
Bispecific antibodies
Four anti-STEAP1 T-BsAbs (named BC259, BC260, BC261, BC262) with different VH and VL sequences were developed using the IgG-[L]-scFv platform31 39 40 by attaching the anti-CD3 humanized OKT3 (huOKT3) single-chain variable fragment (scFv) to the carboxyl end of the rehumanized X120 IgG1 light chain. The amino acid sequences for the VH and VL regions for these bispecific antibodies are summarized in online supplemental table 3. Each BsAb was produced using the Expi293 expression system (Thermo Fisher Scientific RRID:CVCL_D615) or HEK 293 T cell (RRID:CVCL_KS61) and maintained in pH 8.2 50 mM of citrate buffer with 150 mM of NaCl. The lead clone BC261 was expressed in CHO cells (RRID:CVCL_0213). Antibodies were purified with protein A affinity column chromatography, and the stability was monitored by SEC-HPLC weekly for 4 weeks at 40°C.
Tumor cell lines
EFT cell lines, TC-32 (RRID:CVCL_7151), TC71 (RRID:CVCL_2213), SK-ES-1 (RRID:CVCL_0627), SK-N-MC(RRID:CVCL_0530), and A4573 (RRID:CVCL_6245) were purchased from ATCC; SKEAW, SKELP, SKERT were provided by Memorial Sloan Kettering Cancer Center (MSKCC) investigators. Prostate cancer cell lines, CWR22 (RRID:CVCL_3967), LNCaP-AR (CLS Cat# 300265/p761_LNCaP, RRID:CVCL_0395), and VCaP (RRID:CVCL_2235) were purchased from ATCC. Canine osteosarcoma cell lines, D-17 (RRID:CVCL_1916), DAN (RRID:CVCL_1917), DSN (RRID:CVCL_1919), and DSDh (RRID:CVCL_1918) were purchased from ATCC. All cancer cells were authenticated by short tandem repeats profiling using PowerPlex 2.1 System (Promega, Cat# DC8942), and periodically tested for mycoplasma infection using a commercial kit (Lonza, Cat# LT07-318).
Flow cytometry analysis of STEAP1 antibody binding
For STEAP1 density measurement on tumor cells, anti-STEAP1 human IgG1 was chemically conjugated to Alex-Fluor 647 (Thermo Fisher) according to manufacturer’s instructions and used to stain STEAP-1 BsAbs bound to human cancer cells. Confirmatory binding to targets was analyzed using anti-human IgG Fc antibody. For quantitation of target antigens, MFI was translated into antigen density using Quantum MESH microspheres and fluorochrome to protein ratio (Bangs Laboratories, cat# 647). One microgram of STEAP1 IgG monoclonal antibodies or STEAP1 BsAbs were incubated with 1×106 of cancer cells at 4°C for 45 min, washed, reacted with R-phycoerythrin conjugated goat anti-human IgG secondary antibody (SouthernBiotech Cat#2040-09, RRID:AB_2795648), and analyzed by flow cytometry (BD FACSCalibur instrument and Attune NxT Flow Cytometer). Palivizumab or anti-CD33 BsAb was used as a control antibody. Data were analyzed with FlowJo V.10 software (FlowJo, RRID:SCR_008520).
T cell expansion ex vivo
Peripheral blood mononuclear cells (PBMCs) were separated from buffy coats (New York Blood Center) by Ficoll. Naive T cells were purified from human PBMC using Pan T cell isolation kit (Miltenyi Biotec, Cat#130096535) and expanded by CD3/CD28 Dynabeads (Gibco, Cat#11 132D) for 7–14 days in the presence of 30 IU/mL of IL-2 according to manufacturer’s instructions. Unless stated otherwise, these activated T cells were used for all T cell experiments.
Cytotoxicity assay (chromium51 release assay)
In vitro antibody-dependent T cell-mediated cytotoxicity assay was carried out as previously described.35 41 Target cells were cultured in RPMI1640 (Cellgro) supplemented with 10% fetal bovine serum (Life Technologies) at 37°C in a 5% CO2 humidified incubator. Target cells were labeled with sodium 51Cr chromate (51CrNa2CrO4; Amersham, Arlington Heights, Illinois, USA) at 100 µCi/106 cells at 37°C for 1 hour. Percentage of specific lysis was calculated using the formula 100% x× (experimental cpm−background cpm)/(total cpm−background cpm), where cpm represented counts per minute of 51Cr released. Total release was assessed by lysis with 10% SDS (Sigma, St Louis, Missouri, USA), and background release was measured in the absence of effector cells. Glycoprotein A33 (GPA33) or anti-CD33 BsAb was used as a control BsAb.
STEAP1 transduction through lentivirus
Lentiviruses were produced by transfection of 2×1 07 293 T cells using 20 µg of lentiviral vector pLVX-IRES-ZsGreen1 (Takara,Cat#632187), 10 µg of pVSV-G (RRID:Addgene_138479), 20 µg pCMV-dR8.91 (Lifescience Market), and 100 µL PolyJet In Vitro DNA Transfection Reagent (SignaGen, Cat#SL100688, Rockville, MD) as described previously.42 43 After STEAP1 variants transduction, cells were sorted by ZsGreen1 GFP fluorescence.
T cell transduction with luciferase and tdTomato
T cells isolated from peripheral blood were stimulated with Dynabeads Human T-Activator CD3/CD28 for 24 hours. T cells were transduced with retroviral constructs containing tdTomato and click beetle red luciferase in RetroNectin-coated 6-well plates in the presence of IL-2 (100 IU/mL) and protamine sulfate (4 µg/mL) as previously described.44 Transduced T cells were cultured for 8 days before being used in animal experiments.
Bioluminescence imaging
To monitor T cell trafficking into tumors, BsAb armed luciferase transduced T cells were injected intravenously. Mice were anesthetized and imaged after intravenous injection of 3 mg of D-luciferin (Gold Biotechnology, Cat# LUCK-100) on different days post T cell injection. Images were acquired using IVIS Spectrum CT In Vivo Imaging System (Caliper Life Sciences). Bioluminescence images were overlaid with photographs, and regions of interest were drawn based on the location and contour of tumor using Living image 2.60 (Xenogen). Quantification of bioluminescence emission (BLI, photon flux/sec) was performed, monitored by time, and compared with the BLIs of tumors without treatment.
Cytokine release assays
Human Th1 cell-released cytokines were analyzed after EAT injection using LEGENDplex Human Th1 Panel (Biolegend, Cat# 741035). Five human T cell cytokines including IL-2, IL-6, IL-10, IFN-γ, and TNF-α were analyzed using mouse serum after EAT injection.
In vivo studies
All animal experiments were performed in compliance with MSKCC Institutional Animal Care and Use Committee (IACUC) guidelines. BALB/cRag2−/−IL-2Rγc−/−(BRG) mice were purchased from Taconic Biosciences (Hudson, New York, USA) and provided with sulfatrim or amoxicillin food. Two EFT patient-derived tumor xenografts (PDXs), ES03a and ES15a, were established from surgical specimens by the antitumor core facility in MSKCC with MSKCC IRB approval, and prostate cancer PDXs, TM00298 and J000077451, were purchased from The Jackson Laboratory. Tumor cells were suspended in Matrigel Basement Membrane Matrix (Corning, Cat#354234) and implanted in the flank of 6-week-old to 10-week-old BRG mice; only male mice were used for prostate cell line-derived xenografts (CDXs) and PDXs. Treatment was initiated after tumors were established, average tumor volume of 100 mm3 when measured using TM900 scanner (Piera, Brussels, BE) after 8 days to 2 months post subc tumor implantation. Before treatment, mice with small tumors (<50 mm3) or infection signs were excluded from randomization to experimental groups. For treatment, BsAb and human T cells were intravenously (iv) injected separately or together (ex vivo armed T cells) with supplementary subcutaneous IL-2 (1000 IU). Ex vivo armed T cells (EATs) were generated by incubating T cells with T-BsAb for 20 min at room temperature. After incubation, the T cells were washed with PBS twice. The T cell number administered per dose was 2×107 cells based on previous reports.45 For in vivo experiment, anti-GPA33 BsAb was used as a control BsAb. Tumor growth curves and overall survival curves were plotted, and the overall survival was defined as the time from start of treatment to when tumor volume reached 2000 mm3. To define well-being of mice, CBC analyses, changes in body weight, general activity, physical appearance, and graft-versus-host disease (GVHD) scoring were regularly monitored. When tumor growth reached 2000 mm3 or greater, mice were euthanized. All animal experiments were repeated twice more with different donor’s T cells to ensure that our results were reliable.
Statistical analyses
Tumor growth rates and bioluminescence of T cells were analyzed using area under the curve (AUC). The AUC and overall survival were calculated using GraphPad Prism V.8.0. Difference between each treatment group was tested for statistical significance by two-tailed Student’s t-test for two sets of data while one-way analysis of variance with Tukey’s post hoc test was used among three or more groups. All statistical analyses were performed using GraphPad Prism V.8.0 for Windows (GraphPad Software, La Jolla, California, USA, www. graphpad. com). P value <0.05 was considered statistically significant. Asterisks indicate that the experimental p value is statistically significantly different from the associated controls at *p<0.05; **p<0.01; ***p<0.001, ****p<0.0001.
Results
STEAP1 BsAb design and expression in Expi293 cells
Four STEAP1 BsAbs, that is, BC259 (VH1+VL1), BC260 (VH2+VL1), BC261 (VH2+VL2), BC262 (VH5+VL1), were built using the 2+2 IgG-[L]-scFv BsAb format. The representative platform for these 4 STEAP1 BsAbs is described in figure 1A. CH1 through CH3 and CL were based on constant domains of the heavy chain and of kappa light chain of the humanized anti-STEAP1 IgG antibody. The C-terminus of the CL is fused to the scFv of an anti-CD3 humanized OKT3 (huOKT3). The biochemical purity of BC261, representative of four clones, was analyzed by SEC-HPLC (online supplemental figure 2A), where the elution fractions were confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). All four STEAP1 BsAbs remained stable after multiple freeze–thaw cycles and heat stress at 40°C over 4 weeks (online supplemental table 4), and among them, BC261 maintained high purity despite prolonged heat stress.

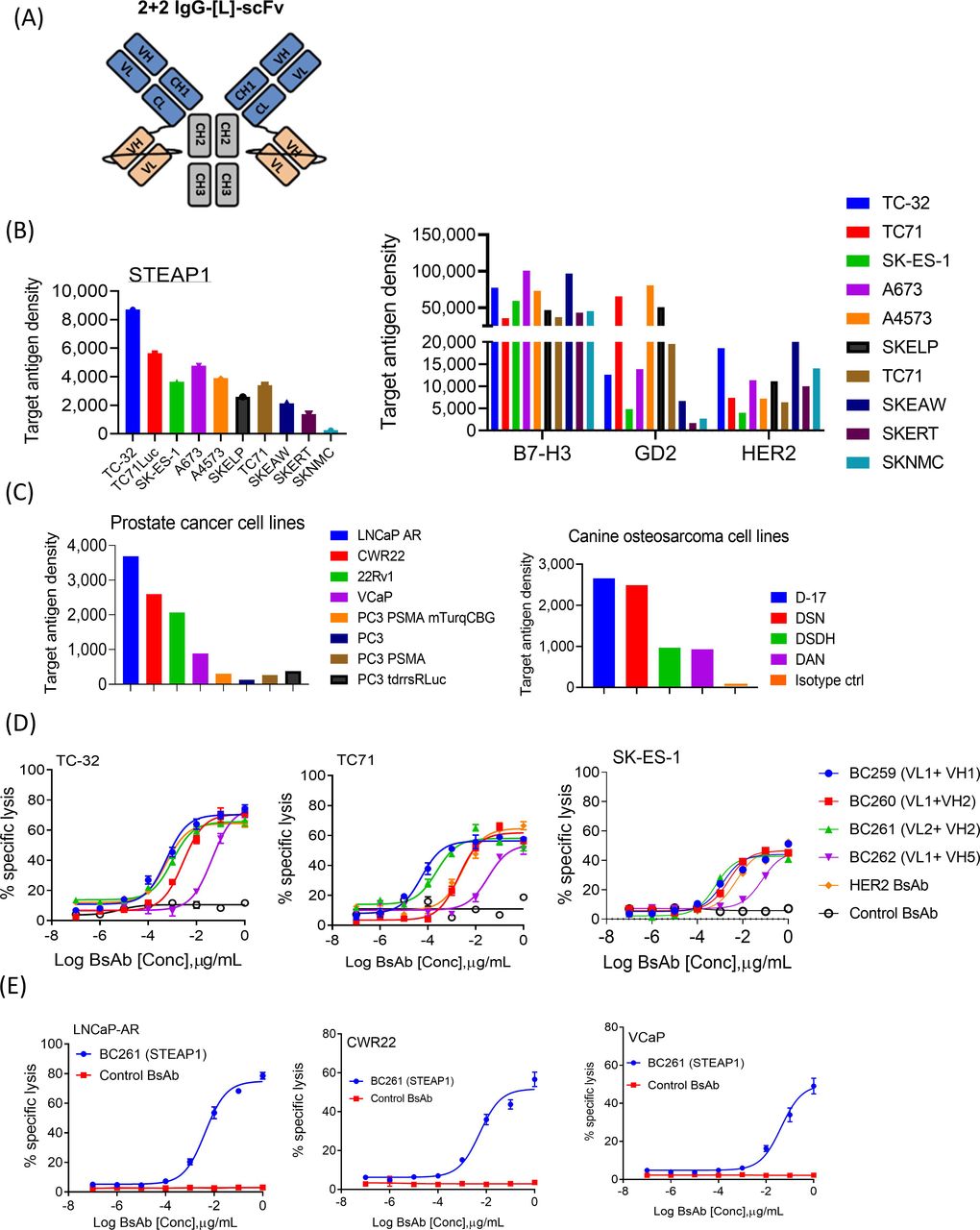
Structure and in vitro antitumor activity of anti-STEAP1 T cell-engaging bispecific antibodies. (A) IgG-[L]-scFv structure of anti-STEAP1 BsAbs. (B) Surface target antigen densities on a panel of Ewing sarcoma family of tumors (EFT) cell lines. EFT cell lines were stained with anti-STEAP1 BsAb BC261, anti-B7-H3 BsAb, anti-GD2 BsAb, or anti-HER2 BsAb, and their binding levels (mean fluorescence intensities (MFIs)) were quantitated using Quantum beads. (C) STEAP1 target antigen density on multiple cancer cell lines including prostate cancer cell lines and canine osteosarcoma cell lines. Cancer cells were stained with BC261, and the MFIs were quantitated using quantum beads. (D) Antibody-dependent T cell-mediated cytotoxicity (ADTC) as a function of increasing doses of anti-STEAP1 BsAbs against EFT cell lines. Effector to target cell ratio (ET ratio) was set to 10:1. (E) In vitro cytotoxicity of BC261 against prostate cancer cell lines. ET ratio was 10:1. CH, constant heavy chain; CL, constant light chain; scFv, single-chain variable fragment; STEAP, six-transmembrane epithelial antigen of prostate; VH, variable heavy chain; VL, variable light chain.
STEAP1 BsAb binding to EFT and prostate cancer cell lines
STEAP1 BsAb binding to EFT cell lines was analyzed by flow cytometry. All four BsAbs showed reactivity against a panel of EFT cell lines (TC-32, TC71, SK-ES-1, and A4573) and prostate cancer cell line (LNCaP-AR) (online supplemental figure 2B). While BC261 MFI was consistently the highest, BC262 MFI was the lowest among the four BsAbs. Immunostaining of EFT by BC261 was expanded to more EFT cell lines (SKEAW, SKERT, SKELP, SKNMC, and TC71Luc), and STEAP1 antigen expression was quantified as molecules per cell, confirming the relatively low density of the STEAP1 compared with other target antigens (figure 1B). After screening a large panel of human or non-human cancer cell lines (online supplemental table 5), STEAP1 BsAb was found to bind prostate cancer cell lines and canine osteosarcoma cell lines as well, and the expression density was quantified (figure 1C).
STEAP1 BsAb-dependent T cell-mediated cytotoxicity
In vitro cytotoxicity of STEAP1 BsAb was tested against a panel of EFT, using HER2-BsAb32 as a positive control and anti-GPA33 BsAb46 as a negative control. BC259 and BC261 showed robust cytotoxicity against TC-32, TC71, and SK-ES-1 cell lines (figure 1D), while BC262 was much less potent. The in vitro potency of STEAP1 BsAb as measured by EC50 of BC261 against A4573, SKEAW, SKELP, SKERT, and SKNMC ranged between 1.1 and 25.8 pM (online supplemental table 6); against prostate cancer cell lines LNCaP-AR, CWR22, and VCaP between 21.1 and 214.8 pM (figure 1E); and against canine osteosarcoma D-17, DSN, DAN, and DSDh between 7.2 and 29.5 pM (online supplemental figure 3A). These EC50s correlated with the levels of STEAP1 expression measured by MFI (online supplemental table 6).
In vivo antitumor effect of STEAP1 BsAbs
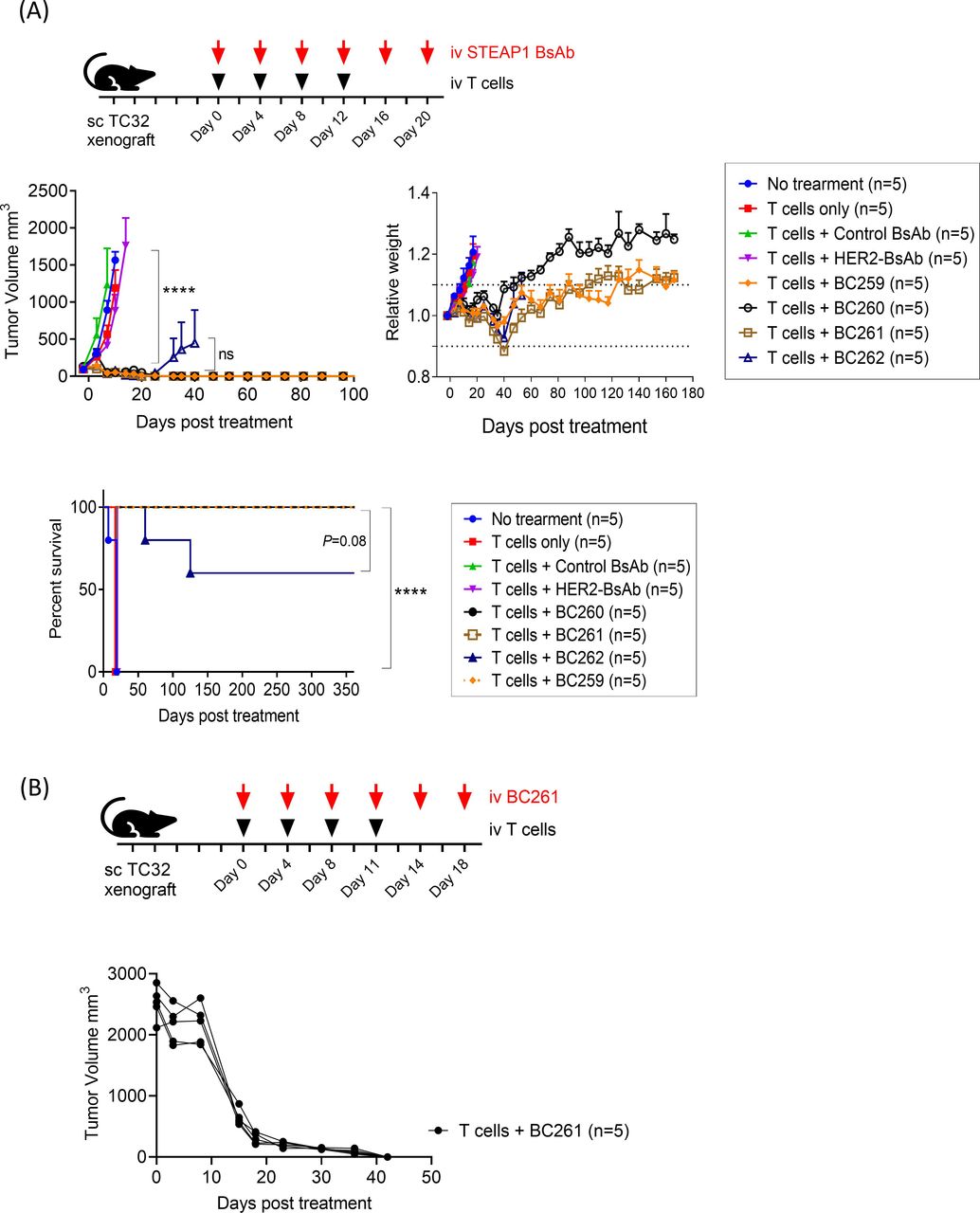
In vivo antitumor efficacy of these 4 STEAP1 BsAbs was evaluated against TC-32 CDXs. Once tumors were established, STEAP1 BsAbs were administered with human T cells twice per week for 2–3 weeks (figure 2A). In contrast to control groups which showed unabated tumor growth, all four STEAP1 BsAbs exerted significant antitumor effects (p<0.0001). The mice developed mild to moderate GVHD between 4 weeks and 8 weeks after treatment start, but recovered spontaneously, showing no obvious toxicity through 1 year after the start of treatment. There were no recurrences except for two mice in the BC262 (VH5+VL1) treated group. Since BC261 (VL2+VH2) had the greatest humanness (>85%), reacted with EFT cell lines with higher MFIs, possessed highest stability at 40°C, and showed the most consistent antitumor effect in vitro and in vivo, it was chosen for further investigations. BC261 achieved exceptional antitumor efficacy against large solid tumors (figure 2B). Even for large established tumors (~2000 mm3), BC261 achieved complete tumor regression, an effect not typically seen with T-BsAbs targeting other tumor antigens (eg, GD2,39 HER2,32 or GPA3346) even when built on the same IgG-[L]-scFv platform.
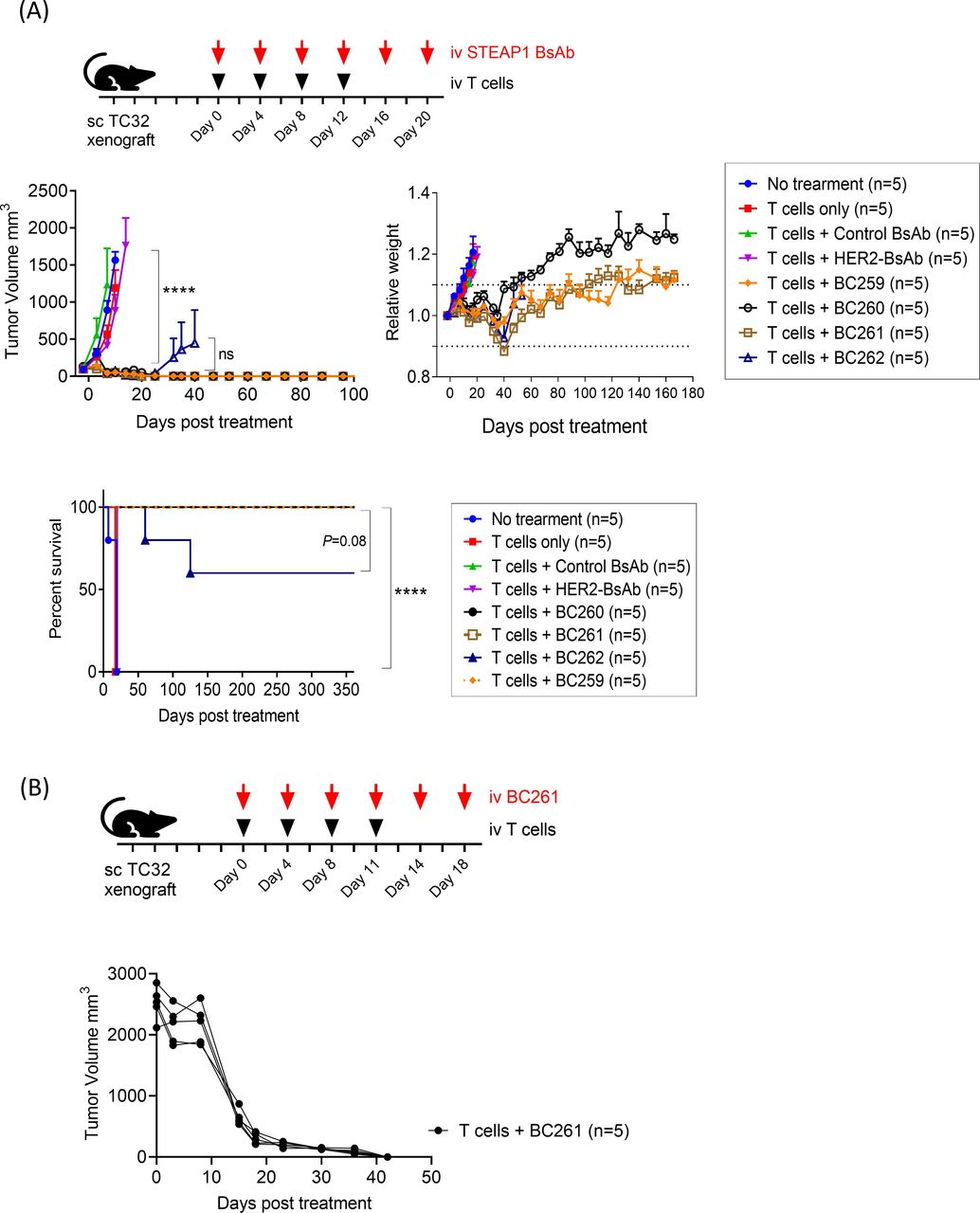
In vivo antitumor activities of STEAP1 bispecific antibodies. (A) In vivo antitumor effect of STEAP1 BsAb in the presence of human T cells against TC-32 cell line xenografts; 3×106 cells of TC32 were subcutaneously implanted into mice, and 10 µg of STEAP1 BsAb and 2×107 of T cells were administered twice per week for 2–3 weeks to treat the tumors. Tumor growth and relative body weight of mice were monitored, and overall survivals of the mice were plotted. (B) In vivo antitumor effect of BC261 against huge established TC-32 tumor xenografts. When tumors reached 2000 mm3, 10 µg of BC261 and 2×107 of T cells were started. Huge EFT xenografts successfully regressed by BC261 treatment. EFT, Ewing sarcoma family of tumors; STEAP, six-transmembrane epithelial antigen of prostate.
Analysis of STEAP1 epitope for BC261
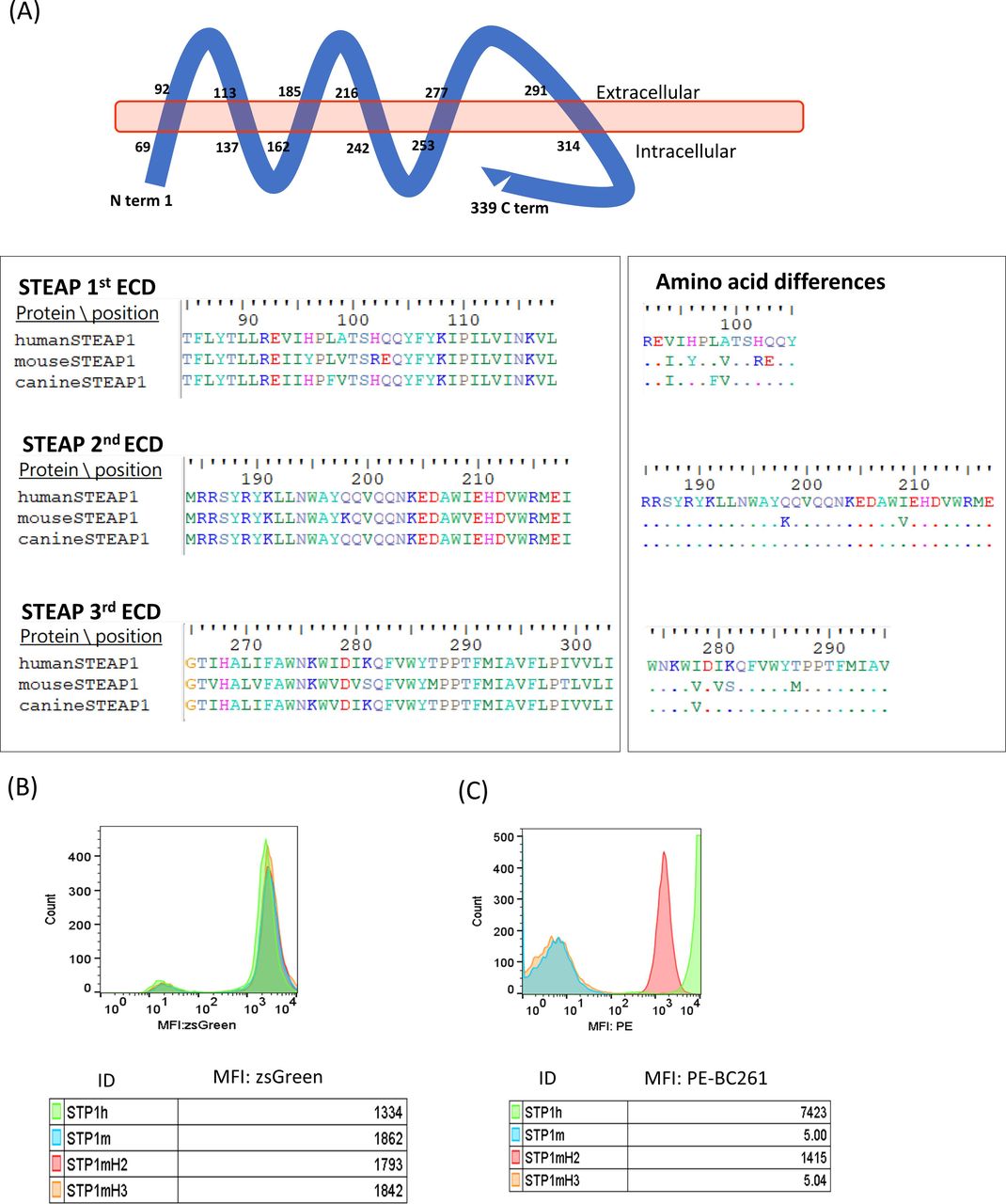
The parental mouse antibody and the humanized anti-STEAP1 IgG did not react with mouse cell lines.37 Given the sequence homology between human and canine STEAP1 of 90.56% and between human and mouse STEAP1 of 82.01% (278/339 aa), analyses of the extracellular domain (ECD) sequences of these STEAP1 orthologs suggested an epitope on the second ECD (figure 3A). Four STEAP1 variants, human STEAP1 (STP1h), mouse STEAP1 (STP1m), mouse STEAP1 with human second ECD (STP1mH2), and mouse STEAP1 with human third ECD (STP1mH3), were constructed and transfected into STEAP1(-) HEK293T cells using lentivirus. After sorting by the expression of ZsGreen1 GFP reporter protein (figure 3B), the STEAP1 transfectants were stained with BC261. Only the STP1mH2 variant showed comparable MFI as STP1h, whereas STP1m and STP1mH3 were non-reactive with BC261 (figure 3C), consistent with the location of the epitope on the second ECD of STEAP1. A BLASTp search (https://blast.ncbi.nlm.nih.gov/) confirmed the presence of this second ECD sequence on STEAP1B, encoded by a gene on human chromosome 7p (in contrast to STEAP1 on 7q). The epitope on the second ECD was also previously reported in two other isoforms, STEAP1B1 and STEAP1B2 in humans47 as well as STEAP1 in cynomolgus monkey.
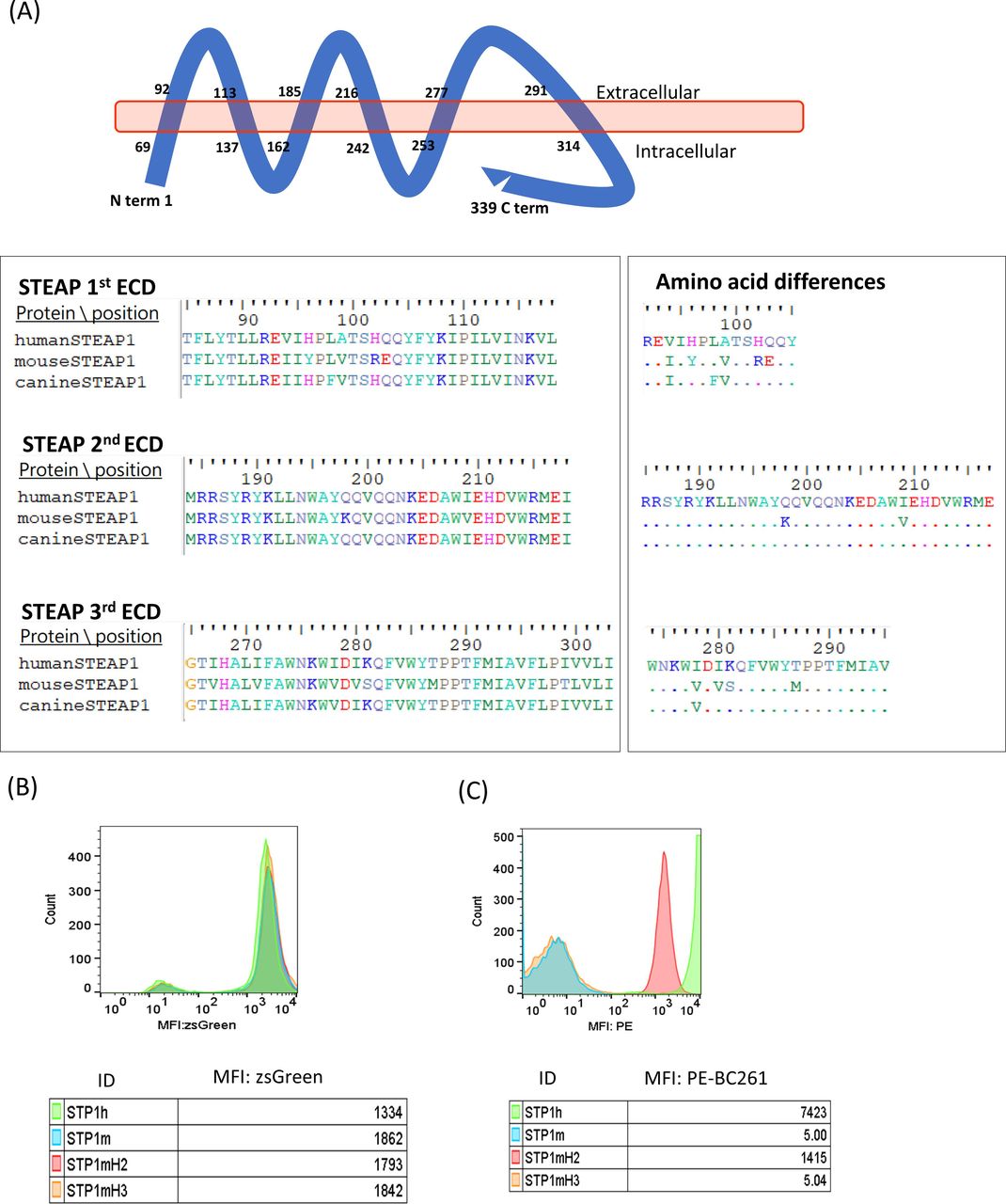
Analysis of STEAP1 epitope for STEAP1 bispecific antibodies. (A) Schematic representation of the structural organization of STEAP1 protein in the cell membrane, highlighting the sequence alignment of the extracellular domain (ECD) of human, mouse, and canine STEAP1. (B) Transfection of HEJ293 cells with viral vector carrying human (STP1h), mouse STEAP1 (STP1m), mouse STEAP1 with human second ECD (STP1mH2), or mouse STEAP1 with human third ECD (STP1mH3), tracked by ZsGreen1. (C) Positive staining of STP1mH2 transfectants by STEAP1 BsAb BC261, while negative staining of STP1m or STP1mH2. MFI, mean fluorescence intensity; STEAP, six-transmembrane epithelial antigen of prostate.
Comparison of antitumor properties of anti-STEAP1 BsAbs built on standard antibody platforms
Next, we constructed five additional anti-STEAP1 BsAbs (figure 4A) using the identical anti-STEAP1 VH2 and VL2 of BC261, and the anti-CD3 VH and VL of huOKT3. They were named BC328 (STEAP1-CD3 tandem scFv as in BiTE48), BC329 (dimeric BiTE using a dimeric tag49), BC330 (BiTE-Fc as in half-life extended BiTE50), BC365 (IgG-[H]-scFv) where the anti-CD3 scFv is attached to heavy chain instead of light chain, and HD148 (IgG heterodimer).35 Cytotoxicity by these alternative BsAb formats were compared with BC261 (IgG-[L]-scFv) in vitro and in vivo. All these BsAbs showed high purity (>90%) by HPLC and high stability after freeze and thaw cycles or heat stress at 40°C for more than 2 weeks (online supplemental table 7). When we compared in vitro potency among STEAP1 BsAb formats, BiTE-Fc and IgG heterodimer were the least effective in killing tumor cells, contrasting with comparable in vitro cytotoxicity by the other four BsAbs (figure 4B).

Antitumor activities of anti-STEAP1 T cell-engaging bispecific antibody. (A) Schematic representation of the six structural formats of STEAP1 BsAb.35 (B) Antibody-dependent T cell-mediated cytotoxicity (ADTC) assay against EFT cell lines (Tc32 ad TC71) using different formats of STEAP1 BsAb. Effector to target cell ratio (ET ratio) was 10:1. (C) Bioluminescence imaging (BLI) of Luc(+) T cells armed with six different formats of STEAP1 BsAb on (a) day 6 and (b) day 18 post treatment and quantitation of bioluminescence intensity in the lesions of tumor. Luciferase transduced T cells (Luc(+) T cells) were armed with various formats of STEAP1 BsAb (10 µg of STEAP1 BsAb/2×107 of T cells) and administered with supplementary IL-2 (1000) IU/dose) to the mice bearing EFT PDXs (ES15a), and the BLIs were followed. (D) TC32 EFT cell line xenografts were harvested on day 7 after two doses of STEAP1 BsAb armed T cell treatment (10 µg of BsAb/2×107 T cells/dose). Paraffin-embedded tumor sections were stained with CD3 (x10): a, no treatment; b, control BsAb; c, BC328; d, BC329; e, BC330; f, BC261; g, BC365; h, HD148. EFT, Ewing sarcoma family of tumors; PDXs, patient-derived tumor xenografts; STEAP, six-transmembrane epithelial antigen of prostate.
Given the different BsAb protein size, plasma pharmacokinetics could confound direct comparison of in vivo potency among BsAbs. We chose to deliver these BsAbs prebound to T cells using a method previously described.51 Luciferase transduced T cells (Luc(+) T cells) were armed with each of the six different formats of anti-STEAP1 BsAb and were injected intravenously into EFT PDX (ES15a) bearing mice, and tumor-infiltrating lymphocytes (TILs) was monitored over time by bioluminescence imaging (figure 4C). Although all 6 BsAbs successfully drove T cells into tumors, BC261 (IgG-[L]-scFv) armed T cells showed the most robust TIL infiltration based on BLIs, with tens of times higher AUC for IgG-[L]-scFv when compared with other formats (online supplemental table 8). The peak BLI for IgG-[L]-scFv was >100-fold higher than those for other BsAb armed T cells, with the most persistence (p<0.0001). The BLI findings were confirmed by the CD3 IHC staining of tumors showing abundant T cell infiltration with BC261 armed T cells (figure 4D).
This superiority of BC261 in increasing TILs and their in vivo persistence translated into substantial and significant differences in antitumor response against EFT PDXs. STEAP1 BsAb armed T cells (10 µg of BsAb/2×107 cells) were administered intravenously on day 0 and day 14 (figure 5A). BC261 (IgG-[L]-scFv) armed T cells were the most effective without overt toxicities (figure 5B). This superior potency of the IgG-[L]-scFv platformed BC261 was confirmed in another EFT PDX (ES3a) mouse model (figure 5C). Here, two doses STEAP1 BsAb armed T cells were administered on day 0 and day 4. BC261 armed T cells induced complete remission of established tumors and significantly prolonged survival (p<0.0001), contrasting with other formats of STEAP1 BsAb armed T cells (figure 5D). The mice treated with BC261 retained their body weight, with no clinical abnormalities throughout the observation period. Among them treated with BC261 armed T cells three mice showed tumor relapse between day 60 and day 90 post-treatment. Interestingly, the relapsed tumors which regrew to 200–2000 mm3 in size responded again to retreatment with two injections of BC261 armed T cells (on day 102 and day 106), and the remission was durable past 200 days (data not shown).
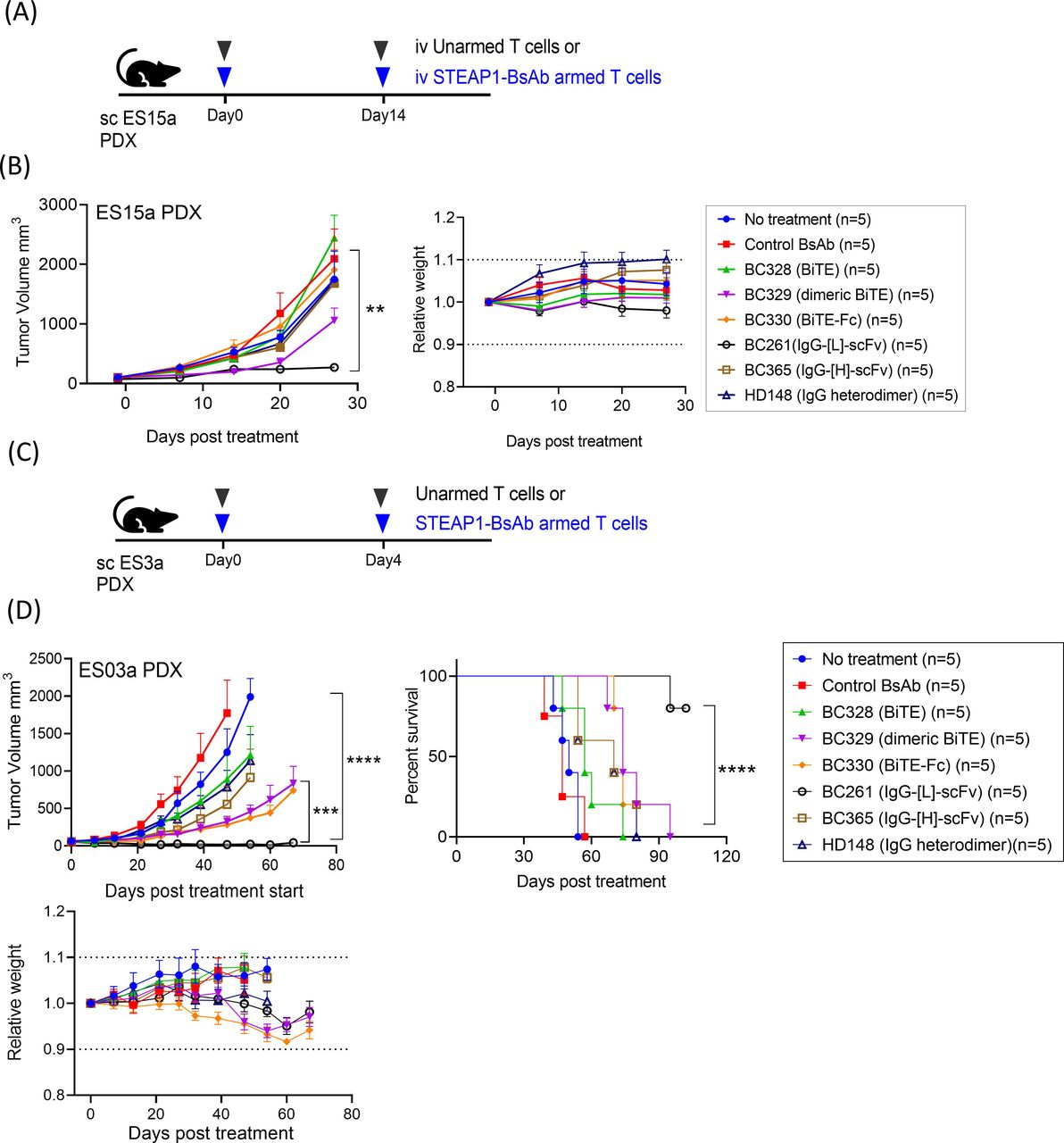
In vivo antitumor effect of STEAP1 bispecific armed T cells. (A) T cells armed with six different formats of STEAP1 BsAb (10 µg of STEAP1 BsAb/2×107 of T cell) were administered to EFT PDX (ES15a) bearing mice. BsAb armed T cells or unarmed T cells were administered on day 0 and day 14, and subcutaneous (sc) IL-2 (1000 IU) was supplemented twice per week. (B) In vivo antitumor effect of six different formats of STEAP1 BsAb armed T cells against EFT PDXs (ES15a) and relative body weight of mice after treatment. (C) T cells were armed with six different formats of STEAP1 BsAb (10 µg of STEAP1 BsAb/2×107 of T cell) and tested for in vivo efficacy against different EFT PDXs (ES3a). BsAb armed T cells or unarmed T cells were administered on day 0 and day 4, and SC IL-2 was supplemented twice per week. (D) In vivo antitumor effect of six different formats of STEAP1 BsAb armed T cells against EFT PDX (ES3a) and relative body weight of mice after treatment. EFT, Ewing sarcoma family of tumors; PDXs, patient-derived tumor xenografts; STEAP, six-transmembrane epithelial antigen of prostate.
In vivo cytokine release was also compared among six different formats of anti-STEAP1 BsAb. Serum TH1 cell cytokine levels were measured at different time points after STEAP1-EAT injection in EFT PDX (ES3a) xenografted mice (online supplemental figure 4A). There was no significant difference in the levels of IL-6, IL-10, and TNF-α among the six different BsAb platforms. When analyzed by AUC of cytokine levels over time (online supplemental figure 4B), only IL-2 and IFN-γ levels differed among six groups of STEAP1 BsAb (p=0.0016, and p=0.0027, respectively), with BC261 (IgG-[L]-scFv) showing the highest levels of IL-2 and IFN-γ.
Dose response of BC261
We titrated the dose of BC261 required for sufficient antitumor effects in vivo. Tumor control was achieved over a range of BsAb doses from 2 µg to 50 µg per dose without toxicities, surpassing the antitumor effect of HER2-BsAb (online supplemental figure 5). For durable remission and long-term survival, 10 µg/dose of BC261 was chosen for the rest of the in vivo experiments.
Anti-tumor effect of BC261 against a panel of EFT xenografts
Antitumor activity of BC261 was tested in multiple EFT xenograft mouse models (figure 6A). While GD2-BsAb failed to suppress TC71 CDX tumor growth, BC261 successfully ablated tumors and improved survival without obvious toxicities (figure 6B). In another EFT cell line SK-ES-1 CDX model, BC261 exerted potent in vivo antitumor effect again, successfully prolonging survival (figure 6C). EFT PDX models, ES03a and ES15a, were also treated with intravenous BC261 and T cells, and durable remission was achieved with BC261 (online supplemental figure 6). Although both PDX tumors were GD2(+) and HER2(+) by immunohistochemical staining, GD2-BsAb was substantially less effective than BC261, whereas HER2-BsAb was comparable to BC261.
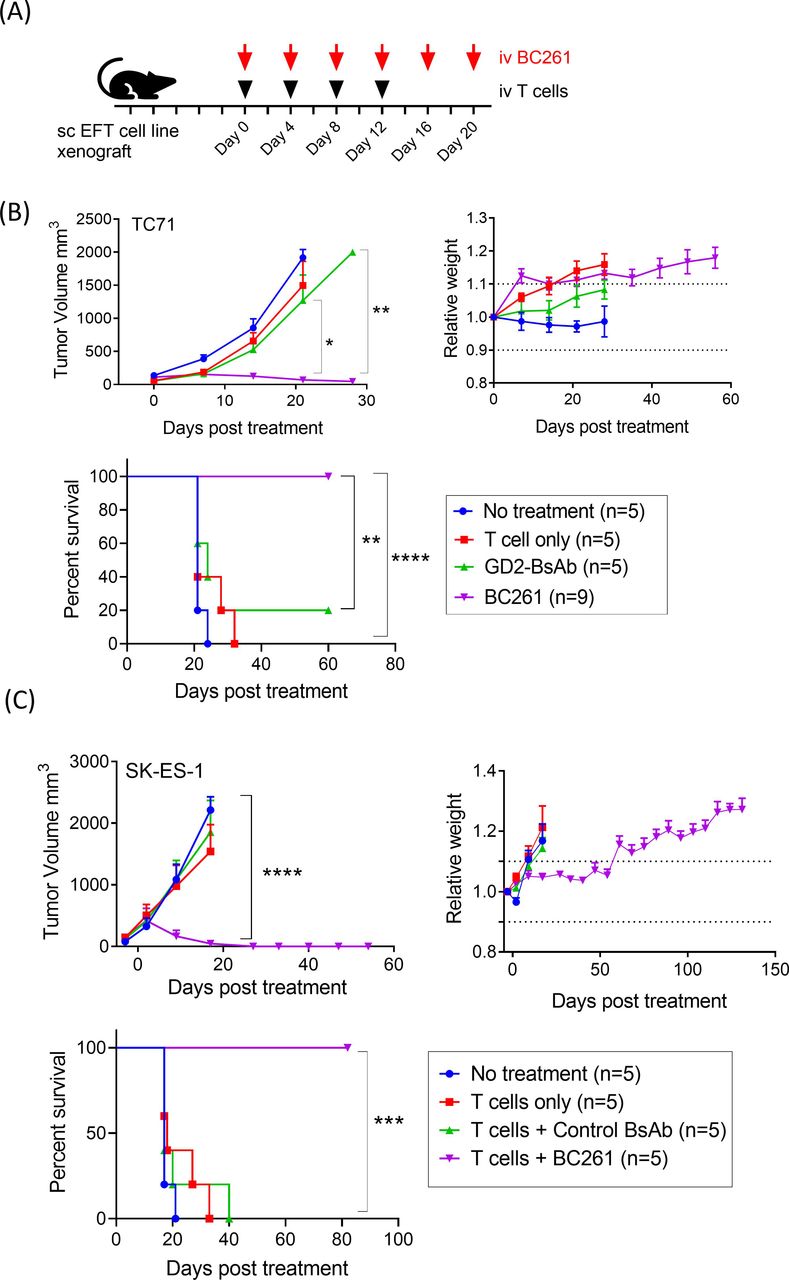
In vivo antitumor effect of BC261 against a panel of STEAP1(+) EFT cell line xenografts (CDXs). (A) Ten micrograms of BC261 was administered with 2×107 of T cells twice per week for 2–3 weeks to treat EFT cell line xenografts. Subcutaneous IL-2 (1000 IU) was supplemented with each T cell injection. (B) In vivo antitumor effect of BC261 against EFT TC-71 cell line xenografts. Tumor growth and relative body weight of mice were monitored, and overall survivals were plotted. (C) In vivo antitumor effect of BC261 against EFT SK-ES-1 cell line xenografts. Tumor growth and relative body weight of mice were monitored, and overall survivals were plotted. EFT, Ewing sarcoma family of tumors; STEAP, six-transmembrane epithelial antigen of prostate.
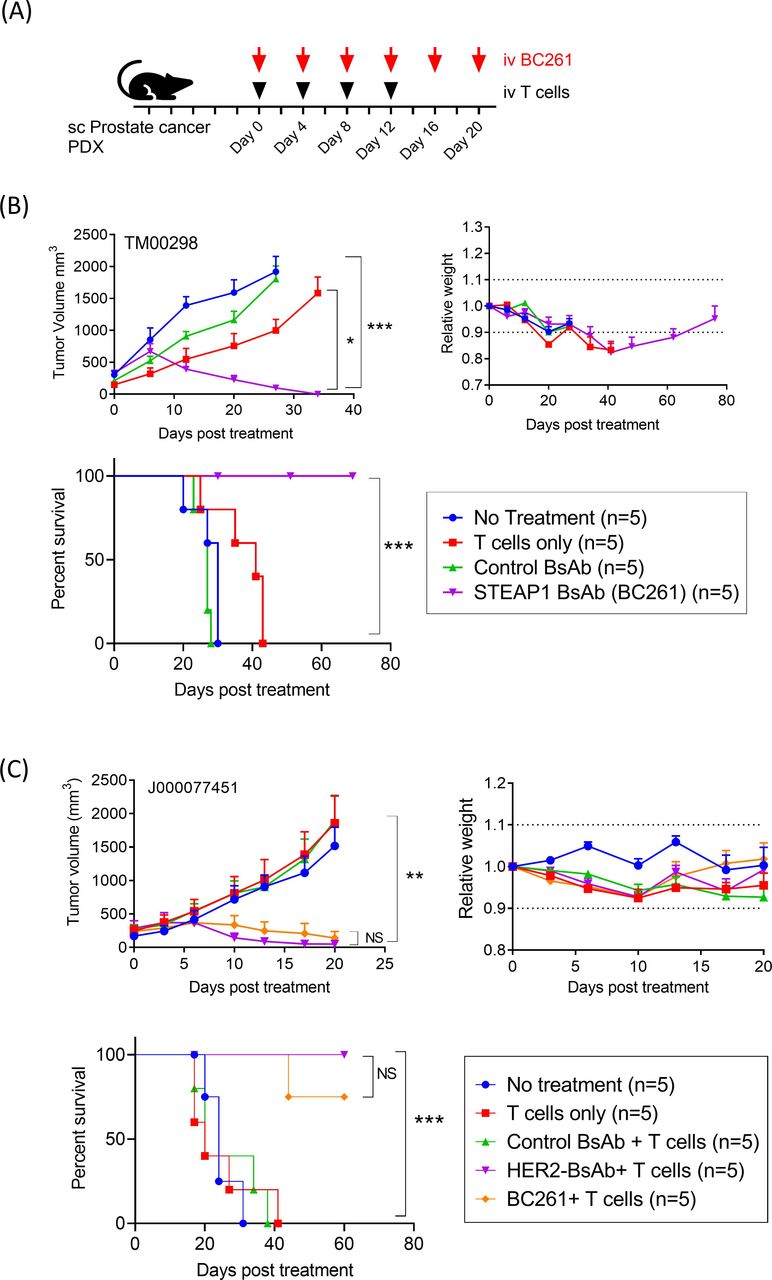
In vivo antitumor response of BC261 against prostate cancer
In addition, since STEAP1 is present on prostate cancer cells (figure 1C), antitumor efficacy of BC261 was evaluated against two prostate cancer PDXs (figure 7A). Despite the rapid tumor growth to >500–1000 mm3 size over 1 week, BC261 successfully ablated all TM00298 PDX tumors, significantly prolonging survival (p=0.0002) (figure 7B). In a second prostate cancer PDX model, J000077451, BC261 again demonstrated potent antitumor efficacy (figure 7C). Mice treated with the BC261 elicited potent tumor regression and prolonged survival without clinical toxicity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vivo antitumor effect of BC261 against STEAP1(+) prostate cancers. (A) Ten micrograms of BC261 was administered with 2×107 of T cells twice per week for 2–3 weeks to treat prostate cancer PDXs. Subcutaneous IL-2 (1000 IU) was supplemented with each T cell injection. (B) In vivo antitumor effect of BC261 against prostate cancer PDX TM00298. (C) In vivo antitumor effect of BC261 against prostate cancer PDX J000077451. Tumor growth, relative body weight, and overall survival after treatment were plotted and compared. PDXs, patient-derived tumor xenografts; STEAP, six-transmembrane epithelial antigen of prostate.
Discussion
This report describes the first discovery and validation of a novel anti-STEAP1 T-BsAb BC261 built on the IgG-[L]-scFv platform with exceptional in vivo antitumor potency. The epitope of STEAP1 BsAb was mapped to the second ECD of STEAP1, an epitope shared among human, canine and cynomolgus monkeys. The BC261 was effective against large established xenografts derived from EFT and prostate cancers despite their modest levels of STEAP1 expression. BC261 showed exceptional antitumor efficacy compared with standard T-BsAb platforms in driving T cells into tumors and suppressing tumor growth, enabling long-term disease-free survival.
Although the superiority of the ’IgG-[L]-scFv’ platform for building T-BsAb has been previously reported for disialogangliosides (GD2),35 36 a carbohydrate antigen anchored in the membrane by a short lipid side chain, the superior potency of this format has never been verified for protein targets. This is the first report to demonstrate this functional superiority of the IgG-[L]-scFv platform for the protein target STEAP1. We compared antitumor effects of representative BsAb formats and confirmed the exceptional antitumor efficacy of STEAP1 BsAb BC261 both as standalone therapeutics and when used for arming T cells targeting a broad spectrum of human cancers.
For T-BsAb, protein size, valency, affinities, and structure can influence their biodistribution,52 clearance, and potency.53 STEAP1 BsAb BC261 was developed using the IgG-[L]-scFv BsAb platform which has longer half-life with the larger protein size (~210 KD)54 and increased CD3 T cell affinity with two anti-CD3 domains. This T-BsAb format maintains bivalency toward the tumor target to maintain avidity, with functional monovalency of CD3 engagement through affinity reduction by mutating cysteine to serine in one of the CDRs. In this format, neonatal Fc receptor [FcR(n)] and protein A binding are retained, while mutations N297A and K322A are introduced to silence Fc functions, thereby reducing complement-mediated cytotoxicity or antibody-dependent cell-mediated cytotoxicity.44 T-BsAbs built using this IgG-[L]-scFv format against GD2, HER2, GPA33, CD33, CD1932 39 46 55–57 and now against STEAP1 have succeeded in driving polyclonal T cells into human tumor xenografts rapidly and quantitatively, producing robust antitumor responses within 2–3 weeks after treatment start, which did not overlap temporally with the xenogeneic GVHD effect which developed between 4 weeks and 8 weeks post-treatment.
The exceptional potency of BC261 could partly be attributed to the properties of target antigen STEAP1. STEAP1 is highly expressed in EFT, and its expression correlates with the EWS-ETS fusion proteins.58 59 STEAP1 is induced by EWS-FLI1 translocation, and its expression promotes proliferation, invasiveness, anchorage-independent colony formation, tumorigenicity, and metastasis of EFT,59 implicating close relationship between STEAP1 and the EWS-ETS translocation. Interestingly, all relapsed tumors after BC261 therapy showed regression when retreated with BC261-EATs (online supplemental figure 5). This sensitivity to retreatment contrasts with most published studies where tumor escape following T cell immunotherapy is typically associated with target antigen loss or downregulation.41 60 If STEAP1 expression is associated with cancer cell stemness, a property driven by the EWS-ETS fusion protein, it could explain the high potency of BC261 targeting STEAP1.61 It is also notable that 50%–70% of prostate cancers have similar chromosomal rearrangements resulting in aberrant expression of an oncogenic ETS family transcription factor, and these ETS factors interact with the Ewing sarcoma breakpoint protein EWS, leading to gene activation, cell migration, transformation, and tumorigenesis.62 Understanding the mechanistic pathways and biology behind STEAP1 and EWS/ETS-related fusions should help future design and testing of immunotherapeutics directed at STEAP1.3
One of the major findings of our study is the discovery of the shared epitope of STEAP1 among human, dogs, and non-human primates (NHP). ‘On-target off-tumor toxicities’ from cellular immunotherapy can induce serious side effects that are difficult to predict in current preclinical mouse models due to the specificity of BsAbs or CARs for human antigens. Although STEAP1 expression is highly restricted to tumor tissues and absent in vital organs, it is found at lower levels in normal tissues such as prostate, bladder, liver, kidney, pancreas, and skeletal muscle.20 27 63 As a non-human preclinical model, dogs and NHP may provide alternatives for evaluating not only the efficacy but also the ‘on-target off tumor toxicity’. Besides, canine osteosarcoma is one of the most common cancers in dogs.64 As an outbred non-human tumor model, it provides an alternative for testing STEAP1 antibody-based therapeutics both for toxicities and for efficacy. Since osteosarcoma is also difficult to cure in canine hosts, STEAP1-targeted antibody-based therapeutics could fulfill another unmet canine need. Even though the STEAP1 protein is highly restricted to tumors, long-term persistence of the cytotoxic drugs or cytotoxic immune effector cells could be harmful. While CAR T cells are life-long and such toxicities could be prolonged and life-threatening, BsAb-driven T cells or EATs have limited functional life expectancy only weeks. As the BsAbs get metabolized, T cells should revert to their non-specific states, reducing or terminating the risk of life-threatening long-term toxicities. In this respect, targeting STEAP1 using T-BsAb and EAT offers safter alternatives.
Taken together, STEAP1 is an appealing target for T cell immunotherapy, and STEAP1 T-BsAb BC261 offers a novel solution to engage T cells to traffic into immunologically ‘cold’ STEAP1(+) malignancies. BC261 induced robust antitumor responses without long-term toxicities for stand-alone BsAb or BsAb armed T cells, warranting clinical development for human solid tumors with major unmet need, such as EFT and prostate cancers.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data generated or analyzed during this study are included in this published article.
Ethics statements
Patient consent for publication
Ethics approval
All animal experiments were approved by the Memorial Sloan Kettering’s Institutional Animal Care and Use Committee (IACUC) and were executed according to the ACUC guidelines. Patient-derived tumor xenografts were established with MSKCC IRB approval.
Acknowledgments
We would like to especially thank Xu Hong for analyzing STEAP1 BsAbs’ affinity to CD3.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @StevenTYLin08
T-YL and JAP contributed equally.
Contributors T-YL, JAP, and N-KVC designed the experiments, interpreted and analyzed the results, and wrote the manuscript. JAP and N-KVC revised the manuscript. AL did experiments with the prostate cancer PDX mouse model. H-FG tested the purity and stability of the STEAP1-BsAbs using HPLC and SDS-PAGE and analyzed STEAP1 antigen expression on multiple cancer cell lines. All authors reviewed this manuscript and approved the final manuscript.
Funding This work was partly supported by Enid A. Haupt Endowed Chair, the Robert Steel Foundation, and Kids Walk for Kids with Cancer. Technical service provided by the MSK Animal Imaging Core Facility, Antitumor Assessment Core Facility, and Molecular Cytology Core Facility were supported in part by the NCI Cancer Center Support Grant P30 CA008748.
Competing interests Both MSK and NKC have financial interest in Y-mAbs, Abpro-Labs and Eureka Therapeutics. NKC reports receiving commercial research grants from Y-mAbs Therapeutics and Abpro-Labs Inc. NKC was named as inventor on multiple patents filed by MSK, including those licensed to Ymabs Therapeutics, Biotec Pharmacon, and Abpro-labs. NKC is a SAB member for Abpro-Labs and Eureka Therapeutics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.