Article Text

Abstract
Background Oncolytic viruses reduce tumor burden in animal models and have generated promising results in clinical trials. However, it is likely that oncolytic viruses will be more effective when used in combination with other therapies. Current therapeutic approaches, including chemotherapeutics, come with dose-limiting toxicities. Another option is to combine oncolytic viruses with immunotherapeutic approaches.
Methods Using experimental models of metastatic 4T1 breast cancer and ID8 ovarian peritoneal carcinomatosis, we examined natural killer T (NKT) cell-based immunotherapy in combination with recombinant oncolytic vesicular stomatitis virus (VSV) or reovirus. 4T1 mammary carcinoma cells or ID8 ovarian cancer cells were injected into syngeneic mice. Tumor-bearing mice were treated with VSV or reovirus followed by activation of NKT cells via the intravenous administration of autologous dendritic cells loaded with the glycolipid antigen α-galactosylceramide. The effects of VSV and reovirus on immunogenic cell death (ICD), cell viability and immunogenicity were tested in vitro.
Results VSV or reovirus treatments followed by NKT cell activation mediated greater survival in the ID8 model than individual therapies. The regimen was less effective when the treatment order was reversed, delivering virus treatments after NKT cell activation. In the 4T1 model, VSV combined with NKT cell activation increased overall survival and decreased metastatic burden better than individual treatments. In contrast, reovirus was not effective on its own or in combination with NKT cell activation. In vitro, VSV killed a panel of tumor lines better than reovirus. VSV infection also elicited greater increases in mRNA transcripts for proinflammatory cytokines, chemokines, and antigen presentation machinery compared with reovirus. Oncolytic VSV also induced the key hallmarks of ICD (calreticulin mobilization, plus release of ATP and HMGB1), while reovirus only mobilized calreticulin.
Conclusion Taken together, these results demonstrate that oncolytic VSV and NKT cell immunotherapy can be effectively combined to decrease tumor burden in models of metastatic breast and ovarian cancers. Oncolytic VSV and reovirus induced differential responses in our models which may relate to differences in virus activity or tumor susceptibility.
- immunotherapy
- oncolytic virotherapy
- dendritic cells
- cytotoxicity
- immunologic
- natural killer T-Cells
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Metastasis from the primary tumor site to other parts of the body is responsible for the majority of cancer-related deaths.1 Current therapeutic approaches, including chemotherapeutics and radiation, are often ineffective and come with off-target effects causing dose-limiting toxicity.2 3 Metastatic clusters form hypoxic and acidic regions, limiting the ability of drugs to target proliferating cells,4 and can also gain treatment resistance via mutations that alter drug uptake and metabolism.5 6 Taken together, these factors highlight the need for novel therapeutics that effectively target metastasis. One option is to use combined immunotherapeutic approaches to harness the power of the immune system.
Natural killer T (NKT) cells are a subset of glycolipid-reactive T lymphocytes that play an important role in tumor immunosurveillance.7 8 Unlike conventional T cells, NKT cells express an invariant TCRα chain rearrangement (Vα14-Jα18 in mice and Vα24-Jα18 in humans) which allows them to recognize endogenous and foreign glycolipids presented by the major histocompatibility-like molecule CD1d.9 NKT cells can be activated therapeutically via administration of dendritic cells (DCs) loaded with the glycolipid antigen α-galactosylceramide (α-GalCer).10 11 Once activated, NKT cells release large quantities of cytokines, including interferon-γ (IFN-γ) and tumor necrosis factor (TNF), allowing them to regulate the function of other immune cells.10 12 Furthermore, NKT cells can directly kill tumor cells via release of cytotoxic molecules.12 Therapeutic activation of NKT cells with glycolipids provides protection from tumor progression,7 8 13 and infiltration of NKT cells is associated with a good prognosis.14 15 Previously, our laboratory demonstrated that NKT cell immunotherapy can effectively target a model of breast cancer metastasis, resulting in complete tumor clearance in 40%–50% of mice.11 Combining NKT cell immunotherapy with the chemotherapeutics cyclophosphamide or gemcitabine further increased survival.16 As chemotherapeutics are associated with dose-limiting toxicities and harsh side effects,2 we wanted to examine the effect of combining NKT cell immunotherapy with another emerging therapeutic modality, oncolytic viruses.
Oncolytic viruses are viruses that preferentially replicate in and kill cancer cells by taking advantage of altered receptor expression, altered metabolism, or defects in antiviral defenses.17–20 While many previous studies focused on direct cancer cell killing by oncolytic viruses, there is growing evidence that they also act by stimulating antitumor immune responses.21 This makes them great candidates to be combined with immunotherapies. Oncolytic vesicular stomatitis virus (VSVΔM51) is an attenuated negative strand RNA virus from the Rhabdoviridae family.19 Oncolytic VSV infects cells using the broadly expressed low-density lipoprotein receptor,17 but is sensitive to type I IFNs, restricting its replication to cancer cells which commonly have defects in type I IFN signaling.19 Oncolytic VSV has shown promise in preclinical trials and can be effectively combined with immunotherapies including checkpoint inhibitors and chimeric antigen receptor T cell therapy.22 23
Another oncolytic virus that exhibits increased tumor control when combined with checkpoint inhibitors is mammalian respiratory enteric orphan virus (reovirus).24 25 Reovirus is a double-stranded segmented RNA virus that has been widely used in clinical studies.26 Cancer cells with an overactive oncogenic RAS signaling pathway are generally more susceptible to reovirus infection.18 Reovirus has be shown to induce antigen-specific T cell responses in a murine prostate cancer model, demonstrating its ability to induce antitumor immunity.27 Here, we examined the ability of oncolytic VSV and reovirus therapy to be combined with NKT cell immunotherapy in mouse models of ovarian and breast cancer metastasis.
Methods
Mice
Female BALB/c and C57BL/6 mice were purchased form Charles River Laboratories. Mice were maintained under specific pathogen-free conditions in the Carleton Animal Care Facility at Dalhousie University and used at 8–12 weeks of age. All experimental procedures were approved by the University Committee on Laboratory Animals following the guidelines of the Canadian Council on Animal Care.
Cell culture
B16-F10 melanoma cells (CRL-6045), 4T1 mammary carcinoma cells (CRL-2539), Lewis lung carcinoma (CRL-1642), and Vero kidney epithelial cells (CCL-81) were obtained from ATCC (Manassas, Virginia, USA). ID8 ovarian cancer cells (generated by Dr K Roby, University of Arkansas) were obtained from Dr Jean Marshall, Dalhousie University. Panc02 pancreatic cancer cells (generated by Dr T H Corbett, Wayne State University) were obtained from Dr John Bell, University of Ottawa. Cells were cultured at 37°C, 5% CO2, in Dulbecco’s Modified Eagle Medium (DMEM; VWR Life Science, Mississauga, ON, Canada) supplemented with 10% fetal bovine serum (FBS), 100 µg/mL streptomycin, and 100 units/mL penicillin (Fisher-HyClone, Ottawa, ON, Canada). Frozen stocks were generated, and aliquots were used in experiments at <8 passages. Mycoplasma testing was performed every 3–6 months using the VenorGem detection kit (Sigma-Aldrich).
Virus purification
VSVΔM51 engineered to express green fluorescent protein was provided by Dr Douglas Mahoney, University of Calgary. Vero cells at ~95% confluency were infected with VSV at a multiplicity of infection (MOI) of ~0.1 in serum-free DMEM for 48 hours. Supernatant was collected, centrifuged at 300×g for 5 min at 4°C and filtered through a 0.45 µm filter. Clarified supernatant was centrifuged at 28,000×g for 1.5 hours at 4°C and the virus pellet resuspended in phosphate buffered saline (PBS), layered on 20% sucrose and centrifuged at 36,000 rpm for 90 min at 4°C. Collected virus was resuspended in PBS containing 15% glucose and stored at −80°C. Reovirus (Dearing strain, T3D) was provided by Dr Patrick Lee, Dalhousie University. Virus titers were determined by plaque assay using Vero cells. UV inactivation was performed using a UVP HL-2000 Hybrilinker (Fischer Scientific) at 100 μJ/CM2 for 15 min.
Bone marrow–derived DCs
To generate DCs, bone marrow was extracted from the femur and tibia of syngeneic donor mice and cultured in 6-well plates with complete RPMI-1640 (10% FBS, 50 µM 2-mercaptoethanol, 2 mM L-glutamine, 1X non-essential amino acids, 1 mM sodium pyruvate, 100 µg/mL streptomycin, and 100 units/mL penicillin) containing 40 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) and 10 ng/mL IL-4 (PeproTech). Media was replenished on day 3. Non-adherent cells were collected and replated in complete RPMI-1640 with 20 ng/mL GM-CSF on day 6. α-GalCer (KRN7000; DiagnoCine) was sonicated for 20 min at 50°C before being added to the DCs at 0.4 µg/mL. DCs were collected the next day and injected intravenously to induce NKT cell activation.
4T1 metastasis model
4T1 cells were harvested in the logarithmic growth phase using trypsin-EDTA (Sigma-Aldrich). Cells were resuspended in saline and 2×105 cells (50 µL volume) were injected subcutaneously into the fourth mammary fat pad of female BALB/c mice. Primary mammary tumors were resected 12 days after tumor cell injection when the primary tumors reached ~200 mm3 in size. Tumor excision was performed aseptically in anesthetized mice (inhaled isoflurane) and the skin was sutured using 5-0 polypropylene suture (Ethicon, Somerville, New Jersey, USA). Mice received a subcutaneous treatment of 0.1 mg/kg buprenorphine (BCM Corporation; Bloomingdale, New Jersey, USA) as an analgesic. On days 13, 15, and 17, mice were treated intravenously with PBS, VSV (5×108 pfu/mL) or reovirus (5×108 pfu/mouse), UV-inactivated VSV, or UV-inactivated reovirus. On day 18, unloaded (control) or α-GalCer-loaded DCs (intravenous 2×105/mouse) were administered to induce NKT cell activation. Survival was monitored over 120 days.
Clonogenic assay
To quantify lung metastasis, lungs were harvested on day 28, dissociated by mechanical dispersion through a sterile 40 micron nylon mesh, and selected in media supplemented with 60 µM 6-thioguanine (Alfa Aesar, Tewksbury, Massachusetts, USA). After 7 days, plates were fixed with methanol and stained with 0.03% methylene blue (BioShop, Burlington, Ontario, Canada). Tumor colonies were enumerated using ImmunoSpot colony-counting software (Cellular Technology Limited, Cleveland, Ohio, USA).
ID8 ovarian cancer model
ID8 cells were harvested in the logarithmic growth phase using trypsin-EDTA (Sigma-Aldrich). Cells were resuspended in saline and 3×106 cells (50 µL volume) were injected intraperitoneally into female C57BL/6 mice. On days 9, 11, and 13, mice were treated intravenously with PBS, VSV (5×108 pfu/mouse), reovirus (5×108 pfu/mouse), or UV-inactivated reovirus. On day 14, unloaded (control) or α-GalCer-loaded DCs (intravenous 6×105/mouse) were administered to induce NKT cell activation. Survival was monitored over 90 days.
Expression of MHC I, MHC II and CD1d
4T1 and ID8 cells were cultured overnight in 96-well plates at 5×103 cells per well. Cells were treated with VSV or reovirus for 24, 48, and 72 hours at an MOI of 1. UV-irradiated viruses (UV-VSV∆M51 or UV-reo) were used as controls. Cells were stained with fluorescein isothiocyanate-labeled major histocompatibility complex (MHC) I antibody (H-2Db; clone 28-14-8), phycoerythrin-labeled CD1d antibody (clone 1B1), or allophycocyanin-labeled MHC II antibody (IA/I-E clone M5/114.15.2) for 30 min at 4°C, washed, and fixed in 2% paraformaldehyde (Fisher Scientific). Analysis was performed using a three-laser FACSCanto and FlowJo software (BD Biosciences).
Surface mobilization of calreticulin
4T1 and ID8 cells were infected with VSV∆M51 or reovirus at an MOI of 10 and incubated for 8 hours. Cells were fixed with 0.25% paraformaldehyde for 5 min, washed in PBS and blocking buffer, and stained with a rabbit anti-calreticulin antibody (Abcam, Cambridge, Massachusetts, USA) for 30 min. Cells were washed and reactivity was detected following 30 min incubation with anti-rabbit Alexa Flour 680 (Fisher Scientific). Analysis was performed using a two-laser FACSCalibur and FlowJo software (BD Biosciences).
Cytokine, ATP, and HMGB1 release assays
Supernatants were harvested 24 hours after VSV or reovirus infection of 4T1 or ID8 cells. ATP was measured using a luciferin-based ATP activity assay (Calbiochem). Chemiluminescence was recorded using a Fluoroskan Ascent FL plate reader (Thermo-Scientific). High mobility group protein 1 (HMGB1) levels were measured by ELISA (IBL International). Blood was obtained by submandibular vein puncture at various time points after NKT cell activation to measure cytokine levels. Serum IL-4 and IFN-γ levels were examined using Ready-Set-Go ELISAs (eBioscience). Absorbance for ELISAs was measured using an Epoch microplate spectrophotometer (BioTek).
RNA isolations, cDNA synthesis and qPCR
4T1 or ID8 cells were infected with VSV∆M51 or reovirus (MOI=1) for 24 hours prior to RNA isolation. Total RNA was isolated using a RNeasy Plus Mini kit (Qiagen) and cDNA was prepared from 200 ng of RNA using the advanced cDNA synthesis kit (Wisent Bio). Quantitative PCRs were performed in duplicate using 1 μL of cDNA and Quantifast SYBR Green (Qiagen). PCR was run with 10 min hold at 95°C before 40 cycles of 15 s at 95°C followed by 45 s at 55°C. Data were collected on an RG-6000 Rotor-Gene (Corbett Research) and analyzed using the 2−ΔΔCT relative quantification technique and expressed relative to gapdh, a validated internal normalizing mRNA. High-stringency primer pairs were used for: ccl3, ccl4, ccl5, ccl8, cxcl2, cxcl9, cxcl10, cxcl11, cxcl16, cx3cl1, gapdh, ifn-α, il-6, m-csf, tap-1, tap-2, tgfβ, and tnf (table 1). Primer performance was validated by melting curve analysis.
Primers used for quantitative PCR
Statistical analysis
Data are expressed as mean±SEM, unless otherwise stated. Statistical comparisons were carried out using GraphPad Prism V.8.02. A non-parametric two-tailed Mann-Whitney U test was used to compare between two data groups. Comparisons between more than two data groups were made using a Kruskal-Wallis non-parametric analysis with Dunn’s post-test. Survival data were analyzed by log-rank (Mantel-Cox) significance test with Bonferroni corrected thresholds. Statistical significance was set at p<0.05.
Results
Oncolytic VSV or reovirus combined with NKT cell immunotherapy increases survival in the ID8 ovarian cancer model
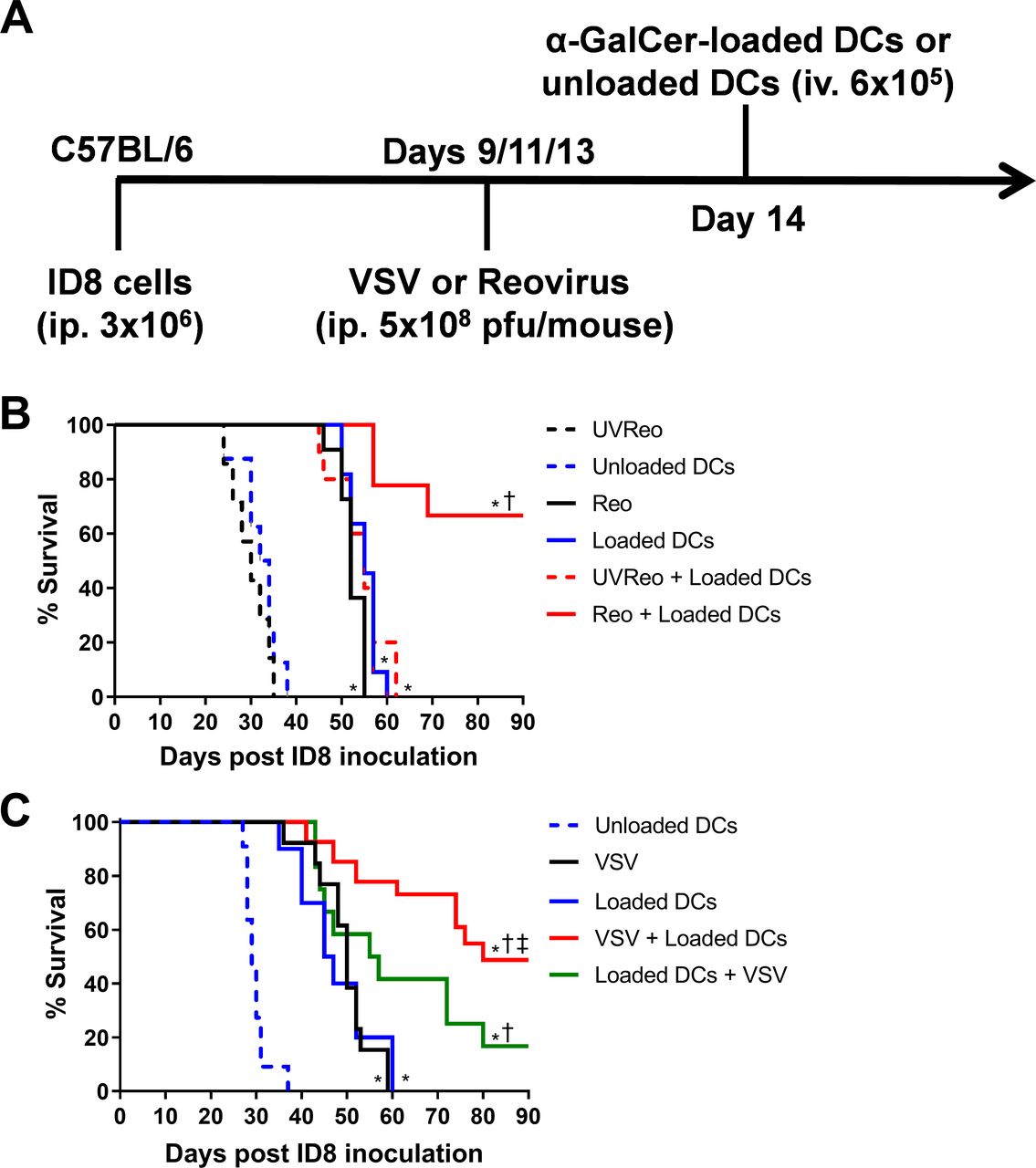
Reovirus has been shown to improve survival in the ID8 ovarian peritoneal carcinomatosis model.28 We compared the effects of combining reovirus or VSV with NKT cell immunotherapy in the ID8 model. ID8 ovarian cancer cells were injected (intraperitoneally 3×105) into C57BL/6 mice. On days 9, 11 and 13 mice were treated intravenously with 5×108 pfu of VSV or reovirus. NKT cells were activated on day 14 via administration of α-GalCer-loaded DCs (figure 1A). Monotherapy treatments with reovirus, VSV, or α-GalCer-loaded DCs significantly increased the overall survival of the mice compared with mice treated with UV-inactivated reovirus or unloaded DCs (figure 1B,C). The combination of reovirus or VSV with NKT cell activation therapy significantly increased survival compared with individual treatments (figure 1B,C). To determine whether the order of treatment impacted survival, additional groups of mice received NKT cell activation therapy followed by oncolytic VSV treatments. While both combined approaches increased survival over individual therapies, treatment with oncolytic VSV before NKT cell activation resulted in superior survival (figure 1C). These results demonstrate that either reovirus or VSV can be combined with NKT cell activation therapy to increase survival in the ID8 model.

Combining natural killer T (NKT) cell activation therapy with oncolytic virus therapy to target ID8 ovarian cancer. (A) Schematic of treatments in the ID8 ovarian model. (B) Survival was assessed following treatment with unloaded dendritic cells (DCs), α-galactosylceramide (α-GalCer)-loaded DCs, reovirus, UV-inactivated reovirus, or α-GalCer-loaded DCs in combination with reovirus (n=7–10 per group). (C) Survival was assessed following treatment with unloaded DCs, α-GalCer-loaded DCs, vesicular stomatitis virus (VSV), UV-inactivated VSV, or α-GalCer-loaded DCs in combination with VSV (n=13–26 per group). *p<0.05 compared with unloaded DCs; †p<0.05 compared with single treatments; ‡p<0.05 compared with reverse order treatment.
Oncolytic VSV but not reovirus combined with NKT cell immunotherapy increases survival and reduces metastasis in the 4T1 breast cancer model
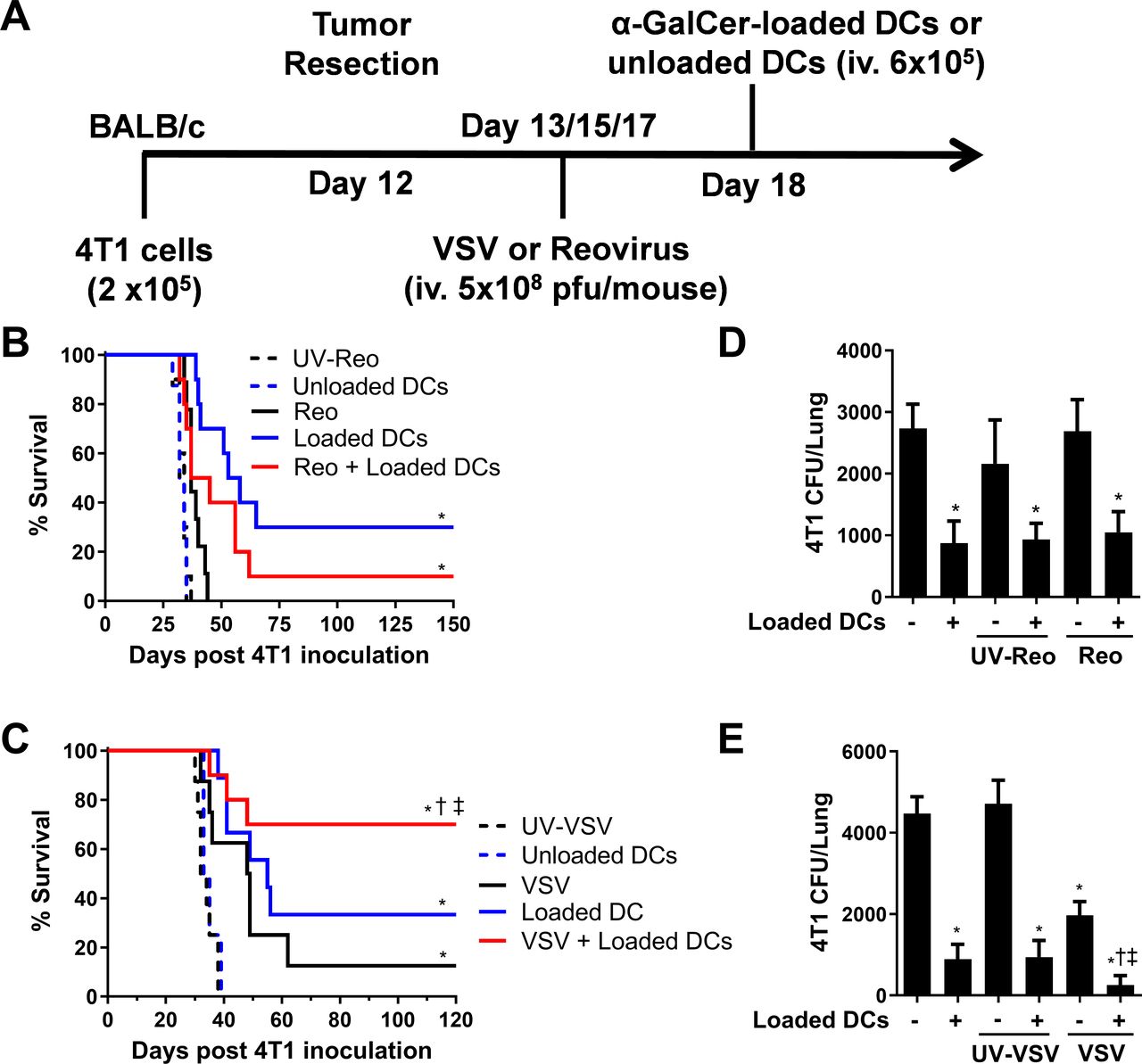
We have previously demonstrated that NKT cell immunotherapy is effective in a metastatic 4T1 breast cancer model, leading to complete tumor clearance in 40%–50% of mice.11 To determine whether oncolytic virotherapy could improve NKT cell immunotherapy in this model, we combined NKT cell activation therapy with either VSV or reovirus treatments. Mice were injected orthotopically into the fourth mammary pad with 2×105 4T1 cells. Primary tumors were resected on day 12 before treating mice with VSV or reovirus (intravenous 5×108 pfu) on days 13, 15, and 17. NKT cells were then activated on day 18 by intravenous injections of α-GalCer-loaded DCs (figure 2A). VSV and reovirus treatments led to modest increases in overall survival compared with control mice treated with UV-inactivated virus (figure 2B,C). However, NKT cell immunotherapy was superior to oncolytic virus monotherapies (figure 2B,C). When oncolytic VSV was combined with NKT cell immunotherapy, there was a significant increase in overall survival compared with individual treatments (figure 2C). In contrast, the combination with reovirus had no increased benefit. While survival appeared worse with combined therapy, it was not significantly different from NKT cell immunotherapy alone (figure 2B). To further examine the effect of our treatments, we examined the metastatic burden of 4T1 cells in the lung on day 28 via a clonogenic plating assay. While treatment with α-GalCer-loaded DCs significantly decreased 4T1 lung metastases, reovirus had no effect on the number of lung metastases on its own or in combination with NKT cell therapy (figure 2D). In contrast, VSV reduced 4T1 metastasis and further reduced the number of metastatic cells when combined with NKT cell immunotherapy (figure 2E). These results demonstrate that the ability of specific oncolytic viruses to increase the efficacy of NKT cell immunotherapy may be cancer type dependent.

Combining natural killer T (NKT) cell activation therapy with oncolytic virus therapy to target metastatic 4T1 mammary carcinoma. (A) Schematic of treatments in the post-surgical 4T1 metastasis model. (B) Survival was assessed following treatment with unloaded dendritic cells (DCs), α-galactosylceramide (α-GalCer)-loaded DCs, reovirus, UV-inactivated reovirus, or α-GalCer-loaded DCs in combination with reovirus (n=10 per group). (C) Survival was assessed following treatment with unloaded DCs, α-GalCer-loaded DCs, vesicular stomatitis virus (VSV), UV-inactivated VSV, or α-GalCer-loaded DCs in combination with VSV (n=9–10 per group). (D, E) Number of 4T1 CFUs present in lung cell suspensions isolated at 28 days post-4T1 injection following treatments incorporating (D) reovirus or (E) VSV (n=9–10 per group). *p<0.01 compared with unloaded DC; †p<0.01 compared with virus alone; ‡p<0.01 compared with α-GalCer-loaded DCs alone.
To determine whether reovirus and VSV differentially impacted NKT cell activation in the 4T1 model, blood was collected 0, 2 and 24 hours after DC injection to examine serum cytokine expression. Neither VSV nor reovirus treatments alone increased serum IFN-γ and IL-4 levels (online supplemental figure 1). As expected, serum IFN-γ and IL-4 levels were increased following NKT cell activation via α-GalCer-loaded DCs (online supplemental figure 1). Oncolytic virus treatments did not alter cytokine release elicited by NKT cell activation (online supplemental figure 1).
Supplemental material
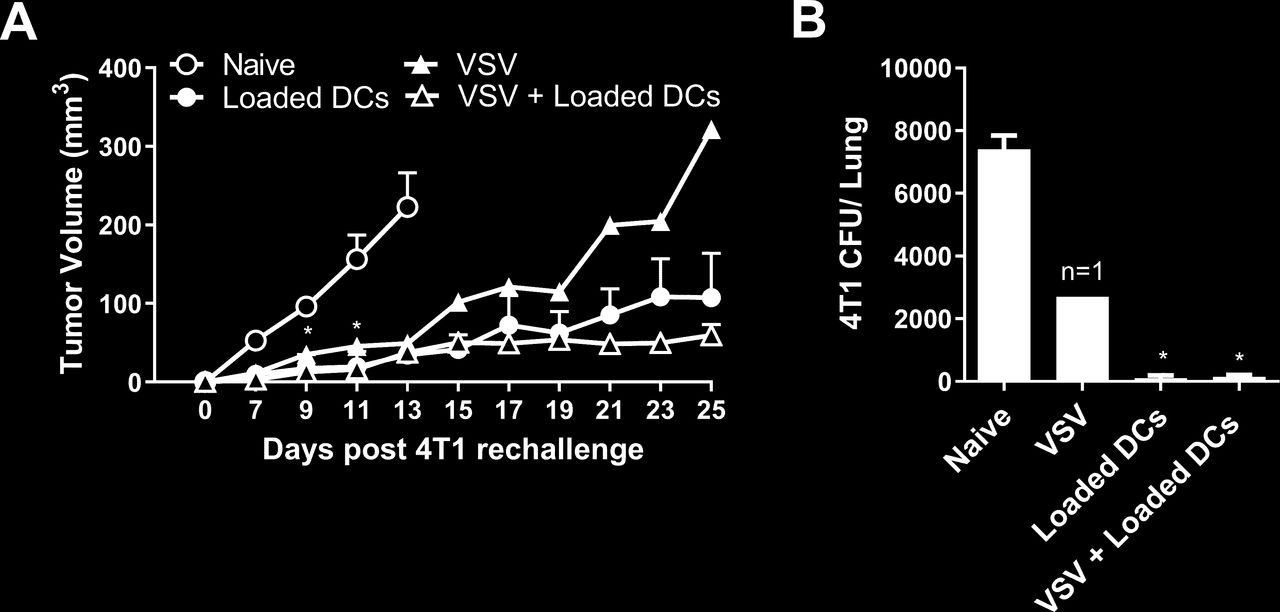
Mice that survived the first 4T1 tumor challenge were rechallenged in the contralateral mammary fat pad with 4T1 cells to assess whether immune memory had developed against the 4T1 cells. All the rechallenged mice exhibited slower tumor growth compared with naïve mice inoculated with 4T1 cells (figure 3A). Rechallenged mice also had significantly reduced metastatic burden at 25 days post tumor rechallenge compared with the naïve mice (figure 3B). While tumor growth was suppressed in all rechallenged mice that had received previous NKT cell therapy, growth was only delayed in the single mouse that survived following VSV therapy alone.
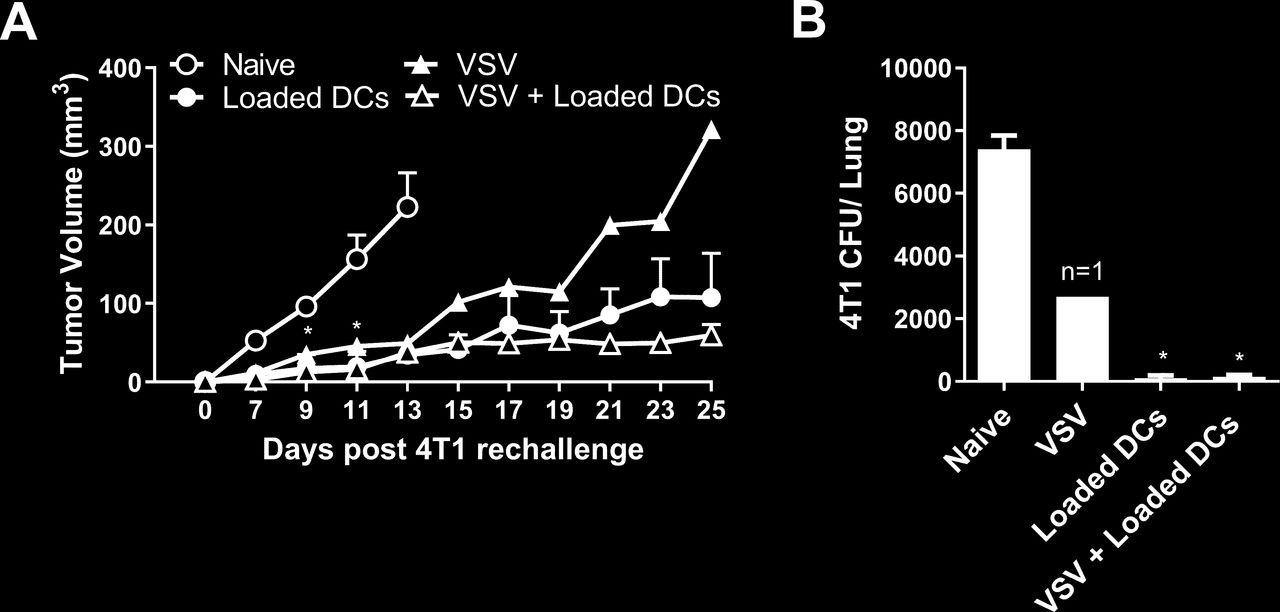
Tumor growth in survivors following rechallenge with 4T1. Tumor resected mice that survived to day 120 after treatment with α-galactosylceramide (α-GalCer)-loaded dendritic cells (DCs), vesicular stomatitis virus (VSV), combination of VSV and α-GalCer-loaded DCs (figure 2) were rechallenged in the contralateral mammary fat pad with 2×05 4T1 cells. (A) Tumor volume was compared with tumors grown in naïve mice (n=4–7 per group, except n=1 for VSV group). (B) Metastatic burden at day 25 was compared with naïve tumor-bearing mice (n=4–7 per group, except n=1 for VSV group). *p<0.05 compared with naïve control.
Oncolytic VSV is better at killing cancer cells than reovirus
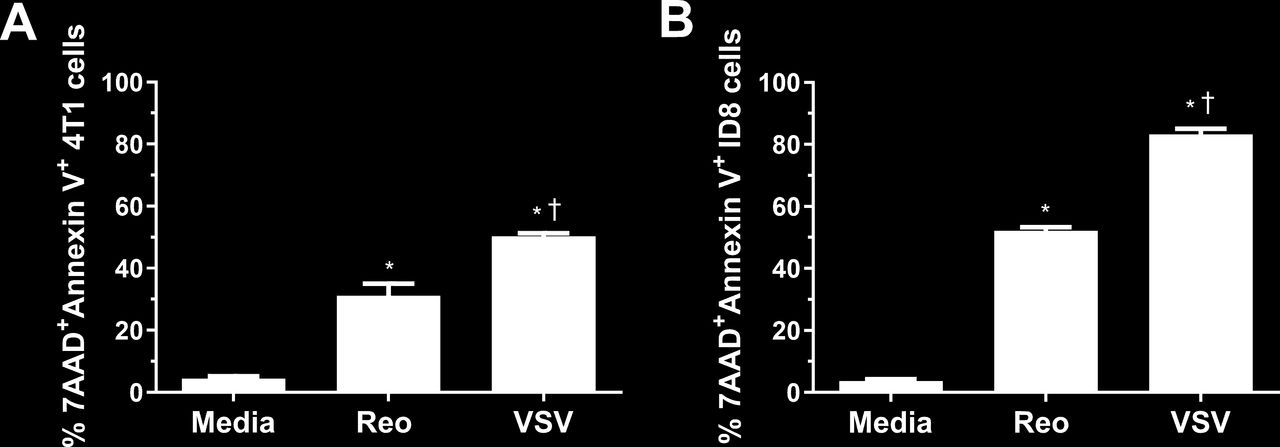
To understand why NKT cell immunotherapy was more effective in combination with VSV than reovirus, we examined the ability of each virus to kill 4T1 and ID8 cells in vitro using an MTT assay. VSV significantly reduced 4T1 viability at a lower MOI and at earlier time points than reovirus (online supplemental figure 2A). Reovirus was able to kill ID8 cancer cells better than 4T1 cells, potentially explaining why it was more successful in the ID8 model; however, the ability of VSV to kill ID8 cells was still greater than reovirus at earlier time points and lower MOI (online supplemental figure 2B). To verify our MTT data, we examined cell death by examining surface expression of 7AAD and Annexin V. VSV and reovirus infection both increased 7AAD and Annexin V surface expression, indicating ability to induce tumor cell death. VSV infection increased 7AAD and Annexin V staining more than reovirus (figure 4), confirming it was better at killing 4T1 and ID8 cells. VSV was also able to kill B16, LLC, and panc02 cells better than reovirus, demonstrating its ability to target a wide array of cancer cells from both the BALB/c and C57BL/6 genetic backgrounds (online supplemental figure 2C–E). Therefore, it is likely that VSV increased survival in the 4T1 model, alone and in combination with NKT cell immunotherapy, at least in part by enhanced killing of tumor cells.
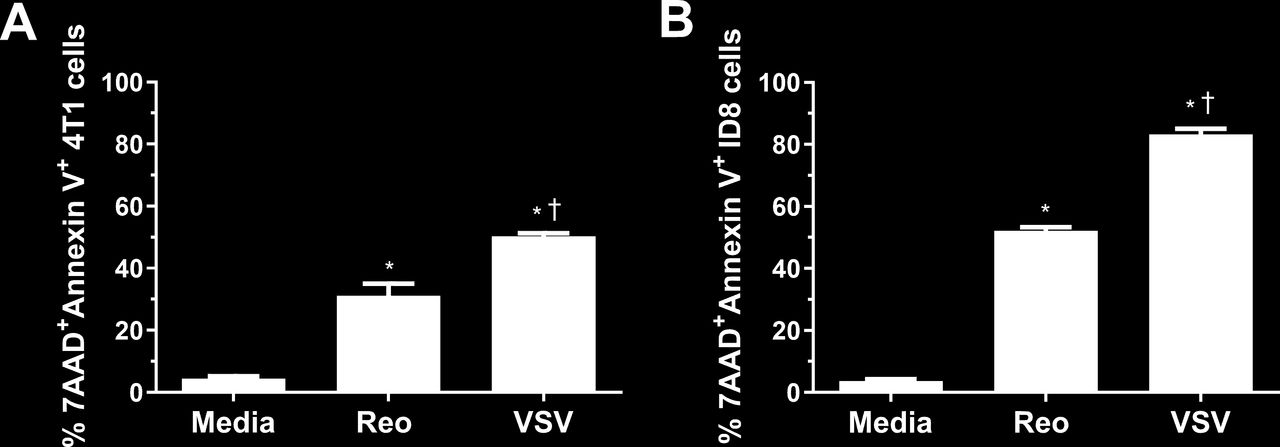
Killing of 4T1 and ID8 cells by VSV∆M51 and reovirus. (A) 4T1 and (B) ID8 cells were infected with vesicular stomatitis virus (VSV) or reovirus at a multiplicity of infection of 1 in vitro. After 24 hours, cells were collected and stained for Annexin V and 7AAD (n=3 per group). *p<0.05 compared with media; † p<0.05 compared with reovirus.
VSV infection increases antigen presentation capacity in 4T1 and ID8 cells
Cancer cells often evade immune detection by downregulating antigen presentation machinery.29 In contrast, oncolytic viruses work in part by increasing antitumor immunity.21 Therefore, we examined the effect of VSV and reovirus infection on the immunogenic potential of 4T1 and ID8 cells. 4T1 and ID8 cells were infected with reovirus, VSV or UV-irradiated viruses at an MOI of ~1 for 24 hours. Cells were then collected for flow cytometric and qRT-PCR analysis. VSV increased surface expression of MHC I on both 4T1 and ID8 cells at 24 hours after virus exposure (figure 5A). Furthermore, VSV upregulated MHC II and CD1d surface expression at 48 and 72 hours (figure 5A), demonstrating that VSV infection can broadly increase antigen presentation capacity of 4T1 and ID8 cells. Reovirus infection did not increase antigen presentation molecules on 4T1 cells until 72 hours, resulting in a small increase in MHC II (figure 5B). Reovirus infection increased antigen presentation earlier in ID8 cells, increasing MHC II surface at 48 hours (figure 5B). Furthermore, MHC I and CD1d were also upregulated in ID8 cells at 72 hours (figure 5B), demonstrating that reovirus can broadly increase antigen presentation in ID8 cells, but not 4T1 cells. At 24 hours after infection with VSV, mRNA transcripts for antigen transporters tap 1 and 2 were upregulated in both 4T1 and ID8 cells (figure 5C,D). Reovirus infection did not enhance tap2 expression and only increased tap1 in the ID8 model (figure 5C,D). These data further support the inability of reovirus to enhance MHC I antigen presentation in 4T1 cells.
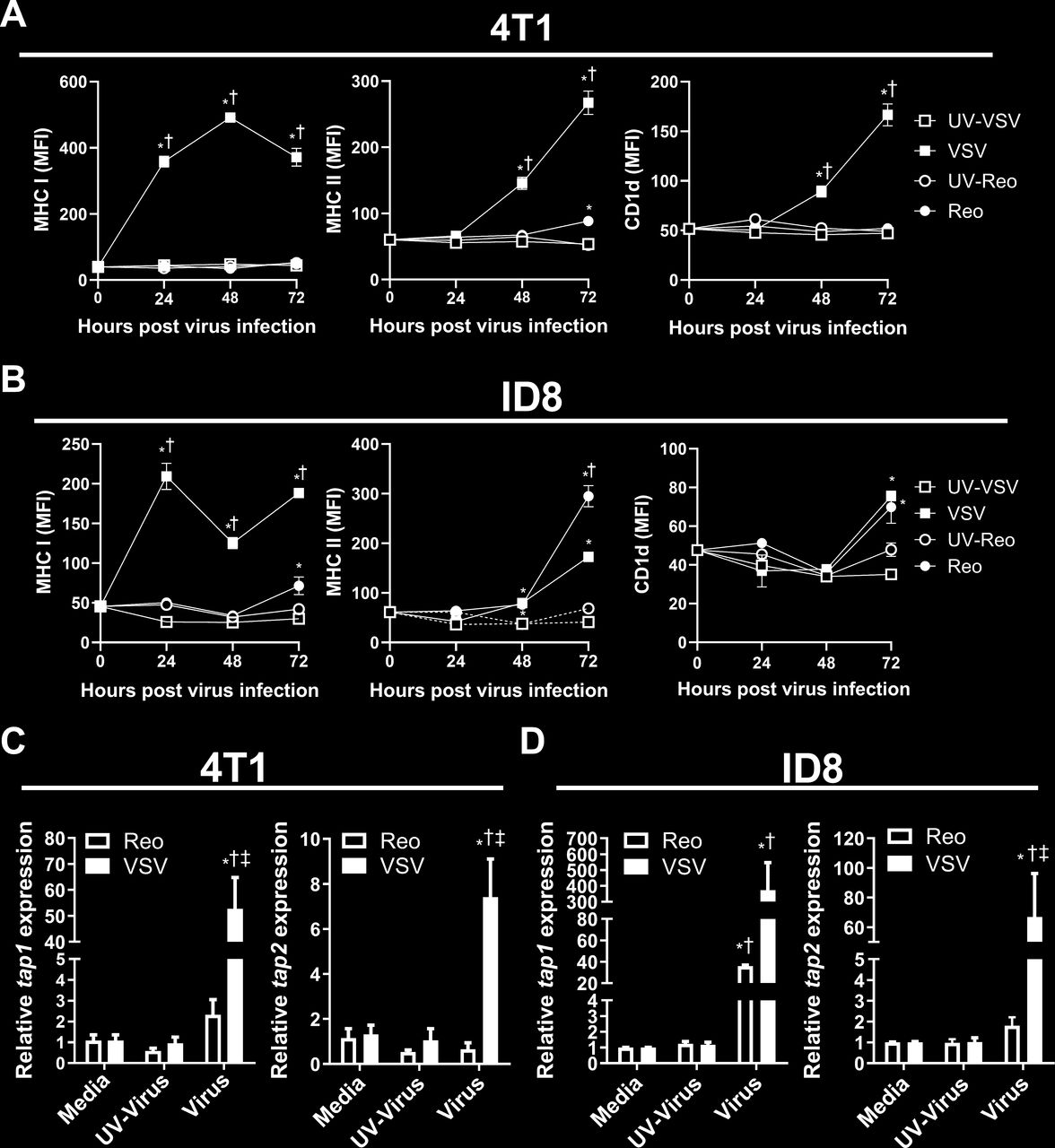
Induction of antigen presentation machinery by 4T1 and ID8 cells infected with VSV∆M51 or reovirus. (A, B) Mean fluorescent intensity (MFI) of major histocompatibility complex (MHC) I, MHC II and CD1d surface expression on cultured (A) 4T1 and (B) ID8 cells 24, 48 and 72 hours after vesicular stomatitis virus (VSV) or reovirus infection at multiplicity of infection (MOI) of 1. UV-inactivated viruses were used as controls (n=4 per group). (C, D) Quantitative PCR examining the expression of tap1 and tap2 in (C) 4T1 and (D) ID8 cells treated with VSV or reovirus for 24 hours. UV-inactivated viruses were used as controls. qPCR was analyzed using the 2−ΔΔCT quantification technique relative to the validated housekeeping gene GAPDH (n=3–6 per group). *p<0.05 compared with media; † p<0.05 compared with UV-inactivated virus; ‡p<0.05 compared with reovirus.
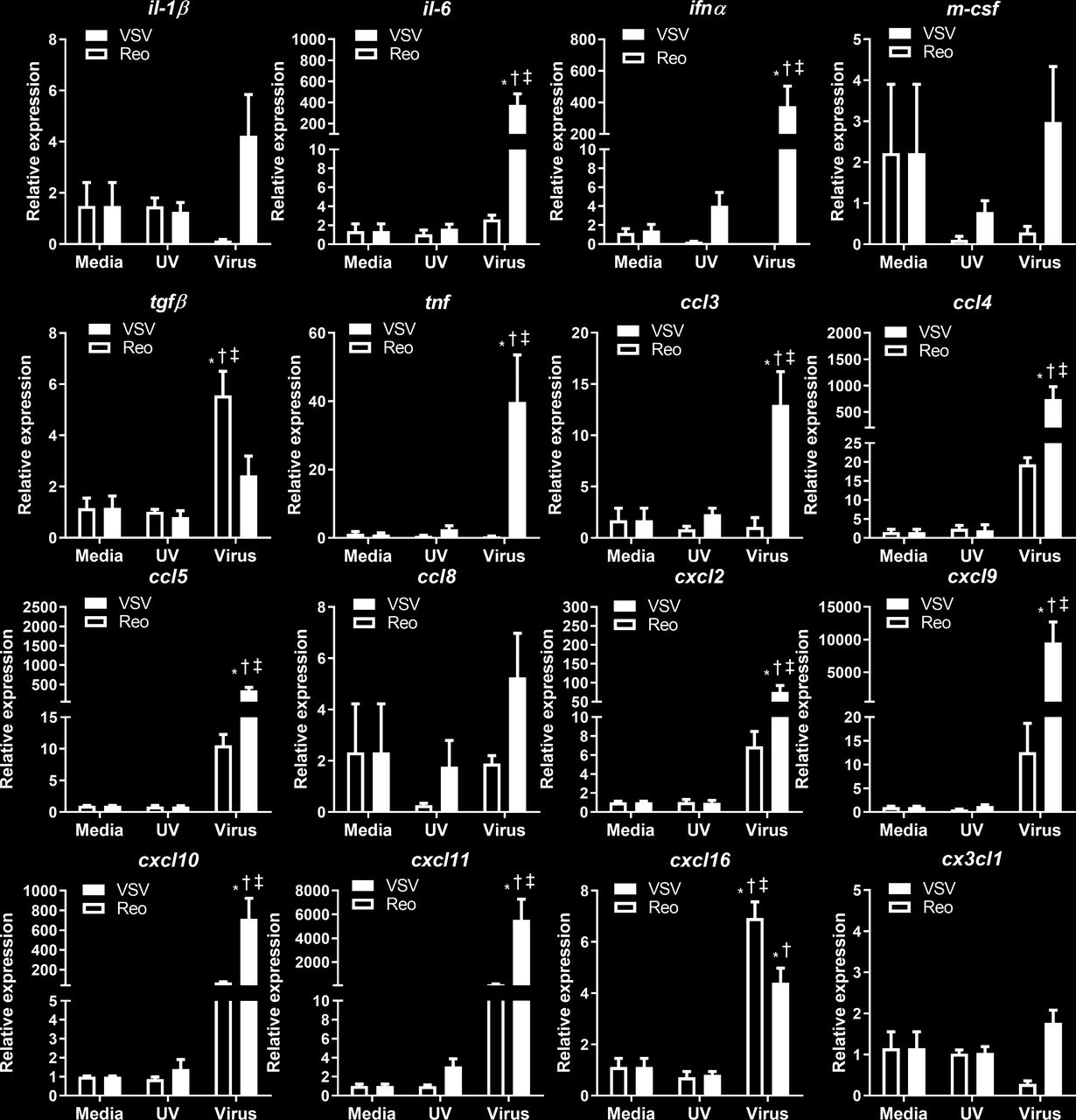
VSV infection alters cytokine and chemokine mRNA expression in 4T1 cells in vitro
We also determined whether VSV and reovirus infection differentially modified the expression of proinflammatory genes associated with an antitumor immune response. VSV infection significantly increased type 1 Ifn-α, Tnf, and il-6 transcripts in 4T1 cells (figure 6). Additionally, VSV infection significantly increased mRNA expression of many chemokines that attract myeloid cells (ccl3, ccl4, cxcl2)30 and lymphocytes (ccl5, cxcl9, cxcl10, cxcl11, cxcl16)30 (figure 6). Reovirus significantly increased transcripts for the chemokine cxcl16 and the immunosuppressive cytokine tgfβ (figure 6). Reovirus infection tended to increase ccl4, ccl8, cxcl2, cxcl9, cxcl10, and cxcl11 transcripts; however, these trends did not reach statistical significance. Differences in inflammatory gene expression indicate that VSV has greater potential to make 4T1 tumors ‘immunologically hot’ and therefore more amenable to immunotherapy.

Cytokine and chemokine mRNA expression in 4T1 cells infected with VSV∆M51 or reovirus. 4T1 cells were treated with reovirus, vesicular stomatitis virus (VSV), or UV-inactivated viruses for 24 hours. qPCR was analyzed using the 2−ΔΔCT quantification technique relative to the validated housekeeping gene GAPDH (n=3–6 per group). *p<0.05 compared with media; † p<0.05 compared with UV-inactivated virus; ‡<0.05 compared with reovirus treatment.
VSV increases NKT cell-mediated antitumor immune activation
NKT cells release large quantities of cytokines, allowing them to regulate the function of other immune cells. Therefore, we examined the effect of VSV infection on immune cell populations when used alone and in combination with NKT cell immunotherapy. Mice were treated as in figure 2A. As we were unable to isolate metastatic foci for analysis of infiltrating immune cells, spleens were harvested and analyzed by flow cytometry on day 19. At this time point, the frequency of T cells, NK cells, Tregs and DCs was not changed in any treatment group. NKT cell immunotherapy alone and in combination with VSV increased the frequency of activated CD69+ and IFN-γ producing NK, CD4+ and CD8+ T cells (online supplemental figure 3). Furthermore, the combination of NKT cell immunotherapy and VSV increased granzyme B production in NK and CD8+ T cells, while NKT cell immunotherapy on its own only increased granzyme B production in NK cells (online supplemental figure 3). NKT cell activation alone and in combination with VSV increased CD80 and CD86 expression on DCs, suggesting an increase in maturation and antigen presentation capacity (online supplemental figure 3). VSV infection alone tended to increase the frequency of activated CD69 positive, IFN-γ producing, and granzyme B positive lymphocytes; however, only CD69 on CD4 T cells and granzyme B production in NK cells reached significance (online supplemental figure 3). Overall, the combination of VSV and NKT cell immunotherapy increased immune activation and cytotoxic molecule production, suggesting induction of a stronger antitumor immune response.
VSV infection increases parameters of immunogenic cell death in 4T1 and ID8 cells
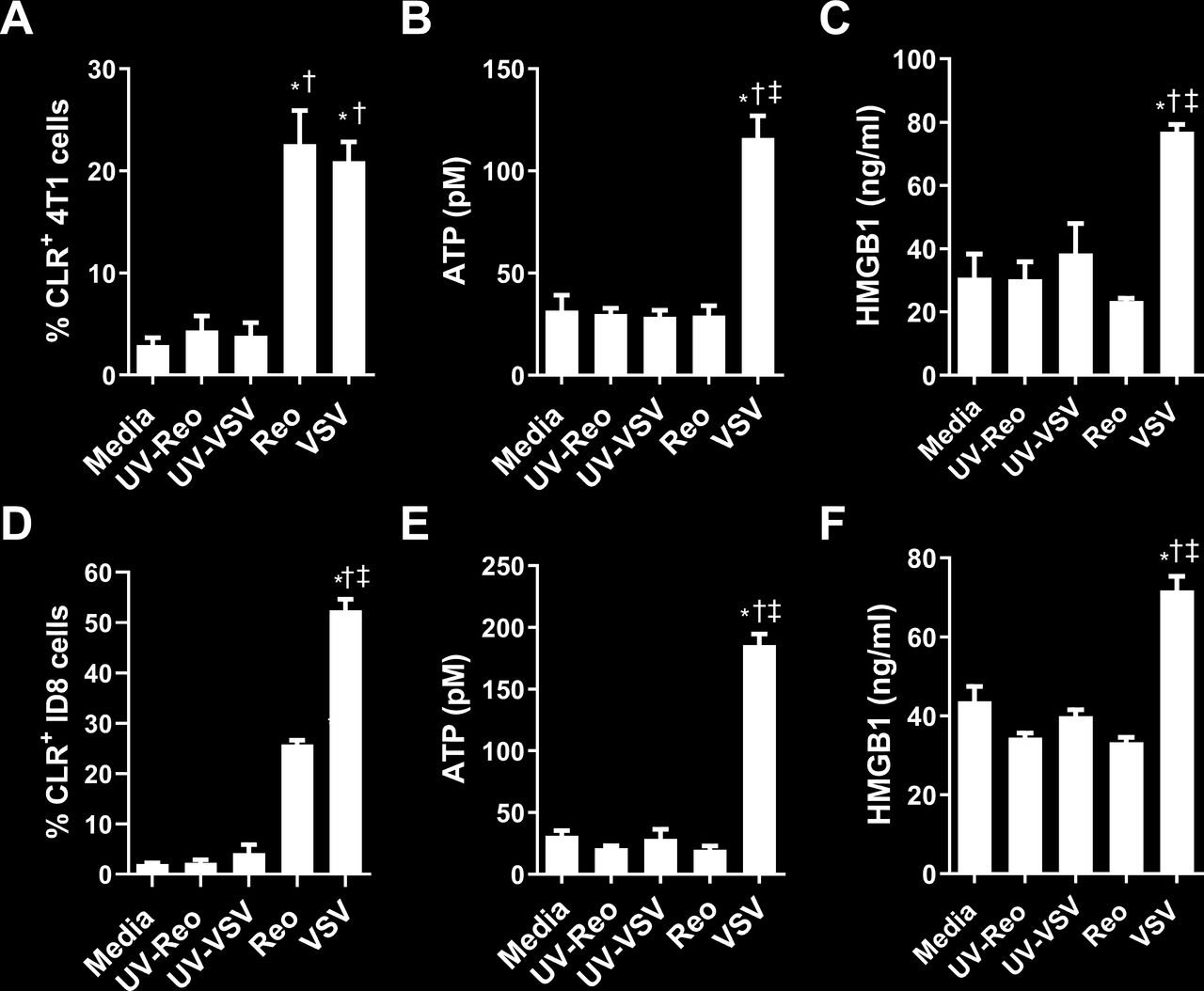
Unlike physiological cell death, which induces a tolerogenic non-inflammatory clearance of dying cells, immunogenic cell death (ICD) generates specific changes in cell signaling that promote and support antitumor immune responses.31 Many therapeutics induce ICD, including radiation, a subset of chemotherapeutics, and some oncolytic viruses.31 Therefore, we examined whether VSV and reovirus infection could differentially induce ICD in 4T1 and ID8 cells. Calreticulin (CLR) mobilization from the endoplasmic reticulum functions as an ‘eat me’ signal and is a critical indicator of ICD.32 4T1 cells infected with VSV or reovirus for 8 hours exhibited increased surface CLR mobilization compared with cells infected with UV-inactivated virus (figure 7A). To verify that the cells were undergoing ICD, we examined additional markers of ICD, release of ATP and HMGB1.33 34 Both were significantly increased in the supernatant of 4T1 cells 24 hours after infection with VSV compared with UV-VSV (figure 7B,C). There was no increase in ATP or HMGB1 release from reovirus-infected cells, indicating reovirus infection was not able to induce the full signature associated with classical ICD. We repeated this analysis in ID8 cells to determine whether reovirus could induce ICD in a model where it was more effective. Reovirus infection increased CLR mobilization (figure 7D) but did not increase ATP or HMGB1 release from ID8 cells (figure 7E,F). In contrast, VSV did increase CLR mobilization plus ATP and HMGB1 release in the ID8 model. These results demonstrate that VSV and reovirus have differing potential to promote antitumor immune responses due to differing capacities to induce ICD.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Virus-induced markers of immunogenic cell death in 4T1 and ID8 cells. Flow cytometric expression of surface calreticulin (CLR) was measured on (A) 4T1 and (D) ID8 cells 8 hours after infection with vesicular stomatitis virus (VSV) or reovirus (n=3 per group). ATP release from (B) 4T1 and (E) ID8 cells was measured by ELISA 24 hours after indicated virus treatment (n=4 per group). HMGB1 release from (C) 4T1 and (F) ID8 cells was measured by ELISA 24 hours following the indicated virus treatments (n=4 per group). *p<0.05 compared with media; †p<0.05 compared with UV-inactivated virus; ‡p<0.05 compared with reovirus treatment.
Discussion
Previously, our laboratory found that NKT cell-mediated immunotherapy increased survival in a metastatic 4T1 mammary carcinoma model,11 and this could be enhanced by combining NKT cell activation with cyclophosphamide or gemcitabine.15 However, cyclophosphamide and gemcitabine come with dose-limiting toxicities and long-term treatment can negatively impact immune function.3 Therefore, it is important to examine the therapeutic benefit of NKT cells combined with therapies that have fewer side effects. Here, we demonstrate that NKT cell immunotherapy can be combined with oncolytic VSV to decrease metastatic burden and increase survival in the 4T1 model. These observations extended to the ID8 model of peritoneal carcinomatosis, where combined VSV and NKT cell activation therapies synergistically enhanced survival. Interestingly, reovirus, which prolongs survival in the ID8 model,28 could combine with NKT cell activation to further enhance survival in this model, but was ineffective in the 4T1 mammary carcinoma model.
The reduced ability of reovirus to kill 4T1 cells could contribute to its ineffectiveness in enhancing survival or controlling lung metastases in mice. More effective tumor cell killing via VSV is likely to reduce metastatic tumor burden, making it easier for NKT cell-mediated immunotherapy to target and control the metastatic disease. Mechanisms underlying differences in the ability of reovirus to target 4T1 and ID8 cells are unclear. Both 4T1 and ID8 cells express high levels of JAM-A and sialic acid, indicating there should be little difference in reovirus’s ability to infect either cell.35 Although increased RAS signaling is a major factor for cancer susceptibility to reovirus,36 ID8 cells do not exhibit increased RAS signaling,37 while 4T1 cells do.36 This suggests an alternative mechanism underlies the differential susceptibility to reovirus. Reovirus can kill cancer cells in a RAS-independent manner; however, no genetic factor has been identified to explain reovirus’s RAS-independent oncolytic activity38 and it is unclear whether this pathway would mediate the differential killing of 4T1 and ID8 cells.
Part of the therapeutic benefit of oncolytic viruses also comes from their potential to increase antitumor immunity.21 VSV infection enhanced antigen presentation capacity of 4T1 and ID8 cells, increasing surface expression of MHC I, MHC II, CD1d, and expression of tap1 and 2 transcripts (figure 4A,B). As tumors often escape detection by downregulating antigen presentation machinery,29 virus-induced antigen presentation would potentially make tumor cells more visible to the immune system, overcoming a major immune evasion strategy. Reovirus infection was not able to increase tap 1 and 2 gene expression in 4T1 cells, suggesting that it is unable to increase presentation that would support immune recognition. In contrast, both reovirus and VSV were able to increase antigen presentation in the ID8 cells, suggesting that 4T1 cells are resistant to the mechanisms by which reovirus works in ID8 cells.
Virus-induced upregulation of CD1d levels on tumor cells may enhance NKT cell-mediated antitumor immunity. Several lines of evidence support the activation of NKT cells by self-lipids displayed on CD1d-positive tumors. For example, B-cell lymphomas and other tumors expressing high levels of CD1d are more susceptible to NKT cell-dependent lysis.39 40 CD1d downregulation by human papillomavirus in infected cervical epithelial cells correlates with progression to cervical carcinoma.41 Similarly, breast cancer cells that downregulate CD1d can evade NKT cell-mediated antitumor immunity and promote metastatic breast cancer progression.42 Collectively, these studies implicate direct CD1d recognition in NKT cell immunosurveillance and may present a mechanism by which oncolytic viruses can enhance NKT cell activation therapy.
VSV infection increased expression of many proinflammatory cytokine genes associated with antitumor immune function, including il-6, tnf and type 1 ifn. These responses were not merely the result of virus binding to pattern recognition receptors as UV-inactivated virus did not induce robust responses. VSV infection also increased gene transcription of chemokines that attract both myeloid and lymphoid cells. Immunotherapies, like NKT cell activation, are likely more effective in eliciting beneficial responses in ‘hot’ tumors that contain immune cell infiltrates amenable to stimulation.43 VSV infection led to increased transcription of the chemokines cxcl9, cxcl10, and cxc11, which are potent chemoattractants for NKT cells.44 The chemokine cxcl16, which regulates NKT cell cytokine responses to promote tumor clearance,45 was also upregulated. Reovirus was also able to increase cxcl9, 10, 11 and 16 transcripts, which could facilitate infiltration of lymphocytes, including NKT cells. However, unlike VSV, reovirus did not increase ccl3 and ccl4, chemoattractants that would recruit myeloid and T cells. Reovirus also increased tgfβ gene expression, a cytokine that can have suppressive effects on tumor immunity.46 This could explain why NKT cell therapy was not effective when combined with reovirus.
To examine the effects of our treatments on immune populations, spleen cells were examined after single and combined immunotherapies. The frequency of DCs, Tregs, T cells and NK cells was not changed following NKT cell immunotherapy, VSV or the combination of both. However, VSV infection led to modest increases in CD69 expression and IFN-γ and granzyme B production in NK and T cells, suggesting enhanced immune cell activation. When used in combination with NKT cell immunotherapy, NK and T cells had greater activation and production of cytotoxic proteins, indicative of a stronger immune response. NKT cell immunotherapy alone and in combination with VSV also increased expression of CD80 on DCs, consistent with increased antigen presentation. There was no change in Treg frequency, indicating the treatment did not impact these immunosuppressive cells. However, we have previously shown that NKT cell activation decreases accumulation of myeloid-derived suppressor cells.11
Some oncolytic viruses have been shown to stimulate antitumor immunity by causing ICD of cancer cells.31 Important mediators of ICD being surface mobilization of CLR and extracellular release of ATP and HMGB1.32–34 Both VSV and reovirus infection increased CLR mobilization to the surface of 4T1 and ID8 cells, a signal that enhances uptake of tumor antigens into antigen presenting cells.31 However, only VSV increased the extracellular release of ATP and HMGB1,32 33 signals that induce migration and maturation of antigen presenting cells. This demonstrates that VSV but not reovirus induced the classical hallmarks of ICD. Newer work identifies CXCL10 as another critical marker of ICD.47 While both VSV and reovirus were able to induce CXCL10, VSV infection led to significantly higher CXCL10 transcript expression than reovirus, further supporting VSV’s increased ability to induce ICD.
While VSV and reovirus enhanced the immunogenicity of the dying cancer cells to varying degrees, antitumor responses must be accompanied by the downregulation of immunosuppressive mechanisms to achieve durable therapeutic outcomes. One such mechanism that can hamper oncolytic virus therapy involves the upregulation of immune checkpoint molecules within the tumor microenvironment. Several preclinical and clinical studies have demonstrated that combining oncolytic virus therapy with anti-PD-1, or anti-CTLA-4, significantly enhances antitumor immunity.23–25 Similarly, NKT cell-mediated control of tumors is enhanced by anti-PD-1/PDL-1 therapies.48 49 Future studies will investigate the potential of combining VSV therapy, NKT cell activation, and immune checkpoint inhibitors.
Our treatments combining oncolytic VSV with NKT cell immunotherapy were effective in two metastasis models, significantly increasing survival over individual therapies. Overall, these therapies have the potential to reduce the off-target effects observed with traditional chemotherapy approaches. While VSV can infect a wide range of cells via recognition of the widely expressed low-density lipoprotein receptor, oncolytic VSV preferentially replicates in cancer cells due to their impaired IFN responses,19 leaving healthy cells unharmed and limiting off-target tissue damage.50 Oncolytic viruses induce transient influenza-like symptoms,21 while NKT cell activation therapies are associated with low-grade adverse effects.13 Overall, our treatments present an effective approach to target metastatic disease while provoking fewer adverse events than current chemotherapeutic approaches.
Acknowledgments
Flow cytometry was supported by infrastructure and expertise in the CORES program at Dalhousie University. We would like to thank Kaitlyn Tanner for her technical assistance on this project.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SG and AN contributed equally.
Contributors Conceptualization: SG, BJ. Methodology: AN, SG. Formal analysis: AN, SG. Investigation: AN, SG, LL, BW, TO. Resources: DM, BJ. Writing—Original draft: AN. Writing—Review and editing: AN, DM, BJ. Visualization: AN, SG, BJ. Supervision: BJ. Funding acquisition: AN, SG, BJ.
Funding This study was supported by operating grants from the Canadian Cancer Society and the Canadian Institutes of Health Research (PJT-153285). AN received a studentship from the Beatrice Hunter Cancer Research Institute with funding from The Terry Fox Research Institute. SG received a studentship from the Canadian Institutes of Health Research.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All experimental procedures were approved by the University Committee on Laboratory Animals following the guidelines of the Canadian Council on Animal Care.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Data available from Dr Brent Johnston (brent.johnston@dal.ca) upon request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.