Article Text

Abstract
Background Chronic inflammation characterised by IgG-producing plasma cell infiltration of colonic mucosa is a histological hallmark of ulcerative colitis (UC); however, whether its function is pathogenic or protective remains unclear.
Objective To explore the contribution of intestinal IgG plasma cells to UC pathogenesis.
Methods We isolated lamina propria mononuclear cells (LPMCs) from intestinal mucosa of UC patients and analysed the characteristics of intestinal plasma cells (expression profiles of differentiation molecules and chemokine receptors). We investigated the involvement of IgG-immune complex (IC)-Fc gamma receptor (FcγR) signalling in intestinal inflammation by examining the cytokine production by LPMCs in response to IgG-IC stimulation.
Results IgG plasma cells that were markedly increased in number in the inflamed mucosa of UC patients showed a distinct expression profile (CD19+CD27low, CCR10lowCXCR4high) compared with IgA plasma cells (CD19+/−CD27high, CCR10highCXCR4−/low). In vitro IgG-IC stimulation activated intestinal CD14 macrophages that were increased in number in the inflamed mucosa of UC patients via FcγRI and FcγRII, and induced the extensive production of pro-inflammatory cytokines such as tumour necrosis factor (TNF) and interleukin-1β (IL-1β), comparable to the effect of commensal bacteria stimulation. Co-stimulation with IgG-IC and commensal bacteria increased TNF and IL-1β production more than stimulation with the latter alone. Furthermore, IgG-IC notably up-regulated the expression of TL1A, whereas commensal bacteria specifically induced IL-23.
Conclusions Collectively, these results demonstrate a novel aspect of UC pathogenesis in which unique IgG plasma cells infiltrate the inflamed mucosa via CXCR4, and critically influence UC pathogenesis by exacerbating mucosal inflammation through the activation of ‘pathogenic’ intestinal CD14 macrophages via IgG-IC-FcγR signalling.
- Ulcerative Colitis
- IBD Basic Research
- Inflammation
- B Cell
- Macrophages
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Ulcerative colitis (UC) is characterised by infiltration of IgG-producing plasma cells into the inflamed colonic mucosa.
-
IgG antibodies can perpetuate several chronic inflammatory disorders; FcγRs (receptors for IgG) are thought to be involved in the pathogenesis of such disorders, including UC.
-
Unique intestinal CD14 macrophages play a central role in the pathogenesis of inflammatory bowel disease via excess production of TNF and IL-23 in response to commensal bacteria.
What are the new findings?
-
IgG plasma cells in the inflamed mucosa of UC patients have a distinct chemokine receptor-expression profile (CCR10lowCXCR4high) compared with IgA plasma cells (CCR10highCXCR4−/low).
-
IgG-immune complex (IC) induces abundant production of pro-inflammatory cytokines such as TNF, IL-1β and TL1A by lamina propria mononuclear cells (LPMCs) from UC patients, demonstrating IgG-IC as another potent inducer of intestinal inflammation besides commensal bacteria.
-
Intestinal CD14 macrophages express FcγRs, and are responsible for IgG-IC-FcγR signalling-induced pro-inflammatory cytokine production by LPMCs.
How might it impact on clinical practice in the foreseeable future?
-
Our results provide a novel mechanistic insight into the disease process of human UC, highlighting the crucial roles of IgG plasma cells and CD14 macrophages in the intestinal inflammation and UC pathogenesis.
-
Modulation of these IgG plasma cells or the signalling pathway might represent a promising therapeutic strategy for the treatment of UC.
Introduction
Inflammatory bowel disease (IBD) is a chronic intestinal inflammatory disorder with two major types: ulcerative colitis (UC) and Crohn's disease (CD). Although the precise aetiologies remain unclear, genetic backgrounds, environmental factors, and immunological disorders are critical factors in the pathogenesis of IBD.1 ,2 Histological observations of UC include infiltration of IgG-producing plasma cells into the inflamed colonic mucosa3 ,4; however, the precise mechanism of IgG plasma cell infiltration, and the roles that IgG antibodies and IgG plasma cells play in the pathogenesis of UC are not fully understood, despite considerable progress made in recent studies.5–7
Plasma cells are the terminal stage of B cell differentiation, and are dedicated to the large-scale secretion of antibodies. Besides bone marrow, lamina propria (LP) is the main reservoir of plasma cells, and most intestinal plasma cells are IgA-producing cells, contributing to the maintenance of gut homeostasis. IgG antibodies are the most abundant serum immunoglobulins involved in the secondary immune response, and their numbers increase in response to infection, chronic inflammation, and autoimmune diseases. Indeed, accumulation of IgG plasma cells in inflamed tissues has been reported not only in UC but also in several autoimmune diseases such as rheumatoid arthritis (RA).8 There is also increasing evidence that autoantibodies produced by IgG plasma cells can perpetuate several autoimmune diseases in humans.9
Fc gamma receptors (FcγRs) play a crucial role in immunity by linking IgG antibody-mediated responses with cellular effector and regulatory functions.10 The three human FcγRs, FcγRI (CD64), FcγRII (CD32) and FcγRIII (CD16), differ in cell distribution, function, and affinity for IgG. FcγRI is a high-affinity receptor for IgG, whereas the low-affinity receptors FcγRII and FcγRIII preferentially bind IgG in the form of the immune complex (IC). FcγRs are critically involved in phagocytosis, IC clearance, degranulation, antibody-dependent cellular cytotoxicity (ADCC), and the release of pro-inflammatory cytokines. Genetic variants of these receptors have previously been identified as risk factors for several chronic inflammatory conditions such as RA, multiple sclerosis (MS) and systemic lupus erythematosus (SLE).11 Recently, FcγRIIA was also identified as a susceptible gene of UC in Japanese and European descent populations12 ,13; FcγRs are therefore thought to play a crucial role in the pathogenesis of several chronic inflammatory disorders including UC. However, no research has focused on the involvement of IgG-FcγR signalling in the intestinal inflammation of patients with UC.
The present study therefore explored the contribution of intestinal IgG plasma cells to chronic intestinal inflammation in UC patients by analysing characteristic cellular features and investigating the involvement of IgG-IC-FcγR signalling in intestinal inflammation. Here we show that IgG plasma cells in the inflamed mucosa of UC patients have a unique immature and chemokine receptor expression phenotype, and suggest that IgG antibodies produced by these cells are key pathogenic effectors of colonic inflammation in UC.
Materials and methods
Tissue samples
Normal intestinal mucosa was obtained from macroscopically and microscopically unaffected areas of patients with colon cancer. Intestinal mucosa was also obtained from surgically resected specimens from patients with UC or CD, diagnosed on the basis of clinical, radiographic, endoscopic and histological findings according to established criteria. The degree of local mucosal inflammation was assessed macroscopically according to the level of superficial erosion, mucosal atrophy and epithelial destruction with ulcerations (−, normal mucosa; +, mild; ++, moderate; +++, severe inflammation). All experiments were conducted according to the Declaration of Helsinki principles, and approved by the institutional review boards of Keio University School of Medicine (Tokyo, Japan) and Yokohama Municipal Citizen's Hospital (Yokohama, Japan). Written informed consent was obtained from all patients (see online supplementary materials and methods for full details).
Results
IgG plasma cells heavily infiltrate the inflamed mucosa of patients with UC
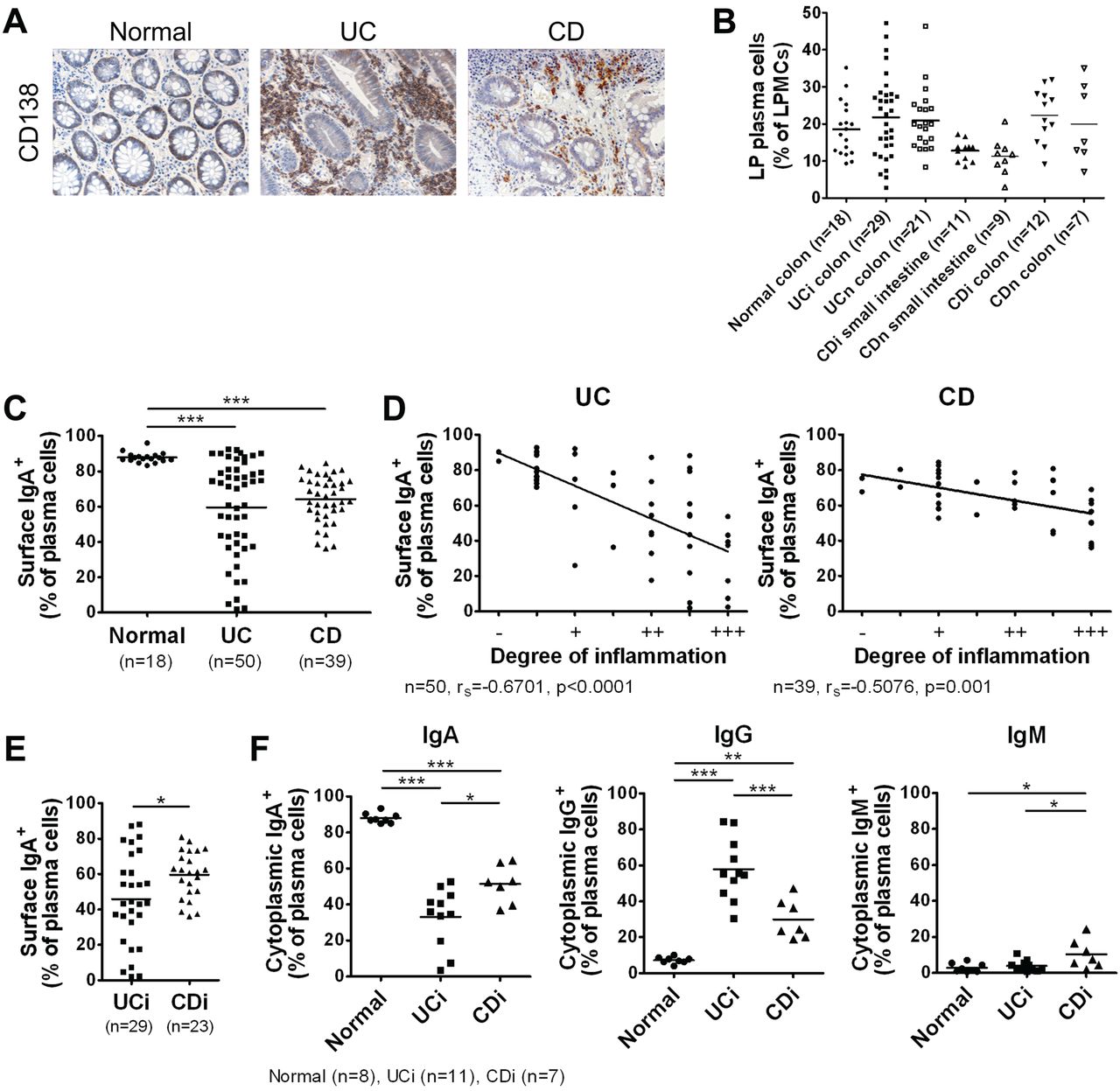
Immunohistochemistry confirmed the previously reported14 massive influx of CD138 plasma cells into the inflamed mucosa of patients with IBD, particularly those with UC (figure 1A). To analyse the characteristics of intestinal plasma cells in detail, we isolated LP mononuclear cells (LPMCs) from the intestinal mucosa of IBD patients or the non-affected colonic mucosa of patients with colon cancer (non-IBD controls; Normal), and analysed them by flow cytometry. The CD38high phenotype is a common specific marker for human plasma cells and is applicable for the identification of intestinal plasma cells (see online supplementary figure S1A).15 We observed no difference in the proportion of LPMCs that were CD38high between Normal and IBD patients (figure 1B). In the Normal group, the vast majority of intestinal plasma cells were IgA-producing (figure 1C); however, this proportion was drastically decreased and negatively correlated with the degree of local mucosal inflammation in patients with IBD (figure 1C,D), especially UC (figure 1E).
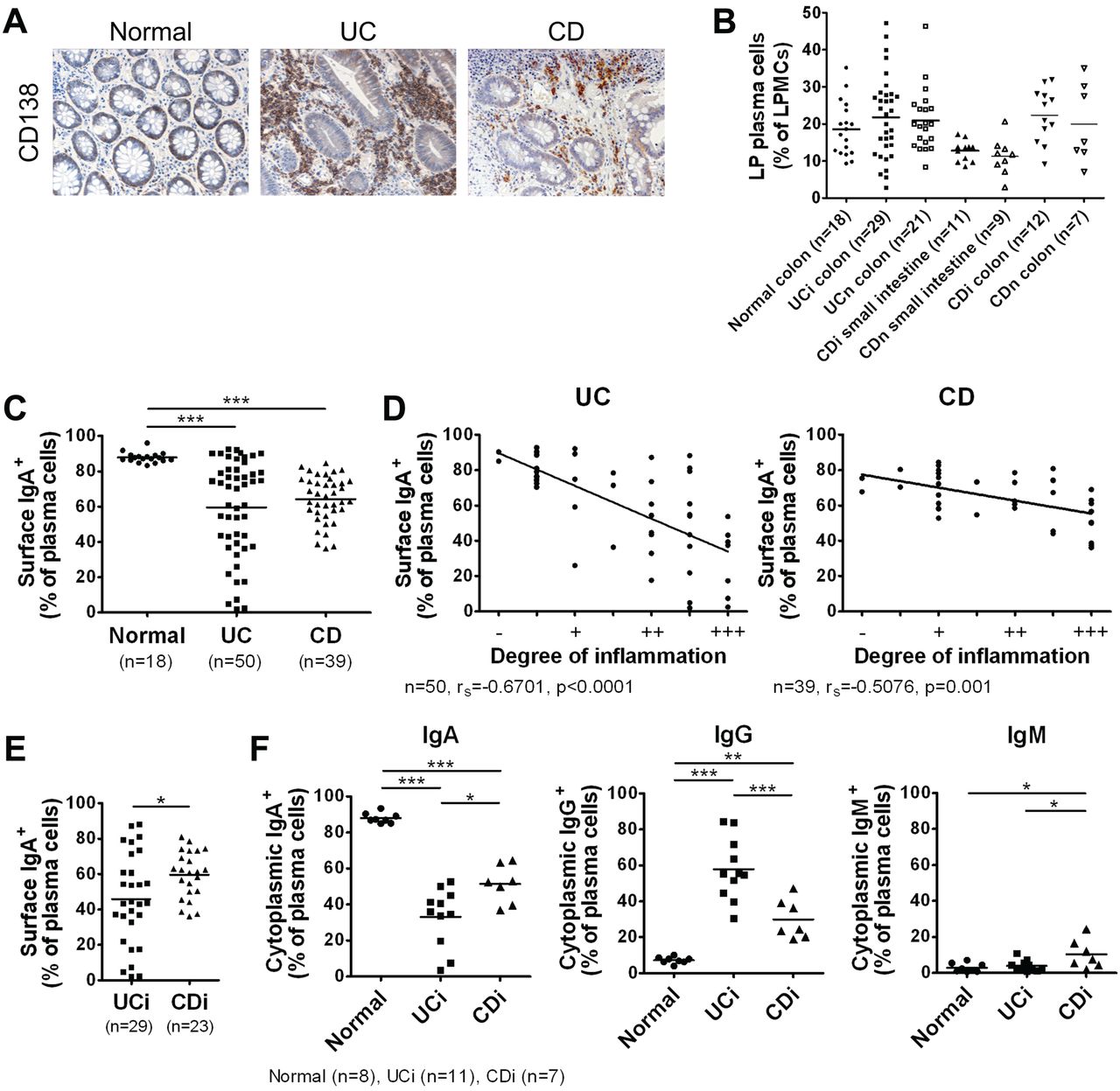
IgG plasma cells heavily infiltrate the inflamed mucosa of ulcerative colitis (UC) patients. (A) Immunostaining images of intestinal mucosa from non-inflammatory bowel disease (IBD) controls (Normal) and IBD patients stained with anti-CD138. Data are representative of three independent specimens. (B) Percentage of lamina propria (LP) CD38high plasma cells among lamina propria mononuclear cells (LPMCs). i, inflamed; n, non-inflamed. Horizontal bars indicate mean values. (C) Percentage of surface IgA+ cells among LP CD38high plasma cells. (D) Negative correlations between the degree of local mucosal inflammation and percentage of surface IgA+ cells among LP CD38high plasma cells. (E) Percentage of surface IgA+ cells among LP CD38high plasma cells from inflamed (moderate-to-severe) mucosa of UC and Crohn's disease (CD) patients. (F) LP CD38high plasma cells from normal intestinal mucosa and inflamed mucosa of IBD patients were analysed by intracellular staining for cytoplasmic IgA, IgG and IgM expression. Shown are percentages of cytoplasmic Ig+ cells among LP plasma cells. *p<0.05, **p<0.01, ***p<0.001.
We also observed an increase of IgG plasma cells in the inflamed mucosa of patients with IBD, especially UC (figure 1F, see online supplementary figure S1B). Further analysis revealed that IgG plasma cells in UC were mainly IgG1-producing, whereas those in CD were mainly IgG1- or IgG2-producing (see online supplementary figure S1C,D), in line with previous findings.16 These results show that IgG-producing plasma cells heavily infiltrate the inflamed mucosa of patients with IBD, especially UC, suggesting their involvement in the intestinal inflammation and pathogenesis of the disorder.
LP plasma cells in the inflamed mucosa of patients with UC have a unique immature phenotype
To reveal the characteristics of intestinal plasma cells in UC patients, we analysed the expression profiles of cell surface-differentiation molecules. We found that plasma cells in the Normal group and in the non-inflamed mucosa of IBD patients had a CD19+/−CD20−CD27high phenotype, whereas plasma cells in the inflamed mucosa of UC patients had a CD19+CD20−CD27low unique immature phenotype (figure 2A,C). These changes were also observed in both IgA and IgG (ie, surface IgA−)-producing plasma cells (figure 2B). These results are partly consistent with an earlier finding that the inflamed mucosa of UC patients contained CD19 immature plasma cells.14

Lamina propria (LP) plasma cells in the inflamed mucosa of ulcerative colitis (UC) patients have a unique immature phenotype. (A) LP CD38high plasma cells from normal intestinal mucosa and inflamed or non-inflamed mucosa of inflammatory bowel disease (IBD) patients were analysed by flow cytometry for IgA, CD19, CD20 and CD27 cell surface expression. CD, Crohn's disease. (B) LP CD38high plasma cells from normal intestinal mucosa and inflamed mucosa of IBD patients were analysed by flow cytometry for IgA and CD19/CD27 cell surface expression. (C) Mean fluorescence intensities (MFI) of CD19 and CD27 expression levels on LP CD38high plasma cells from normal controls and IBD patients. (D) LPMCs and isolated LP plasma cells (PCs) from inflamed mucosa of UC patients were analysed for CD38 and CD19 expression. (E) Microarray analysis of LP plasma cells isolated from normal intestinal mucosa and inflamed mucosa of UC patients, presented as a heat map of five major transcription factors involved in B cell to plasma cell differentiation (presented as a ratio of normalised intensity). (F) Quantitative real-time RT-PCR of basal mRNA expression levels in isolated LP plasma cells from normal intestinal mucosa and inflamed mucosa of IBD patients.*p<0.05, **p<0.01, ***p<0.001, N.S., not significant.
For further characterisation, we isolated intestinal plasma cells from the LPMCs of Normal and UC patients (figure 2D, see online supplementary figure S2), and performed microarray gene-expression analysis. As shown in figure 2E, among the five major transcription factors involved in B cell to plasma cell differentiation,17 plasma cells in UC patients expressed BCL6 more highly than those of the Normal group; the expression of other transcription factors such as PRDM1, which encodes Blimp-1 and is necessary for plasma cell survival,9 did not differ between the two groups. This result was confirmed by quantitative real-time RT-PCR analysis (figure 2F).
We also showed that plasma cells of the inflamed mucosa of CD patients had a CD19+CD20−CD27high phenotype, and that the expression level of BCL6 was comparable to Normal plasma cells (figure 2A–C,F). As BCL6 functions as a transcription factor that maintains the B cell phenotype and proliferation,17 these data support the idea that plasma cells in the inflamed mucosa of UC patients have a unique immature phenotype that differs in part from that of CD patients.
LP IgG plasma cells in the inflamed mucosa of patients with UC have a different chemokine receptor-expression profile
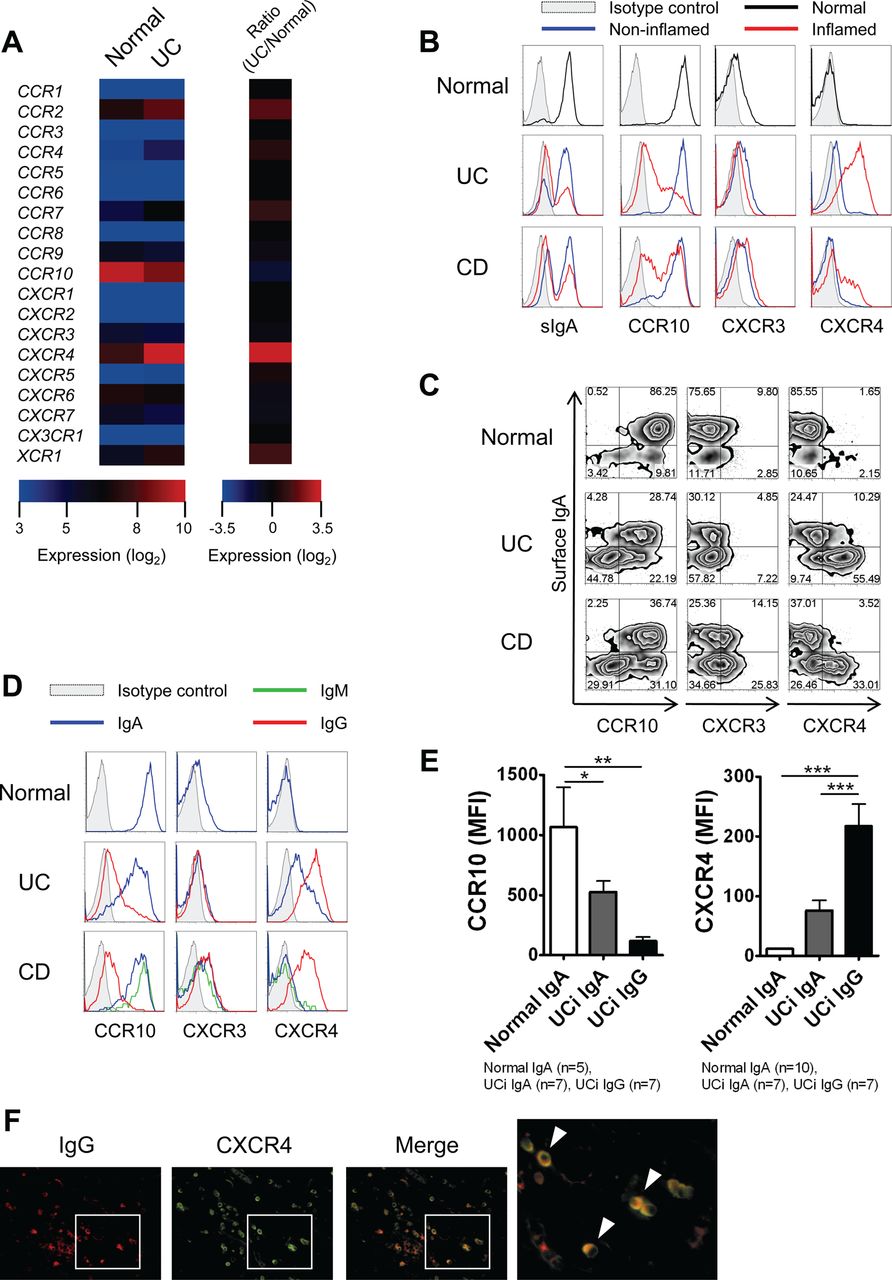
Chemokines, together with tissue-specific adhesion molecules, coordinate the trafficking of plasma cells.18 Therefore, to dissect the mechanism of IgG plasma cell accumulation in the inflamed mucosa of UC patients, we examined the expression profiles of chemokine receptors in intestinal plasma cells. Normal intestinal IgA plasma cells express the chemokine receptor CCR10, which is important for the homing mechanism to the mucosal tissues.18 Among the panel of chemokine receptors tested, we found that the expression of CXCR4 was higher in UC plasma cells than in Normal plasma cells, and that the former had relatively low CCR10 expression (figure 3A). Thereafter, the expression profiles of these chemokine receptors were confirmed by flow cytometry.

Lamina propria (LP) IgG plasma cells in the inflamed mucosa of ulcerative colitis (UC) patients have a different chemokine receptor-expression profile. (A) Microarray analysis of LP plasma cells isolated from normal intestinal mucosa and inflamed mucosa of UC patients, presented as a heat map of chemokine receptors (presented as a normalised intensity (log2) and a ratio of normalised intensity). (B) LP CD38high plasma cells from normal intestinal mucosa and inflamed or non-inflamed mucosa of inflammatory bowel disease (IBD) patients were analysed by flow cytometry for IgA, CCR10, CXCR3 and CXCR4 cell surface expression. CD, Crohn's disease. (C) LP CD38high plasma cells from normal intestinal mucosa and inflamed mucosa of IBD patients were analysed by flow cytometry for IgA and CCR10/CXCR3/CXCR4 cell surface expression. (D) LP IgA, IgG and IgM plasma cells from normal intestinal mucosa and inflamed mucosa of IBD patients were analysed by flow cytometry for CCR10, CXCR3 and CXCR4 cell surface expression. IgA, surface IgA+ CD38high plasma cells; IgM, surface IgM+ CD38high plasma cells; IgG, surface IgA− and surface IgM− CD38high plasma cells. (E) Mean fluorescence intensities (MFI) of CCR10 and CXCR4 expression levels on LP CD38high plasma cells from normal controls and UC patients. *p<0.05, **p<0.01, ***p<0.001. (F) Immunofluorescent double staining of colonic mucosa from UC patients stained with anti-IgG (red) and anti-CXCR4 (green). Double-positive cells displaying a classical plasma cell morphology are observed in yellow (arrowheads). Data are representative of three independent specimens.
We also examined the expression of CXCR3, which is involved in the homing to inflamed tissues and the development of several autoimmune diseases.19 As shown in figure 3B, the Normal group plasma cells and those of non-inflamed mucosa of IBD patients had a CCR10highCXCR3+CXCR4−/low phenotype, whereas plasma cells of inflamed mucosa of UC patients had a CCR10lowCXCR3+CXCR4high phenotype. Notably, we showed that the up-regulation of CXCR4 and the down-regulation of CCR10 were more evident in IgG (ie, surface IgA−) plasma cells than IgA plasma cells in the inflamed mucosa of UC patients (figure 3C).
A more detailed analysis showed that IgA and IgG plasma cells in the inflamed mucosa of UC patients showed different chemokine receptor-expression profiles: a CCR10highCXCR4low phenotype for IgA plasma cells, similar to Normal IgA plasma cells, and a CCR10lowCXCR4high phenotype for IgG plasma cells (figure 3D,E). In the inflamed mucosa of CD patients, IgA and IgM plasma cells had a CCR10highCXCR4−/low phenotype, whereas IgG plasma cells had a CCR10lowCXCR4high phenotype (figure 3D). We used immunohistochemistry to confirm the infiltration of IgG+CXCR4+ cells displaying a classical plasma cell morphology in the inflamed mucosa of UC patients (figure 3F). Overall, these results suggest that IgG, not IgA, plasma cells selectively infiltrate the inflamed mucosa of UC patients via CXCR4.
IgG-IC stimulation induces abundant pro-inflammatory cytokine production by LPMCs from patients with UC
The mechanism by which IgG plasma cells contribute to the UC pathogenesis remains unclear. We therefore evaluated the potential involvement of IgG-IC-FcγR signalling in intestinal inflammation. Our hypothesis was that IgG antibodies produced by IgG plasma cells form an IgG-IC with their specific antigens, thereby activating a particular immune cell subset via FcγRs and exacerbating intestinal inflammation. To test this, we examined the cytokine production by LPMCs from the inflamed mucosa of UC patients in response to IgG-IC stimulation, using plate-immobilised IgG to mimic complexed IgG.20 As expected, we found that IgG-IC stimulation, but not soluble human IgG, induced the production of pro-inflammatory cytokines such as tumour necrosis factor (TNF) and interleukin-1β (IL-1β) by UC LPMCs in a dose-dependent manner (figure 4A).
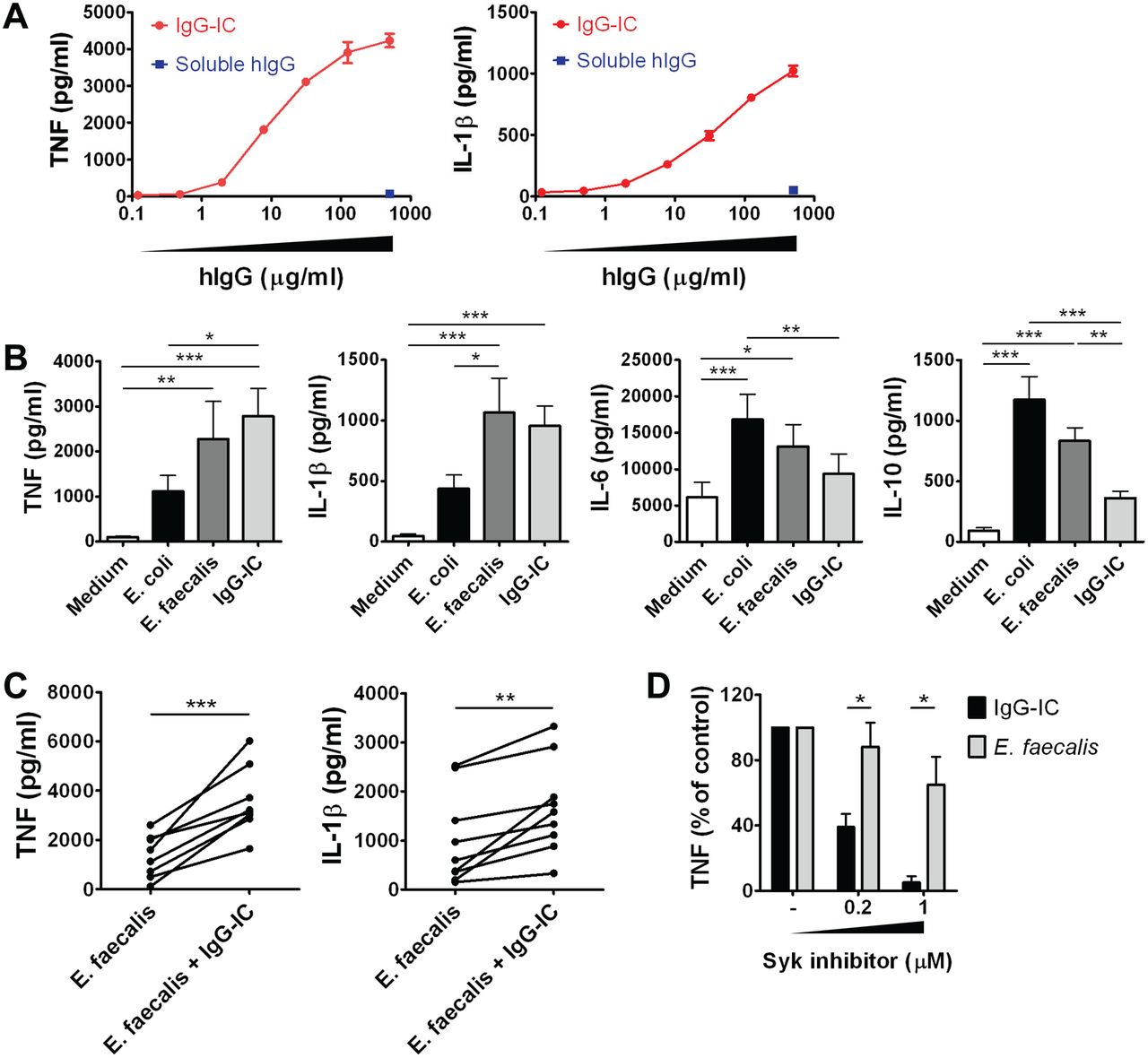
IgG-immune complex (IC) stimulation induces abundant pro-inflammatory cytokine production by lamina propria mononuclear cells (LPMCs) from ulcerative colitis (UC) patients. (A) Cytokine production by LPMCs from the inflamed mucosa of UC patients stimulated with IgG-IC or soluble human IgG (hIgG). Data represent means±SD and are representative of three independent experiments. (B) Cytokine production by LPMCs from the inflamed mucosa of UC patients stimulated with heat-killed commensal bacteria (E coli or E faecalis) or IgG-IC. Data represent means±SEM (n=11); statistical analysis was performed using repeated measures ANOVA and Tukey's multiple comparison test. (C) Cytokine production by LPMCs from the inflamed mucosa of UC patients stimulated with E faecalis alone or with IgG-IC (paired t test, n=9). (D) Effect of Syk inhibitor on IgG-IC or E faecalis-induced TNF production by LPMCs from the inflamed mucosa of UC patients. Data represent means±SEM (n=4) and statistical analysis was performed using paired t test. *p<0.05, **p<0.01, ***p<0.001.
The cytokine production induced by IgG-IC stimulation was compared with that induced by commensal bacteria (heat-killed Escherichia coli and Enterococcus faecalis) stimulation, which plays an important role in the pathogenesis of IBD.1 ,2 Notably, we found that pro-inflammatory cytokine (TNF and IL-1β) production was stimulated to similar levels by IgG-IC and commensal bacteria (E faecalis), whereas the amount of other cytokines (IL-6 and IL-10) produced by IgG-IC was relatively low (figure 4B). In addition, IgG-IC stimulation could act additively with commensal bacterial stimulation, as they co-stimulated LPMCs to produce many more pro-inflammatory cytokines than commensal bacteria stimulation alone (figure 4C). Furthermore, FcγR signalling inhibitor (spleen tyrosine kinase (Syk) inhibitor) selectively inhibited IgG-IC-induced TNF production in a dose-dependent manner, demonstrating that IgG-IC stimulation-induced pro-inflammatory cytokine production is transmitted through the activation of FcγR(s) and its downstream kinase Syk (figure 4D). These findings demonstrate that IgG-IC activates UC LPMCs through an independent signal transduction pathway and is another potent inducer of intestinal inflammation, comparable to commensal bacteria. We obtained similar results in CD patients (see online supplementary figure S3A).
IgG-IC stimulation and commensal bacteria stimulation induce the production of different pro-inflammatory cytokines by LPMCs from patients with UC
Although the amount of pro-inflammatory cytokines produced by both IgG-IC and commensal bacteria stimulation was similar, we postulated that the cytokine expression profiles must differ between these two stimulations. For example, it was previously reported that TL1A (TNFSF15) expression is strongly induced by IgG-IC, but not TLR ligand, stimulation in human monocytes and dendritic cells.20
We focused here on two key pro-inflammatory cytokines, TL1A and IL-23, which were reported to be largely involved in intestinal inflammation and the pathogenesis of IBD.21 We stimulated UC LPMCs with commensal bacteria (E faecalis) or IgG-IC, and then assessed the mRNA expression of TL1A, IL-23p19 and IL-12/23p40. As shown in figure 5A, the expression of TL1A was strongly induced by IgG-IC stimulation, whereas the expression of IL-23p19 and IL-12/23p40 was specifically induced by commensal bacteria stimulation. To determine whether induction of these cytokines led to protein expression, TL1A and IL-23 levels were measured in supernatants following stimulation. Although the production of IL-23 was detected in commensal bacteria (E faecalis)-stimulated samples, soluble TL1A production was not detected in all samples (figure 5B), suggesting that the expression of the membrane-bound form of TL1A was induced by IgG-IC stimulation.22 Thus IgG-IC stimulation is thought to be involved in intestinal inflammation through distinct signal transduction pathways, resulting in differential cytokine induction from commensal bacteria stimulation.

IgG-immune complex (IC) stimulation and commensal bacteria stimulation induce the production of different pro-inflammatory cytokines by lamina propria mononuclear cells (LPMCs) from ulcerative colitis (UC) patients. (A) mRNA expression of TL1A, IL-23p19 and IL-12/23p40 by LPMCs from the inflamed mucosa of UC patients after stimulation with heat-killed commensal bacteria (E faecalis, blue dotted line) or IgG-IC (red solid line). Data are representative of two independent experiments. (B) Cytokine production by LPMCs from the inflamed mucosa of UC patients stimulated with heat-killed commensal bacteria (E coli or E faecalis) or IgG-IC. Data represent means±SEM (n=11); statistical analysis was performed using repeated measures ANOVA and Tukey's multiple comparison test. **p<0.01, ***p<0.001, N.D., not detected.
Intestinal CD14 macrophages are responsible for IgG-IC-FcγR signalling-induced pro-inflammatory cytokine production by LPMCs from patients with UC
Finally, to determine which subset of immune cells is responsive to IgG-IC stimulation, LPMCs from the inflamed mucosa of UC patients were analysed for the expression of FcγRs by flow cytometry. As shown in figure 6A, some CD16 (FcγRIII) cells were detected in the CD14− population, which was considered to be composed of neutrophils. CD32 (FcγRII) cells were partially found to be CD14 cells, and were also detected in the CD14− population, which mainly comprised plasma cells (data not shown). CD64 (FcγRI) cells were largely CD14 cells. We therefore focused on these CD14CD33 cells as the subset that expressed FcγRI and FcγRII (figure 6B). Previously, we reported that this subset was composed of unique intestinal macrophages that are increased in number in the inflamed mucosa of IBD, and produce large quantities of TNF and IL-23 in response to commensal bacteria, contributing to the pathogenesis of IBD.23

Intestinal CD14 macrophages are responsible for IgG-immune complex (IC)-FcγR signalling-induced pro-inflammatory cytokine production by lamina propria mononuclear cells (LPMCs) from ulcerative colitis (UC) patients. (A) LPMCs from the inflamed mucosa of UC patients were analysed by flow cytometry for CD14 and CD16 (FcγRIII)/CD32 (FcγRII)/CD64 (FcγRI) cell surface expression. Data are representative of three independent patients. (B) Flow cytometry for CD16 (FcγRIII)/CD32 (FcγRII)/CD64 (FcγRI) cell surface expression in CD14CD33 macrophages. The shaded histograms show staining with isotype controls. (C) IgG-IC-induced TNF production by isolated CD14 macrophages from the inflamed mucosa of UC patients (201.9±65.3 vs 8157.8±1463.5 pg/ml, n=3). (D) IgG-IC-induced TNF production by LPMCs or LPMCs depleted of CD14 cells from the inflamed mucosa of UC patients (n=5). (E) Effect of FcγRI or FcγRII blocking antibody on IgG-IC-induced TNF production by LPMCs from the inflamed mucosa of UC patients (n=3). Statistical analysis was performed using repeated measures ANOVA and Dunnett's multiple comparison test (vs isotype control). (F) Quantitative real-time RT-PCR of basal mRNA expression levels in isolated CD14 macrophages from the inflamed mucosa of inflammatory bowel disease patients. AU, arbitrary unit. *p<0.05, **p<0.01, N.S., not significant.
To analyse whether these CD14 macrophages are responsible for the IgG-IC-FcγR signalling-induced pro-inflammatory cytokine production by UC LPMCs, we isolated or depleted CD14 cells from LPMCs, and stimulated them with IgG-IC. We found that isolated CD14 macrophages produced large quantities of TNF following IgG-IC stimulation (figure 6C), whereas LPMCs depleted of CD14 macrophages did not (figure 6D). These results demonstrate that CD14 macrophages are responsible for the IgG-IC-FcγR signalling-induced pro-inflammatory cytokine production by UC LPMCs. We also obtained similar results in CD patients (see online supplementary figure S3B,C).
To establish which FcγR(s) was involved in triggering pro-inflammatory cytokine production, LPMCs were pretreated with anti-FcγR blocking antibodies or related isotype control antibodies before IgG-IC stimulation. FcγRI blocking antibodies inhibited IgG-IC-induced TNF production in a dose-dependent manner (figure 6E). Unexpectedly, however, IgG-IC-induced TNF production was significantly increased by FcγRII blocking antibodies. As human FcγRII has three isoforms with opposite functions, and the balance between these receptors allows the IgG-IC to mediate opposing effects on cell function,24 we confirmed the expression of these activating and inhibitory receptors on CD14 macrophages by quantitative real-time RT-PCR analysis. As shown in figure 6F, although the balance was different between UC and CD, intestinal CD14 macrophages from the inflamed mucosa of IBD patients expressed both activating (FcγRIIA and FcγRIIC) and inhibitory (FcγRIIB) isoforms. Therefore, the observed increase of cytokine production by treatment with FcγRII blocking antibodies appeared to be caused by blocking inhibitory FcγRIIB. These results demonstrate an involvement of FcγRI and FcγRII (at least inhibitory FcγRIIB) in the regulation of IgG-IC-mediated CD14 macrophage activation and cytokine production.
Discussion
This study explored the contribution of intestinal IgG plasma cells to chronic inflammation in UC patients, and revealed a novel aspect and crucial roles for IgG antibodies and IgG plasma cells in its pathogenesis. We confirmed the known inflammatory condition-correlated massive influx of IgG plasma cells in the intestinal mucosa of IBD, and found that UC plasma cells had a unique immature phenotype. Notably, these characteristics were more evident or were only observed in UC, not CD, plasma cells. Thus, these results are possibly based on the differences in pathophysiological conditions between UC and CD, indicating that UC plasma cells are, at least partially, qualitatively different from CD plasma cells, and also suggesting their differential contribution to the pathogenesis of each disease.
We showed that chemokine receptor-expression profiles differed between IgG (CCR10lowCXCR4high) and IgA (CCR10highCXCR4low) plasma cells in the inflamed mucosa of UC patients. These differences can explain the mechanism of selective infiltration of IgG plasma cells into the inflamed mucosa of UC patients. Although we did not analyse the expression of chemokines in the intestinal mucosa, previous studies reported the up-regulation of CXCR3 ligand chemokines (CXCL9/MIG, CXCL10/IP-10 and CXCL11/I-TAC)25 and CXCR4 ligand chemokine (CXCL12/SDF-1α)26 in the inflamed mucosa of IBD patients. We observed no difference in CXCR3 expression between IgA and IgG plasma cells in the inflamed mucosa of UC patients, which does not support the hypothesis that IgG plasma cells selectively infiltrate the inflamed mucosa via CXCR3. Therefore, we suggest that CXCR4 is crucially involved in the selective infiltration of IgG plasma cells into the inflamed mucosa of UC patients.
Recent studies have described the relationship between the CXCL12-CXCR4 axis and the pathophysiology of several autoimmune diseases such as RA, MS and SLE.27–29 For example, Nanki et al reported that synovial tissue CD4 memory T cells highly express CXCR4, and that the CXCL12 concentration is relatively high in the synovial fluid of RA patients.27 Dotan et al reported that the expression of CXCL12 is up-regulated in the intestinal mucosa (specifically more inflamed mucosa) of patients with IBD, especially UC, and showed a chemotactic effect of CXCL12 on LP T cells in IBD patients.26 In addition, Mikami et al reported that CXCR4 expression on peripheral T cells was increased in patients with active UC, and showed that administration of a CXCR4 antagonist decreased the severity of dextran sodium sulphate (DSS)-induced mice colitis and the colonic inflammation of IL-10-knockout mice.30 These reports suggest that the CXCL12-CXCR4 axis plays a crucial role in cell trafficking under inflammatory conditions, and might be a common pathway in IBD and other autoimmune diseases for the homing of T cells and IgG plasma cells to inflamed tissues.
Although the infiltration of IgG plasma cells into the inflamed mucosa is a well-known characteristic feature of UC, its contribution to the UC pathogenesis is controversial. It has been reported that colonic plasma cells in UC patients express multiple β-defensins, possibly contributing to host defence,6 whereas intestinal plasma cells in UC patients were shown to express large amounts of matrix metalloproteinase-3, suggesting a crucial role in tissue destruction.7 As the classical role of plasma cells is defined by antibody secretion, we previously showed that IgG autoantibodies against colonic epithelial cells produced by intestinal lymphocytes in UC patients bind the epithelial cell surface and mediate ADCC.31 The present study evaluated the potential involvement of IgG-IC-FcγR signalling in intestinal inflammation and identified IgG-IC as another potent inducer of intestinal inflammation besides commensal bacteria. Moreover, among UC LPMCs, we identified CD14 macrophages as an immune cell subset responsible for IgG-IC-FcγR signalling-induced pro-inflammatory cytokine production. Intestinal CD14 macrophages were therefore found to be key pathogenic immune cells that respond not only to commensal bacteria but also to IgG-IC, further highlighting their crucial roles in intestinal inflammation and IBD pathogenesis.
The present study showed that commensal bacteria and IgG-IC activate intestinal CD14 macrophages through distinct signal transduction pathways and induce the expression of pro-inflammatory molecules with different profiles. Consistent with our results, it has been reported that both commensal bacteria and IgG-IC can initiate TL1A expression, yet commensal bacteria are less efficient in TL1A induction, and the continued activation of TL1A expression requires FcγR signalling in human CD14 monocytes.32 Importantly, we and others previously reported that TL1A and IL-23 synergistically enhance T cell function.21 ,22 Therefore, IgG-IC signalling induces not only the production of pro-inflammatory cytokines but also the expression of the T cell co-stimulatory molecule TL1A, and possibly exacerbates the disease through T cell-mediated inflammation. In addition, TL1A and IL-23R have been identified and confirmed as IBD susceptibility genes,33 ,34 and TL1A is also implicated in severe UC.35 Therefore, these distinct potent inducers of the inflammatory response might mutually accelerate intestinal inflammation and synergistically contribute to the establishment of the complicated pathophysiological conditions of IBD.
Using blocking antibodies, we revealed the involvement of activating FcγRI and inhibitory FcγRIIB in the regulation of IgG-IC-mediated CD14 macrophage activation. We could not clarify the involvement of activating FcγRIIA, as no isoform-specific blocking antibodies are commercially available. As mentioned above, FcγRIIA has been identified as a susceptible gene of UC, and it is suggested that individuals with the His131 variant of FcγRIIA, the susceptibility loci of UC, might have a higher capacity for IC, which could lead to hyperactivation of immune cells.12 Therefore, although further studies focusing on the involvement of FcγRIIA and the effects of susceptibility loci in the regulation of IgG-IC-mediated CD14 macrophage activation are needed, our proposed concept agrees with this hypothesis and suggests a novel mechanism of IgG-IC-mediated intestinal inflammation.
Several previous reports have described the relationship between plasma cell infiltration and IBD clinical events. For example, basal plasmacytosis, defined as a dense infiltration of plasma cells extending into the lower third of the LP, is associated with a shorter time to relapse in UC patients.36 In addition, in a mouse model of colitis, Kobayashi et al reported that passive administration of anti-bacterial IgG (anti-flagellin IgG) promotes the development of intestinal inflammation in DSS-induced colitis, suggesting that antibacterial IgG antibodies are themselves pathogenic.37 Together, these reports suggest that intestinal plasma cells and IgG antibodies have the potential to exacerbate intestinal inflammation, consistent with our present results.
Several unresolved issues remain, one of which is the identification of antigens that react with IgG antibodies produced by intestinal IgG plasma cells to form IgG-IC. We presume that a number of heterogeneous antigens are involved, and that components of commensal bacteria and auto-antigens are potent candidates for such antigens. Previous reports support this hypothesis: marked elevation of IgG antibodies specific for bacterial antigens is a characteristic hallmark of human IBD38; and IgG auto-antibodies against colonic epithelial cells (including anti-human tropomyosin isoform 5 antibodies) are also observed in UC patients.39
The present study does not show direct evidence that IgG antibodies produced by intestinal IgG plasma cells can form IC with their specific antigens, and activate intestinal CD14 macrophages through FcγRs in vivo. A recent study reported that DSS-treated RORγt-deficient mice showed severe colonic inflammation, characterised by infiltration of IgG plasma cells, which was mitigated by treatment with intravenous IgG (IVIG).40 As the anti-inflammatory effect of IVIG treatment is FcγRs-dependent, the authors hypothesised that the severe inflammation is a consequence of the formation of IC consisting of bacteria and specific IgG, which activates FcγR-expressing inflammatory cells. Collectively, these data suggest that IgG plasma cells accumulating in intestinal mucosa trigger IgG-IC-FcγR signalling-mediated intestinal inflammation in mice, and possibly in humans.
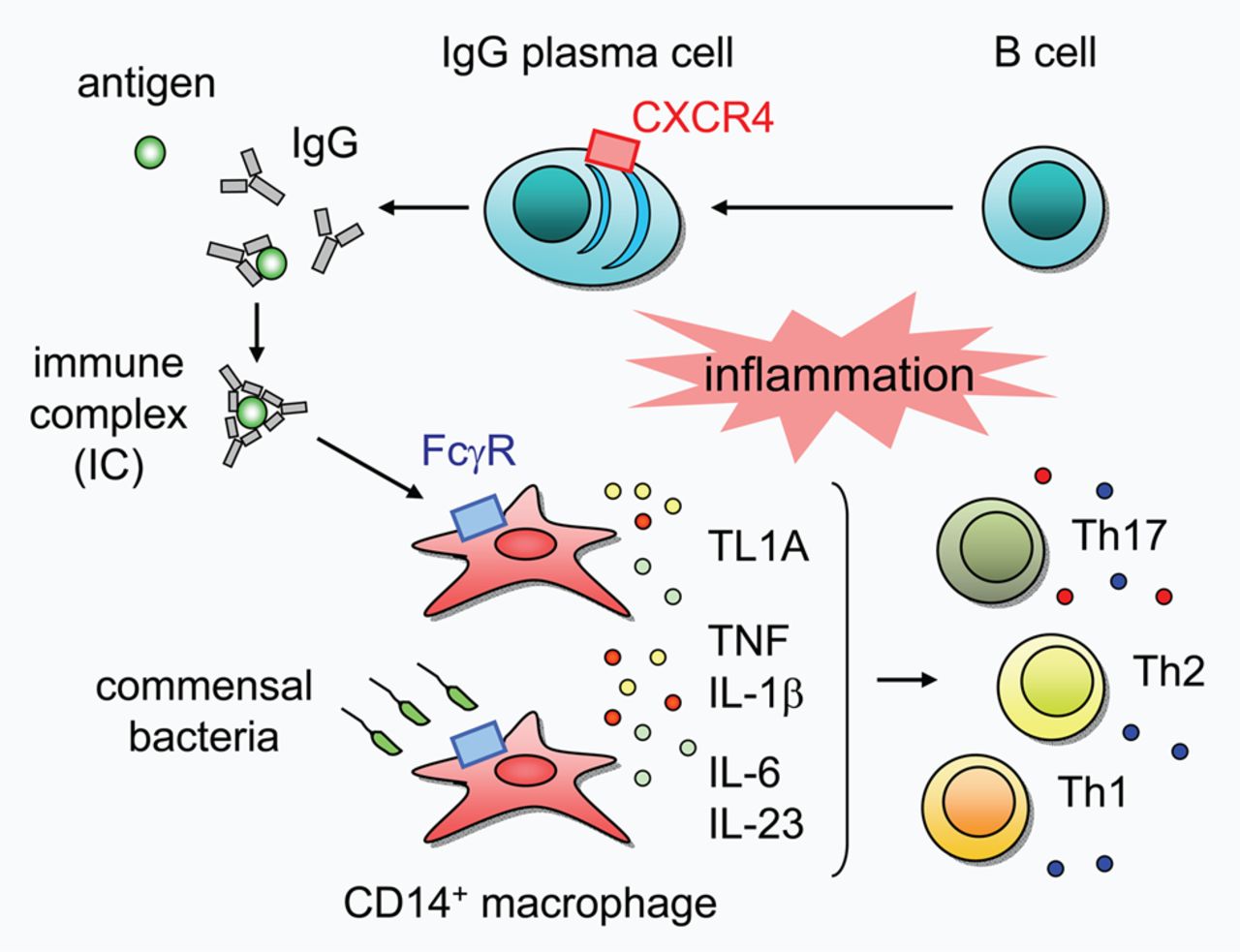
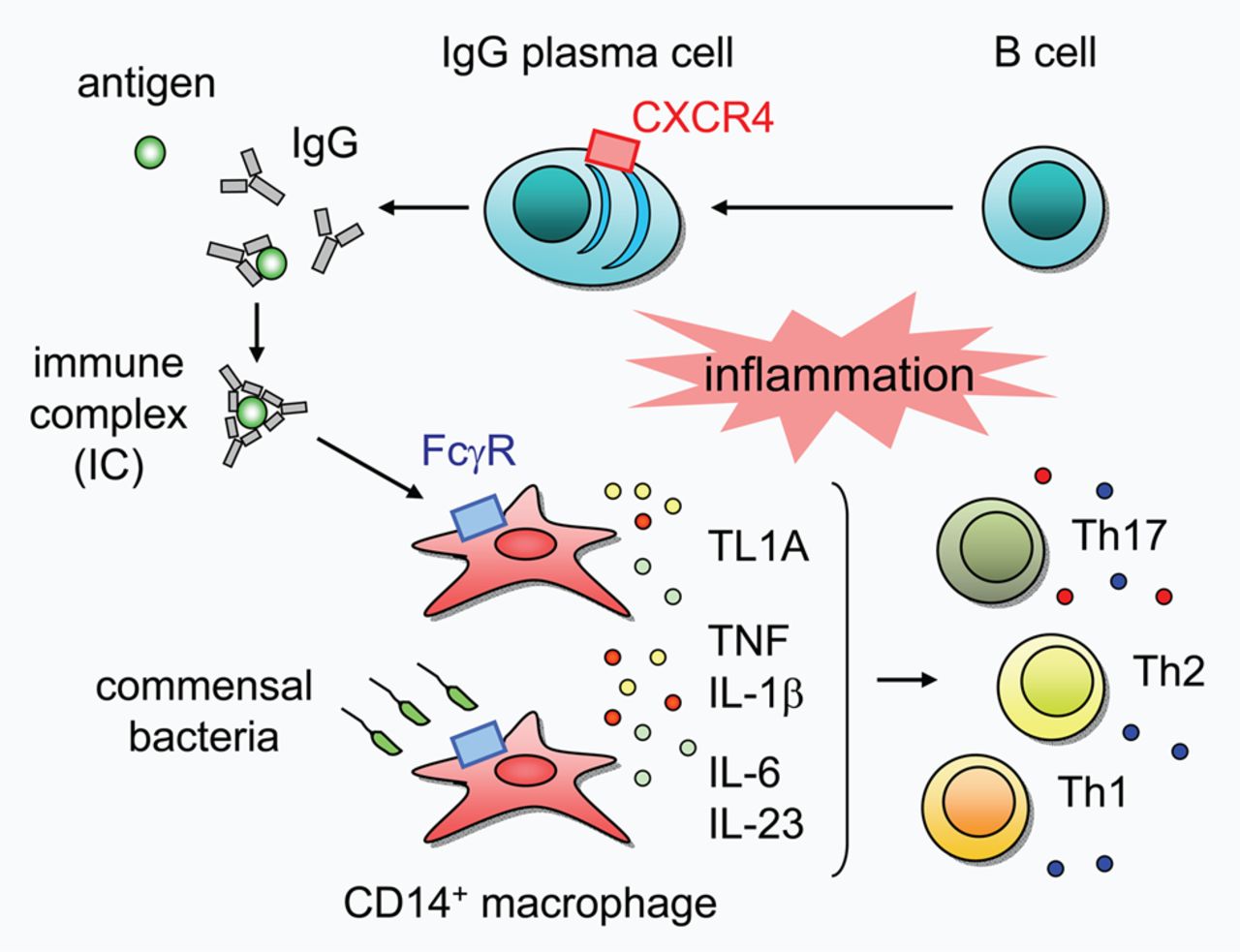
In summary, our results reveal a novel aspect of UC pathogenesis in which unique IgG plasma cells infiltrate the inflamed mucosa via CXCR4, and critically influence UC pathogenesis by exacerbating mucosal inflammation through the activation of ‘pathogenic’ intestinal CD14 macrophages via IgG-IC-FcγR signalling (figure 7). Modulation of these IgG plasma cells or the signalling pathway might therefore represent a promising therapeutic strategy for the treatment of UC.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the pathogenic role of intestinal CXCR4+ IgG plasma cells in human ulcerative colitis (UC). Unique IgG plasma cells might infiltrate the inflamed mucosa via CXCR4, and IgG antibodies produced by IgG plasma cells form immune complex (IC) with their specific antigens, thereby activating intestinal CD14 macrophages via FcγR. IgG-IC-activated CD14 macrophages produce large quantities of TNF and IL-1β, and notably up-regulate the expression of TL1A. Commensal bacteria also activate CD14 macrophages and induce the production of TNF, IL-1β, IL-6 and IL-23. Collectively, these distinct potent inducers of inflammatory response might mutually accelerate intestinal inflammation and synergistically contribute to the establishment of the complicated pathophysiological conditions of UC.
Acknowledgments
The authors thank Drs N. Kamada, T. Yajima, T. Kobayashi, T. Takayama, R. Ichikawa, Y. Wada, K. Saeki, T. Sujino, Y. Mikami, T. Doi, K. Shimamura (Keio University), Y. Ikenoue, H. Eda and M. Shiozaki (Ajinomoto Pharmaceuticals Co. Ltd) for helpful discussions and critical comments, H. Naruse and S. Ando (Keio University) for technical assistance, and N. Kobayashi (Inagi Muncipal Hospital, Inagi, Japan) for providing materials.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors MU, TH (Hisamatsu): conceived and designed the study, wrote the manuscript. MU: conducted most of the experiments, analysed data. JM, DK, KY, MK, AS, KK: provided materials and performed experiments. MM, KM, TK: helped design the study and interpretation of data. TH (Hibi): directed and supervised the study.
-
Funding This study was supported by Grants-in-Aid from the Japanese Ministry of Education, Culture, Sports, Science and Technology; the Japanese Ministry of Health, Labor and Welfare; the Young Investigator's Scholarship Committee/Awards Committee, the Japanese Society of Gastroenterology; Keio University Medical Fund.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.