Article Text

Abstract
Background Pancreatic ductal adenocarcinoma (PDA) is characterised by a robust desmoplasia, including the notable accumulation of immunosuppressive cells that shield neoplastic cells from immune detection. Immune evasion may be further enhanced if the malignant cells fail to express high levels of antigens that are sufficiently immunogenic to engender an effector T cell response.
Objective To investigate the predominant subsets of immunosuppressive cancer-conditioned myeloid cells that chronicle and shape the progression of pancreas cancer. We show that selective depletion of one subset of myeloid-derived suppressor cells (MDSC) in an autochthonous, genetically engineered mouse model (GEMM) of PDA unmasks the ability of the adaptive immune response to engage and target tumour epithelial cells.
Methods A combination of in vivo and in vitro studies were performed employing a GEMM that faithfully recapitulates the cardinal features of human PDA. The predominant cancer-conditioned myeloid cell subpopulation was specifically targeted in vivo and the biological outcomes determined.
Results PDA orchestrates the induction of distinct subsets of cancer-associated myeloid cells through the production of factors known to influence myelopoiesis. These immature myeloid cells inhibit the proliferation and induce apoptosis of activated T cells. Targeted depletion of granulocytic MDSC (Gr-MDSC) in autochthonous PDA increases the intratumoral accumulation of activated CD8 T cells and apoptosis of tumour epithelial cells and also remodels the tumour stroma.
Conclusions Neoplastic ductal cells of the pancreas induce distinct myeloid cell subsets that promote tumour cell survival and accumulation. Targeted depletion of a single myeloid subset, the Gr-MDSC, can unmask an endogenous T cell response, disclosing an unexpected latent immunity and invoking targeting of Gr-MDSC as a potential strategy to exploit for treating this highly lethal disease.
- Pancreatic Cancer
- Immune Response
- Growth Factors
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
-
Pancreatic ductal adenocarcinoma (PDA) is characterised by a robust desmoplastic reaction with multiple immune suppressive elements.
-
T cell responses to antigens overexpressed by pancreas cancer can be detected and/or elicited in a substantial fraction of patients.
-
Production of granulocyte macrophage-colony stimulating factor by PDA allografts or by preinvasive murine ductal cells enables the establishment and/or progression of transplantable tumour cell growth.17 ,18
What are the new findings?
-
Systemic and intratumoral accumulation of distinct subsets of immunosuppressive cancer-conditioned myeloid cells, each with their own kinetics, chronicles PDA progression.
-
Primary and metastatic PDA cells secrete factors involved in the induction, recruitment and survival of myeloid cells, leading to accumulation of myeloid-derived suppressor cells (MDSC).
-
Targeted in vivo depletion of the granulocytic MDSC (Gr-MDSC) subset resulted in a subsequent expansion of a distinct monocytic MDSC (Mo-MDSC) subset, yet was sufficient to induce the intratumoral accumulation of endogenous CD8 T cells and tumour cell apoptosis in autochthonous PDA.
How might it impact clinical practice in the foreseeable future?
-
Modulation of cancer-conditioned myeloid cells may provide therapeutic benefit alone or in combination with other modalities. Successful translation of such strategies will benefit from in-depth investigation in rational models that reflect the natural pathobiology, antigenic diversity and evolution of the cognate human disease.
-
Developing strategies to target the plastic and inter-related myeloid cell lineages should take into account the potential for homeostatic regulation between distinct MDSC subsets.
Introduction
Pancreatic ductal adenocarcinoma (PDA) is a highly lethal cancer with a rising incidence and unabated mortality.1 It is associated with the highest 1-year and 5-year mortalities of any cancer, reflecting the ability to evade detection until advanced stages, and also an unusual proclivity for metastatic spread. Combined with a notorious resistance to conventional chemical and radiotherapies, the diagnosis of PDA inevitably portends a poor prognosis.
A robust fibroinflammatory reaction is essentially pathognomonic for PDA and renders tumour epithelial cells a minor component of the overall cellular mass.2 The tumour stroma, comprising inflammatory cells, pancreatic stellate cells and endothelial cells embedded within a dense extracellular matrix (ECM), predominates in PDA and influences the response to treatment.3–5 Our previous work examined the content and kinetics of the immune reaction in a genetically engineered mouse model (GEMM) of PDA.6 An impressive immune presence at even the earliest stages of preinvasive disease could be identified, with a specific chronology of distinct immune subsets evolving during disease progression.7 The unique temporal kinetics suggest that distinct immunosuppressive cells, accumulating from a very early time point, may mask tumour epithelial cells from immune detection at inception and invasion. Strategies that target specific immune cell subsets may therefore be required at each stage of cancer progression to surmount barriers to effective immunity.
An additional challenge to eliciting an endogenous T cell response may be a lack of sufficiently immunogenic antigens. Despite the widespread and complex genomic instability of PDA,7 this disease appears to have few coding mutations.8 ,9 This is in contrast to the mutational profiles of melanoma10 and lung cancer,11 malignancies that have been shown to respond to immunotherapies.12 ,13 The dependence on modulation of endogenous immunity in PDA may be more challenging owing to what appears to be a rather limited neoantigenic spectrum. Indeed, similar immunomodulatory approaches in PDA have thus far exhibited limited but provocative clinical effects.14–16 Thus, a better understanding of the impact of immunosuppressive cells on the recognition of PDA by the endogenous immune system is necessary for designing potentially useful immune-based treatments.
Recent studies have suggested that the establishment of transplanted preinvasive murine ductal epithelial cells17 or of PDA allografts18 could be inhibited by decreasing granulocyte macrophage-colony stimulating factor (GM-CSF) production in the transplant before implantation. The inhibition of tumour growth was reflected by a decrease in myeloid cells at the tumour site and was dependent on endogenous CD8 T cells.18 However, since the grafts comprised cell lines from a heterologous (mixed) genetic background, or from cells virally transduced to exogenously express foreign proteins, the antitumour activity of endogenous CD8 T cells in this setting was probably due, at least in part, to recognition of minor histocompatibility alloantigens or foreign epitopes. These findings are consistent with previous work demonstrating that myeloid-derived suppressor cells (MDSC) can prevent graft rejection,19–22 but leave unaddressed the potential therapeutic benefit of this strategy in a clinical oncology setting. Furthermore, it remains unclear if disrupting cancer-associated myeloid cells would have an impact on established, autochthonous pancreas cancers.
Here, we investigate the role of cancer-conditioned myeloid cells in a GEMM of autochthonous PDA, in which invasive disease arises in situ via the stochastic development and progression of ductal precursor lesions. As a consequence, the only antigens available for recognition by the endogenous immune system are naturally occurring tumour antigens. We find that two distinct subsets of MDSC, granulocytic (Gr-MDSC) and monocytic (Mo-MDSC), expand and chronicle PDA progression, and that selective targeting of Gr-MDSC is sufficient to induce the activation and proliferation of systemic and intratumoral CD8 T cells. The influx of activated CD8 T cells is associated with an increase in tumour epithelial cell apoptosis and remodelling of the stroma. These results suggest that depletion of MDSC is an attractive, if not essential, approach to potentiate classical cytotoxic and/or adoptive immunotherapy technologies and may represent a critical component of a comprehensive platform to treat this formidable disease.
Materials and methods
Mouse strains
All animal studies were approved by the Institutional Animal Care and Use Committee of Fred Hutchinson Cancer Research Center. The KrasLSL-G12D/+; Cre (KC)23 and KrasLSL-G12D/+; Trp53LSL-R172H/+; Cre (KPC)24 genetically engineered models of PDA have been previously described.
Isolation of mononuclear cells from tissues
Pancreatic lymph nodes were removed from primary PDAs using a dissecting microscope before digestion of tumours with collagenase (Sigma-Aldrich C2674, 1 mg/mL in Dulbecco's modified Eagle's medium (DMEM)/F12) for 30 min at 37°C in a rotating incubator. Cells were washed in DMEM/F12/10% fetal bovine serum (FBS) before cell counting and staining. Erythrocytes were lysed from blood, spleen and bone marrow mononuclear preparations using ACK lysing buffer (Life Technologies). Single-cell suspensions were established in DMEM/F12/10% FBS for analyses.
Immune profiling and sorting
Mononuclear cells isolated from the spleens and tumours of KPC mice were incubated with fluorescently conjugated monoclonal antibodies as follows: CD45 (Ly5 1:200), CD11b (M1/70 1:200), Gr-1 (RB6-8C5 1:200), Ly6C (HK1.4 1:200), Ly6G (1A8 1:200), CD8α (53-6.7 1:200), CD69 (R1-2 1:100), CD25 (PC61 1:100) and Ki67 (B56 1:100) (BD Biosciences). Intracellular staining for Ki67 was performed using the eBioscience Foxp3/transcription factor staining buffer set. Annexin-V (BD Biosciences) staining was performed according to the manufacturer's recommendations. Flow cytometric analysis of immune cells was performed by gating on live CD45 cells using a BD Biosciences FACSCanto II. CD45+CD11b+Gr-1high Ly6Cint cells were purified from the bone marrow, spleen and tumour of KPC mice with invasive PDA by cell sorting using a BD Biosciences FACSAria II to >90% purity.
Histopathology and immunofluorescence
For histopathological analysis, tissues were fixed in 10% formalin for 96 h, embedded in paraffin and 4–5 μm sections were stained with haematoxylin and eosin, Masson's trichrome or Movat's pentachrome. For immunofluorescence, OCT tissue sections (7 μm) were fixed in acetone at −20°C, blocked with phosphate-buffered saline (PBS)/1% bovine serum albumin (BSA) and incubated with the following primary antibodies: cleaved caspase-3 (Cell Signalling D175, 1:200), CD8α (BD Biosciences 53-6.7, 1:25), Gr-1 (eBioScience RB6-8C5, 1:50), Ly6G (Bioxcell 1A8, 1:50), PanCK-FITC (Sigma-Aldrich F3418, 1:200), SMA-1 (DAKO 1A4, 1:100), CD31 (BD Biosciences 390, 1:50), GM-CSF (Biolegend, 1:50) or granzyme B (R&D Systems, 1:50). Sections were washed with PBS/1% BSA, labelled with species-specific Alexa-conjugated antibodies (Invitrogen) and washed with PBS/1% BSA followed by PBS. The sections were mounted using Prolong gold anti-fade reagent with 6-diamidino-2-phenylindole to label nuclei (Invitrogen).
T cell suppression assay
To measure CD8 T cell proliferation, 96-well round-bottom plates were precoated with 100 μL anti-CD3ε (BD Biosciences 145-2C11, 1 μg/mL) and anti-CD28 (BD Biosciences 37.51, 10 μg/mL) and incubated at 4°C for 24 h. Splenic CD8 T cells were purified using Dynabeads Untouched Mouse CD8 Cell isolation kit (Invitrogen) according to the manufacturer's protocol. Purified CD8 T cells were labelled with 1 μM 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen) at 37°C for 20 min in serum-free RPMI. Excess dye was removed by washing the labelled cells with RPMI 1640 supplemented with 2 μM glutamine, 100 U/mL penicillin/streptomycin, 10% fetal calf serum and 30 μM 2-mercapatoethanol (complete media). Sorted Gr-MDSC (CD45+CD11b+ Gr-1highLy6Cint), Mo-MDSC (CD45+CD11b+Gr-1intLy6Chigh) or TAM (CD45+CD11b+Gr-1intLy6Cint) were incubated at titrating numbers with 1×105 CFSE-labelled T cells in complete media. After 48 h, cell cultures were stained for surface markers with CD8α-e450 (BD Biosciences 53-6.7, 1:200) and Thy1.1 (CD90.1)-PercP (BD Biosciences OX-7, 1:200) in PBS/2.5% FBS, followed by Annexin-V-APC (BD Biosciences) staining according to the manufacturer's protocol and immediately analysed on a FACSCanto II. The percentages of apoptotic (Annexin-V) or proliferating (CFSE dilution) T cells were determined by gating on CD8 Thy1.1 cells.
Proteome analysis
Conditioned media (CM) isolated from 24 h supernatants of purified preinvasive (n=2), invasive (n=2) and paired metastatic (n=2) KPC carcinoma cells were incubated on Mouse Cytokine Array Panel A nitrocellulose membranes (R&D Systems ARY006) according to manufacturer's recommendations. The mean pixel density for each capture antibody spot in duplicate was calculated and proteins that were positive in two or more independent invasive cell preparations were quantified by real-time PCR.
Gene expression analysis
Total RNA was extracted (RNeasy Miniprep Kit, Qiagen) from primary cultures of KPC tumour epithelial cells and paired metastatic cells to the livers of the same animals (n=3 each) and also from preinvasive pancreatic ductal epithelial cells. RNA was converted to cDNA using a high capacity reverse transcriptase kit (Applied Biosystems). Quantitative PCR was performed using SYBR Green mastermix, and triplicate samples were run on a C1000 thermal cycler (BioRad). Primers (see online supplementary table S1) were based on published literature or designed using Primer-BLAST software. Quantifications were normalised to endogenous cycA, and fold-change gene expression in invasive and metastatic cells compared with preinvasive cells was calculated using the ΔΔCT method.
MDSC depletion
KPC mice were serially imaged by high-resolution ultrasound (Vevo 2100, Visualsonics) and enrolled when the primary tumour reached 2–5 mm in diameter. The monoclonal antibody (mAb) 1A8 (Bioxcell, 400 μg in sterile saline) was administered intraperitoneally on days 0, 5 and 10 to deplete Ly6G+ Gr-MDSC. Control KPC mice were treated with saline or isotype (rat IgG2a) control. For immunomodulation experiments, tumour diameters were assessed in a blinded manner by high-resolution ultrasound at the time of enrolment and on days 8 and 12 of treatment.
MDSC proliferation and apoptosis assays
CD11b+Gr-1high Ly6Cint MDSC were isolated to >90% purity and incubated with 1 μM CFSE (Invitrogen) at 37°C for 20 min in serum-free media. Excess dye was removed by washing labelled cells with CM. A total of 1×105 Gr-MDSC were incubated with basic media (DMEM/F12/10% FBS) or with CM isolated from the supernatants of primary PDA cells (PDA-CM) or metastatic PDA cells (Met-CM); the recombinant murine cytokines, GM-CSF, granulocyte-colony stimulating factor (G-CSF), or monocyte-colony stimulating factor (M-CSF; R&D Systems, 50 ng/mL); or blocking antibodies to GM-CSF, G-CSF, or M-CSF (R&D Systems, 10 µg/mL). After 48 h, cells were stained for Gr-1-e450 (BD Biosciences RB6-8C5, 1:200) and CD11b-PE (BD Biosciences M1/70, 1:200), and Annexin-V-APC and CFSE dilution were measured by gating on live Gr-1+CD11b+ cells to assess for apoptosis and proliferation, respectively.
Quantification of vessel diameter in tumours
Frozen sections were dual stained with anti-mouse CD31 (BD Biosciences 390, 1:50) followed by species-specific Alexa-conjugated secondary antibody (Invitrogen, 1:1000) and PanCK-FITC (Sigma-Aldrich F3418, 1:200). Vessel diameters were measured using NIS-Element imaging software from three to five non-overlapping ×20 fields in PDA sections from control and mAb 1A8-treated KPC mice (n=3 animals each). Typically between five and 10 vessels were measured for each high-powered field.
Statistical analyses
Analyses were performed with Graphpad Prism software. Data are provided as mean±SEM, unless indicated otherwise. For comparisons between two groups, statistical analyses were performed using a Student t test, and p<0.05 was considered significant. For comparisons involving three groups or more, a one-way analysis of variance using the Kruskal–Wallis test was first performed (p<0.05 was considered significant), followed by a Dunn's multiple comparison test (unless otherwise indicated). Symbols indicating statistical significance are as follows: *p<0.05; **p<0.005; ***p<0.0005.
Results
Systemic and intratumoral accumulation of myeloid cells chronicles PDA progression
Targeted endogenous expression of oncogenic KrasG12D and point mutant Trp53R172H to progenitor cells of the murine pancreas24 induces the development of a clinical, histopathologic and molecular syndrome of disease that faithfully recapitulates human PDA.25 Disease initiates in preinvasive ductal precursors, termed pancreatic intraepithelial neoplasias and progresses spontaneously to invasive and metastatic ductal adenocarcinoma. As described previously in KC mice,6 we found that discrete immune cell populations infiltrate at distinct stages in evolving KPC PDA. CD4+Foxp3+ regulatory T cells (Treg) and tumour associated macrophages (TAM) infiltrated predominantly at the preinvasive stage, while Gr-1+CD11b+ cells increased most markedly in the transition from preinvasive to invasive disease (figure 1A).
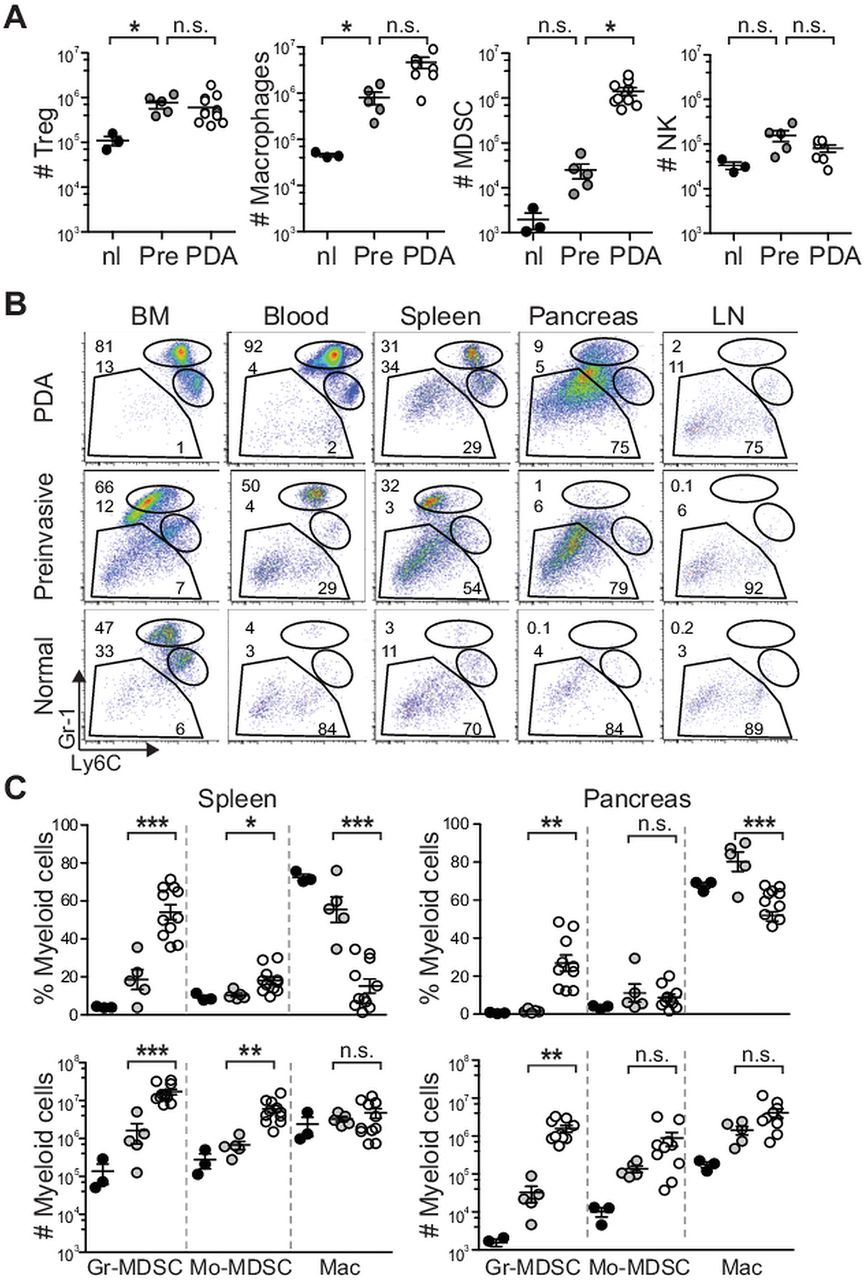
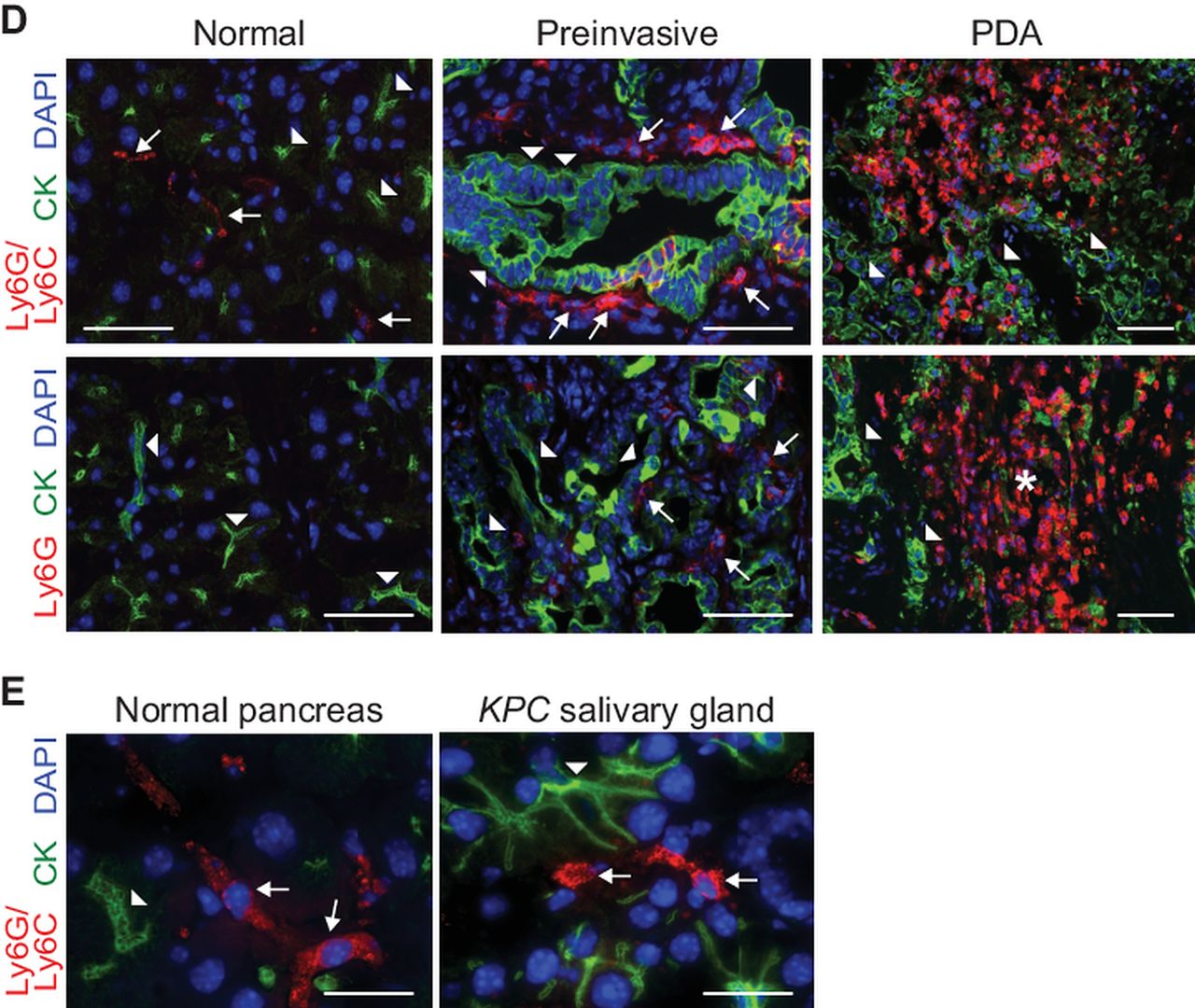
Cancer-conditioned myeloid cells chronicle the evolution of pancreatic ductal adenocarcinoma (PDA) in KPC mice. (A) The number of pancreatic Treg (CD45+CD4+FoxP3+), macrophages (CD45+CD11b+F4/80+), myeloid-derived suppressor cells (MDSC) (CD45+CD11b+RB6-8C5 [Ly6G/Ly6C]+) and NK cells (CD45+NK1.1+) for each pancreas were quantified from normal pancreas (nl), 6–8-week-old KPC pancreata with confirmed preinvasive disease (Pre) and invasive tumours (PDA). Significant differences were detected in the number of Treg, tumour-associated macrophages and MDSC during disease progression. (B) Evolving profiles of three distinct populations of myeloid cells (gated on CD45+CD11b+) were seen in various organs based on expression patterns of Gr-1 and Ly6C. BM, bone marrow; LN, lymph node. (C) The percentages (top panel) and absolute numbers (bottom panel) of CD45+CD11b+ myeloid populations in the spleen and pancreas in normal (black filled circles) and preinvasive (grey filled circles) and invasive (open circles) disease settings. Data are plotted as mean±SEM and each data point represents an individual mouse. Granulocytic MDSC (Gr-MDSC)=CD45+CD11b+Gr-1highLy6Cint; monocytic MDSC (Mo-MDSC)=CD45+CD11b+Gr-1intLy6Chigh and macrophage (Mac)=CD45+CD11b+Gr1intLy6Cint. (D) Specific immunofluorescence reveals rare Ly6G/Ly6C+ (RB6-8C5) cells in normal pancreas, focal accumulation in pancreata with preinvasive disease and diffuse infiltration in invasive PDA. Specific Ly6G immunofluorescence demonstrates that Gr-MDSC are absent from normal pancreas, rare in preinvasive disease and abundant in invasive PDA. The majority of myeloid cells in normal pancreas and surrounding preinvasive lesions appear to be macrophages. Arrowheads, epithelial cells; arrows, myeloid cells; asterisk, Gr-MDSC. Scale bars, 50 µm. (E) Ly6G/Ly6C (RB6-8C5) staining in normal pancreas and KPC salivary gland. Arrowheads, epithelial cells; arrows, myeloid cells. Scale bars, 10 µm. *p<0.05; **p<0.005; ***p<0.0005.
Since Gr-1 cells increased precipitously from preinvasive to invasive disease to become a dominant immune cell population infiltrating PDA, we sought to further clarify the nature of these cells during malignant progression. The mAb RB6-8C5 was initially described to bind to granulocyte receptor 1 (Gr-1), historically synonymous with Ly6G.26 It is now clear, however, that RB6-8C5 binds to two distinct cell surface antigens: Ly6G, which is specifically expressed on granulocytes, and Ly6C which is expressed on a variety of cell types including CD11b inflammatory monocytes,27 plasmacytoid dendritic cells,28 ,29 and CD8 T cells.30 Therefore, to distinguish potentially distinct cancer-conditioned myeloid cell populations, we used three different reagents: mAb αGr-1 (clone RB6-8C5), which binds both Ly6G and Ly6C; mAb αLy6G (clone 1A8); and mAb αLy6C (clone HK1.4). Of relevance, CD11b inflammatory monocytes in normal mice (also referred to as Mo-MDSC in tumour-bearing mice) bind RB6-8C5 owing to high levels of Ly6C expression, but do not express Ly6G.31 The predominant subset of CD11b myeloid cells that was induced systemically stained brightly with RB6-8C5 (Gr-1high) and expressed intermediate levels of Ly6C (Ly6Cint) and these CD11b+Gr-1high Ly6Cint cells are hereafter referred to as Gr-MDSC (figure 1B). As expected, Gr-1high cells also stained positively for Ly6G as detected by mAb 1A8, while other CD11b myeloid subsets, including Gr-1int Ly6Chigh cells did not express Ly6G (data not shown). Significant increases in the percentage of Gr-MDSC in bone marrow (p=0.0036), blood (p=0.0033), spleen (p=0.0238) and pancreas (p=0.0167) were seen in mice with PDA compared with control littermates. Furthermore, Gr-MDSC were rarely detected in pancreatic draining lymph nodes from non-tumour bearing mice, but were increased in those associated with PDA (figure 1B, see online supplementary figure S1). The absolute number of Gr-MDSC was significantly higher in the spleens and primary tumours of mice that had progressed to invasive disease than in KPC mice with preinvasive disease only (figure 1C). The absolute number of Mo-MDSC (CD11b+ Gr-1int Ly6Chigh) increased significantly in the spleens of mice with invasive PDA as compared with mice with preinvasive disease only, but this population was not significantly increased within the autochthonous tumour itself (figure 1C). The marked rise in Gr-MDSC and Mo-MDSC in the circulation during disease progression also correlated with a reduction in the percentage, though not absolute number, of Gr-1neg/low F4/80+ cells within the CD11b compartment in the spleen, probably reflecting dilution from extramedullary haematopoiesis and associated splenomegaly. CD11b+ Ly6Cint F4/80+ Ly6G− cells were common in PDA, stained positive for RB6-8C5 owing to Ly6C expression and were adherent in vitro (not shown), further distinguishing them from the two MDSC subpopulations.
The localisation of Gr-MDSC within the pancreas during disease evolution was determined by dual immunofluorescence for ductal epithelial cells (Pan-CK) and for myeloid populations using 1A8, which specifically detects Gr-MDSC and not Mo-MDSC, or RB6-8C5, which detects both of these populations and TAM as well. Immunoreactivity with RB6-8C5 was seen on a small fraction of cells in the normal pancreas and notably in rings of cells encircling pancreatic intraepithelial neoplasias, identifying tissue resident Ly6C+ macrophages (note that these cells are not identified by 1A8) (figure 1D).32 RB6-8C5 staining was also detected in a small fraction of cells in the normal salivary gland (figure 1E). In contrast, there were scant Gr-MDSC (as determined by 1A8 reactivity) in the normal pancreas and only a modest infiltration in pancreata with preinvasive lesions. Ly6G+ Gr-MDSC were abundant and widely dispersed throughout invasive PDA, consistent with the flow cytometry data (figure 1D).
Distinct morphological spectra distinguish MDSC subsets
The nuclear morphology of phenotypic Gr-MDSC revealed cells with segmented nuclei, consistent with polymorphonuclear cells,33 as well as more immature forms that had ring- or horseshoe-shaped nuclei indicative of immature myeloid progenitors (so-called ‘band’ cells; see online supplementary figures S2A, B). Phenotypic Mo-MDSC isolated from the spleen contained these immature forms as well as monocytes, but segmented cells were rarely detected (see online supplementary figure S2A). Although ring cells were identified in both subsets, these cells have been suggested to be functionally distinct, as Gr-1high ring cells include mature cells of the granulocyte lineage, while Gr-1low ring cells include a more heterogeneous mononuclear population.34 Gr-MDSC also exhibited higher side scatter by fluorescence activated cell sorting (FACS) analyses, reflecting increased granularity as compared with Mo-MDSC and were more uniform in size as compared with the Mo-MDSC population (see online supplementary figures S2C). Splenic and intratumoral Gr-MDSC expressed the differentiation markers F4/80 and major histocompatibility complex (MHC) class II, which were not detected on polymorphonuclear cells isolated from normal mice (data not shown). The presence of these additional markers was restricted to Gr-MDSC isolated from the spleen and tumours and not found on CD11b+Gr-1high cells in the blood or bone marrow of animals with PDA (see online supplementary figure S2D), suggesting evolution through additional maturation stages from marrow to periphery during malignancy. The fact that tumour-associated Gr-MDSC can express F4/80 and MHC class II molecules also indicates that some of these markers in malignancy are imprecise and may fail to clearly distinguish macrophages from MDSC.
Gr-MDSC inhibit T cell proliferation and induce T cell death after activation
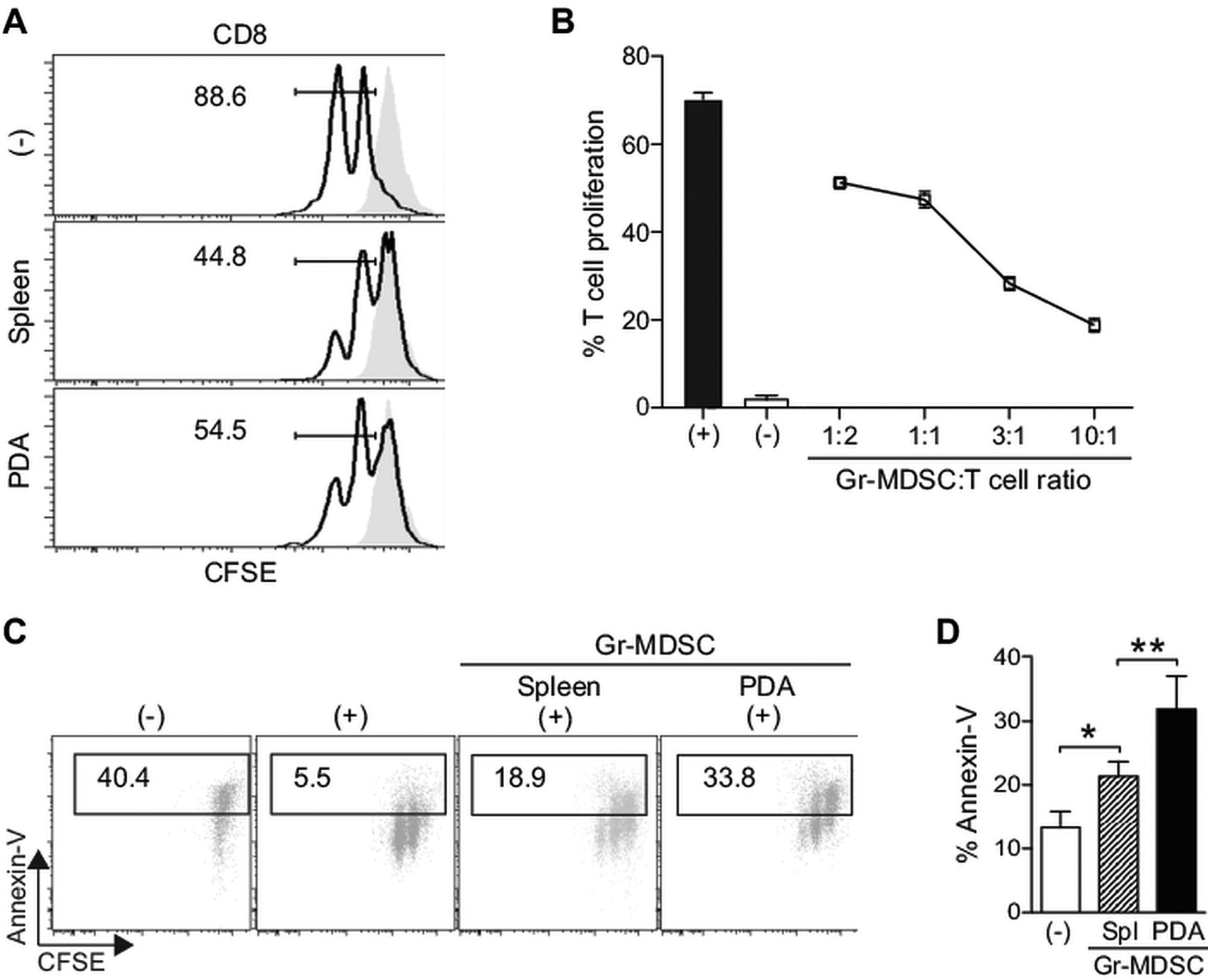
Pooled Gr-1+ cells from tumour-bearing mice, which contain both the monocytic and granulocytic subsets, have been shown to suppress T cell proliferation.6 ,18 To determine the suppressive potential of each of these subpopulations, sorted Gr-MDSC (CD11b+ Gr-1high Ly6Cint), Mo-MDSC (CD11b+ Gr-1int Ly6Chigh) and TAM (CD11b+ Gr-1int Ly6Cint) from KPC mice were incubated with T cells in both proliferation and apoptosis assays. At a 1:1 myeloid-to-T cell ratio, Gr-MDSC moderately decreased naïve T cell proliferation (figure 2A). However, at the higher myeloid-to-T cell ratios seen in vivo in mice with PDA, Gr-MDSC exhibited a pronounced ability to suppress T cell proliferation and the degree of suppression was dose dependent (figure 2B). Mo-MDSC also significantly suppressed T cell proliferation in a dose-dependent manner and TAM potently blocked T cell cycling (see online supplementary figure S3A).

Gr-myeloid-derived suppressor cells (Gr-MDSC) isolated from the spleen and pancreatic ductal adenocarcinoma (PDA) are immunosuppressive. (A) Naïve T cell proliferation in response to anti-CD3/CD28 is suppressed by Gr-MDSC (assays performed at 1:1 Gr-MDSC:T cell ratio). Numbers indicate the percentage of CD8 T cells that have undergone one or more cell division after 48 h. Grey filled histograms indicate T cells in the absence of CD3/CD28. (−) indicates no Gr-MDSC added. (B) The suppressive capacity of Gr-MDSC increases at higher Gr-MDSC:T cell ratios. Representative data from one of four independent experiments are plotted as mean±SD (n=3 replicates each). (−)=no CD3/CD28 stimulation. (C) Gr-MDSC induce the apoptosis of activated T cells. Representative fluorescence activated cell sorting profiles (of four independent experiments) performed at 1:1 Gr-MDSC:T cell ratio and gated on CD8+ Thy1.1+ cells. (−)=no stimulation; (+)=CD3/CD28 stimulation. (D) Gr-MDSC isolated from spleen (Spl) and PDA of KPC mice induce apoptosis of activated CD8 T cells. (−)=T cells only (no MDSC). Data represent mean±SEM from four independent experiments. CFSE, 5,6-carboxyfluorescein diacetate succinimidyl ester.
Gr-MDSC, Mo-MDSC and TAM also induced apoptosis of activated T cells (figure 2C,D and see online supplementary figure S3B). Gr-MDSC isolated from PDA were 1.5-fold more potent at inducing T cell apoptosis than their splenic or bone marrow counterparts (figure 2C,D and data not shown). Thus, Gr-MDSC can both suppress T cell proliferation and promote T cell death, and the tissue milieu from which these cells are recovered appears to influence their immunosuppressive capacities. Since Gr-MDSC undergo the most dramatic and significant increase in number from preinvasive to invasive PDA, we chose to study this subset in more detail.
Tumour conditioned media and GM-CSF promote Gr-MDSC survival
We observed that neither splenic nor tumour-derived Gr-MDSC survived after 24 h of culture in standard media as indicated by the collapse of cellular integrity and the appearance of pyknotic nuclei and cell debris (figure 3A). This was in clear contrast to the abundant, large, viable, birefringent Gr-MDSC clusters in media that had been conditioned by either primary or metastatic PDA cells (figure 3A and data not shown). CM from cultured PDA cells prevented the majority of Gr-MDSC from undergoing apoptosis (figure 3B).
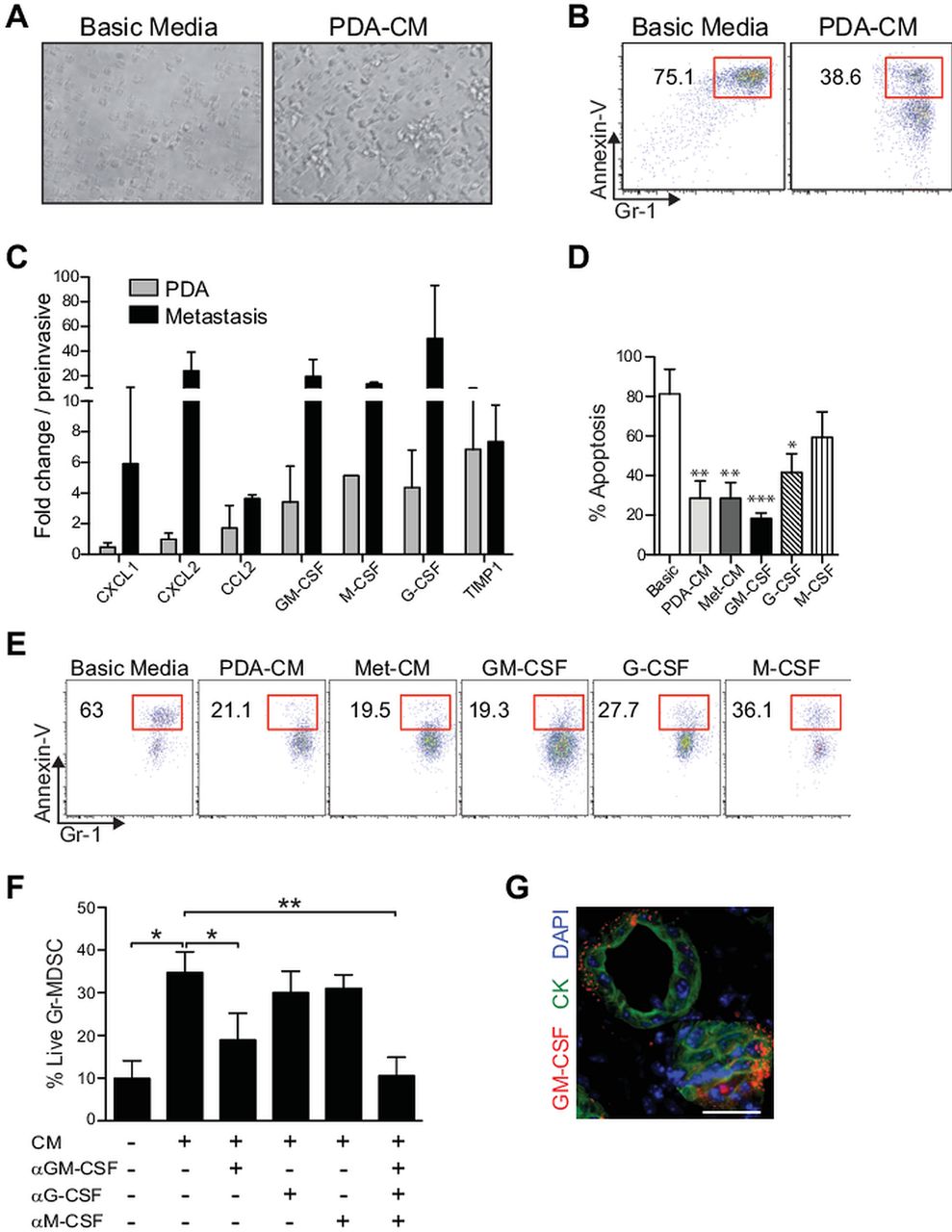
Conditioned media from primary pancreatic ductal adenocarcinoma (PDA-conditioned media (CM)) or metastatic epithelial cells (Met-CM) promotes the survival of Gr-myeloid-derived suppressor cells (Gr-MDSC). (A) Sorted Gr-MDSC cultured in basic media or PDA-CM for 48 h. (B) PDA-CM protects a cultured Gr-MDSC from apoptosis (48 h). (C) Relative gene expression of select cytokines and chemokines as determined by quantitative PCR (data represent mean±SEM of three independently derived primary invasive tumour (PDA) and paired metastatic cell preparations and are normalised to expression in preinvasive cells). (D) Impact of conditioned media and cytokines on apoptosis of splenic-derived Gr-MDSC. Bar graphs represent mean±SEM of four independent experiments. Treated samples were compared with the basic media (Basic) control and significance was determined by using a one-way analysis of variance followed by a Dunnett's multiple comparison test. (E) Sorted splenic Gr-MDSC from KPC mice with PDA were incubated with the indicated media or cytokines and the percentage of Annexin-V+ Gr-MDSC at 48 h determined by fluorescence activated cell sorting (FACS; representative results of four independent experiments). (F) Sorted splenic Gr-MDSC were incubated with PDA-CM±the indicated blocking monoclonal antibody for 48 h. The percentage of live cells was determined by FACS analyses of Annexin-Vneg Gr-MDSC. (G) Clusters of PDA cells contain GM-CSF at their stromal surface. CK, cytokeratin; G-CSF, granulocyte-colony stimulating factor; GM-CSF, granulocyte macrophage-colony stimulating factor; M-CSF, monocyte-colony stimulating factor. Scale bar, 25 µm. *p<0.05; **p<0.005; ***p<0.0005.
To investigate the molecular mechanisms underlying the enhanced survival of Gr-MDSC in response to CM, we profiled the soluble factors expressed by purified carcinoma cells in culture. The secretory profile of invasive and metastatic cells showed a substantial commitment to synthesis of growth factors and chemokines involved in both granulocyte (eg, CXCL1, CXCL2) and monocyte (eg, CCL2) trafficking (see online supplementary figure S4A–C). GM-CSF, G-CSF and M-CSF were also upregulated in tumour epithelial cells as compared with preinvasive ductal cells. Relative gene expression by quantitative PCR confirmed the increase in these factors in invasive versus preinvasive cells (figure 3C). Of these three myelopoietic cytokines,35 GM-CSF had the most pronounced effect on promoting Gr-MDSC survival in vitro (figure 3D,E). Furthermore, blockade of GM-CSF in CM significantly decreased the percentage of live (non-apoptotic) Gr-MDSC (figure 3F), implicating GM-CSF as a primary tumour cell secreted factor promoting Gr-MDSC survival. Of note, G-CSF produced by PDA also consistently increased Gr-MDSC survival (figure 3D–F). However, neither CM nor these cytokines were sufficient to promote appreciably the proliferation of Gr-MDSC in vitro (not shown), suggesting either that additional factors sustain MDSC proliferation in vivo or accumulation is instead due to the prevention of apoptosis and/or the induction or expansion of precursors. The chemokines CXCL1, CXCL2 and CCL2 had no impact on Gr-MDSC survival or proliferation in vitro (data not shown). Finally, we confirmed that preinvasive and invasive PDA epithelial cells secrete GM-CSF in vivo and note that GM-CSF-containing vesicles within the cells are orientated toward the parenchyma rather than the lumen of duct-like structures (figure 3G). These data support a model in which primary tumour epithelial cells secrete factors into their microenvironment that promote the survival and recruitment of Gr-MDSC, contributing to their in vivo accumulation during cancer progression.
Depletion of Gr-MDSC increases CD8 T cell accumulation and activation in autochthonous PDA
In order to investigate the clinical potential of targeting MDSC, we performed experiments to deplete MDSC in animals with autochthonous invasive and metastatic PDA. KPC mice with a defined primary pancreatic tumour burden of 2–5 mm in diameter by high-resolution ultrasound were injected with the αLy6G mAb 1A836 to deplete the Gr-MDSC, while not reducing the number of Ly6G− Mo-MDSC (figure 4A,B and data not shown). We developed and refined our treatment protocol to maintain effective depletion of intratumoral Gr-MDSC for over 2 weeks. We also established the ability of RB6-8C5 to deplete MDSC (data not shown); however, since this antibody has been shown to induce signalling,37 and is also less specific, we focused on 1A8 for the more detailed studies presented here.

Systemic administration of 1A8 (αLy6G) specifically depletes Gr-myeloid-derived suppressor cells (Gr-MDSC) in autochthonous pancreatic ductal adenocarcinoma (PDA). (A) Representative myeloid cell profiles in peripheral blood mononuclear cells from a normal mouse and untreated (KPC) and 1A8-treated KPC (KPC + 1A8) mice. Numbers indicate the percentage of each subset gated on CD45 mononuclear cells. We note that the gates for the discrete subpopulations defined in the blood were then applied to the tissue-specific analyses. (B) Percentage of Gr-MDSC (squares) and monocytic MDSC (Mo-MDSC; circles) in blood after 1A8 treatment. Data represent mean±SD from three independently treated animals. (C) Representative fluorescence activated cell sorting (FACS) profiles of CD45+ CD11b+ splenocytes from control (−) and 1A8-treated (+) KPC mice (4–6 animals per group). (D) Both the percentage and number of splenic Gr-MDSC at endpoint of 1A8 treatment (day 12) are significantly decreased (**, p=0.005). (E) Representative FACS profiles of intratumoral myeloid cells in control (−) and 1A8-treated (+) KPC mice at day 12 of Gr-MDSC depletion. (F) The percentage and number of Gr-MDSC in PDA at the endpoint are significantly decreased in 1A8-treated mice compared with control KPC mice.
Administration of 1A8 markedly reduced the percentage and number of Gr-MDSC in the circulation (figure 4A,B), and also in the spleen (figure 4C,D) and PDA (figure 4E,F). Intriguingly, a corresponding 4–5-fold increase was seen in the number of Mo-MDSC in the spleen (1.4±0.24×107 control vs 6.3±1.9×107 in 1A8-treated, p=0.0019) and PDA of 1A8-treated mice (5.0±1.1×106 control vs 2.6±0.52×107 in 1A8-treated, p<0.0001), suggesting a homeostatic relationship between these two myeloid subpopulations (not shown). Tumour masses did not generally decrease in size during this treatment period and, in the majority of 1A8-treated mice, the tumours instead grew (not shown). Although this might be interpreted as reflecting a lack of efficacy, immune therapies have frequently been shown to cause acute increases in tumour size because of the recruitment and infiltration of immune cells, rather than the progressive growth of the cancer cells.38 Consistent with this possibility, the number of intratumoral CD45 immune cells in 1A8-treated mice increased about twofold compared with untreated PDA, although this did not reach statistical significance (1.04±0.16×107 in controls vs 2.4±0.9×107 in 1A8-treated mice; n=4 each; p=0.1557).
The influx of immune cells in Gr-MDSC-depleted animals prompted us to examine the composition of the infiltrate in greater detail and to analyse additional cellular and molecular endpoints. Depletion of Gr-MDSC significantly increased the percentage and absolute number of CD8 T cells in autochthonous PDA (figure 5A,B), as well as the fraction that were proliferating (figure 5C,D). Consistent with this proliferative signature, a higher proportion of CD8 T cells in the spleen and invasive PDA expressed the activation marker CD69, indicative of antigen recognition and signalling by the T cell receptor 39 ,40 (figure 5E,F). CD8 T cell accumulation in PDA after Gr-MDSC depletion was also detected in situ by specific immunofluorescence (figure 5G). Tumour-infiltrating CD8 T cells were found in the stroma, and also immediately adjacent to tumour epithelial cells (figure 5G). Cytokeratin staining was more punctate and less uniform in areas adjacent to CD8 T cells than in immune-privileged tumour areas, suggesting a potential effect on epithelial cell integrity (and see below). An additional indication of T cell activation is transient release of the serine protease granzyme B that can be captured in a subset of cells at any given time. The number of granzyme B+ cells was significantly increased in Gr-MDSC-depleted mice (see online supplementary figures S5A,B). Thus, Gr-MDSC depletion also affects the functional activity of tumour-infiltrating T cells.

Gr-myeloid-derived suppressor cells (Gr-MDSC) depletion increases CD8 T cell number, proliferation, activation and tumour-specific infiltration in autochthonous pancreatic ductal adenocarcinoma (PDA). The percentage (A) and number (B) of CD8 T cells significantly increased in pancreatic tumours, but not the spleen of 1A8-treated compared with untreated KPC mice. (C) Representative fluorescence activated cell sorting (FACS) plots of proliferating CD8 T cells in the spleen and pancreatic tumours of control and treated KPC mice as determined by Ki67 expression (4–6 mice per cohort). (D) The percentage of Ki67+ CD8 T cells in 1A8-treated mice (n=4) compared with control KPC mice (n=6 mice) in the spleen and tumours was significantly increased. (E, F) Gr-MDSC depletion increases the percentage of CD8 T cells in both the spleen and PDA that express the activation marker CD69. (G) CD8 T cells (arrow) are rare in control PDA and increase significantly (asterisk) after Gr-MDSC depletion (1A8). Control, open bars; 1A8-treated, filled bars. Scale bars, 50 µm.
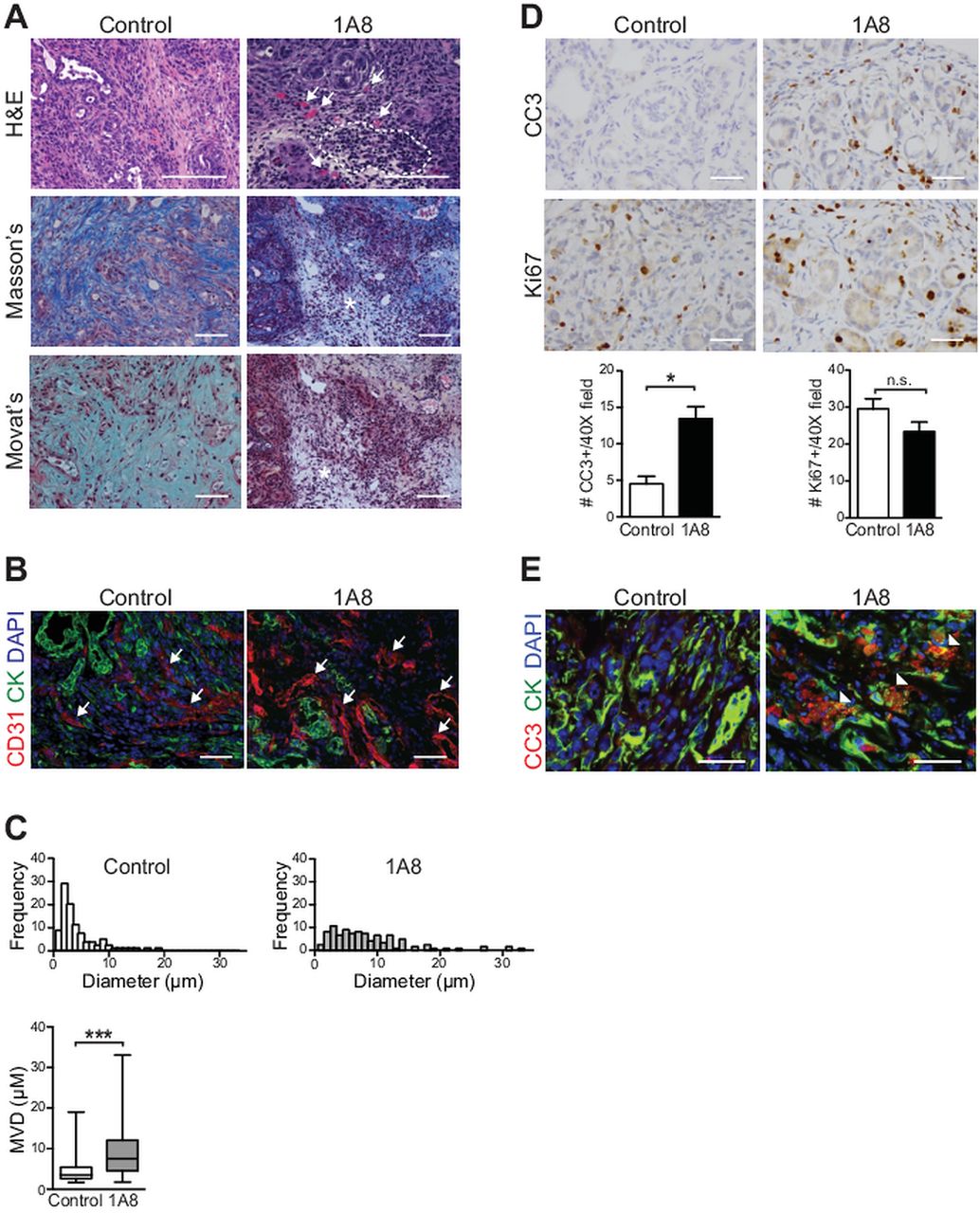
Depletion of Gr-MDSC increases tumour epithelial apoptosis in autochthonous PDA
Standard histological and histochemical studies immediately revealed that targeted depletion of Gr-MDSC had influenced the stromal architecture and integrity of the tumours in treated mice, particularly in regions with concentrated mononuclear cells (figure 6). These areas were also characterised by decreased ECM deposition and the appearance of patent blood vessels (figure 6A), with increased vessel diameters (figure 6B,C), which are otherwise typically compressed and not readily visible in PDA.3–5 Depletion of Gr-MDSC also resulted in a threefold increase in cleaved caspase-3-positive tumour cells, but did not change the number of proliferating cells (figure 6D). Compound immunofluorescence assays confirmed that a significant fraction of apoptotic cells represented tumour epithelium (figure 6E). Apoptosis was not appreciably increased in activated myofibroblasts (see online supplementary figure S5C). Taken together, these results support the hypothesis that cancer-conditioned Gr-MDSC may impede the recognition of tumour antigens by CD8 T cells and the acquisition of effector T cell activity in PDA, and that targeted depletion of Gr-MDSC with systemic mAb therapy can potentially unmask PDA to endogenous immunity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Targeted depletion of Gr-myeloid-derived suppressor cells (Gr-MDSC) remodels the stroma and increases tumour epithelial cell apoptosis. (A) Administration of 1A8 increases mononuclear cell infiltrates in autochthonous pancreatic ductal adenocarcinoma (PDA) (outlined) and patency of blood vessels (arrows) and depletes extracellular matrix content and integrity (asterisks). Representative fields are shown for histology (H&E), collagen content (Masson's trichrome) and combined collagen and glycosaminoglycan content (Movat's pentachrome). Scale bars, 50 µm. (B) CD31 immunofluorescence of PDA reveals a shift toward larger vessel diameters (arrows) after 1A8 treatment (quantified in (C)). Scale bars, 25 µm. (C) Administration of 1A8 significantly increases the mean vessel diameter (MVD) in PDA. (D) Apoptosis of tumour epithelial cells, as assessed by cleaved caspase-3 (CC3), was significantly increased in 1A8-treated mice (n=4) compared with control (n=6) KPC mice, while the number of proliferating tumour epithelial cells (Ki67) was not significantly different. (E) CC3 and CK double-positive cells indicate tumour epithelial cells undergoing apoptosis (arrowheads). Scale bars, 25 µm.
Discussion
Cancer implies a failure of immunity. Indeed, in cancers like PDA, it may even suggest immune complicity. Preinvasive cells can persist in the context of a fully functional immune system for various reasons. Since tumours are derived from self, they benefit from multiple mechanisms of self-tolerance. Common driver mutations in cancer cells may not encode strongly immunogenic epitopes, and malignant cells are notoriously defective in antigen processing and presentation, further contributing to immune evasion.41 Neoplastic cells have additional elaborate mechanisms that subvert antitumour T cell activity, including the induction of host immunosuppressive cells. Although endogenous tumour-reactive T cells can, in principle, recognise cancer antigens expressed on preinvasive cells and delay malignant progression,42 concomitant induction of T cell tolerance can abrogate expression of an effector response.43
This study is the first to report that depletion of a single immunosuppressive myeloid cell subset can induce endogenous CD8 T cell accumulation and tumour cell death in autochthonous ductal adenocarcinomas of the pancreas. That adaptive immunity appears to be induced without providing additional signals to positively engage an antigen-specific T cell response is especially surprising in PDA, which has seemed to be less immunogenic than other cancers.8 ,9 Our data demonstrate that the endogenous immune response does have some capacity to recognise native PDA antigens and that Gr-MDSC present a critical barrier to eliciting such immune activity. Furthermore, in contrast to the hypothesis that intratumoral fluid pressure and subsequent vessel compression (which we have shown can inhibit passive transport of molecules into PDA),4 also limits immune cell infiltration,44 our data indicate that T cells readily infiltrate tumours, as would be expected for cells which can employ active, ATP-dependent mechanisms to overcome passive barriers.
MDSC are induced in a variety of pathological settings and have even been shown to prevent allograft rejection.19–22 A recent study showed that inhibiting GM-CSF production by tumour cells reduced MDSC and inhibited the ability to establish PDA-derived allografts.18 It is difficult, however, to draw definitive conclusions about PDA immunobiology from transplantable tumours, particularly allografts differing in histocompatibility antigens. Moreover, those studies did not distinguish among MDSC subpopulations. As shown here, depletion of Gr-MDSC in advanced stages of invasive autochthonous PDA is sufficient to induce epithelial tumour cell apoptosis and stromal remodelling. Our observation of increased systemic activation of CD8 T cells in this setting further supports the hypothesis that Gr-MDSC may limit the priming and/or boosting of tumour-reactive T cells in secondary lymphoid organs. Additionally, intratumoral MDSC may limit T cell accumulation in PDA by inducing apoptosis of the tumour-infiltrating T cells.
The remodelling of the stroma was an unexpected consequence of Gr-MDSC depletion and may result from the killing of tumour epithelial cells by activated T cells. Tumour epithelial cells can contribute to stromal architecture and composition through the direct secretion of ECM components and via paracrine signalling to myofibroblasts.45 Although not our primary intent when investigating immunomodulatory strategies to render PDA more permissive to T cell activity, Gr-MDSC depletion might have the added benefit of improving chemotherapy access into tumours by increasing tumour vessel diameters. However, combining immunomodulatory strategies to stimulate T cell function with cytotoxic agents could be challenging and even counterproductive, as the latter might undo the effects of the former by killing newly proliferative T cells.
The targeted depletion of Gr-MDSC was associated with a corresponding increase in a monocytic subset (Mo-MDSC) with comparable in vitro suppressive activity. It is somewhat surprising, therefore, that endogenous T cells are activated and infiltrate PDA despite the compensatory rise in Mo-MDSC. Mo-MDSC may be unable to substitute fully for Gr-MDSC to confer an equivalent immunosuppressive environment in vivo; however, it is also possible that targeted depletion of Gr-MDSC provides a ‘window of opportunity’ to induce an activated CD8 T cell response.
The human corollary of the murine Gr-MDSC described here are the CD11b+ CD33int Lin−/low cells that are increased in patients with PDA.46–48 Additionally, the granulocytic markers CD15 and CD66b have also been used to define Gr-MDSC in human malignancies.48–50 Thus, targeting this subpopulation of cells might be an attractive therapeutic strategy. Some caution with this approach is warranted, however, as the monocytic subset of MDSC (CD11b+ CD33high CD14+/dull HLA-DRneg/low) has also been defined in a variety of human malignancies,46 ,47 ,51 and we have shown here that targeting Gr-MDSC can induce a corresponding expansion of Mo-MDSC. A recent study found that the percentage of circulating inflammatory monocytes, which share many of the same markers as Mo-MDSC in both mice and humans, correlated with a higher incidence of lymph node metastasis in patients with PDA and inversely with patient survival.52 Therefore, additional studies are necessary to elucidate the ideal approach to modulate the highly plastic and interdependent cancer-conditioned myeloid cell subsets, and the success of such strategies may be cancer subtype-specific.
A number of mechanisms may be exploited to disrupt the immunosuppressive capability of Gr-MDSC, including targeting their induction from progenitor cells, preventing trafficking into tumours, preventing their accumulation, inhibiting their immunosuppressive activity or inducing their maturation (eg, into cells that promote T cell activation).53 Since the phenotypically defined myeloid subsets may reflect a continuum of cellular differentiation governed by homeostatic regulation, it might be possible to shift the respective balance to advantage. As an example, vaccination in combination with the administration of all-trans-retinoic acid, which can have both immunogenic and inhibitory effects including inducing immature myeloid cells to differentiate into mature dendritic cells, was shown to promote the development of functional tumour antigen-specific T cells in patients with small cell lung cancer.54
That tumour-produced GM-CSF and G-CSF may figure prominently in the induction and survival of MDSC suggests that a reassessment of the use of this cytokine as part of cancer treatment might be warranted. Providing growth factor support is common in oncology practice as a means of restoring or bolstering haematopoiesis in patients receiving cytotoxic chemotherapies.55 Some of these agents are also used as adjuvants in many antitumour vaccine strategies.16 Of course, growth factors can themselves exert pleiotropic effects on immunity and tumour biology, and these effects depend both on the specific complement of cells in question and the stage of disease progression. The potential role of GM-CSF in facilitating the invasive and metastatic programme in PDA suggests that it might be prudent to re-examine its use in at least certain cancer treatments.
The limitations encountered thus far in applying immunomodulatory strategies such as αCTLA4 and αPD1/PDL1 in PDA to stimulate an endogenous T cell response may be the result of the profoundly suppressive effects of MDSC. Combining targeted disruption of MDSC with additional immune-based strategies might help to harness the full potential of the immune system to recognise and eradicate malignancies. Adoptive T cell technologies employed with a platform to abrogate MDSC function may prove especially potent in PDA and other solid tumours.
Acknowledgments
We thank members of FHCRC Comparative Medicine and Experimental Histopathology Shared Resources for assistance, Paolo Provenzano for assistance with analysing expression data, Michael Ports for help with preliminary experiments, Ashley Dotson for assistance with animal husbandry and care, Shelley Thorsen for expert assistance with manuscript and figure preparation and John Potter for helpful discussions and comments on the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online table
Footnotes
-
Contributors IMS, SB, KI, MAC, CC and RMS performed the experiments; IMS, PDG and SRH designed the study and wrote the manuscript.
-
Funding This work was supported by the National Cancer Institute grants CA161112 and CA114028 to SRH and support to SRH from the Giles W and Elise G Mead Foundation and the Safeway Foundation, National Cancer Institute grant CA033084 to PDG and the Fred Hutchinson Cancer Research Center/University of Washington Cancer Consortium Cancer Center Support Grant CA015704 (to PDG and SRH). The Irvington Institute Fellowship Program of the Cancer Research Institute and the Jack and Sylvia Paul Fund to support collaborative immunotherapy research provided support for IMS.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.