Article Text

Abstract
Objective Chemokine pathways are co-opted by pancreatic adenocarcinoma (PDAC) to facilitate myeloid cell recruitment from the bone marrow to establish an immunosuppressive tumour microenvironment (TME). Targeting tumour-associated CXCR2+neutrophils (TAN) or tumour-associated CCR2+ macrophages (TAM) alone improves antitumour immunity in preclinical models. However, a compensatory influx of an alternative myeloid subset may result in a persistent immunosuppressive TME and promote therapeutic resistance. Here, we show CCR2 and CXCR2 combined blockade reduces total tumour-infiltrating myeloids, promoting a more robust antitumour immune response in PDAC compared with either strategy alone.
Methods Blood, bone marrow and tumours were analysed from PDAC patients and controls. Treatment response and correlative studies were performed in mice with established orthotopic PDAC tumours treated with a small molecule CCR2 inhibitor (CCR2i) and CXCR2 inhibitor (CXCR2i), alone and in combination with chemotherapy.
Results A systemic increase in CXCR2+ TAN correlates with poor prognosis in PDAC, and patients receiving CCR2i showed increased tumour-infiltrating CXCR2+ TAN following treatment. In an orthotopic PDAC model, CXCR2 blockade prevented neutrophil mobilisation from the circulation and augmented chemotherapeutic efficacy. However, depletion of either CXCR2+ TAN or CCR2+ TAM resulted in a compensatory response of the alternative myeloid subset, recapitulating human disease. This was overcome by combined CCR2i and CXCR2i, which augmented antitumour immunity and improved response to FOLFIRINOX chemotherapy.
Conclusion Dual targeting of CCR2+ TAM and CXCR2+ TAN improves antitumour immunity and chemotherapeutic response in PDAC compared with either strategy alone.
- pancreatic cancer
- cytokines
- immunoregulation
- inflammatory mechanisms
- immune response
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Bone marrow-derived myeloid cells, including tumour-associated neutrophils (TAN) and tumour-associated macrophages (TAM) are recruited by pancreatic adenocarcinoma (PDAC) and facilitate immune evasion.
PDAC production of CCL2 promotes CCR2+ monocyte efflux from the bone marrow to the tumour microenvironment, where they become immunosuppressive TAM.
Treatment with a small molecule CCR2 inhibitor, in combination with FOLFIRINOX, prevented TAM accumulation and demonstrated a promising treatment response rate in PDAC compared with chemotherapy alone.
The ELR+/CXCR2 chemokine axis has been implicated in PDAC-mediated recruitment of TAN, and CXCR2 blockade has shown promise in preclinical tumour studies.
What are the new findings?
Treatment with a CCR2 inhibitor results in a compensatory influx of CXCR2+ TAN in patients with human PDAC.
A systemic increase in CXCR2+ neutrophils correlates with poor prognosis in PDAC.
CXCR2 blockade, both alone and in combination with FOLFIRINOX chemotherapy, prevents TAN mobilisation from the peripheral blood and increases effector tumour-infiltrating T cells in an orthotopic PDAC model.
Myeloid substitution is recapitulated in an orthotopic PDAC model, and combining CCR2 plus CXCR2 blockade reduces total tumour-infiltrating myeloid cells, enhancing antitumour immunity and chemotherapeutic responses compared with either therapy alone.
How might it impact on clinical practice in the foreseeable future?
Targeting the predominant tumour-infiltrating myeloid lineages in combination may represent an optimal approach to reverse the immunosuppressive TME and enhance chemotherapeutic responses in PDAC.
Introduction
Pancreatic adenocarcinoma (PDAC) is the fourth leading cause of cancer-related deaths, with incidence expected to increase over the coming decade.1 2 Despite advances using conventional chemotherapy in PDAC, 5-year survival remains a dismal 8%, and the immunosuppressive tumour microenvironment (TME) remains a significant barrier to effective treatment. This fibrotic stromal compartment is infiltrated by tumour-associated macrophages (TAM) and tumour-associated neutrophils (TAN) that are recruited from the bone marrow to facilitate PDAC immune evasion.3 4 Thus, therapies disrupting myeloid trafficking represent an attractive modality to restore antitumour immunity.
Neutrophils are the most abundant myeloid population, and a systemic granulocytic expansion has been reported in cancer, with intratumoural accumulation of TAN (CD11b+, CD14−, CD15+ and CD66b+) correlating with poor prognosis.5 6 CXCR2 and its ligands in the ELR+ chemokine family, including CXCL1, CXCL2, CXCL3, CXCL5, CXCL7 and CXCL8, are responsible for recruiting neutrophils under normal physiological conditions and have been implicated in tumour-mediated TAN mobilisation.7 8 Tumour-bearing animals also demonstrate a systemic increase in TAN (CD11b+, Gr1High, Ly6CLow and Ly6GHigh), and targeting TAN has been shown to increase tumour-infiltrating lymphocytes (TIL) and improve response to checkpoint blockade in preclinical studies.9–13
Our previous work found PDAC expression of CCL2 results in mobilisation of CCR2+ inflammatory monocytes from the bone marrow to the tumour, where they become immunosuppressive TAM.14 A dense TAM (CD11b+, CD115+, CD14+, CD15− and CD68+) infiltrate has been associated with decreased survival in multiple malignancies and targeting CCR2+ TAM (CD11b+, Gr1Low, Ly6Glow, Ly6CMid and F4/80High) reduces tumour progression in murine cancer models.14–18 Based on these findings, we conducted a clinical trial (NCT01413022) in patients with non-metastatic PDAC using the orally dosed, small molecule CCR2 inhibitor (CCR2i) PF-04136309 in combination with FOLFIRINOX that demonstrated a favourable clinical response rate compared with chemotherapy alone.19 However, tumour-mediated recruitment of bone marrow-derived cells is an elaborate process, and disruption by immunomodulatory therapies is not without a compensatory response. Several studies have found that depleting TAM results in a compensatory TAN influx, which may contribute to treatment failure following CCR2 blockade.20 21 Thus, it remains unclear if disrupting a single myeloid subset is the most effective approach to targeting the immunosuppressive TME.
Here we report that targeting CCR2+ TAM results in a compensatory neutrophil influx in patients with PDAC, providing the first reported evidence that immunomodulating therapies may result in dynamic myeloid responses at the primary tumour, with the potential to limit intended treatment efficacy. This phenomenon is recapitulated in tumour-bearing animals, and synergistic targeting of both CXCR2+ TAN and CCR2+ TAM prevented a compensatory influx of myeloid cells, along with evidence of a more robust antitumour immune response. These data build on emerging therapies targeting bone marrow-derived cells in human cancer and suggests that targeting both TAM and TAN collectively may represent the optimal therapeutic combination.
Materials and methods
Analysis of preoperative neutrophil to lymphocyte ratio (NLR)
An institutional review board (IRB)-approved, prospectively maintained database of PDAC resections (n=561) performed at Barnes Jewish Hospital (St. Louis, Missouri, USA) between 1997 and 2012 was used, and the NLR was calculated by dividing the absolute neutrophil and lymphocyte values from routine preoperative labs obtained within 30 days prior to the index pancreatectomy. Subjects with leucocytosis (>11 000 cells/dL) (n=51), without a differential count (n=64) or postoperative death within 30 days (n=7) were excluded. The NLR, as a continuous variable, was used as the test state and overall survival as the control variable in a standard receiver operator curve (ROC) to determine the optimal cut-off point for patient stratification.
Collection of blood, bone marrow and tumour from human patients
Human studies were conducted with informed consent under IRB-approved protocol at Washington University School of Medicine. Unfractionated whole blood and bone marrow aspirates were collected from patients with PDAC and healthy controls in EDTA vacuum tubes (BD Bioscience) and underwent red blood cell (RBC) lysis per manufacturer’s instructions (BioLegend). Resected tumour or normal pancreas from organ donors without malignancy were processed at time of collection. Baseline and post-treatment tumour biopsies from a clinical trial were processed as previously described.19 Tissue was allocated for cryopreservation, fixation or enzymatically digested to create a single cell suspension using GentleMACS (Miltenyi).
PDAC tumour tissue microarray (TMA)
A TMA of treatment-naive PDAC tumours was constructed from 1 mm punch biopsies of well-demarcated tumours from paraffin-embedded tissue blocks by a pathologist. An Aperio Scan-Scope XT Slide Scanner (Aperio Technologies) was used to acquire 20× images. A previously validated tumour-specific nuclear algorithm (IHC-MARK) was used to quantify protein expression.14 22 Positive staining was normalised to total tissue surface area and the mean intensity or cellular density per cm2 was used to classify samples into high, mid and low cohorts and overall survival following PDAC resection compared (online supplementary figure S1).
Supplementary file 1
Animal studies and cell lines
C57BL/6, CCR2−/− (B6.129S4-Ccr2tm1Ifc/J) and Nur77 GFP (Nur77-GFPCre) mice were purchased from The Jackson Laboratory and maintained in a pathogen-free facility under an approved animal studies protocol at Washington University School of Medicine (St. Louis, Missouri, USA). Mice were observed daily, and survival events were recorded when animals lost >15% total body weight or per absolute survival event. The murine PDAC cell line KCKO was the kind gift of Dr Pinku Mukherjee.23 KPC cells derived from p48-CRE/LSL-Kras/p53flox/flox mice were provided by Dr David DeNardo. All cell lines tested negative for mycoplasma and verified to be of C57BL/6J origin using CellCheck Plus (IDEXX BioResearch). Mice aged 8–10 weeks were anaesthetised and injected in the tail of the pancreas with 2.5×105 cells suspended in a 1:1 PBS to Matrigel (Corning) mixture and randomised to treatment 96 hours following tumour inoculation. The CXCR2 inhibitor (CXCR2i) SB225002 (Tocris) was administered intraperitoneally (IP) at a dose of 10 mg/kg daily. The CCR2i RS504393 (Tocris) was given subcutaneously (SQ) at a dose of 5 mg/kg twice per day or PF-04136309 (Pfizer) at a dose of 100 mg/kg given SQ twice daily. FOLFIRINOX (5-FU (25 mg/kg), irinotecan (50 mg/kg) and oxaliplatin (5 mg/kg)) was administered intravenously weekly. For depletion experiments, 250 µg of Ly6G (A18) or CD8 (2.43) antibodies was administered IP every 4 days (BioXCell). Treatment was continued until designated time points, at which time mice were euthanised and perfused. Tumours were collected along with blood and bone marrow in heparinised capillary tubes followed by RBC lysis (BioLegend).
Flow cytometry
Single cell suspensions were assessed for viability with Trypan Blue and manually counted, then incubated with Fc receptor blocking solution followed by fluorophore-conjugated antibodies (BioLegend). For intracellular staining, cells were permeabilised and stained with appropriate antibody per manufacturer’s instructions (eBioscience). Flow cytometry was performed on an LSRII (BD Biosciences) and analysed using FloJo Version X (Tree Star). See online supplementary materials for list of antibodies.
RNA isolation and PCR
Snap-frozen samples were homogenised in Trizol (Thermo Scientific) with a Tissue Lyser LT (Qiagen). Total RNA was extracted with the RNeasy Mini Kit and treated with DNase (Qiagen). RNA was reverse transcribed into cDNA and quantitative real-time PCR performed using TaqMan PCR Master Mix and predesigned TaqMan Gene Expression Assays (Thermo Scientific). Gene expression was normalised to lyceraldehyde-3-phosphate dehydrogenase (GAPDH), hypoxanthine phosphoribosyltransferase (HPRT1) and β-actin using the comparative CT (ΔΔCT) method.
Immunofluorescence and immunohistochemistry
Five micrometre tumour sections were cut from embedded tissue using an HM 325 microtome (Thermo Scientific) and baked at 70°C for 30 min. Tissue sections were dewaxed in xylene followed by rehydration and antigen retrieval using citrate buffer (10 mM sodium citrate, 0.05% Tween 20), then washed and blocked with serum-free protein block (DAKO). Primary antibodies were diluted in antibody diluent (DAKO) and incubated overnight at 4°C then washed and stained with fluorescently conjugated secondary antibodies diluted 1:200 for 30 min at room temperature. For immunohistochemistry, endogenous peroxidase activity was blocked by incubation with 3% H2O2 in PBS, followed by primary and biotinylated secondary antibodies diluted 1:200. Antigen staining with VECTASTAIN Standard ABC Kit (Vector Laboratories) and Liquid DAB+ Substrate Chromagen System (DAKO) was performed, and slides were counterstained with Gill’s Hematoxylin (Fisher Scientific). Images were acquired on an Olympus BX43 microscope with a DP80 camera and analysed with manufacture’s software (Olympus).
Statistical analysis
SAS V.9.1 and GraphPad Prism V.4 (GraphPad Software) were used for statistical analysis using a significance level of α=0.05. A two-sided Student’s t-test or Mann-Whitney test was used for normally and non-Gaussian distributed data, respectively. Matched samples were compared with a two-sided paired t-test. A one-way analysis of variance using the Tukey post-test was used for multiple comparisons. Survival analysis was performed using the log-rank test.
Results
Preoperative NLR correlates with survival following PDAC resection
Using standard ROC analysis the optimal NLR cut-off was determined to be 2.1, with patients (n=439) stratified into high (>2.1) and low (≤2.1) cohorts. Overall, a high NLR (n=286) was associated with a worse prognosis and 5-year survival of 17.3% compared with 26.5% in the low NLR group (n=153) following PDAC resection (figure 1A). Patients with a high NLR had a median survival of 18 months compared with 23 months in the low NLR group (HR=0.74 (95% CI 0.58 to 0.93)). Multiple logistic regression using baseline patient and tumour characteristics reveals that an elevated preoperative NLR is independently associated with a shorter mean survival (online supplementary table S1).

Systemic elevation of CXCR2+ neutrophils in the peripheral blood and bone marrow correlates with overall survival in patients with human PDAC. (A) Standard ROC analysis (P<0.0001; AUC=0.74 (95% CI 0.71 to 0.78)) was used to ascertain the preoperative neutrophil to lymphocyte ratio (NLR) optimal cut-off point of 2.1 with a sensitivity of 66.2% (95% CI 61.2 to 70.6) and specificity of 73.2% (95% CI 68.9 to 77.3). All patients (n=439) were stratified into high (>2.1; n=286) and low (≤2.1; n=153) cohorts and a Kaplan-Meier survival analysis following PDAC resection was performed, with alive patients censored at date of last follow-up. (B) Representative flow cytometry plots gated on CD45+ cells depicting CD15+, CXCR2+ neutrophils in the peripheral blood (top) and bone marrow (bottom) from healthy controls (left) and patients with PDAC (right). (C) Analysis of CXCR2+ neutrophils in the peripheral blood of healthy controls (n=6) and patients with PDAC (n=17) represented as a percentage of total cells (left). Comparison of CXCR2+ neutrophils in the peripheral blood of patients with human PDAC surviving (n=11) or deceased (n=6) at 1 year following surgical resection (middle). Pearson correlation (two tailed) of peripheral blood CXCR2+ neutrophils and survival following surgical resection (right). (D) Analysis of CXCR2+ neutrophils in the bone marrow of healthy controls (n=5) and patients PDAC (n=24) represented as a percentage of total cells (left). Comparison of CXCR2+ neutrophils in the bone marrow of patients with human PDAC surviving (n=18) or deceased (n=6) at 1 year following surgical resection (middle). Pearson correlation (two tailed) of bone marrow CXCR2+ neutrophils and survival following surgical resections (right). Two-sided Mann-Whitney test was used to determine P values for comparisons between groups. AUC, area under the curve; PDAC, pancreatic adenocarcinoma; ROC, receiver operator curve.
Neutrophils expressing CXCR2 are elevated in the blood and bone marrow of patients with human PDAC
Flow cytometry analysis reveals that neutrophils in both the blood and bone marrow express CXCR2 (figure 1B). Patients with PDAC demonstrated a significant increase in CXCR2+ neutrophils in the blood compared with healthy controls (figure 1C). The circulating CXCR2+ neutrophil burden was higher in patients surviving less than 1 year and inversely correlated with survival (P=0.019; Pearson’s rho=−0.66 (95% CI −0.90 to −0.14)). Likewise, patients with PDAC had increased CXCR2+ neutrophils in bone marrow compared with controls (figure 1D). Similar to the peripheral blood, this increase was associated with worse 1-year survival and the percentage of bone marrow CXCR2+ neutrophils inversely correlated with survival (P=0.014; Pearson’s rho=−0.50 (95% CI −0.76 to −0.11)). This indicates that PDAC results in a systemic elevation of CXCR2+ neutrophils in the blood and bone marrow, correlating with worse clinical outcomes.
Increased expression of ELR+ chemokines correlates with the prevalence of tumour-infiltrating CXCR2+ TAN
Consistent with prior reports, we found that human PDAC tumours overexpress GM-CSF and G-CSF, but not M-CSF, relative to normal pancreas (figure 2A).24 25 PDAC tumours had increased expression of the ELR+ chemokines CXCL1, CXCL2, CXCL5 and CXCL8 compared with normal tissue (figure 2B). Fluorescent microscopy shows neoplastic ductal cells, rather than stromal elements, to be the primary source of CXCL1, CXCL2, CXCL5 and CXCL8 (figure 2C). In particular, we focused on CXCL5 as it demonstrated the greatest fold increase in PDAC expression among the ELR+ chemokines and found a positive correlation between CXCL5 expression and both tumour-infiltrating CD15+ granulocytes (Pearson rho=0.837 (95% CI 0.73 to 0.90); figure 2D) and neutrophil elastase positive (NE+) granulocytes (Pearson rho=0.635 (95% CI 0.43 to 0.70); figure 2E). Overall, this suggests that PDAC expression of ELR+ chemokines, in particular CXCL5, may be important in facilitating recruitment of CXCR2+neutrophils.
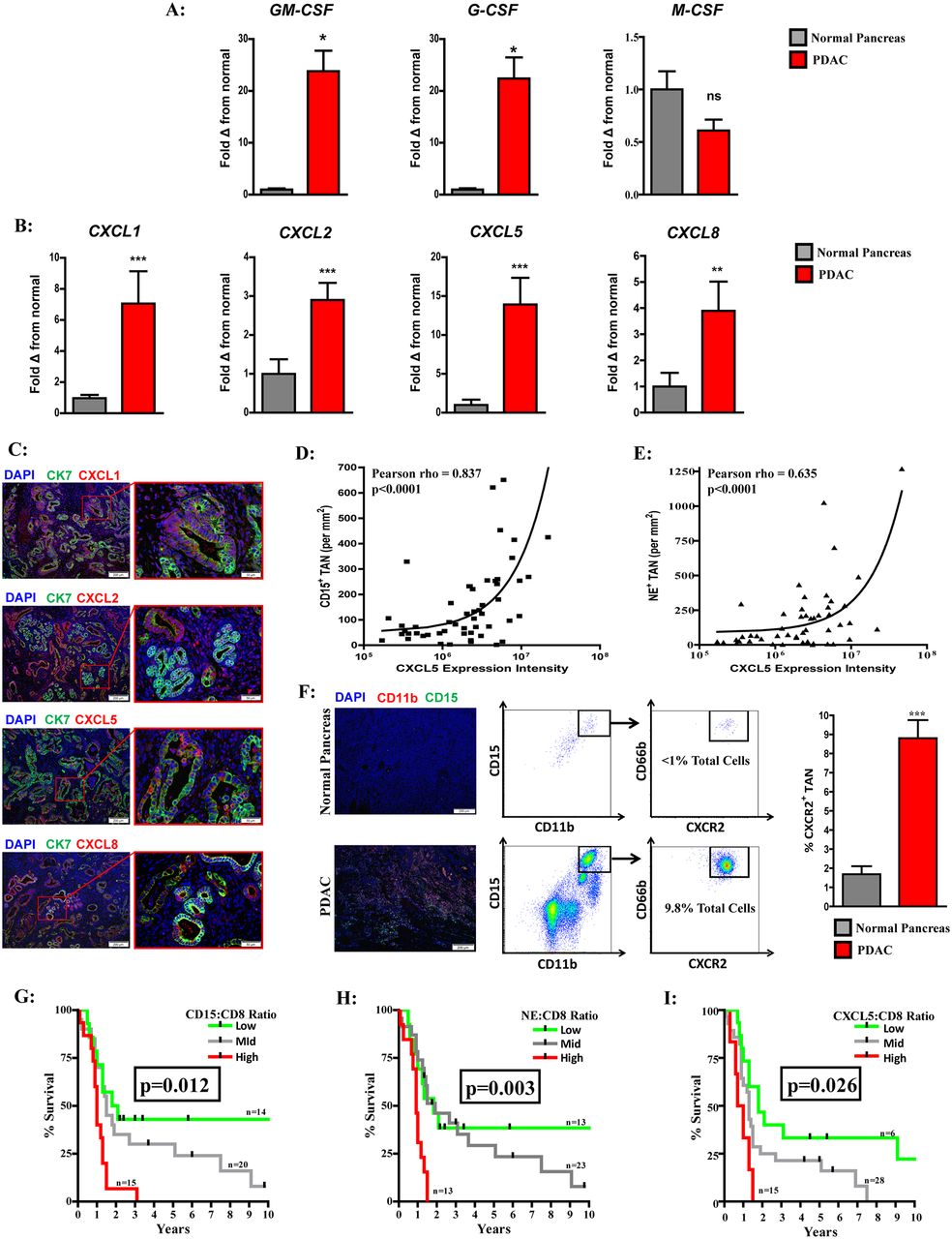
Elevated ELR+ chemokine expression correlates with CXCR2+TAN and worse prognosis in human PDAC. (A) Quantitative real-time PCR analysis of GM-CSF, M-CSF and G-CSF expression from human PDAC tumours (n=11) relative to normal pancreas (n=7). (B) Quantitative real-time PCR analysis of the ELR+ chemokine CXCL1, CXCL2, CXCL5 and CXCL8 expression from human PDAC tumours (n=11) relative to normal pancreas (n=7). (C) Fluorescent microscopy images demonstrating CXCL1, CXCL2, CXCL5 and CXCL8 (red) expression with DAPI nuclear (blue) and cytokeratin 7 (CK7; green) counterstain in human PDAC tumours. (D) Pearson (two tailed) correlation of CXCL5 expression intensity and CD15+ TAN infiltrate in human PDAC tumours (n=54). (E) Pearson correlation (two tailed) of CXCL5 expression intensity and NE+ TAN infiltrate in human PDAC tumours (n=54). (F) Representative immunofluorescence showing CD11b+, CD15+ TAN (yellow) in normal pancreas (top) and PDAC tumours (bottom). Representative flow cytometry plots illustrating CD11b+, CD15+, CD66b+, CXCR2+ TAN in normal pancreas (top) and PDAC tumour (bottom) gated on CD45+ cells. Graph depicts CXCR2+ TAN as percentage of total cells by flow cytometry from normal pancreas (n=7) and human PDAC tumours (n=11). (G) Survival analysis of intratumoural CD15+ to CD8+ ratio from resected human PDAC tumour microarray (n=49). (H) Survival analysis of intratumoural NE+ to CD8+ ratio from resected human PDAC tissue microarray (n=49). (I) Survival analysis of intratumoural CXCL5 to CD8+ ratio from resected human PDAC tissue microarray (n=49). Two-tailed unpaired t-test or Kaplan-Meier survival analysis was used to calculate P values. *P<0.05; **P<0.01; ***P<0.001. G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; M-CSF, macrophage colony-stimulating factor; PDAC, pancreatic adenocarcinoma; TAN, tumour-associated neutrophils.
PDAC tumours are infiltrated by CXCR2+ TAN with prognostic implications in human disease
Compared with normal pancreas, PDAC is heavily infiltrated by CXCR2+ TAN (figure 2F). Similar to our finding that the NLR was prognostic in the peripheral blood, we found PDAC tumours with a low, mid or high CD15+ TAN to CD8+ lymphocyte ratio had median survival of 2, 1.5 and 1 years, respectively (figure 2G). A high ratio was associated with worse overall survival compared with both the mid (P=0.009; HR=0.44 (95% CI 0.14 to 0.76)) and low (P=0.006; HR=0.35 (95% CI 0.12 to 0.70)) groups. Furthermore, PDAC patients with a high NE to CD8 intratumoural ratio demonstrated decreased survival compared with those in either the mid (P=0.008; HR=0.35 (95% CI 0.07 to 0.51)) or high (P=0.006; HR=0.34 (95% CI 0.10 to 0.68)) cohorts (figure 2H). Consistent with our finding that CXCL5 expression correlated with TAN infiltrate, an elevated CXCL5 to CD8+ intratumoural ratio was also associated with a worse prognosis, with a median survival of 0.65 years compared with the 1.3 years (P=0.019; HR=0.39 (95% CI 0.05 to 0.75)) or 1.8 years (p=0.005; HR=0.47 (95% CI 0.03 to 0.51)) observed in the mid and low groups, respectively (figure 2I). In summary, this suggests that the CXCR2+ TAN are a predominant immune infiltrate and have clinical significance in PDAC.
CXCR2 inhibition sequesters neutrophils in the blood and prevents TAN accumulation in orthotopic PDAC tumours
Orthotopically implanted, syngenic KCKO pancreatic tumours demonstrated increased expression of GM-CSF, G-CSF and the CXCR2 ligands CXCL1, CXCL2 and CXCL5 relative to normal pancreas (figure 3A and figure 3B). In addition, we found a systemic elevation of CXCR2+ neutrophils in the bone marrow and peripheral blood of tumour-bearing mice relative to controls (figure 3C). CXCR2i treatment increased the absolute number of neutrophils in the periphery, but not the bone marrow, compared with vehicle treated animals. In addition, we explored the impact of FOLFIRINOX, which also demonstrated an increase in circulating, but not bone marrow, granulocytes following CXCR2i plus myelosuppressive chemotherapy (online supplementary figure S2).

Targeting CXCR2 sequesters neutrophils in the peripheral blood and prevents TAN accumulation in PDAC tumour-bearing animals. (A) Quantitative real-time PCR was performed for expression of GM-CSF, M-CSF and G-CSF on normal murine pancreas (n=5) and orthotopically implanted KCKO PDAC tumours (n=9). (B) Quantitative real-time PCR was performed for expression of ƩCXC+ chemokines on normal murine pancreas (n=5) and orthotopically implanted PDAC tumours (n=9). (C) Representative flow cytometry plots of CXCR2+ neutrophils in the peripheral blood (top) and bone marrow (bottom) from non-tumour controls and tumour-bearing animals receiving treatment with vehicle or CXCR2i. Graphs depict absolute number of CXCR2+ neutrophils (×105) per 100 µL of blood (n=9–12 per group) and bone marrow from the femur (n=4 per group) of non-tumour-bearing controls and KCKO tumour-bearing animals receiving treatment with vehicle or CXCR2i (D) Representative IHC and analysis of Ly6G+ TAN from orthotopic PDAC tumours of animals treated with vehicle or CXCR2i (n=6–9 per group). (E) Tumour growth curve of subcutaneously implanted KCKO tumours from mice treated with vehicle or CXCR2i. Treatment initiated following randomisation of palpable tumours on postimplantation day 10. (F) Tumour weights from established orthotopic KPC tumours treated with CXCR2i or Ly6G specific depleting antibody for 20 days (n=6 per group). Data reflect analysis from at least two repeated experiments. Two-tailed unpaired t-test or ANOVA was used to calculate P values. *P<0.05; **P<0.01; ***P<0.001. ANOVA, analysis of variance; PDAC, pancreatic adenocarcinoma; TAN, tumour-associated neutrophils.
The primary tumour was characterised by an abundant infiltrate of polynuclear Ly6G+ cells, which was abrogated with CXCR2 inhibition (figure 3D). Targeting CXCR2+ TAN resulted in a significant reduction in subcutaneous KCKO tumour progression (figure 3E). To confirm these findings, we examined orthotopic tumours from another syngenic PDAC cell line derived from the KPC genetically engineered model (p48-CRE/LSL-KrasG12D/p53flox/flox), revealing CXCR2i also prevented TAN accumulation in this model (online supplementary figure S3). Furthermore, treatment with either CXCR2i or a Ly6G-specific depleting antibody (A18) demonstrated a similar reduction in established KPC disease burden compared with controls (figure 3F). Collectively, this suggests that CXCR2 blockade prevents neutrophil trafficking from the blood, reducing TAN with similar therapeutic efficacy to systemic depletion.
Targeting a myeloid subset results in tumour compensatory recruitment of alternative bone marrow-derived cell populations
Neutrophil substitution for CCR2+ TAM has been observed in tumour models, and targeting CCR2 has shown promising results in a clinical trial treating borderline resectable and locally advanced PDAC.19–21 Thus, we examined if CXCR2+ TAN compensation occurred in human patients by analysing baseline and post-treatment tumour biopsies following 4 weeks of treatment with CCR2i, which revealed a significant increase in CXCR2+ TAN following CCR2 blockade (figure 4A). Both tumour-bearing CCR2−/− and CCR2i-treated wild-type mice demonstrated a compensatory replacement of TAM by CXCR2+ granulocytes, which also occurred when treated in combination with FOLFIRINOX (figure 4B,C). Conversely, depletion of Ly6G+ neutrophils has been reported to result in elevated TAM in a spontaneous PDAC murine model, which is similar to our findings following CXCR2i treatment (figure 4C,D)(figure 4D).12 One possible explanation for this phenomenon is the observation that factors associated with TAM production and recruitment, namely CSF1 and CCL2, were increased following blockade of CXCR2+ TAN. Similarly, CCR2i resulted in an increased expression of CXCL1, CXCL2 and CXLC5 as well as G-CSF (online supplementary figure S4). Nonetheless, combined CCR2i plus CXCR2i treatment, both alone and in combination with FOLFIRINOX, prevented this compensatory effect and reduced tumour-infiltrating myeloid cells (figure 4E).

Human and murine PDAC tumours demonstrate myeloid substitution following bone marrow cell targeted therapies, which is overcome by combined CCR2 plus CXCR2 blockade. (A) Comparison total PDAC-infiltrating CXCR2+ TAN was assessed by flow cytometry from matched FNA tumour biopsies at baseline following treatment with an orally dosed, small molecule CCR2i (blue) in combination with FOLFIRINOX (n=6 matched specimens). (B) Absolute number of CXCR2+ TAN per gram of tissue (×105) was assessed by flow cytometry from CCR2−/− and wild-type mice treated with vehicle alone (left) or in combination with FOLFIRINOX (right) 25 days following orthotopic KCKO tumour implantation (n=5–6 mice/group). (C) Absolute number of CXCR2+ TAN (Left) and CCR2+ TAM (Right) per gram of tumour (×105) was assessed by flow cytometry following 25 days of treatment with chemokine blockade alone (n=12 mice/group) . (D) Absolute number of CXCR2+ TAN (Right) and CCR2+ TAM (Left) per gram of tumour (×105) was assessed by flow cytometry following 25 days of treatment with chemokine blockade in combination with FOLFIRINOX chemotherapy (n=9 mice/group). (E) Total tumour-infiltrating myeloids was assessed by flow cytometry and represented as percentage change from vehicle treated controls following chemokine receptor blockade alone and in combination with FOLFIRINOX (n=9–12 mice/group). (F) Orthotopic KCKO tumour weights following 25 days of treatment with chemokine receptor blockade alone (left; n=12 mice/group) and in combination with FOLFIRINOX chemotherapy (right; n=9 mice/group). (G) Survival analysis of KPC orthotopic tumour-bearing mice treated with FOLFIRINOX alone and in combination with PF-04136309 (CCR2i) or CXCR2i (n=10–15 mice per group). One-way ANOVA and two-tailed paired, for matched samples, or unpaired t-test were used to calculate P values. For survival analysis, P values were obtained by the log-rank (Mantel-Cox) test. *P<0.05; **P<0.01; ***P<0.001. ANOVA, analysis of variance; PDAC, pancreatic adenocarcinoma; TAM, tumour-associated macrophages; TAN, tumour-associated neutrophils.
Combined CCR2 plus CXCR2 blockade enhances chemotherapeutic efficacy and improves survival in tumour-bearing mice
While CCR2i and CXCR2i were effective at reducing established orthotopic tumour burden as single agents, using a dual CCR2 plus CXCR2 blockade strategy resulted in significantly smaller KCKO tumours (figure 4F). Furthermore, this reduction in tumour burden was enhanced when myeloid blockade was combined with FOLFIRINOX. Administration of PF-04136309, a small molecule CCR2i used in a human PDAC clinical trial, in combination with FOLFIRINOX, reveals that chemotherapy increased survival compared with vehicle treatment (P<0.001; median survival: 15 vs 19 days), but the addition of either CCR2i (P=0.013; median survival: 19 vs 25 days) or CXCR2i (P=0.013; median survival: 19 vs 24 days) further improved survival in mice with orthotopic tumours derived from the KPC genetically engineered model (figure 4G). Furthermore, dual CCR2 plus CXCR2 blockade with FOLFIRINOX increased survival compared with either CCR2i (P=0.016; median survival: 25 vs 33 days) or CXCR2i (P=0.015; median survival: 24 vs 33 days). Overall, this suggests that targeting myeloid cell recruitment with both CCR2 and CXCR2 simultaneously enhances chemotherapeutic responses compared with either strategy used alone.
Reduction of tumour-infiltrating myeloids increases effector T cells in PDAC-bearing mice
Targeting CCR2+ TAM and CXCR2+ TAN in combination resulted in a significantly greater influx of both CD8+ and CD4+ TIL compared with either strategy alone, which was also found when tested in combination with FOLFIRINOX (figure 5A,B). Examination of TIL subsets found that myeloid-targeting strategies decreased immunosuppressive FoxP3+ and CD25+ regulatory T cell infiltrate, with no further clear benefit found when CCR2i and CXCR2i were combined (figure 5C). Confirming our prior findings, fluorescent microscopy shows that CD8+ TIL are increased following myeloid blockade (online supplementary figure S5). Next, we assessed markers of CD8+ TIL activation status, which revealed an increase in intracellular IFNγ and CD44+, CD69+, CD62L− surface marker expression on CD8+ TIL following combined CCR2i plus CXCR2i treatment (figure 5D,E). To confirm that this increase in activation status faithfully reflected antigen recognition by the T cell receptor (TCR), we next examined the green fluorescent protein (GFP) expression in CD8+ lymphocytes extracted from PDAC tumours in Nur77 GFP reporter mice, which upregulate GFP in response to TCR stimulation.26 27 This demonstrated an increase in GFP expressing CD8+ TIL following dual CCR2i plus CXCR2i treatment (figure 5F). Overall, these results indicate that the corresponding reduction of tumour-infiltrating myeloid cells and influx of effector TIL following dual chemokine receptor inhibition may represent the ideal treatment strategy to augment chemotherapeutic responses in PDAC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Depleting PDAC-infiltrating CCR2+ macrophages and CXCR2+ neutrophils in combination improves antitumour T cell responses. (A) Absolute number of CD8+ TIL per gram of tumour (×105) was assessed by flow cytometry from orthotopically implanted KCKO tumours following 25 days of treatment with chemokine blockade alone (left; n=12 mice/group) and in combination with FOLFIRINOX chemotherapy (right; n=9 mice/group). (B) Absolute number of CD4+ TIL per gram of tumour (×105) was assessed by flow cytometry from orthotopically implanted KCKO tumours following 25 days of treatment with chemokine blockade alone (left; n=12 mice/group) and in combination with FOLFIRINOX chemotherapy (right; n=9 mice/group). (C) Regulatory T cells (FoxP3+ and CD25+) expressed as percentage of total CD4+ TIL was assessed by flow cytometry from orthotopically implanted KCKO tumours following 25 days of treatment with chemokine blockade alone (left; n=9–12 mice/group) and in combination with FOLFIRINOX chemotherapy (right; n=6 mice/group). (D) Intracellular IFNγ-positive CD8+ TIL expressed as percentage of total CD8+ T cells was assessed by flow cytometry from orthotopically implanted KCKO tumours following 25 days of treatment with chemokine blockade alone (left; n=6 mice/group) and in combination with FOLFIRINOX chemotherapy (right; n=6 mice/group). (E) CD69+, CD44+, CD62L−and activated CD8+ TIL expressed as percentage of total CD8+ TIL were assessed by flow cytometry from orthotopically implanted KCKO tumours following 25 days of treatment with chemokine blockade alone (left; n=9 mice/group) and in combination with FOLFIRINOX chemotherapy (right; n=6 mice/group). (F) GFP expressing CD8+ TIL assessed by flow cytometry from Nur77 GFP T cell report mice with KCKO tumours 21 days following treatment with chemokine receptor inhibitor (n=4–6 mice/group). (G) Intratumoural effector to suppressor cell ratio per gram of tumour from orthotopically implanted KCKO tumours following 25 days of treatment with chemokine blockade alone (left; n=9–12 mice/group) and in combination with FOLFIRINOX chemotherapy (right; n=9 mice/group). (H) Orthotopic KCKO tumour gene expression following 25 days of treatment with chemokine blockade alone (left; n=5 mice/group) and in combination with FOLFIRINOX chemotherapy (right; n=5 mice/group). (I) Orthotopic KCKO tumour weights following chemokine receptor inhibitor with CD8 depleting antibody or IgG control (n=5–6 mice/group). One-way ANOVA was used to calculate P values. *P<0.05; **P<0.01; ***P<0.001. ANOVA, analysis of variance; PDAC, pancreatic adenocarcinoma; TIL, tumour- infiltrating lymphocytes.
Dual CCR2 plus CXCR2 blockade restores antitumour immunity in established PDAC tumours
The effector to suppressor cell ratio in established, orthotopically implanted PDAC tumours was increased following treatment with either CCR2i or CXCR2i compared with controls (figure 5G). Nonetheless, simultaneous targeting of both CCR2+ TAM and CXCR2+ TAN further enhanced this effect, which was magnified when combined with FOLFIRINOX. This corresponded with reduced expression of immunosuppressive genes and increase in factors associated with antitumour immunity and cytolytic T cell responses (figure 5H). As our results suggest that antitumour activity following myeloid blockade is mediated through an adaptive immune mechanism, we performed CD8 depletion studies that showed the therapeutic efficacy of myeloid blockade is lost in the absence of CD8+ lymphocytes (figure 5I). In summary, these data highlight the critical importance of bone marrow-derived cells in mediating PDAC immune evasion and provides a clinical rationale for concurrent targeting of both CCR2+ TAM and CXCR2+ TAN.
Discussion
Herein we demonstrate that myeloid dynamics within the TME facilitates a compensatory increase in an alternative subset of bone marrow-derived cells following isolated depletion of either CCR2+ TAM or CXCR2+ TAN. Furthermore, we provide the first evidence of this phenomenon occurring in human cancer, with post-treatment tumour biopsies from patients with non-metastatic PDAC receiving a small molecule CCR2i plus FOLFIRINOX demonstrating a significant increase in TAN compared with matched baseline samples. This increase may have clinical implications as we found that CXCR2+ neutrophils are elevated in the blood, bone marrow and tumour of patients with human PDAC and correlates with poor overall survival. Likewise, tumour-bearing mice demonstrate an increase in CXCR2+ neutrophils following therapeutic targeting of CCR2+ TAM. Conversely, we also show that preventing CXCR2+ neutrophil mobilisation in PDAC results in an increase in TAM, and synergistic targeting of both CXCR2+ TAN and CCR2+ TAM improves antitumour immunity and reduces established tumour burden compared with either therapy alone. In summary, these data build on emerging therapies targeting myeloid cells in human cancer and suggests that targeting both TAN and TAM collectively may present the optimal therapeutic combination in combating the immunosuppressive microenvironment.
An emerging body of evidence supports the role of TAN in facilitating tumour immune evasion and progression.28 Patients with cancer demonstrate a systemic increase in neutrophils, and the NLR is a biomarker in several tumours, including PDAC, and elevated neutrophils in the circulation correlated with tumour-infiltrating TAN in patients with glioblastoma.29–33 Recruitment of CXCR2+ neutrophils via tumour production of ELR+ chemokines has been observed in multiple cancers, with CXCL5 correlating with increased TAN infiltrate and worse prognosis in hepatocellular carcinoma and PDAC.8 9 34 Orally dosed small molecule inhibitors of CXCR2 are currently in clinical development, with trials demonstrating evidence of target engagement and a favourable toxicity profile in non-malignant conditions, and targeting CXCR2+ TAN has demonstrated efficacy in preclinical tumour models.35 36 Jamieson et al found CXCR2 blockade prevented TAN migration to colorectal tumours and increased CD8+ effector TIL, reducing tumour progression. In rhabdomyosarcoma, Highfill et al found that CXCR2 blockade potentiated anti-PD1 immunotherapy in tumour-bearing mice.10 13 In pancreatic cancer, Stromnes et al 12 reported that TAN depletion with a Ly6G-specific antibody leads to enhanced CD8+ TIL influx and an increase in cancer cell apoptosis in KPC mice. Likewise, strategies using CXCR2 blockade have also been associated with an influx of TIL and augmented checkpoint therapy in PDAC.9 11 Collectively, both our work and that of others strongly suggests that targeting CXCR2-mediated mobilisation of neutrophils may present a promising therapy for cancer.
Immunosuppressive TAM are also abundant in cancer and correlate with poor prognosis.37 Strategies targeting TAM, including CCR2 and CSF1 receptor blockade, repolarise the immunosuppressive microenvironment, improve responses to chemotherapy and reduce metastasis in preclinical models.14 16–18 38 The translation of these therapies to the clinic has also demonstrated promise with our group previously reporting that PF-04136309, an orally dosed CCR2i, in combination with FOLFIRINOX, demonstrated a favourable clinical response rate compared with chemotherapy alone in patients with borderline resectable and locally advanced PDAC.19 Correlative studies from this clinical trial showed CCR2i reduced TAM and increased TIL, suggesting that CCR2 blockade favourably reprogrammes the immunosuppressive TME. Based on these findings, larger clinical trials exploring CCR2 blockade in metastatic PDAC in combination with gemcitabine plus nab-paclitaxel (NCT02732938) and FOLFIRINOX (NCT02345408) are currently in progress. Likewise, strategies targeting CSF1 receptor have shown evidence of TAM reduction and promising response rates in patients with tenosynovial giant cell tumours, suggesting such strategies may be beneficial in the treatment of a variety of human cancers.39 40
While TAM targeted therapeutics have demonstrated evidence of safety and tolerability in cancer, inhibition of neutrophil recruitment has been met with more resistance, particularly in combination with chemotherapy regimens associated with myelosuppression and neutropaenia. Yet, systemic neutrophilia correlates with poor clinical outcomes in cancer, with evidence that chemotherapy-induced neutropaenia is protective in multiple solid tumours.41–43 Furthermore, elevated neutrophils have been associated with resistance to conventional therapies.44 45 Kurihara et al 46 reported similar findings in advanced PDAC, where neutropaenia conveyed a survival benefit following gemcitabine treatment. Importantly, a study in ovarian cancer found the baseline neutrophil count to correlate with survival, but carboplatin dose-escalation based on nadir neutrophil counts did not improve clinical outcomes, suggesting that chemotherapy does not overcome tumour-mediated granulocyte expansion.47 We used the FOLFIRINOX regimen in this study as it has shown an improvement in survival in PDAC compared with gemcitabine but is associated with an increase in treatment-related toxicities, including neutropaenia.48 Our results indicate that conventional chemotherapy does not sufficiently target TAN and suggests that CXCR2 inhibition may represent a synergistic treatment combination.
Despite successes targeting a single myeloid subset, the dynamics of bone marrow-derived cell recruitment in response to such immunomodulatory strategies has not been fully evaluated. Sawanobori et al 21 revealed that CCR2−/− mice had an influx of tumour-infiltrating neutrophils in both Lewis lung carcinoma and B16 melanoma tumours. Pahler et al 20 have reported similar findings in cervical cancers from CCR2−/− mice, in which the neutrophil influx was responsible for treatment resistance. Neutrophil substitution for macrophages has also been reported to other sites of inflammation in CCR2−/− mice infected with Listeria monocytogenes or viral-induced arthritis.49 50 Conversely, there is evidence that neutrophil depletion results in a compensatory increase in TAM. Stromnes et al showed that targeted depletion of neutrophils resulted in a systemic accumulation of myelomonocytic cells in both the periphery and tumour in a genetically engineered model of PDAC, suggesting that tumour-infiltrating myeloid compensation may occur following therapies targeting TAN as well.12 Collectively, these findings demonstrate both the critical nature of tumour-mediated recruitment of bone marrow-derived cells and support the concept of tumour-infiltrating myeloid substitution to counteract therapeutic strategies targeting an isolated myeloid cell population. Thus, further understanding of the mechanisms behind this phenomenon and how it may contribute to treatment resistance is paramount in the clinical application of targeting myeloid cells in cancer.
The dynamic communication between the tumour and bone marrow demonstrates the importance of precisely defining the immune composition of the TME and recognition of the potential mechanisms that tumours may use to combat immunomodulatory therapies. Future studies should expand beyond the current isolation of selective targeting of either TAM or TAN and progress to multimodality strategies aimed at elucidating the complex interactions among all inflammatory cells within the TME. Importantly, the ability of tumours to recruit additional myeloid populations should be considered as a complementary target to enhance antitumour therapies.
Acknowledgments
The authors would first and foremost like to express their appreciation to the patients who participated for contributing to further the scientific understanding of this devastating disease. The authors would like to personally thank Lori Worley and Dr Steven M Strasberg for assistance with specimen collection as well as Dr Daniel Kreisel and Dr Timothy Fleming for providing editorial feedback on the manuscript. The small molecule CCR2 inhibitor PF-04136309 was generously supplied by Pfizer.
References
Footnotes
Contributors TMN, BAB, SPG and DCL designed the study. TMN, BAB, DRC, RZP, BJH, DES, RCJ, JY and AAP performed experiments and conducted analysis. RCF, WGH and DCL assisted in operative specimen collection. RCF, WEG, DGD, WGH and DCL provided funding support, assistance with experimental design and edited the manuscript. TMN wrote the manuscript with input from all authors.
Funding TMN, DRC, RZP and DES recognise support from the NIH T32 CA 009621 training grant in Surgical Oncology (awarded to WEG). BAB, BJH, JY, AAP and DCL acknowledge funding from the NIH 5R01CA168863 and the Pancreatic Cancer Action Network (PANCAN) Translational ResearchGrant (15-65-25 LINE). TMN, DRC, SPG and WGH also recognise support from the Pancreatic Action Network (16-65-Hawk) Translational Research Grant (16-65-Hawk). TMN, DRC, RCF, DGD, SPG and WGH recognise support from the Siteman Cancer Center Frontier Fund. TMN, BAB, DRC, WEG, DGD, DCL and WGH recognise funding from the NCI/NIH Specialized Programs of Research Excellence (SPORE) grant (1P50CA196510). Work supported in part by a research grant to DCL through the Pfizer/Washington University School of Medicine Biomedical Collaborative.
Competing interests None declared.
Ethics approval IRB.
Provenance and peer review Not commissioned; externally peer reviewed.