


Abstract
Guanylyl cyclases are a family of enzymes that catalyze the conversion of GTP to cGMP. The family comprises both membrane-bound and soluble isoforms that are expressed in nearly all cell types. They are regulated by diverse extracellular agonists that include peptide hormones, bacterial toxins, and free radicals, as well as intracellular molecules, such as calcium and adenine nucleotides. Stimulation of guanylyl cyclases and the resultant accumulation of cGMP regulates complex signaling cascades through immediate downstream effectors, including cGMP-dependent protein kinases, cGMP-regulated phosphodiesterases, and cyclic nucleotide-gated ion channels. Guanylyl cyclases and cGMP-mediated signaling cascades play a central role in the regulation of diverse (patho)physiological processes, including vascular smooth muscle motility, intestinal fluid and electrolyte homeostasis, and retinal phototransduction. Topics addressed in this review include the structure and chromosomal localization of the genes for guanylyl cyclases, structure and function of the members of the guanylyl cyclase family, molecular mechanisms regulating enzymatic activity, and molecular sequences coupling ligand binding to catalytic activity. A brief overview is presented of the downstream events controlled by guanylyl cyclases, including the effectors that are regulated by cGMP and the role that guanylyl cyclases play in cell physiology and pathophysiology.
I. Introduction
Guanylyl cyclases have evolved to synthesize cGMP in response to diverse signals, such as nitric oxide (NO),2 peptide ligands, and fluxes in intracellular Ca2+([Ca2+]i). These signals use specific guanylyl cyclase-coupled receptors and cofactors to initiate the conversion of the cytosolic purine nucleotide GTP to cGMP. Intracellular cGMP ([cGMP]i) regulates cellular physiology by activating protein kinases, directly gating specific ion channels, or altering intracellular cyclic nucleotide concentrations through regulation of phosphodiesterases (PDEs). The structure and function of the family of guanylyl cyclases, the molecular mechanisms regulating their activities, and the downstream effectors that underlie the physiology of cGMP-dependent processes are summarized in this review.
II. Guanylyl Cyclases
A. Molecular Biology
1. Identification of the Members of the Guanylyl Cyclase Family.
It was established by the mid-1970s that guanylyl cyclase activity was found in both the soluble and particulate fractions of most cells (Hardman and Sutherland, 1969; Ishikawa et al., 1969;Schultz et al., 1969; White and Aurbach, 1969), and that these activities were due to different proteins (Garbers and Gray, 1974;Kimura and Murad, 1974; Chrisman et al., 1975). However, only with the development of molecular cloning techniques more than a decade later could the breadth of this enzyme family be fully explored (Tables 1 and2). Purification of guanylyl cyclase from the cytosolic compartment revealed the soluble isoform was a heterodimer composed of α- and β-subunits. The β-subunit had a molecular mass of ∼70 kDa, whereas the α-subunit was reported to be 73 to 82 kDa (Gerzer et al., 1981c; Kamisaki et al., 1986). Soluble guanylyl cyclase (sGC) was purified to apparent homogeneity from bovine or rat lungs (Koesling et al., 1988, 1990; Nakane et al., 1988, 1990). Degenerate oligonucleotide probes based on the structure of purified subunits were used to screen cDNA libraries and thereby clone α1- and β1-subunits. The C-terminal region of both subunits had a high degree of sequence identity with cloned adenylyl and particulate guanylyl cyclases (pGCs), suggesting this was the catalytic domain. Sodium nitroprusside (SNP)-sensitive guanylyl cyclase activity was expressed when the cloned cDNAs for α1 and β1 were cotransfected into a heterologous cell system, but not when transfected individually (Harteneck et al., 1990;Nakane et al., 1990). These data demonstrated both subunits of sGC are required for basal and nitrovasodilator-stimulated catalytic activity.
Soluble guanylyl cyclase isoform chromosomal localization and tissue distribution
Particulate guanylyl cyclase isoforms, ligand and cofactor specificities, chromosomal localization, and tissue distribution
Studies of pGCs suggested a new paradigm for signal transduction. Sea urchin sperm is one of the richest sources of pGC. In echinoderms, peptides secreted by eggs activate pGC of sperm in a species-specific manner (Suzuki et al., 1984; Ramarao and Garbers, 1985). Moreover, radiolabeled egg peptides could be chemically cross-linked to a sperm cell surface protein of the same size as that recognized by antiserum against guanylyl cyclase (Shimomura et al., 1986). These observations suggested that pGC might also serve as a receptor for peptide ligands. While these studies were being conducted in the sea urchin, atrial natriuretic peptide (ANP) was demonstrated to activate guanylyl cyclase and to increase [cGMP]i in mammalian tissues (Hamet et al., 1984; Waldman et al., 1984; Winquist et al., 1984). Subsequently, ANP binding and guanylyl cyclase activity were copurified, strongly suggesting the two activities reside in a single molecule (Kuno et al., 1986; Paul et al., 1987; Shimonaka et al., 1987;Meloche et al., 1988). In 1988, pGC was first cloned from a sea urchin testis cDNA library using probes based on tryptic peptides obtained from the purified protein (Singh et al., 1988). This clone provided the necessary probe for isolating mammalian cDNAs encoding pGCs. The natriuretic peptide receptors, guanylyl cyclase A (GC-A) and B (GC-B), were the first pGCs cloned from mammalian tissues (Chang et al., 1989;Chinkers et al., 1989; Lowe et al., 1989; Schulz et al., 1989). The deduced primary sequences of the natriuretic peptide receptors predicted a protein with a single transmembrane domain that divides an extracellular ligand-binding domain from an intracellular domain. Deletion mutagenesis studies have demonstrated that the intracellular domain serves regulatory, dimerization, and catalytic functions (Chinkers and Garbers, 1989). This regulatory domain has sequence similarity with protein kinases, particularly the protein tyrosine kinases, which are also single transmembrane domain receptors (Singh et al., 1988). The sequences of the C-terminal catalytic domains are highly homologous to those of the α- and β-subunits of sGC and have limited identity with the two catalytic domains of adenylyl cyclases (Krupinski et al., 1989; Thorpe and Garbers, 1989).
Development of the polymerase chain reaction (PCR) facilitated the search for new members of the guanylyl cyclase family. Degenerate PCR primers based on conserved amino acid sequences in the catalytic domains of both sGCs and pGCs were used to preferentially amplify guanylyl, as opposed to adenylyl, cyclases and yielded sequences of a second α- and a second β-subunit of sGC and five unique pGC sequences (GC-C to GC-G) (Yuen et al., 1990; Harteneck et al., 1991). A third pair of sGC subunits, cloned by screening a human cDNA library with rat cDNA clones, is most likely the human ortholog of α1/β1 (Giuili et al., 1992). Guanylyl cyclase C (GC-C) is the receptor for the bacterial heat-stable enterotoxins (STs) (Schulz et al., 1990; de Sauvage et al., 1991), and for the endogenous peptides guanylin and uroguanylin (Currie et al., 1992; Hamra et al., 1993). The remaining cloned mammalian pGCs are orphan receptors without known extracellular ligands. Guanylyl cyclase D (GC-D) is expressed in the olfactory neuroepithelium in a zonal pattern resembling that of the seven-transmembrane domain odorant receptors (Fülle et al., 1995). Two other members of the sensory tissue subfamily of guanylyl cyclases, guanylyl cyclase E (GC-E, retGC-1) and guanylyl cyclase F (GC-F, retGC-2), are expressed in retina (Shyjan et al., 1992; Lowe et al., 1995; Yang et al., 1995). GC-E also is expressed in the pineal gland (Yang et al., 1995). Although these enzymes are orphan receptors, their extracellular domains are homologous to that of GC-D and share a similar arrangement of cysteine residues in the extracellular domain with the other pGCs. This suggests they may have an extracellular ligand, although the catalytic activity of the retinal cyclases is regulated by [Ca2+]ithrough guanylyl cyclase-activating proteins (GCAPs). The recently cloned GC-G most closely resembles the natriuretic peptide receptors, although it is not activated by natriuretic peptides (Schulz et al., 1998b). Apparently, the family of mammalian guanylyl cyclases is relatively small because low stringency library screening and degenerate PCR have not yielded an abundance of unique cDNAs. In contrast, Caenorhabditis elegans has approximately 30 genes encoding guanylyl cyclase-like sequences and is seemingly rich in cGMP-coupled pathways (Yu et al., 1997).
2. Structure and Location of Guanylyl Cyclase Genes.
The chromosomal loci of the genes encoding isoforms of guanylyl cyclase and their ligands have been mapped in the human and/or the mouse (Tables 1,2) and are unlinked and scattered throughout the genome, with notable exceptions. Thus, the genes encoding the natriuretic peptide ligands for GC-A, ANP and brain natriuretic peptide (BNP) are organized in tandem in both the human and the mouse (Huang et al., 1996; Tamura et al., 1996b). Similarly, guanylin and uroguanylin, the endogenous activators of GC-C, are encoded by closely linked genes (Whitaker et al., 1997). Retinal guanylyl cyclase activity is regulated by GCAPs, which are calcium-binding proteins. To date, three members of the GCAP family have been identified. GCAP1 and GCAP2 are found in a tail-to-tail arrangement on human chromosome 6, whereas GCAP3 is located on chromosome 3 (Subbaraya et al., 1994; Haeseleer et al., 1999).
The genes encoding human sGC subunits α3 (equivalent to α1) and β3 (equivalent to β1) have been mapped to chromosome 4q32 (Giuili et al., 1993). Because both subunits are required in a 1:1 stoichiometry for activity, their common chromosomal locus may imply a coordinated regulation of gene expression. The genes encoding α1 and β1 sGC subunits in the medaka fish are organized in tandem within a 34-kb span (Mikami et al., 1999). The activity of the 5′-upstream region of each of the medaka fish genes was analyzed using green fluorescent protein reporter constructs expressed in medaka embryos (Mikami et al., 1999). Although the α1 upstream region promoted expression of green fluorescent protein, the β1 5′ region was insufficient, suggesting expression of the α1- and β1-genes is coordinated. However, the α2-subunit, which also can form an active dimer in vitro with β1, is encoded by a gene on chromosome 11 (Yu et al., 1996). That α2- and β1-subunits dimerize under physiological conditions argues against the requirement for coordinated regulation of expression of α- and β-subunits (Russwurm et al., 1998).
The structure of several genes for pGC has been determined, and the organization of their domains is reflected in the conservation of the intron/exon arrangement. This arrangement is most highly conserved in the portion of the gene encoding the catalytic and kinase homology domains. The extracellular domains of the guanylyl cyclases are conserved among, but not between, subfamilies and the structure varies most in those parts of the genes. Genes for GC-A and -B are similar in size (16.5–17.5 kb) and structure, with 22 exons and virtually identical intron/exon boundaries (Yamaguchi et al., 1990; Rehemudula et al., 1999). However, the size of introns is not conserved between these genes. Similarly, the guanylyl cyclases in sensory tissue share a conserved gene structure and have only 20 exons (Yang et al., 1996). The gene for GC-C is much larger (>50 kb) than genes encoding the other guanylyl cyclases and has a unique intron/exon arrangement (S. Schulz, J. Park, and S. A. Waldman, unpublished data). The structures of the genes for sGC subunits have not yet been reported.
Little is known regarding the regulation of expression of the genes for guanylyl cyclase. The 5′ regulatory regions of genes that have been sequenced (GC-A, -C, -E) have no typical TATA box and an absent or inverted CAAT box. While consensus binding sites for many general transcription factors are present, the elements controlling tissue-specific expression are only now beginning to be explored. The GC-A gene promoter has at least three consensus binding sites for Sp1, a transcription factor that is implicated in the expression of a number of genes in the vasculature (Liang et al., 1999). Assays using electromobility shift and reporter gene techniques have demonstrated all three sites bind Sp1 and are essential for basal transcription of the GC-A gene (Liang et al., 1999). Expression of the gene for GC-A also is regulated by its ligand, ANP. Levels of GC-A mRNA were suppressed by ANP in a time- and concentration-dependent manner in cultured aortic smooth muscle cells (SMCs) and primary cultures of inner medullary collecting duct cells (Cao et al., 1995, 1998). A cell-permeable analog of cGMP also inhibited transcription of GC-A, suggesting the second messenger, rather than the natriuretic peptide, is responsible for modulating gene activity (Cao et al., 1995, 1998). The ANP/cGMP-responsive element in the promoter for GC-A has not been identified.
Whereas GC-A is expressed in a variety of cell types and in many tissues, expression of GC-C in the adult human appears to be confined to the intestinal epithelium and primary and metastatic colorectal cancers (Carrithers et al., 1996). In the marsupial North American opossum, a guanylyl cyclase-coupled ST receptor, possibly the opossum ortholog of GC-C, is expressed in epithelial cells of the kidney, liver, testis, trachea, and intestine (Forte et al., 1989; London et al., 1999). The mRNA for GC-C and binding of radiolabeled ST are detectable in neonatal and weanling mouse liver, and in fetal, neonatal, and regenerating rat liver (Laney et al., 1992, 1994;Scheving and Russell, 1996; Swenson et al., 1996). Although the sensitive reverse transcription-PCR technique has been used to amplify the mRNA for GC-C in a number of tissues, production of cGMP in response to ST has only been observed outside the intestine in rodent stomach and inner ear (Krause et al., 1997; London et al., 1997).
An initial characterization of the 5′ flanking region of the gene for GC-C, using reporter gene constructs, suggested intestine-specific transcriptional activity lies within the proximal 128 bp (Mann et al., 1996a). An analysis of this region, which is conserved between the human and the mouse, revealed potential binding sites for several transcription factors. Hepatocyte nuclear factor-4 (HNF-4) binds to a specific element in the proximal promoter for GC-C and stimulates expression of GC-C when transfected into a cell line that normally expresses neither GC-C nor HNF-4 (Swenson et al., 1999). Mutation of the HNF-4 binding site abolished activity of the promoter for GC-C in intestinal cells, demonstrating that HNF-4 is necessary for basal gene expression (Swenson et al., 1999).
Recent observations suggest the transcription factor Cdx2 mediates the intestine-specific expression of GC-C. Cdx2 is a member of the homeodomain family of transcription factors related to caudal, aDrosophila protein, and is required for the selective expression of several other genes in intestinal tissues (Traber and Silberg, 1996). Deletion, or mutation, of a Cdx2 consensus binding site in the proximal GC-C gene promoter reduced the activity of a reporter gene construct expressed in intestinal cells to the level observed in extraintestinal cells (Park et al., 2000).
3. Genetic Disorders Associated with Guanylyl Cyclases.
The only human diseases mapped to a gene for guanylyl cyclase involve retinal dystrophies. Leber's congenital amaurosis (LCA1), dominant cone-rod dystrophy (CORD6), cone dystrophy (CORD5), and central areolar choroidal dystrophy have been mapped to chromosome 17p12-p13, the interval containing the gene for GC-E (Balciuniene et al., 1995;Perrault et al., 1996; Hughes et al., 1998; Kelsell et al., 1998). In LCA1, the gene for GC-E contains mutations, including frameshifts, which result in truncated proteins that lack the kinase-like and catalytic domains due to premature termination of translation or a missense mutation in the kinase-like domain (Perrault et al., 1996). Expression of GC-E with this missense mutation in a heterologous cell line demonstrated that the mutant protein is stable but not activated by GCAP1 (Duda et al., 1999). In CORD6, GC-E contains mutations in the intracellular dimerization domain (Kelsell et al., 1998). It was postulated these mutations might cause a steric change in the protein that affects both mutant/mutant and mutant/wild-type dimers and thereby results in the dominant phenotype of CORD6. Indeed, one of the mutants has an increased affinity for GCAP-1, producing an enzyme that is stimulated at higher [Ca2+]ithan wild-type GC-E (Tucker et al., 1999). Thus, an abnormal increase in [cGMP]i in dark-adapted photoreceptor cells may be the cause of their degeneration. When the gene encoding GC-E was eliminated in mice by targeted disruption, cones disappeared by 5 weeks of age (Yang et al., 1999). Although the numbers and morphology of rods from GC-E null mice were similar to those from wild-type mice and the dark current was normal, retinas from null mice had a decreased response to light. The reason for the paradoxical rod behavior is not known.
Genetic alterations in other members of the guanylyl cyclase family are not associated with any described disease phenotype in humans. Several of the genes encoding guanylyl cyclases have been functionally eliminated in mice by targeted disruption. This approach can provide insight into the normal physiological role of a gene product. Targeted disruption of the GC-A gene resulted in mice with salt-resistant hypertension (Lopez et al., 1995; Oliver et al., 1997). GC-A null mice were unable to respond either to an infusion of ANP or to acute volume expansion, stimuli that induced diuresis and natriuresis in their wild-type littermates (Kishimoto et al., 1996). Null mice developed cardiac hypertrophy. In one study, all GC-A null male mice died of congestive heart failure or aortic dissection by 6 months of age (Oliver et al., 1997; Franco et al., 1998). Thus, the GC-A null mice exhibit many of the features of human essential hypertension and may prove to be a valuable model in which to study and develop treatment for this disease. Alterations in the sGC signaling pathway also may contribute to the development of hypertension. In the spontaneously hypertensive rat, an animal model for hypertension, the expression of both α1- and β1-subunits of sGC and the expression of cGMP-dependent protein kinase (PKG) I are reduced in the aorta (Ruetten et al., 1999). The reduction in expression was observed even in young (normotensive) spontaneously hypertensive rats, suggesting this is an early event in the pathogenesis of the disease.
The gene encoding GC-C has also been subjected to targeted disruption (Mann et al., 1997; Schulz et al., 1997). Null mice were viable and healthy and were resistant to infection with the enterotoxigenic bacteria that cause diarrhea and death in wild-type mice. The normal physiological role of GC-C therefore remains undefined.
B. Membrane-Bound Guanylyl Cyclases
1. Introduction.
Seven eutherian mammalian pGC isoforms (GC-A to GC-G) have been identified (Table 2). They exhibit highly conserved domain structures, including (1) an extracellular binding domain at the N terminus that in some cases binds defined ligands (GC-A, -B, -C), (2) a single transmembrane domain, (3) a cytoplasmic juxtamembrane domain, (4) a regulatory domain that shares significant homology with protein kinases, (5) a hinge region, and (6) a C-terminal catalytic domain (Fig. 1). Isoforms expressed in intestinal mucosal cells (GC-C) and in sensory organs (GC-D, -E, -F) also possess a C-terminal tail.

Domain structure of guanylyl cyclases. The cognate domains of ANPCRs, pGCs, and sGCs are compared. ANPCRs are homodimeric truncated guanylyl cyclases that possess extracellular ligand binding, transmembrane, and juxtamembrane domains but lack kinase homology, hinge, and catalytic domains. The pGC illustrated is a homodimer modeled after GC-A and -B and possesses a single ligand-binding site formed by two extracellular amino terminal domains. In addition to the domains present in ANPCRs, pGCs also possess kinase homology domains, hinge regions, and catalytic domains that form two functional catalytic sites. GC-C, -D, -E, and -F possess a carboxyl terminal tail that is not depicted here. sGCs are heterodimers possessing amino terminal regulatory domains containing a heme prosthetic group with a ferrous (Fe2+) core that forms an imidazole axial bond with His105 of the β-subunit. In addition, sGCs possess dimerization domains and carboxyl terminal catalytic domains that form one active and one inactive (flat blue) catalytic site. Colors identify domains with significant homology.
Based on their ligand specificities, pGCs have been classified as (1) natriuretic peptide receptors, (2) intestinal peptide-binding receptors, and (3) orphan receptors (Table 2). GC-A and GC-B bind to and are activated by natriuretic peptides, including ANP, BNP, and C-type natriuretic peptide (CNP). GC-C originally was characterized as the receptor for the family of homologous STs that produce secretory diarrhea. Recently, endogenous mammalian peptides, including guanylin, uroguanylin, and lymphoguanylin, were demonstrated to bind to and activate GC-C (Currie et al., 1992; Hamra et al., 1993; Forte et al., 1999). GC-D, -E, -F, and -G are orphan receptors for which ligands remain to be identified.
PGCs are expressed in almost all tissues in placental mammals (Table2). GC-A mRNA is highly expressed in kidney, adrenal, and adipose tissue, and at lower levels in terminal ileum and human placenta (Lowe et al., 1989). GC-B mRNA is abundant in brain, lung, and kidney (Schulz et al., 1989). The mRNA for GC-C has been identified not only in intestinal mucosal cells in adult placental mammals, but also in many epithelia in marsupials (Forte et al., 1988; Schulz et al., 1990;Carrithers et al., 1996). In the central nervous system, GC-D is expressed in a subpopulation of olfactory sensory neurons, GC-E is expressed in retinal rod and cone cells, whereas GC-F is expressed only in retinal rod cells (Fülle et al., 1995; Yang et al., 1995). GC-G is predominantly expressed in the lung, the intestine, and skeletal muscle (Schulz et al., 1998b). Thus, the natriuretic peptide receptor-like cyclases, including GC-A, -B, and -G, are broadly expressed in many tissues. In contrast, GC-C and the sensory organ cyclases, GC-D, -E, and -F, all possess a C-terminal tail and are expressed in a tissue-specific fashion.
2. Isotypes of Particulate Guanylyl Cyclases.
a. Natriuretic Peptide Receptors.
Mammalian atrial cardiomyocytes produce ANP, which mediates a pleiotropic response designed to maintain cardiovascular homeostasis in the face of a pressure or volume challenge (de Bold, 1985). Thus, ANP induces natriuresis, diuresis, and hypotension and inhibits secretion of renin and aldosterone (de Bold et al., 1981; Atarashi et al., 1984a,b). Indeed, ANP appears to mediate short- and long-term control of blood pressure and fluid and electrolyte balance (de Bold, 1985; John et al., 1995, 1996; Lopez et al., 1995; Kishimoto et al., 1996; Oliver et al., 1997; Franco et al., 1998). In addition to ANP, other natriuretic peptides identified to date include BNP and CNP. ANP is synthesized as a prepro-polypeptide of 151 residues in which the C-terminal portion contains the biologically active sequences (de Bold, 1985). The circulating form of this hormone is the mature 28-amino acid peptide consisting of a 17-amino acid loop stabilized by a single intrachain disulfide bridge and N- and C-terminal extensions, all of which are required for biological activity. BNP is a 26-amino acid peptide that was first isolated from acid extracts of porcine brain (Sudoh et al., 1988). Subsequently, this peptide also was identified in heart and blood (Aburaya et al., 1989). The disulfide-stabilized 17-amino acid ring and C-terminal sequence, essential for ANP bioactivity, are conserved in BNP. The genes encoding ANP and BNP are organized in tandem, and in humans, the BNP gene is located upstream of ANP (Huang et al., 1996; Tamura et al., 1996b). CNP is a 22-amino acid peptide that was first identified in acid extracts of porcine brain. CNP contains the disulfide-stabilized 17-residue ring structure found in ANP and BNP. In contrast, CNP lacks a C-terminal extension and the N-terminal region is not homologous with that of ANP and BNP. Although CNP induces natriuresis, diuresis, and vascular smooth muscle relaxation, it is significantly less potent than ANP or BNP (Sudoh et al., 1990).
The principal signaling mechanism by which natriuretic peptides induce their physiological effects involves activation of guanylyl cyclase-coupled receptors and accumulation of [cGMP]i. ANP has a selective affinity for GC-A, compared with GC-B, with a concentration for half-maximal response that is 1,000 times lower for the former than the latter (Schulz et al., 1989). ANP binding to GC-A has been demonstrated by both ligand binding analysis and affinity cross-linking studies (Leitman et al., 1988;Chinkers et al., 1989; Lowe et al., 1989; Jewett et al., 1993). ANP increases [cGMP]i in a concentration- and time-dependent fashion in a variety of cells and tissues (Chinkers et al., 1989; Schulz et al., 1989). Similarly, ANP activates guanylyl cyclase in cell-free membranes prepared from various cells and tissues (Hamet et al., 1984; Waldman et al., 1984; Leitman et al., 1987). The concentration-dependence of guanylyl cyclase activation and [cGMP]i accumulation induced by ANP compares favorably with that of receptor binding (Wong et al., 1995). Heterologous expression of GC-A in cells lacking endogenous expression of this protein results in the development of the ability of ANP to specifically bind to those cells, increase their pGC activity, and induce intracellular accumulation of [cGMP]i(Chinkers et al., 1989). ANP does not bind to tissues from animals that are homozygous null mutants for GC-A. These animals do not undergo natriuresis or diuresis in response to ANP and their aortic rings do not relax in response to that peptide (Kishimoto et al., 1996; Lopez et al., 1997). Furthermore, ANP directly activates GC-A purified from mammalian tissues (Kuno et al., 1986; Inagami et al., 1991; Waldman et al., 1991).
The endogenous receptor for BNP also appears to be GC-A, but BNP is 10-fold less potent than ANP in stimulating that receptor (Goeddel, 1991). BNP increases guanylyl cyclase activity and accumulation of [cGMP]i in cells and tissues in a pattern that mimics that of ANP (Goeddel, 1991). Similarly, BNP binds to heterologously expressed GC-A and increases guanylyl cyclase activity and [cGMP]i accumulation in those cells (Schulz et al., 1989). Aortic rings from animals that are homozygous null mutants for GC-A do not relax in response to BNP (Lopez et al., 1997).
The primary ligand for GC-B is CNP. Although ANP and BNP bind to this receptor with low affinity, CNP has a 50- to 500-fold greater affinity for GC-B than the other natriuretic peptides (Koller et al., 1991). CNP stimulates guanylyl cyclase activity and increases [cGMP]i in cells and tissues that express GC-B (Moriwaki et al., 1998; Chrisman and Garbers, 1999; Tao et al., 1999). Also, CNP binds to cells in which GC-B is heterologously expressed and increases activity of guanylyl cyclase and accumulation of [cGMP]i in those cells (Koller et al., 1991;Chrisman et al., 1993). CNP induces physiological effects in animals that are homozygous null mutants for GC-A (Lopez et al., 1997).
b. Intestinal Peptide Receptor Guanylyl Cyclase.
GC-Coriginally was identified and cloned from mRNA extracted from intestinal mucosal cells (Schulz et al., 1990). Although GC-C possesses the conserved domain structure characteristic of the guanylyl cyclase family (Fig. 1), it does not serve as a receptor for natriuretic peptides. The first ligand identified for GC-C was ST, and binding of ST to GC-C activates guanylyl cyclase and increases [cGMP]i in intestinal cells (Schulz et al., 1990). ST is produced by bacteria that colonize the intestine, including Escherichia coli, Enterobactersp., Klebsiella sp., and Yersinia enterocolitica (Rao et al., 1979; Thorne et al., 1979). This peptide contains six cysteines that form three intrachain disulfide bridges which stabilize the tertiary structure and confer the heat-stability characteristic of ST (Guerrant et al., 1980;Gariépy et al., 1987). Reduction of the disulfide bridges eliminates the ability of ST to bind to receptors, stimulate transmembrane signaling, and induce intestinal secretion and diarrhea (Staples et al., 1980). GC-C appears to be the only identified receptor for ST that is expressed in adult placental mammals (Guerrant et al., 1980). ST binds to GC-C in a concentration- and time-dependent manner (Guarino et al., 1987). Similarly, GC-C is activated by ST in a concentration-dependent manner and is not influenced by natriuretic peptides (Schulz et al., 1990; Krause et al., 1994a). Specific association of ST and GC-C has been demonstrated by ligand binding analysis and affinity cross-linking (Guarino et al., 1987; Schulz et al., 1990; Hugues et al., 1992). Interaction of ST and GC-C activates guanylyl cyclase in cell-free membranes prepared from intestinal cells (Gazzano et al., 1991a; Hugues et al., 1991). Similarly, ST induces accumulation of [cGMP]i in cells derived from intestine (Guerrant et al., 1980; Rao et al., 1980). Heterologous expression of GC-C confers specific ST binding to cells (Schulz et al., 1990; de Sauvage et al., 1991; Deshmane et al., 1995). Similarly, ST activates guanylyl cyclase and induces [cGMP]i accumulation in cells heterologously expressing GC-C (Schulz et al., 1990; de Sauvage et al., 1991). Disruption of the gene encoding GC-C results in mice that are resistant to ST-induced secretory diarrhea (Mann et al., 1997;Schulz et al., 1997). Homogenates of intestinal membranes from GC-C null animals specifically bind ST at very low levels, potentially identifying a binding protein other than GC-C (Mann et al., 1997). However, that novel binding protein has not yet been identified or characterized. Thus, GC-C remains the only identified receptor for ST and directly mediates the pathophysiological consequences of ligand-receptor interaction, including secretory diarrhea. A detailed discussion of the postreceptor mechanisms mediating ST-induced intestinal secretion is presented in a later section.
Unlike natriuretic peptide receptors, significant expression of GC-C appears limited to intestinal mucosal cells found from the duodenum to the rectum in adult placental mammals (Krause et al., 1994a). Expression of GC-C in intestinal, but not extraintestinal, cells has been identified by specific ST binding, stimulation of guanylyl cyclase, accumulation of cGMP, and detection of specific mRNA (Guerrant et al., 1980; Guarino et al., 1987; Schulz et al., 1990; Hugues et al., 1992; Krause et al., 1994a). Similarly, intestinal mucosal cells continue to express functional GC-C after undergoing neoplastic transformation to colorectal adenocarcinomas. Detection of this protein or its specific mRNA in extraintestinal sites appears to be a sensitive and specific marker for detecting metastatic colorectal cancer cells (Carrithers et al., 1996; Waldman et al., 1998; Cagir et al., 1999). Functional GC-C also has been detected in regenerating rat liver after surgical or chemical hepatectomy, but the functional significance of this observation remains to be elucidated (Laney et al., 1994). In contrast to placental mammals, GC-C in marsupials is expressed in the epithelia of the gut, gall bladder, lung, renal, and testis (Forte et al., 1988, 1989; White et al., 1989).
ST-like peptides that bind to and stimulate GC-C have been isolated from mammals. Guanylin and uroguanylin were isolated from intestinal mucosa; uroguanylin also has been isolated from urine (Currie et al., 1992; Hamra et al., 1993). These peptides share significant homology with ST, and their tertiary structure stabilized by two intrachain disulfide bonds is essential for biological activity (Currie et al., 1992; Hamra et al., 1993). The precise physiological importance of guanylin and uroguanylin remains to be elucidated. They may play a role in the regulation of fluid and electrolytes in the intestine (Hamra et al., 1993). However, mice that are homozygous null mutants for GC-C appear to develop normally, with normal intestinal function (Mann et al., 1997; Schulz et al., 1997). Uroguanylin, guanylin, and ST appear to induce natriuresis, diuresis, and kaliuresis in rodent kidney. These peptides may play a role in regulating fluid and electrolytes in the intestinal-renal axis (Greenberg et al., 1997; Fonteles et al., 1998). However, it is notable that analysis by sensitive nested reverse transcription-PCR does not detect expression of mRNA for GC-C in the kidney (Carrithers et al., 1996). Recently, a novel mRNA transcript encoding lymphoguanylin, a polypeptide related to guanylin and uroguanylin, was identified in spleen and lymphoid tissues from opossum (Forte et al., 1999). In these studies, the cDNA of lymphoguanylin was produced from isolated RNA and used to synthesize a putative endogenous peptide. Although ST, guanylin, and uroguanylin possess multiple disulfide bonds, synthetic lymphoguanylin contains only a single intrachain disulfide bond yet is biologically active (Forte et al., 1999). Because these studies did not isolate an endogenous peptide from tissues, the significance of this finding remains to be established.
c. Orphan Receptor Guanylyl Cyclases.
PGCs for which endogenous ligands have not yet been identified are categorized as orphan receptors. Ligands known to activate other guanylyl cyclases do not activate GC-D, -E, -F, or -G (Fülle et al., 1995; Yang et al., 1995; Schulz et al., 1998b). The primary structures of GC-D, -E, and -F extracellular domains are homologous, and the expression of these pGCs is restricted to sensory tissues (Yang et al., 1995). The ligand of GC-G may resemble the natriuretic peptides because the extracellular domain of this pGC is homologous to that of the natriuretic peptide receptors (Schulz et al., 1998b).
3. Structure of Particulate Guanylyl Cyclases.
a. Extracellular Domain.
Extracellular domains of pGCs exhibit the least homology among the members of the family. This diversity in structure presumably reflects the functional specificity of binding and induction of transmembrane signaling by different ligands. The precise molecular mechanisms mediating interaction of ligand with the extracellular domain and coupling of ligand binding with activation of the catalytic domain remain to be defined. In GC-B, Glu332 appears to be required for CNP binding and signaling because its removal or substitution with His or Lys results in complete loss of cyclase activity (Duda et al., 1994). However, it is not known whether Glu322 is essential for proper folding of receptor protein required for ligand binding, or if it is located in the binding site for the ligand (Duda et al., 1994). Similarly, amino acids 387 to 393 in the extracellular domain of GC-C interact directly with and are required for binding to ST (Hasegawa et al., 1999).
i. Glycosylation of Receptors.
All mammalian pGCs, except GC-F, contain at least one N-linked glycosylation site in the extracellular domain, although the extent of glycosylation varies among receptors (Chinkers et al., 1989). Glycosylation results in heterogeneity in the size of guanylyl cyclase receptors. Thus, pulse-chase studies revealed two species of GC-A that are synthesized during the first 7.5 h of incubation, but only one species in incubations longer than 7.5 h. The two species identified in shorter incubations were heterogeneously glycosylated GC-A, and the less glycosylated receptor served as a precursor for the fully glycosylated protein (Lowe and Fendly, 1992). Similarly, affinity labeling of GC-C with radiolabeled ST revealed multiple, specifically labeled proteins that could be resolved by SDS-polyacrylamide gel electrophoresis (PAGE) (Thompson and Giannella, 1990; Hugues et al., 1992). Heterologously expressed GC-C also yielded multiple receptor proteins specifically labeled with ST, demonstrating they were derived from a single transcript by post-translational processing (de Sauvage et al., 1992; Vaandrager et al., 1993a,b). Receptors of different sizes were converted to a single molecular weight by treating membranes with endoglycosidase, which removes N-linked carbohydrates (Vaandrager et al., 1993a). Similarly, cells grown in tunicamycin, an inhibitor of glycosylation, generated a single receptor species (Vaandrager et al., 1993a).
Glycosylation appears to play a role in the binding of ligand to the extracellular domain of pGCs. In a study performed in human embryonic kidney cells that were stably transfected with GC-A, fully glycoslyated GC-A specifically bound ANP, but removal of carbohydrate residues with endoglycosidase prevented binding of ANP to this receptor (Lowe and Fendly, 1992). In other studies performed in rat glioma cells that were stably transfected with GC-A, inhibition of N-glycosylation of GC-A by tunicamycin did not alter the ability of ANP to bind to cells and activate guanylyl cyclase (Heim et al., 1996). However, tunicamycin pretreatment inhibited the response of GC-A to ANP analogs, such as urodilatin [ANP (95–126)] (Heim et al., 1996). The different results obtained in these two studies might reflect differences in the type of ANP (native or analog) used. Similarly, when membranes prepared from cells that heterologously express GC-B were treated with endoglycosidase to remove carbohydrate from glycosylated sites in the protein, binding of CNP to the membranes was lost (Fenrick et al., 1996). Also, the activity of GC-B that had been deglycosylated was lower under stimulated conditions compared with the activity of fully glycosylated receptor (Fenrick et al., 1996). In close agreement, deglycosylation with endoglycosidase eliminated ST binding from the extracellular domain of GC-C (Hasegawa et al., 1999b). Studies in which each of the N-linked glycosylation sites was replaced with Ala demonstrated that Asn379, which is close to the transmembrane domain, was required for ligand binding and catalytic activity (Hasegawa et al., 1999a). Although glycosylation is important for binding of ligand, it does not appear to be required for distribution of receptors to the cell surface (Lowe and Fendly, 1992;Fenrick et al., 1996; Hasegawa et al., 1999b).
GC-D contains two potential N-linked glycosylation sites, whereas GC-E only contains one (Fülle et al., 1995; Yang et al., 1995). The extracellular domain of GC-G contains five potentialN-linked glycosylation sites and is about 40% homologous with the extracellular domain of GC-A (Schulz et al., 1998b). Whether glycosylation of these orphan receptors is essential for expression on the cell surface or for transducing a signal has not yet been determined.
ii. Cysteines and Oligomerization of Receptors.
All mammalian pGCs have two conserved cysteine residues at the N terminus and one midway in the extracellular domain (Chinkers et al., 1989; Schulz et al., 1989, 1990, 1998b; Fülle et al., 1995; Yang et al., 1995;Foster et al., 1999). All pGCs, except GC-C, also contain two conserved cysteine residues at the C-terminus of the extracellular domain proximal to the membrane-spanning domain (Chinkers et al., 1989; Schulz et al., 1989, 1990, 1998b; Fülle et al., 1995; Yang et al., 1995). GC-C shares two cysteine residues that are located midway in the extracellular domain with GC-A, but shares only one cysteine residue with other pGCs (Chinkers et al., 1989; Schulz et al., 1989, 1990,1998b; Fülle et al., 1995; Yang et al., 1995). Historically, it has been presumed that the cysteines in the extracellular domain form intrachain disulfide bonds important for stabilizing the tertiary structure of the receptor, similar to their function in other members of the superfamily of growth factor receptors (Itakura et al., 1994;Stults et al., 1994). The ANP clearance receptor (ANPCR), a truncated isoform of GC-A that lacks the cytoplasmic domain beyond the juxtamembrane domain, possesses five cysteines in the extracellular domain (Lowe et al., 1990a) (Fig. 1). Site-specific mutagenesis demonstrated the first four cysteines are joined sequentially, forming Cys104-Cys132 and Cys209-Cys257 intrachain disulfide bridges (Itakura et al., 1994; Iwashina et al., 1994). The precise role of these intrachain disulfide bridges in the function of the extracellular domain and in transmembrane signaling remains to be defined.
Cysteine residues in the extracellular domain appear to mediate ligand-independent oligomerization of receptor monomers (Chinkers and Wilson, 1992). In ANPCR, the fifth cysteine in the extracellular domain (Cys469) is proximal to the membrane-spanning domain and forms an interchain disulfide bridge that stabilizes a dimeric structure (Itakura et al., 1994). Heterologously expressed human GC-A migrates as high molecular weight oligomeric complexes when subjected to SDS-PAGE under nonreducing conditions. These oligomeric complexes can be converted to monomers after SDS-PAGE under reducing conditions (Lowe, 1992). Truncated mutants of GC-A possessing only the extracellular domain also form oligomeric complexes, supporting the suggestion that the extracellular domain contributes the cysteines that form the interchain disulfide bonds mediating oligomerization (Lowe, 1992). GC-A from bovine adrenal gland migrates as a tetrameric 550-kDa complex when subjected to SDS-PAGE under nonreducing conditions but as a 140-kDa monomer when exposed to reducing conditions (Iwata et al., 1991). GC-C migrates as complexes of high molecular weight when subjected to SDS-PAGE under nonreducing conditions, but these complexes were converted to monomers on exposure to reducing conditions (Ivens et al., 1990). Heterologously expressed GC-C also forms higher order complexes that are converted to monomers on exposure to reducing conditions (Vaandrager et al., 1993a,b).
These data support the suggestion that, in the absence of ligands, pGCs spontaneously form complexes that are stabilized by disulfide bonds in the extracellular domain. Those cysteines that contribute to the formation of interchain disulfide bridges remain to be defined for each isoform of guanylyl cyclase. GC-A mutants, in which Cys423 near the membrane-spanning domain was replaced, spontaneously formed interchain disulfide bonds and underwent dimerization, presumably mediated by Cys432 that was unpaired in the mutant protein (Labrecque et al., 1999). However, the relevance of this observation to native receptors is unclear because dimerization in this study was associated with constitutive activity (Labrecque et al., 1999). Furthermore, GC-C, which also undergoes ligand-independent oligomerization, does not possess these conserved cysteines proximal to the membrane-spanning domain (Hasegawa et al., 1999b).
b. Transmembrane Domain.
All pGCs have a single transmembrane domain similar to that in other members of the superfamily of growth factor receptors. The α-helix found in the transmembrane domain creates a hydrophobic region that permits insertion into the hydrophobic membrane lipid bilayer. Deletion of hydrophobic amino acids in the transmembrane domain of the epidermal growth factor receptor (EGFR) did not alter ligand binding or dimerization, suggesting this domain is required for localization to the membrane, but not for signal transduction (Kashles et al., 1988). In contrast, deletions in the transmembrane domain of the transforming growth factor-β receptor alter its ability to mediate transmembrane signaling (Zhu and Sizeland, 1999). In addition to membrane insertion, transmembrane domains may facilitate receptor oligomerization through α helix-helix interactions (Lemmon and Engelman, 1994; Lemmon et al., 1994). The precise role of the transmembrane domain in pGCs beyond membrane localization remains to be defined. However, it is notable that truncated mutants of GC-A and GC-C, which contain the extracellular domain but lack the transmembrane domain, are capable of forming dimers and binding ligand (Chinkers and Wilson, 1992; Hasegawa et al., 1999b).
c. Juxtamembrane domain.
The juxtamembrane domain is a short region of approximately 25 amino acids distal to the transmembrane domain in the cytoplasmic region of the protein. Although a precise function has not been ascribed to this domain, it may mediate alternate signaling mechanisms involving pGCs. This region in pGCs contains a consensus sequence that exists in other single transmembrane domain receptors and is important for their coupling to heterotrimeric G proteins (G proteins) and their downstream effectors.
Traditionally, G proteins are activated by the heptahelical family of receptors (Gudermann et al., 1995). However, members of the single transmembrane domain growth factor receptor superfamily, of which pGCs are members, also activate G proteins and signal through their downstream effectors. These receptors include EGFR, the insulin-like growth factor receptor, and the insulin receptor (Okamoto et al., 1990;Okamoto and Nishimoto, 1991; Ramirez et al., 1995; Krieger-Brauer et al., 1997). All the G protein-coupled single transmembrane domain receptors contain a consensus sequence in their juxtamembrane domains. This consensus sequence interacts with and activates G proteins. The consensus sequence ranges in length from 14 to 20 amino acids, contains two basic residues in the N-terminal end and a BBXXB motif in the C-terminal end (B is a basic residue and X is a nonbasic, nonaromatic residue). Interestingly, GC-A, -B, and -C contain this consensus sequence in a homologous position in their juxtamembrane domains.
Similarly, ANPCR possesses the above consensus sequence in its juxtamembrane domain (Fuller et al., 1988; Lowe et al., 1990a). This truncated guanylyl cyclase has a short cytoplasmic domain of 37 amino acids (Lowe et al., 1990a) (Fig. 1). ANPCR binds ANP, BNP, and CNP, and its primary function appears to be clearance of natriuretic peptides from the circulation through constitutive ligand-independent endocytosis (Nussenzveig et al., 1990). However, this receptor also regulates a variety of physiological processes (Anand-Srivastava and Trachte, 1993), including inhibition of adenylyl cyclase in rat and human platelets (Anand-Srivastava et al., 1991; Marcil et al., 1996), atrial and ventricular cardiocytes (Anand-Srivastava and Cantin, 1986), rat heart (Anand-Srivastava et al., 1996), and pheochromocytoma cells (Drewett et al., 1992). ANPCR also regulates cellular growth in hepatoblastoma cells (Rashed et al., 1993), proliferation and invasion of matrix by endothelial cells (Pedram et al., 1997), [Ca2+]i in adrenal glomerulosa cells (Isales et al., 1992), activation of endothelial NO synthase (eNOS) in gastric SMCs (Murthy et al., 1998), activation of phospholipase C-β3 in tenia coli SMCs (Murthy and Makhlouf, 1999), and inhibition of MAP kinase in astrocytes (Prins et al., 1996).
Of significance, the juxtamembrane domain of ANPCR is required for receptor signaling (Murthy and Makhlouf, 1999). Antibodies directed against the cytoplasmic domain of ANPCR prevent signaling by this domain (Anand-Srivastava et al., 1996). Also, the juxtamembrane domain interacts with Go in membranes isolated from PC12 cells, presumably mediating inhibition of catecholamine secretion by ANP (Takida et al., 1999). Similarly, heterologously expressed ANPCR was coupled to activation of coexpressed eNOS through Gi in gastric SMCs (Murthy et al., 1998). In tenia coli SMCs, interaction of ANPCR with Gi and activation of phospholipase C-β3 by the βγ-subunit of Gi was mediated by the juxtamembrane domain consensus sequence. Mutation of the sequence eliminated coupling between the clearance receptor and activation of phospholipase C-β3 (Murthy and Makhlouf, 1999).
These studies demonstrate that the G protein-regulating consensus sequence within the context of the juxtamembrane domain of a member of the guanylyl cyclase family can couple with G proteins and regulate downstream effectors in a ligand-dependent manner. This consensus sequence mediates alternate transmembrane signaling in other single transmembrane receptors including EGFR, insulin-like growth factor receptor, and insulin receptor. These observations suggest the intriguing possibility that pGCs may signal through different pathways, including synthesis of cGMP through activation of the catalytic domain and regulation of G protein-coupled effectors through the consensus sequence in the juxtamembrane domain.
d. Kinase Homology Domain.
i. Structure.
All pGCs possess between the juxtamembrane and catalytic domains a ∼250-residue kinase homology domain (KHD) that is absent in sGCs. The KHDs of pGCs are ∼30% homologous with a wide range of protein kinases (Koller et al., 1992). Generally, protein kinases contain 11 conserved subdomains and 33 invariant amino acids that are critical for kinase activity (Hanks et al., 1988). Within the natriuretic peptide receptor-like cyclases, GC-A, GC-B, and GC-G, the KHD possesses 9, 9, or 8 of the conserved subdomains and 28, 27, or 22 of the invariant residues, respectively. The intestinal receptor cyclase GC-C contains 8 of the conserved subdomains and 25 of the 33 invariant residues and is 30% conserved relative to GC-A and GC-B (Koller et al., 1992). The sensory organ cyclases, GC-D, -E, and -F contain 9, 7, or 8 of the conserved subdomains and 22, 21, or 22 of the invariant residues, respectively. An invariant aspartate residue found in subdomain VI of protein kinases, which functions as the catalytic base, is substituted in all pGCs (Knighton et al., 1991; Taylor et al., 1992). Similarly, the glycine-rich region of subdomain I (GXGXXG) that mediates nucleotide binding to protein kinases is present in GC-A and -B, but absent in GC-C. This structural difference may underlie some of the functional differences in regulation of these receptors by adenine nucleotides (Koller et al., 1992).
ii. Kinase Activity.
All protein kinases contain an HRD consensus sequence in subdomain VI in which the acidic Asp mediates the transfer of a phosphate group from ATP to the appropriate substrate (Hanks et al., 1988). The HRD consensus sequence is absent from the JH2 domain of the JAK protein kinase family, which does not exhibit kinase activity (Foster et al., 1999). Similarly, mutation of the invariant Asp to Asn in c-Kit and v-Fps results in loss of kinase activity (Moran et al., 1988; Tan et al., 1990). This catalytic Asp residue is replaced in all pGCs, commonly by Ser, Arg, or Asn. Thus, the prevailing hypothesis is that pGCs do not possess kinase activity, reflecting the absence of the catalytic Asp residue in subdomain VI (Potter and Hunter, 1998b). Although no other guanylyl cyclase possesses protein kinase activity, retinal pGC exhibits intrinsic kinase activity (Aparicio and Applebury, 1996). Thus, ATP binds to purified retinal guanylyl cyclase at a site distinct from the catalytic GTP substrate-binding site. The protein kinase activity is magnesium-dependent, autophosphorylates serine residues, and transfers phosphate groups to myelin basic protein. TheK m for ATP is 81 μM, and the kinase activity is unaffected by cyclic nucleotides, phorbol 12-myristate 13-acetate (PMA), l-α-phosphatidylserine, or Ca2+, but is inhibited by staurosporine. These properties are distinct from other Ser/Thr kinases identified in rod outer segments including protein kinase A, protein kinase C (PKC), and rhodopsin kinase. This observation of intrinsic kinase and autophosphorylation activity in a pGC remains an isolated result, and its functional significance continues to be unclear.
iii. Phosphorylation.
Receptor guanylyl cyclases exist as phosphoproteins in the basal state. Upon binding of ligand, dephosphorylation of the receptor occurs, resulting in desensitization and a reduction in ligand-induced guanylyl cyclase activity. GC-A possesses six phosphorylation sites within the KHD, including Ser497, Thr500, Ser502, Ser506, Ser510, and Thr513, that are necessary for ligand-dependent activation of GC-A (Potter and Garbers, 1992; Koller et al., 1993; Potter and Hunter, 1998b). Mutation of these sites individually to Ala reduced GC-A activation by ANP, and replacement of five of the sites simultaneously yielded a receptor that was unresponsive to ligand. Similarly, GC-B exists as a phosphoprotein in the basal state when no ligand is bound to the receptor. GC-B possesses five residues within the KHD, including Ser513, Thr516, Ser518, Ser523, and Ser526, that are phosphorylated (Potter and Hunter, 1998a). As in GC-A, the phosphorylation state of GC-B at these residues correlates with catalytic responsiveness, and dephosphorylation results in desensitization. Mutation of these five residues to Ala results in a 90% decrease in CNP-dependent cyclase activity, demonstrating that phosphorylation of these sites is necessary for ligand-induced activation of GC-B.
e. Hinge Region.
The hinge region is a 43-residue domain in GC-A between the KHD and catalytic domain. This region mediates dimerization of catalytic subunits and is required for the expression of enzymatic activity. Truncated mutants of GC-A possessing only the catalytic domain migrate on SDS-PAGE as monomers that are devoid of enzymatic activity (Wilson and Chinkers, 1995). However, truncated mutants inclusive of the hinge region migrate as homodimers on SDS-PAGE and possess guanylyl cyclase catalytic activity (Thompson and Garbers, 1995; Wilson and Chinkers, 1995). The primary structure of the hinge domain is consistent with a coiled coil configuration that favors specific protein-protein interactions (Thompson and Garbers, 1995; Wilson and Chinkers, 1995). Indeed, incorporating this sequence into a yeast two-hybrid system demonstrated the hinge domain mediates spontaneous formation of protein multimers (Wilson and Chinkers, 1995). The hinge region mediates dimerization of catalytic subunits. Dimerization is generally required for catalytic activity of sGC, pGCs, and adenylyl cyclases (Thompson and Garbers, 1995; Wilson and Chinkers, 1995). In addition, this region may play a larger role in mediating holoreceptor dimerization because truncation mutants of GC-A lacking cytoplasmic domains failed to form dimers or higher-order structures in one study (Lowe, 1992). Whether this domain is necessary for dimerization of catalytic subunits or plays a role in mediating receptor oligomerization in guanylyl cyclases other than GC-A remains to be elucidated.
f. Catalytic Domain.
i. Dimerization of Catalytic Domains Is Required for Enzymatic Activity.
Guanylyl cyclases must undergo dimerization to express catalytic activity. Heterodimeric sGCs require coexpression of both α- and β-subunits for catalytic activity, and expression of either subunit individually yields catalytically inactive protein (Harteneck et al., 1990; Buechler et al., 1991). Expression of truncated α and β catalytic domains in Sf9 cells yielded catalytically active heterodimers (Wedel et al., 1995). Mammalian adenylyl cyclases contain two catalytic domains in a single polypeptide chain (Krupinski et al., 1989). The hinge region in GC-A, discussed above, is absolutely required for GC-A catalytic subunits to dimerize and express catalytic activity (Thompson and Garbers, 1995; Wilson and Chinkers, 1995). Similarly, GC-C forms oligomers in a ligand-independent fashion that are important for producing catalytically active protein (Hasegawa et al., 1999b). Interestingly, coexpression of the α1-subunit of sGC and the C-terminal catalytic domain of adenylyl cyclase (type I, II, or V), each of which is inactive when expressed alone, produced a catalytically active adenylyl cyclase (Weitmann et al., 1999). This chimeric enzyme was regulated by P-site inhibitors but was not stimulated by Gα s or forskolin. These data support the suggestion that two catalytic domains are required for expression of nucleotide cyclase activity. Also, they support the suggestion that the catalytic domains of adenylyl and guanylyl cyclase are structurally and functionally homologous.
ii. Determinants of Purine Specificity.
The primary structure of the catalytic domain is highly conserved in both pGCs and sGCs and closely related to the catalytic domain of adenylyl cyclases (Krupinski et al., 1989). Thus, insights into the function of guanylyl cyclase catalytic domains were obtained using the solution structure of the X-ray crystal of the rat type II adenylyl cyclase C2catalytic domain. This provided critical information about substrate binding to the catalytic domain (Tesmer et al., 1997). Three invariant residues (Lys, Asp, Gln) present in the active site of adenylyl cyclase interact with the purine ring, which determines substrate specificity. The catalytic subunits of rat sGC (α1β1) contain three invariant residues (Glu, Cys, Arg) in positions homologous to those of the three invariant residues in adenylyl cyclase (Fig. 2). Mutations of adenylyl and guanylyl cyclase, in which these three residues were exchanged, resulted in the exchange of nucleotide substrate specificity: the guanylyl cyclase mutant specifically utilized ATP as substrate, whereas the adenylyl cyclase mutant became a nonselective purine nucleotide cyclase (Sunahara et al., 1998). Both enzymes retained their ability to be regulated by activators that were specific for the parent nucleotide cyclase. Thus, cAMP production by the mutant guanylyl cyclase was regulated by SNP, whereas cyclic nucleotide production by the mutant adenylyl cyclase was activated by Gα s. Similarly, mutation of retinal guanylyl cyclase (Ret GC 1) based on the structural model of adenylyl cyclase, in which Glu925 was substituted with Lys and Cys995 was substituted with Asp, reversed substrate specificity from GTP to ATP (Tucker et al., 1998). This mutant retained the characteristic ability of retinal guanylyl cyclases to be regulated by GCAP-1 and GCAP-2.

Catalytic mechanism of guanylyl cyclase activity. GTP binds to a single catalytic site in soluble and two catalytic sites in pGCs. The purine ring is specifically bound to residues in the β-subunit (red), and the divalent cation cofactor (Me2+), which stabilizes the β- and γ-phosphates of the nucleotide substrate, binds to acidic residues in the α-subunit (green). Both subunits contribute residues to the catalytic center that mediates cleavage of the α-phosphoanhydride bond through a single direct displacement reaction. The products of that catalysis are cGMP and pyrophosphate (PPi).
iii. Configuration of the Catalytic Site.
A model of the catalytic mechanism of adenylyl and guanylyl cyclases was developed based on the crystal structure of the rat type II adenylyl cyclase C2 catalytic domain (Liu et al., 1997b) (Fig. 2). This model predicts that heterodimeric cyclases such as sGCs and adenylyl cyclases have a single active site formed by two catalytic subunits that can bind one substrate molecule per dimer (Figs. 1, 2). Homodimeric cyclases such as pGCs have two sites within a single cleft and are capable of binding two substrate molecules per dimer (Fig. 1). Three residues are required to form a catalytic center, an Asp from one catalytic domain and an Asn/Arg pair from the other. In heterodimeric cyclases, the two domains form one catalytic center, with one domain providing the Asp and the other domain providing the Asn/Arg pair (Fig.2). In homodimeric cyclases, each domain contributes both an Asp and an Asn/Arg pair forming two catalytic centers. This model of catalytic center formation is consistent with the characteristic kinetic behavior of guanylyl cyclases (Waldman and Murad, 1987). Purified sGCs exhibit linear Michaelis plots with Hill coefficients of 1.0, consistent with a single class of substrate binding sites that are not interactive (Chrisman et al., 1975; Garbers, 1979; Wolin et al., 1982). In contrast, purified pGCs exhibit curvilinear Michaelis plots with Hill coefficients >1.0, consistent with multiple substrate-binding sites that interact in a positively cooperative fashion (Wong et al., 1995).
In GCA, Glu974 represents an invariant residue in all cloned guanylyl cyclases and a conserved residue in most adenylyl cyclases (Wedel et al., 1997). Mutation of this residue to Ala in GC-A yielded an enzyme that was constitutively activated and unresponsive to ANP and ATP. These effects were independent of the KHD because replacing Glu974 with Ala constitutively activated a truncated mutant of GC-A lacking the extracellular domain, transmembrane domain, and KHD. The precise role of Glu974 in mechanisms regulating pGCs remains to be defined.
g. Carboxyl Terminal Tail.
GC-C and the sensory organ guanylyl cyclases GC-D, -E, and -F contain a C-terminal tail that extends beyond the catalytic domain (Schulz et al., 1990; Fülle et al., 1995; Yang et al., 1995). The precise function of the tail remains to be defined, although deletion of a portion of this domain eliminated the ability of ST to stimulate production of cGMP by GC-C (Wada et al., 1996). PMA potentiates activation of GC-C by ST, and this is associated with increased phosphate incorporation into receptor protein (Weikel et al., 1990; Crane and Shanks, 1996; Wada et al., 1996). However, PMA did not potentiate ST activation of, or phosphate incorporation into, a truncated mutant lacking the C-terminal 22 residues or a substitution mutant in which Ser1029 was replaced with Ala. These observations suggest that PMA regulates ST-dependent activity by inducing PKC-mediated phosphorylation of Ser1029 in the C-terminal tail of GC-C.
There is speculation the C-terminal tail may be involved in associating guanylyl cyclase receptors with the cytoskeleton. GC-C, -E, and -F are resistant to detergent solubilization, compared with GC-A and -B (Fleischman and Denisevich, 1979; Fleischman et al., 1980; Waldman et al., 1986; Hakki et al., 1993). Indeed, chaotropic agents are required to optimally solubilize guanylyl cyclases from membrane preparations of intestinal epithelial cells (GC-C) or retinal rod outer segments (GC-E, GC-F) (Fleischman et al., 1980; Waldman et al., 1986). This relative insolubility may reflect association of those receptor cyclases with the cytoskeleton, which may be mediated by the C-terminal tail, rendering them refractory to solubilization by detergents. It is interesting to note that GC-C in intestinal brush border membranes and GC-E and GC-F in retinal rod outer segments are localized in membrane specializations with common origins (modified ciliary processes) that are stabilized by an intricate and well-developed cytoskeleton (Wiederhold, 1976).
The C-terminal tail may mediate internalization of guanylyl cyclase receptors. GC-C undergoes ST-dependent endocytosis in intestinal cells (Urbanski et al., 1995). Receptors that undergo ligand-dependent receptor-mediated endocytosis typically contain cytoplasmic domain consensus sequences that interact with the endocytotic apparatus. One such consensus sequence is YXXZ (Z is one of the following hydrophobic amino acids: L, I, V, M, C, A) (Johnson et al., 1990; Canfield et al., 1991; Thomas and Roth, 1994). GC-C contains this consensus sequence in the C-terminal tail, which is presumed to mediate ligand-dependent endocytosis in intestinal cells (Urbanski et al., 1995).
4. Receptor-Effector Coupling and Particulate Guanylyl Cyclase Function.
The mechanisms by which ligand-receptor interaction is translated into catalytic activation and a cellular signal, and the termination of that signal have not been completely elucidated for pGCs. However, the details of receptor function, receptor-effector coupling, effector activation, and signal termination are outlined below.
a. Interaction of Ligand and Receptor.
Initial studies of the equilibrium binding characteristics of GC-C in membranes from intestinal cells demonstrated that ST associates with a single class of receptor with a nanomolar K d (Hugues et al., 1991). However, further examination with a broad range of ST concentrations under well-defined equilibrium conditions revealed two populations of ST binding sites. One population of receptors exhibits a low capacity (∼5% of the total receptors) and high affinity, whereas the other population of receptors exhibits a high capacity (>95% of the total receptors) and low affinity (Crane et al., 1992b). Although high affinity ST receptors exhibit robust ligand binding, they are not coupled to activation of guanylyl cyclase, and their functional significance remains to be defined (Crane et al., 1992b). The molecular mechanism underlying the appearance of high versus low affinity receptors remains unclear, but may reflect receptor oligomerization, glycosylation, or ligand heterogeneity (de Sauvage et al., 1992;Vaandrager et al., 1993a; Schulz et al., 1998a; Hasegawa et al., 1999b).
Low affinity ST receptors (GC-C) appear to be homologous to functionally coupled ANP receptors (GC-A). Interestingly, both GC-A and GC-C undergo a ligand-induced shift in affinity, which appears to be important in transmembrane signaling. Thus, ST binding induces a time-dependent shift from higher (0.1 nM) to lower (1.0 nM) affinity, and the lowest affinity state of GC-C appears to be the receptor subtype functionally coupled to catalytic activation (Crane et al., 1992b). Similarly, ANP binding to GC-A results in a time-dependent shift in affinity. At equilibrium, 70% of ANP receptors exist in the lower affinity state (K d = 2.5 nM), whereas 30% remain in the higher affinity state (K d = 0.3 nM) (Larose et al., 1991; Jewett et al., 1993).
The ligand-induced shift in receptor affinity appears to be mediated by the KHD. Deletion of this domain yields GC-A and GC-C mutants “locked” in the higher affinity state, unable to undergo transition to the lower affinity state (Crane et al., 1992b; Jewett et al., 1993;Rondeau et al., 1995). ATP potentiates the shift of GC-A from higher to lower affinity induced by ANP binding. Potentiation of the affinity shift by ATP is blocked by amiloride, which competitively inhibits ATP binding to protein kinases (Heim et al., 1988; Jewett et al., 1993;Rondeau et al., 1995). The shift from higher to lower affinity is associated with coupling of ligand-receptor interaction to activation of guanylyl cyclase. GC-C locked in the higher affinity state is insensitive to activation by ST, whereas occupancy of the lowest affinity state is specifically associated with ligand-sensitive activation (Crane et al., 1992a). These observations suggest a model in which ligand binding to the extracellular domain of pGCs induces an alteration in the cytoplasmic domain that permits ATP to bind to the KHD. Adenine nucleotide association with the KHD derepresses the catalytic domain, resulting in activation and a subsequent decrease in ligand affinity for the extracellular domain (Potter and Hunter, 1998b). This paradigm, wherein ligand-receptor interaction activates downstream effectors in a nucleotide-dependent fashion that is associated with reduced ligand affinity, is a characteristic that pGCs share with G protein-coupled receptors and effectors. In the latter system, receptor, effector, and nucleotide regulatory functions reside on separate proteins, whereas in pGCs they are functions of individual domains within a single polypeptide.
b. Oligomerization of Receptors.
Receptor guanylyl cyclases exhibit a general structure that is similar to the tyrosine kinase family of receptors. The accepted model for tyrosine kinase activation includes a requirement for ligand-induced dimerization of receptor monomers. Interestingly, the mechanism underlying guanylyl cyclase activation deviates from this model. Thus, pGCs appear to exist as preformed oligomers in the basal state, and ligand-receptor interaction does not alter receptor oligomerization. The requirement for preformed receptor oligomers in the absence of ligand may reflect, in part, the requirement of nucleotide cyclases for two catalytic subunits to convert nucleotide triphosphates to cyclic nucleotides (Harteneck et al., 1990; Thompson and Garbers, 1995; Yan et al., 1996). Among the pGCs, GC-A, -B, -C, and -E exist as oligomers (Iwata et al., 1991;Chinkers and Wilson, 1992; Rondeau et al., 1995; Hasegawa et al., 1999b; Yu et al., 1999).
Oligomerization of GC-A has been the most extensively characterized of the pGCs. Covalent cross-linking and gel filtration experiments demonstrated the presence of both monomers and higher molecular weight complexes consistent with receptor oligomers (Ishido et al., 1986;Iwata et al., 1991; Lowe, 1992). GC-A immunoprecipitated in the absence of ANP yielded oligomeric complexes supporting the suggestion that this receptor self-associates in a ligand-independent fashion. Constructs lacking both the kinase homology and catalytic domains were monomers in the basal state, but formed oligomers in the presence of ANP, suggesting that cytoplasmic domains of GC-A contribute to oligomerization (Lowe, 1992). However, mutants lacking the extracellular domain did not coimmunoprecipitate with full-length GC-A, suggesting that the extracellular domain also is important in ligand-independent receptor oligomerization (Chinkers and Wilson, 1992).
Although GC-C forms oligomers in a ligand-independent fashion, the receptor appears to undergo ligand-dependent disulfide-stabilized dimerization (Almenoff et al., 1993; Vaandrager et al., 1993b, 1994). Receptor labeling and ligand-receptor cross-linking demonstrated that GC-C exists in an oligomerized state independent of ST (Vaandrager et al., 1993b, 1994). Coimmunoprecipitation of differentially tagged GC-C expressed in COS cells confirmed that oligomerization is independent of ligand stimulation (Rudner et al., 1995). Incubation with ST resulted in the formation of GC-C dimers stabilized by interchain disulfide bonds (Vaandrager et al., 1993b). Thus, GC-C may exist in the basal state as an inactive homotrimer that undergoes ligand-dependent internal rearrangement to form a catalytically active disulfide cross-linked dimer (Vaandrager et al., 1994). Interestingly, GC-C mutants lacking cytoplasmic domains formed trimers only in the presence of ligand, in close agreement with results obtained with homologous GC-A mutants (Lowe, 1992; Hasegawa et al., 1999b). Again, these observations support a role for cytoplasmic domains in mediating ligand-independent oligomerization of pGCs.
c. Regulation by Adenine Nucleotides.
i. Allosteric Activation of Guanylyl Cyclases by Nucleotides.
ATP potentiates the activation of GC-A and -B by natriuretic peptides 2- to 3-fold by increasing maximum enzyme velocity (V max) without altering substrate affinity (Kurose et al., 1987; Chang et al., 1990; Gazzano et al., 1991b). The EC50 for ATP potentiation of ligand activation is ∼0.1 mM, which is within the physiological range of cellular concentrations of this nucleotide (Kurose et al., 1987; Chang et al., 1990; Gazzano et al., 1991b). The rank order of potency of nucleotides to potentiate natriuretic peptide receptor activation is adenosine-5′-O-(3-thiotriphosphate) (ATPγS) > ATP > adenylylimidodiphosphate (Chang et al., 1990; Chinkers et al., 1991; Gazzano et al., 1991b; Foster and Garbers, 1998). The superior efficacy of ATPγS and ATP likely reflects their ability to serve both as: (1) allosteric modulators of guanylyl cyclases, and (2) substrates for protein kinases that mediate the phosphorylation of these receptors (Foster and Garbers, 1998). In contrast, adenylylimidodiphosphate is a nonhydrolyzable analog of ATP, which is not a kinase substrate. Potentiation of ligand activation by adenine nucleotides reflects direct interaction of nucleotides with guanylyl cyclases, rather than nucleotide-dependent accessory proteins or enzymes. Thus, in addition to the nonhydrolyzable analogs of ATP, other nucleotides that are not substrates for nucleotide or protein kinases potentiate activation, including ADP, adenosine-5′-O-(3-thiodiphosphate, and adenosine-5′-O-(3-thiomonophosphate) (AMPS) (Kurose et al., 1987; Chang et al., 1990; Gazzano et al., 1991b; Parkinson et al., 1994). Also, adenine nucleotides potentiate ANP activation of GC-A purified to apparent homogeneity (Larose et al., 1991; Wong et al., 1995). Indeed, GC-A expressed in baculovirus or purified to homogeneity could not be activated by ANP in the absence of ATP, demonstrating that adenine nucleotides are absolutely required for receptor-effector coupling by natriuretic peptide receptors (Chinkers et al., 1991;Larose et al., 1991; Wong et al., 1995).
In addition to their effects on receptor-effector coupling, adenine nucleotides also allosterically regulate the affinity of natriuretic peptide receptors for ligands. GC-A undergoes a ligand-induced time-dependent shift from higher (0.1 nM) to lower (1 nM) affinity (K d) (Larose et al., 1991; Jewett et al., 1993). ATP potentiated this shift in affinity that was associated with nucleotide effects on activation. Amiloride, an antagonist of ATP binding to kinase catalytic domains, blocked the effects of ATP on ligand binding and catalytic activation (Jewett et al., 1993; Rondeau et al., 1995). Indeed, amiloride “locked” GC-A in a high affinity state that was unresponsive to ANP. These data suggest that allosteric regulation of natriuretic peptide receptors by adenine nucleotides coordinately modulates ligand-receptor interaction and receptor-effector coupling.
Adenine nucleotides also potentiate the activation of GC-C by ST 2- to 3-fold with an EC50 of 0.1 mM (Gazzano et al., 1991a; Vaandrager et al., 1993a). The rank order of potency of adenine nucleotides for potentiating ligand activation of GC-C is similar to that for the natriuretic peptide receptors, and the superior potency of ATPγS and ATP, compared with the hydrolysis-resistant nucleotides, likely reflects their ability to serve both as allosteric modulators and substrates for protein kinases. However, in contrast to natriuretic peptide receptors, GC-C activation by ST does not absolutely require ATP (Gazzano et al., 1991a; Vaandrager et al., 1993a). Also, ATP does not alter the V max or substrate affinity of GC-C (Vaandrager et al., 1993a). Rather, adenine nucleotides potentiate ST activation of GC-C by stabilizing the activated form of the enzyme, preventing its time-dependent desensitization. GC-E and -F are similar to GC-C in that they do not require ATP for catalytic activation, but are potentiated by adenine nucleotides (Tucker et al., 1997). Like GC-C, potentiation of GC-E and -F by adenine nucleotides may reflect protection against catalytic inactivation (Vaandrager et al., 1993a; Tucker et al., 1997).
Allosteric regulation of catalytic activation and receptor binding of pGCs by adenine nucleotides appears to be mediated by the KHD. Mutants of GC-A and GC-B lacking the KHD are unresponsive to adenine nucleotides and ANP (Chinkers et al., 1991; Koller et al., 1992; Jewett et al., 1993). These mutants are “frozen” in the high-affinity state, unable to undergo the ligand-dependent shift to the lower affinity state (Jewett et al., 1993). Indeed, the binding characteristics of these mutants mimics wild-type receptors treated with amiloride (Jewett et al., 1993; Rondeau et al., 1995). GC-A and -B possess in their KHD a glycine-rich subdomain with the consensus sequence GXGXXXG, which mediates nucleotide binding in protein kinase catalytic domains by immobilizing the terminal phosphate of ATP (Hanks et al., 1988). Mutants of GC-A and -B in which this glycine-rich domain was altered were refractory to the effects of ligand and adenine nucleotides (Duda et al., 1993). These data suggest that allosteric regulation of GC-A and -B by adenine nucleotides occurs through the interaction of adenine nucleotide with the KHD, initiated by ligand-receptor interaction and mediated by the glycine-rich region of the KHD. Nucleotide-KHD interaction transmits information distally to the catalytic domain and is required for ligand-dependent catalytic activation. In addition, information is transmitted proximally across the membrane to effect a shift in the affinity of the receptor domain for ligand. Interestingly, GC-C lacks the glycine-rich subdomain of the KHD, ATP is not required for ligand activation of this protein, adenine nucleotides do not alter the kinetics of catalysis, and those nucleotides do not regulate ligand-receptor interaction (Vaandrager et al., 1993b; Deshmane et al., 1997). It is notable that the role of adenine nucleotides in guanylyl cyclase receptor-effector coupling is analogous to that of guanine nucleotides in coupling heptahelical receptors to their downstream effectors. Indeed, the KHD of pGCs and G proteins appear to subserve analogous functions in mediating purine nucleotide regulation of receptor-effector coupling in their respective systems.
ii. Allosteric Inhibition of Guanylyl Cyclases by Nucleotides.
Adenine nucleotides substituted in the 2-position of the purine ring, including 2-chloroATP and 2-methylthioATP, inhibited basal and ST-stimulated GC-C with a K i of 1 μM (Parkinson et al., 1994, 1997; Parkinson and Waldman, 1996). Inhibition was associated with a ∼90% reduction inV max, but it had only minor effects on the affinity of the enzyme for substrate. 2-Substituted nucleotides did not alter ST-induced cGMP accumulation in intact intestinal cells but blocked this effect in permeabilized cells, suggesting that allosteric inhibition was not mediated by purinergic receptors. Nucleotide inhibition was mediated by a site on the receptor that was separate and distinct from that mediating nucleotide activation (Parkinson et al., 1994). Interestingly, the guanylyl cyclase substrate GTP increased the potency of 2-substituted nucleotides to inhibit GC-C in a concentration-dependent fashion (Parkinson and Waldman, 1996). In addition, the hydrolysis-resistant guanine nucleotide analog guanosine-5′-O-(3-thiotriphosphate) was more potent in supporting 2-substituted nucleotide inhibition of GC-C compared with GTP. Furthermore, high concentrations of GTP mimicked the effects of 2-substituted nucleotides and directly inhibited GC-C. These data are consistent with a model in which inhibition of GC-C by 2-substituted nucleotides may be mediated by an accessory guanine nucleotide binding protein possessing intrinsic GTP hydrolase activity (Parkinson and Waldman, 1996). Although the endogenous ligands regulating this allosteric inhibitory pathway remain undefined, this mechanism can be exploited to block ST-induced fluid and electrolyte secretion by intact intestinal cells (Zhang et al., 1999).
d. Kinase Homology Domain.
The KHD is a key regulatory component coupling ligand-receptor interaction with effector activation in pGCs. Indeed, in this respect it subserves a function that is analogous to G proteins, which couple heptahelical receptors to their downstream effectors. Deletion of the KHD from GC-A, -B, and -C resulted in constitutive activation of those enzymes (Chinkers and Garbers, 1989; Koller et al., 1992; Rudner et al., 1995). Also, mutant receptors lacking the KHD were insensitive to stimulation by adenine nucleotides and ligand. In addition, GC-A and -B lacking the KHD were “locked” in the high affinity state, insensitive to the effects of adenine nucleotides on receptor affinity, and unable to undergo the ligand-dependent shift to lower affinity characteristic of these receptors (Jewett et al., 1993). These data are consistent with a model in which the KHD behaves as a repressor of the catalytic domain in the basal state (Chinkers and Garbers, 1989; Koller et al., 1992; Jewett et al., 1993; Rudner et al., 1995). Ligand-receptor interaction results in an alteration in the KHD that permits adenine nucleotide binding, derepression of the catalytic domain reflected as activation, and a decrease in the affinity of the receptor for ligand. Of note, KHDs from individual isoforms of pGCs appear to be functionally specific for each isoform. Thus, the KHDs of GC-A and -B, which are structurally homologous proteins with similar functions, can be exchanged without altering the ability of these receptors to respond to adenine nucleotides and natriuretic peptides (Koller et al., 1992). However, exchange of the KHD of GC-A with that from GC-C or the kinase domain of the EGFR produced enzymes that were unresponsive to natriuretic peptides (Koller et al., 1992). These data suggest that KHDs are structurally and/or functionally “matched” to specific pGC isoforms. The molecular mechanisms underlying this component compatibility remain to be elucidated.
e. Phosphorylation and Homologous and Heterologous Desensitization.
Early studies demonstrated that sea urchin sperm pGCs underwent activation followed by rapid desensitization upon interacting with their cognate egg peptides (Garbers, 1989). These receptors were highly phosphorylated in the basal state, and ligand-receptor interaction resulted in massive dephosphorylation. Activation of these receptors by ligand required that they be fully phosphorylated. Thus, treatment of these preparations with phosphatases yielded receptors that were unresponsive to ligand. In addition, dephosphorylation of receptors associated with ligand-receptor interaction resulted in desensitization of receptors. Similarly, GC-A and -B require receptor phosphorylation for ligand-dependent catalytic activation (Potter and Garbers, 1992, 1994; Koller et al., 1993). Purified preparations of GC-A in which the enzyme is phosphorylated retain full sensitivity to natriuretic peptides and adenine nucleotides (Foster and Garbers, 1998). Also, phosphatase treatment of GC-A results in desensitization and an inability of ligand to activate the catalytic domain (Potter and Garbers, 1992). These data suggest that phosphorylation is an important mechanism regulating pGCs. In the basal state, pGCs are phosphorylated, which is required for ligand-induced activation. Ligand-receptor interaction associated with catalytic activation initiates a cascade leading to dephosphorylation of the receptor resulting in homologous desensitization. It is interesting to note that this mechanism of homologous desensitization is the converse of that regulating heptahelical G protein-coupled receptors, which are unphosphorylated in the basal state but desensitized by kinase-mediated phosphorylation, induced by ligand-receptor interaction (Gudermann et al., 1995).
Phosphorylation sites that are important for ligand activation and homologous desensitization have been identified in the KHD of GC-A and -B (Duda et al., 1993; Potter and Hunter, 1998a,b). However, the kinase(s) responsible for maintaining phosphorylation of GC-A and -B in the basal state have not been identified. Similarly, the phosphatase(s) responsible for ligand-induced dephosphorylation of GC-A and -B mediating homologous desensitization have not been identified, although evidence for a guanylyl cyclase-associated phosphatase was presented previously (Chinkers, 1994). Although this mechanism appears to be generalizable to sea urchin sperm pGCs and the natriuretic peptide receptor cyclases, homologous desensitization by dephosphorylation has not been demonstrated for other pGC isoforms.
GC-A and -B also undergo heterologous desensitization by a number of ligands that activate PKC, including PMA, endothelin, vasopressin, and angiotensin (Potter and Garbers, 1994). Activation of PKC uncouples GC-A and -B from ligand-induced cGMP production (Jaiswal et al., 1988;Potter and Garbers, 1994). However, unlike other receptor systems, GC-A and -B appear to be desensitized by PKC-mediated dephosphorylation (Potter and Garbers, 1994). Indeed, PMA, a direct activator of PKC, induced the dephosphorylation of natriuretic peptide receptors that was associated with receptor desensitization (Potter and Garbers, 1994). A specific inhibitor of PKC blocked dephosphorylation and desensitization induced by PMA but not by ANP (Potter and Garbers, 1994). Furthermore, PMA treatment resulted in dephosphorylation of receptor residues that did not overlap with those residues dephosphorylated by ANP. These data suggest that GC-A and -B undergo heterologous desensitization mediated by dephosphorylation of residues distinct from those involved in homologous desensitization (Potter and Garbers, 1994). The precise mechanisms underlying heterologous desensitization and whether PKC stimulation results in the inhibition of a pGC kinase or the activation of a pGC phosphatase remain to be defined.
f. Accessory Protein Regulation.
Retinal pGCs (GC-E, -F) are modulated by a family of calcium-regulated accessory proteins, GCAPs. In the vision process, phototransduction of light into electrical impulses occurs in the retina (Lolley and Lee, 1990). pGCs located in outer segments of the retinal membrane are central in the cascade mediating phototransduction (Lolley and Lee, 1990). Light-activated rhodopsin is coupled to a G protein that activates a cGMP-specific PDE (Yarfitz and Hurley, 1994). This PDE hydrolyzes cGMP, resulting in the closure of cGMP-gated channels that mediate Ca2+ influx. Reduced [Ca2+]i mediates GCAP activation of GC-E and -F and production of cGMP (Shyjan et al., 1992; Lowe et al., 1995; Nakatani et al., 1995). The resulting accumulation of cGMP reopens cGMP-gated channels reestablishing the resting state (dark) current.
GCAPs share significant homology with the calmodulin family of calcium-binding proteins (Palczewski et al., 1994). Thus far, three mammalian GCAPs have been identified (Palczewski et al., 1994; Dizhoor et al., 1995; Gorczyca et al., 1995; Haeseleer et al., 1999). These GCAPs exhibit guanylyl cyclase specificity: GCAP-1 only activates GC-E, whereas GCAP-2 activates both GC-E and GC-F (Gorczyca et al., 1995;Otto-Bruc et al., 1997a,b). In general, GCAPs activate guanylyl cyclase when [Ca2+]i is <300 nM and inhibit activity when [Ca2+]i is >500 nM (Dizhoor and Hurley, 1996; Dizhoor et al., 1998). Although GCAPs modulate guanylyl cyclase activity in a calcium-dependent manner, the association of GCAPs with guanylyl cyclases is calcium-independent (Dizhoor and Hurley, 1996; Laura and Hurley, 1998).
GCAP activation of retinal guanylyl cyclases has been suggested to occur by two mechanisms: enhancement of dimerization of retinal guanylyl cyclase catalytic subunits through dimerization of GCAPs, and/or GCAP-mediated stabilization of GTP bound to the catalytic subunit (Olshevskaya et al., 1999; Sokal et al., 1999). The dimerization model suggests: (1) GCAPs bind to retinal guanylyl cyclases independently of [Ca2+]i, (2) at high [Ca2+]i, GCAPs bind Ca2+ and have a low affinity for self-association, (3) at low [Ca2+]i, GCAPs dissociate from Ca2+ and homodimerize, and (4) GCAP homodimerization promotes pGC dimerization, and therefore enhances catalytic activity (Olshevskaya et al., 1999). This proposed mechanism offers a reasonable explanation for the calcium-sensitive regulation of guanylyl cyclases by GCAPs, however further work defining the association of GCAP and guanylyl cyclase dimerization is required.
Although multiple potential GCAP binding sites have been identified, thus far only a binding site on the retinal guanylyl cyclase catalytic domain has been characterized (Sokal et al., 1999). Interestingly, this binding site has homology to a region in the adenylyl cyclase catalytic domain that mediates activation by Gsα (Tesmer et al., 1997; Sokal et al., 1999). Gsαactivates adenylyl cyclase at the catalytic site by stabilizing the transition state with ATP (Tesmer et al., 1997). Similarly, it has been suggested that GCAPs may modulate catalytic activity by stabilizing the guanylyl cyclase-GTP transition state (Sokal et al., 1999). Although there is substantial homology between the structure of retinal and other pGCs, accessory proteins regulating nonretinal guanylyl cyclases remain to be identified.
g. Model for Coupling of Particulate Guanylyl Cyclase Receptor and Effector.
The above discussion suggests the following general model of receptor-effector coupling for ligand regulation of pGCs (Fig.3). In the inactive state, pGC exists as self-associated homo-oligomerized complexes in the absence of ligand. Each monomer within the complex is phosphorylated on key serine or threonine residues surrounding the glycine-rich region of the ATP binding site within the KHD. Oligomerization and phosphorylation are requirements for the extracellular binding domains to exist in the high affinity state and for the cytoplasmic domains to be competent to respond to ligand-receptor interaction. Under these conditions, ligand binding to high affinity sites is translated through the plasma membrane by an undefined mechanism into an alteration in the KHD, resulting in the binding of ATP to that domain. Association of ATP with the KHD initiates three important processes. First, the KHD represses the catalytic domain of pGC in the basal state, and adenine nucleotide interaction with the KHD results in derepression, reflected as activation of the catalytic domain and an increase in theV max of cGMP production. Derepression of the catalytic domain may be associated with internal rearrangement of homo-oligomers with formation of interchain disulfide bonds, but not with overall disruption of oligomers to monomers. Second, association of ATP with the KHD is translated back through the plasma membrane into a reduction in the affinity of the extracellular binding domain for ligand. Third, there is a time-dependent dephosphorylation of the KHD resulting in homologous desensitization of receptors to adenine nucleotides and ligand. The precise trigger initiating dephosphorylation, including ligand-receptor interaction, adenine nucleotide binding to the KHD, and/or activation of the catalytic domain remains unclear. Signal termination reflects three processes, including ligand dissociation from the extracellular domain, the reduction in binding affinity of the extracellular domain initiated by adenine nucleotide binding to the KHD, and homologous desensitization of the receptor by dephosphorylation of the KHD. In this model, requirements for receptor-effector coupling include receptor oligomerization, phosphorylation of the KHD, ligand occupancy of the extracellular binding domain, and adenine nucleotide binding to the KHD. Failure to fulfill any of these requirements results in uncoupling of receptors and effectors. Although the above process is offered as a general model of receptor-effector coupling for pGCs, it should be noted that it was specifically generated from studies of natriuretic peptide receptors. Indeed, GC-C does not possess a glycine-rich ATP binding domain in the KHD, ATP is not required for GC-C receptor-effector coupling, and there is no apparent role for KHD dephosphorylation and homologous desensitization for this receptor. In addition, the other known pGCs are orphan receptors, with no defined ligands. Although the above process is specifically referable to natriuretic peptide receptors, it serves as a working model to study the other members of the family.
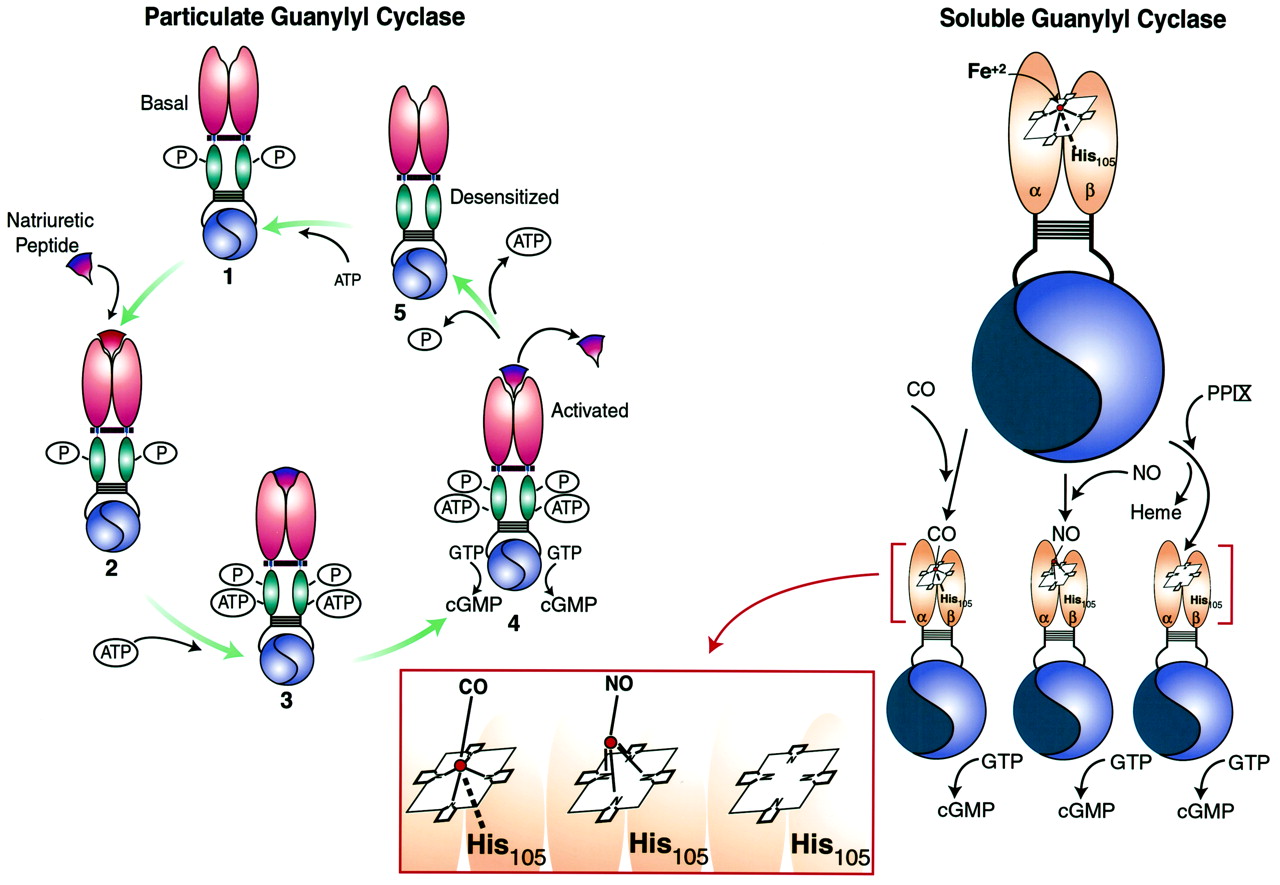
Regulation of particulate and soluble guanylyl cyclases by their ligands. Particulate guanylyl cyclases: the model of ligand regulation of pGCs depicted is based on regulation of GC-A by natriuretic peptides. In the basal unliganded state, pGC exists as a self-associated homo-oligomerized complex (1). Each monomer within the complex is phosphorylated on key serine or threonine residues within the KHD. Oligomerization and phosphorylation are requirements for the extracellular binding domains to exist in the high affinity state and for the cytoplasmic domains to be competent to respond to ligand-receptor interaction. Under these conditions, natriuretic peptide binding to a single high affinity site formed by a homodimer (2) is translated through the plasma membrane into an alteration in the KHD, resulting in the binding of ATP to that domain (3). In the basal state, the KHD represses the catalytic domain of pGC, and ATP interaction with that domain results in derepression, reflected as activation of two functional catalytic domains and an increase in theV max of cGMP production (4). However, association of ATP with the KHD also reduces the affinity of the extracellular binding domain for natriuretic peptides and induces dephosphorylation of the KHD, resulting in dissociation of ATP and ligand, leading to desensitization (5). Regeneration of receptors in the basal state that are responsive to ligand presumably requires rephosphorylation of sites in the KHD. Soluble guanylyl cyclases: sGCs are heterodimers that possess a heme prosthetic group with a pentacoordinated ferrous (Fe2+) core associated with an imidazole axial ligand at His105 of the β-subunit. NO activates sGC by directly binding to the sixth position of the ferrous core, breaking the bond between iron and histidine, dissociating it from that axial imidazole ligand, and displacing the iron from the plane of the porphyrin ring. This creates a structure resembling PPIX, a potent activator of sGC. Termination of activation presumably results from dissociation of NO from the axial ligand position of the heme and return of the iron core to the plane of the porphyrin ring. CO binds to the heme group of sGC, yielding a hexacoordinated complex in which the iron core is associated with imidazole and CO axial ligands simultaneously. The inability of CO to dissociate the imidazole axial ligand to form a pentacoordinated complex does not permit displacement of the heme iron from the plane of the porphyrin ring. Consequently, CO is far less efficacious as an activator of sGC, compared with NO. The precise mechanism by which PPIX activates sGC remains unclear. In one model (depicted), PPIX displaces heme from the sGC, resulting in activation. Alternatively, sGC-bound heme may exchange Fe2+with free PPIX, resulting in free heme, occupancy of the regulatory domain of the enzyme by PPIX, and activation.
C. Soluble Guanylyl Cyclase
sGC is expressed in the cytoplasm of almost all mammalian cells and mediates a wide range of important physiological functions, such as inhibition of platelet aggregation, relaxation of smooth muscle, vasodilatation, neuronal signal transduction, and immunomodulation (Collier and Vallance, 1989). This enzyme is a heterodimeric protein consisting of α- and β-subunits, and expression of both subunits is required for catalytic activity (Kamisaki et al., 1986; Harteneck et al., 1990; Buechler et al., 1991). Each subunit has an N-terminal regulatory domain and a C-terminal catalytic domain that shares sequence homology with the corresponding domains in particulate guanylyl and adenylyl cyclases (Chinkers et al., 1989; Krupinski et al., 1989; Thorpe and Morkin, 1990).
1. Subunit Structure and Isotypes of Soluble Guanylyl Cyclase.
Analysis of sGC from different tissues demonstrated multiple isotypes with different subunit compositions. The most abundant subunits are α1 and β1, which are found in many tissues (Braughler et al., 1979; Garbers, 1979; Lewicki et al., 1980). These subunits were first cloned from rat and bovine lung (Koesling et al., 1988, 1990; Nakane et al., 1988, 1990). Expression of α1 and β1 individually yields a protein that does not exhibit catalytic activity, whereas coexpression of these subunits yields sGC, which can be activated by NO (Buechler et al., 1991). The β2-subunit, cloned from rat kidney, contains 86 additional amino acids in its C-terminal region compared with β1 (Yuen et al., 1990). This additional sequence contains a consensus sequence (CVVL) that is involved in post-translational modifications, such as isoprenylation and carboxymethylation, suggesting that β2 might serve to localize sGC to membranes. This ∼76-kDa subunit is most abundant in kidney and liver. Although the β2-subunit can form a heterodimer with α1, this holoenzyme exhibits lower specific activity compared with α1/β1. Thus, NO-stimulation of COS-7 cells cotransfected with α1/β1 resulted in three times more cGMP production than in cells transfected with α1/β2 (Gupta et al., 1997). Coexpression of β2 with α1/β1 decreased the formation of the α1/β1 heterodimer, presumably due to competition between β1 and β2 for binding to α1. These data support the hypothesis that expression of β2 may serve to regulate α1/β1 sGC activity. In fact, expression of β2 has been suggested to play a role in the pathogenesis of hypertension in the Dahl rat (Gupta et al., 1997). The ∼82-kDa α2 cloned from human fetal brain forms heterodimers with β1 or β2, but has lower affinity for β1. Indeed, α2/β1 has a lower specific activity than α1/β1 (Harteneck et al., 1991). Two other subunits of human sGC, α3 (82 kDa) and β3 (70 kDa), have been cloned from adult brain. Although α3 and β3 have limited homology in their N-terminal regions, there is 72% homology between their 310-residue C-terminal regions (Giuili et al., 1992). The presence of homologous regions could reflect the existence of a common ancestor for these subunits (Giuili et al., 1992).
RNA splicing also contributes to the heterogeneity of sGC subunits. A variant of the α2-subunit, α2i, was identified in a number of cell lines and tissues by PCR using primers based on conserved sequences in the catalytic domain of mammalian guanylyl cyclase (Behrends et al., 1995). The subunit α2i is produced through alternative splicing of RNA that adds 31 amino acids to the catalytic domain, with homology to a region within the catalytic domain of adenylyl cyclases. Previous studies demonstrated sGC catalyzed the conversion of ATP to cAMP (Mittal and Murad, 1977). The region of homology in α2i was postulated to increase the ability of this isoform of sGC to utilize ATP as a substrate and produce cAMP (Behrends et al., 1995). However, coexpression of the α2i/β1 in Sf9 cells abolished the ability of sGC to produce cAMP (Behrends et al., 1995). In addition, Sf9 cells transfected with α2i/β1 are devoid of guanylyl cyclase activity, whereas coexpression of α2/β1 results in production of cGMP in these cells, suggesting the α2i-subunit can compete with α2 for binding to β1 and act as a dominant negative inhibitor. Expression of this subunit may serve as a mechanism to regulate sGC activity in specific cells (Behrends et al., 1995). Additionally, two forms of the β-subunit of human sGC, HSGC-1 and HSGC-2, were detected in lung using PCR with oligonucleotide primers corresponding to conserved sequences from rat and bovine sGC (Chhajlani et al., 1991). HSGC-1 is identical with the smaller subunit in bovine and rat lung, whereas HSGC-2 contains a 33-amino acid deletion. Splice variants of sGC may reflect tissue specific expression of the enzyme, as well as mechanisms regulating activity.
A novel isoform of sGC that does not require heterodimer formation to express catalytic activity has been isolated from the nervous system of the tobacco hornworm moth Manduca sexta. This novel isoform, MsGC-β3, is closely related to the rat β1-subunit (Nighorn et al., 1999). However, MsGC-β3 contains an additional 315 amino acids at the C-terminus, which are not homologous with any known protein. MsGC-β3 lacks those conserved sequences present in other sGCs that are important for activation of the enzyme by NO. The catalytic activity of MsGC-β3 expressed in COS-7 cells is less than that of MsGC-α1 and MsGC-β1 isoforms. MsGC-β3 is activated weakly in the presence of SNP, which generates NO. However, like all other known cyclases, MsGC-β3 exhibits higher enzyme activity when Mn2+, rather than Mg2+, is used as the metal substrate cofactor (Nighorn et al., 1999).
In addition to MsGC-β3, a second new class of guanylyl cyclases has been isolated from the soluble fraction of cells in the nervous system of Manduca sexta. MsGC-I is similar to pGCs, with the greatest homology to GC-B. However, it does not contain a signal sequence or the ligand binding, transmembrane, and kinase homology domains characteristic of mammalian pGCs. Although the location of this enzyme is postulated to be in the cytoplasm of the cell, MsGC-I has no similarities to the regulatory domains of α- and β-subunits of sGC. It contains a sequence of 149 amino acids extending beyond the catalytic domain that has no homology to any known protein. MsGC-I expressed in COS cells forms homodimers with high levels of basal guanylyl cyclase activity, but cannot be activated by SNP. The fact that an extracellular ligand or NO cannot activate this enzyme suggests the existence of a new regulatory mechanism for this guanylyl cyclase (Simpson et al., 1999).
Although naturally occurring homodimers have not been isolated, extensive homology between sGCα and sGCβ subunits suggests that possibility (Wilson and Chinkers, 1995). Glutathione S-transferase (GST)-tagged recombinant human α1- and β1-subunits can form both homodimeric GST-α1/α1 and GST-β1/β1 complexes in Sf9 insect cells. Cotransfection of complementary subunits results in catalytically active heterodimers, although homodimers are detectable. Both homodimers are catalytically inactive and are detected in lower amounts than heterodimers. The preference for heterodimer formation in Sf9 cells may reflect a higher affinity between complementary subunits. These results suggest the possibility of a physiological equilibrium between homo- and heterodimers that could regulate sGC activity in cells (Zabel et al., 1999). Although the presence of both complementary subunits is necessary for catalytic activity, these subunits may not be expressed with the same temporal pattern. In rat brain, α1 is expressed earlier than β1 during fetal brain development. This observation suggests the existence of an uncharacterized β-subunit that substitutes for β1 during this developmental period (Smigrodzki and Levitt, 1996).
2. Domain Structure.
Each subunit of sGC can be divided into three functional domains: heme-binding, dimerization, and catalytic (Fig. 1). The heme-binding domain is located at the N terminus of each subunit. The presence of the heme prosthetic group is required for activation of sGC by NO (Craven and DeRubertis, 1978, 1983; Gerzer et al., 1982; Ignarro et al., 1982a; Ohlstein et al., 1982). Heme is a five-membered nitrogen-containing ring wherein four nitrogen atoms are coordinated with a central iron that can be either Fe2+(ferrous or the reduced form) or Fe3+ (ferric or the oxidized form) (Fig. 1). The fifth member of the ring in sGC is an imidazole axial ligand coordinated by the β1-subunit at His105 (Stone and Marletta, 1994). Mutation of this histidine, located near the N terminus of the β1-subunit, results in the inability of sGC to bind heme and produces an enzyme that is unresponsive to NO. Mutation of other conserved histidines does not affect the ability of the enzyme to bind heme (Wedel et al., 1994). The enzyme mutated at His105 can be purified and reconstituted with heme, but it remains unresponsive to NO.
Both α1- and β1-subunits are required to express basal catalytic activity and activation of sGC by NO (Buechler et al., 1991). This observation is consistent with the hypothesis that both subunits play a role in the association of the heme with the enzyme. To define the role of each subunit in the binding of heme, deletions of the less conserved N-terminal sequence of either α1 (deletion of 131 residues) or β1 (deletion of 64 residues) were performed. Deletions in the β-subunit resulted in a loss of sensitivity to NO, confirming the importance of this subunit in the activation of the enzyme by NO (Foerster et al., 1996). Deletions in the α1-subunit did not alter the NO responsiveness of sGC (Foerster et al., 1996). However, heme binding by sGC requires the presence of both full-length subunits implying a role for the α-subunit in coordinating heme (Stone and Marletta, 1994).
The presence of Cys78 and Cys124 in the β-subunit is important for coordinating the heme group of sGC (Foerster et al., 1996). This is in agreement with the observed importance of Cys residues in other heme-containing proteins. In cytochrome C, heme is covalently bound to the protein through thioether bonds to two Cys residues, consistent with the suggestion that heme in sGC also may be bound through a Cys residue in the β-subunit.
In sGC, each heterodimer contains approximately one heme with a high affinity for NO (Gerzer et al., 1981a). This is in contrast to heme in other proteins, such as hemoglobin and myoglobin, which has a high affinity for oxygen and in the aerobic environment binds oxygen preferentially to form a ferrous-oxy species, rather than a ferrous-nitrosyl species. Even in an aerobic environment, sGC prefers to bind NO rather than oxygen. If sGC binds oxygen, it forms a ferrous-oxy species that must exchange oxygen with NO to form a ferrous nitrosyl complex (Gerzer et al., 1981a,b,c). Oxidation of the heme group to the ferric state results in the loss of enzyme activity and often a complete loss of the heme moiety from the protein. Ferricyanide oxidizes sGC to the ferric state, which is insensitive to NO stimulation. Thus, reducing agents such as thiols, ascorbate, or dithiothreitol enhance enzyme activation, presumably by maintaining the iron of the metalloporphyrin in the ferrous state that is sensitive to NO.
3. Regulation of Soluble Guanylyl Cyclase by Ligands.
a. Nitric Oxide.
NO is a free radical that activates sGC by binding directly to heme to form a ferrous-nitrosyl-heme complex (Fig.3). The half-life of the ferrous-nitrosyl heme is between 4 min and 3 h at 20°C (Hille et al., 1979; Sharma and Ranney, 1978). NO binds to the sixth position of the heme ring, breaks the bond between the axial histidine and iron, and forms a bond with iron (Fig. 3). This results in a 5-coordinated ring where NO is now in the fifth position. Removal of heme results in loss of NO responsiveness. Carbon monoxide (CO), another activator of sGC, also is capable of binding to the heme group of sGC, yielding a 6-coordinated complex (Fig. 3). However, NO is a more potent activator of sGC than CO, and the purified enzyme is activated 100- to 200-fold by NO, but only about 4-fold by CO (Stone and Marletta, 1994).
Binding of NO to sGC appears to fit a model based on two populations of heme. The minor population, which contains 28% of the heme, initially forms a 6-coordinate complex with NO and then is rapidly converted to a 5-coordinate nitrosyl complex. NO dissociates from this population with a rate of ∼ 20 s−1. The second population, which contains 72% of the heme, also forms a 6-coordinate nitrosyl complex, but the conversion to a 5-coordinate complex is much slower. NO dissociates from this population with a rate of 0.1 to 1.0 s−1, which is too slow to deactivate the enzyme. This slow dissociation from the ferrous heme in the second population is due to reassociation of the histidine with the iron in the heme. The dimerization state of sGC is not affected by binding of NO to heme. Both the 5-coordinate ferrous enzyme and the 5-coordinate nitrosyl form of the enzyme exhibited the same molecular mass of ∼200 kDa (Stone and Marletta, 1996).
Electron paramagnetic resonance spectroscopy of sGC suggests a model in which the formation of a 5-coordinated nitrosyl-heme complex results from the breaking of a bond between the axial imidazole ligand and the heme iron. This creates a conformational change in the structure of the protein that activates sGC. The iron is displaced from the plane due to steric hindrance, creating a structure that resembles protoporphyrin IX (PPIX) with an open central core (Fig. 3). The presence of an open central core in the structure of the protoporphyrin ring is crucial for activation of the enzyme (Stone et al., 1995).
b. Protoporphyrin IX.
PPIX, the precursor of heme, is a naturally occurring compound synthesized from glycine that activates heme-deficient, heme-free, and heme-containing forms of sGC in a heme- and NO-independent manner. PPIX binds to sGC at low concentrations (K d = 1.4 nM) and forms a stable complex that does not dissociate during gel filtration or dialysis (Ignarro et al., 1982b; Wolin et al., 1982). There are two parts of the porphyrin ring that are important for binding of heme to sGC. Two vinyl groups at positions 2 and 4 are essential for creating a hydrophobic interaction between the porphyrin ring and the enzyme. If these vinyl groups are substituted with less hydrophobic or polar groups, sGC has reduced enzyme activity due to a weaker interaction between the porphyrin ring and the enzyme. Also, the negatively charged COOH groups of the vicinal propionic acid residues at position 6 and 7 form electrostatic bonds with basic residues, such as arginine, in the sGC apoprotein (Ignarro et al., 1984).
The effects of PPIX on the kinetic parameters of sGC are similar to those of NO. In the presence of Mg2+-GTP, PPIX increases the specific activity of heme-containing bovine lung sGC from 0.1–0.2 to 2–8 μmol cGMP/min/mg protein and decreases theK m from 100 to 56 μM. In the presence of Mn2+-GTP, PPIX increases the specific activity slightly (from 0.3–0.6 to 1–1.4 μmol cGMP/min/mg protein), but the K m remains unchanged (Ignarro et al., 1982b; Wolin et al., 1982). PPIX alters enzyme catalysis, in part, by increasing the affinity of the enzyme for Mg2+-GTP or free Mg2+, because divalent cations readily form complexes with PPIX. This direct interaction between PPIX and divalent cations may be critical for activation of sGC by this porphyrin. Enzyme-associated PPIX binds Mg2+-GTP or uncomplexed Mg2+, but not GTP alone (Ignarro et al., 1984). Divalent cations (Mg2+, Mn2+) that complex with GTP are absolutely required as substrate cofactors to support guanylyl cyclase catalytic activity.
An iron atom can be incorporated after the protoporphyrin has been assembled into the protein to form heme (ferroprotoporphyrin with Fe2+) or hematin (ferriprotoporphyrin with Fe3+). Although heme is the required prosthetic group mediating NO activation of sGC, ferro-PPIX is a competitive inhibitor of PPIX (K i = 350 nM) (Ignarro, 1994). Hematin inhibits basal activity in a noncompetitive manner in the presence of either Mg2+-GTP (K i = 2.8 μM) or Mn2+-GTP (K i = 8.3 μM). Hematin concentrations below 1.5 μM competitively inhibit PPIX without altering guanylyl cyclase activity (K i = 0.35 μM). This indicates that ferroprotoporphyrin and ferriprotoporphyrin compete for the same binding site on sGC (Wolin et al., 1982). However, hematoporphyrin IX, a ferriprotoporphyrin with two hydroxyethyl groups instead of the vinyl groups found in protoporphyrin, activates sGC. Hematoporphyrin IX does not occur naturally and is a less potent activator than PPIX. Alterations in the structure of PPIX produce analogs, such as uroporphyrin I, coproporphyrin I, and the dimethyl ester of PPIX, that have no effect on sGC activity (Ignarro et al., 1982b).
c. Catalytic Mechanism.
Catalytic domains present in the C-terminus of both α- and β-subunits share significant homology with the C1 and C2 catalytic domains of adenylyl cyclase and the catalytic domains of pGCs. As discussed above, coexpression of α- and β-subunits, which form a heterodimer possessing two catalytic domains, is required for expression of enzymatic activity. Although two catalytic domains are present, each contributes specific residues to a single substrate binding and catalytic site (Liu et al., 1997b) (Fig. 1). Dimerization is mediated by a specific region that is homologous to the dimerization domain of pGCs and is located proximal to the catalytic domains of α- and β-subunits (Fig. 1). Indeed, coexpression of the C-terminal domains of α1 and β1 that possess dimerization and catalytic regions is sufficient for basal cGMP production (Wedel et al., 1995). NMR spectroscopy of sGC purified from rat liver demonstrated that cGMP and pyrophosphate are the sole products of catalysis of GTP (Tsai et al., 1980). Analyses of pGC purified from sea urchin sperm established the α-phosphoanhydride bond as the site of cleavage during catalysis (Walseth et al., 1981) (Fig. 2). Examination of the stereochemical course of the catalytic reaction of sGC purified from bovine lung using [α-18O]GTP established that formation of cGMP is a single direct displacement reaction (Senter et al., 1983) (Fig. 2).
d. Divalent Cations.
Both pGCs and sGCs require divalent cations as substrate cofactors and allosteric modulators to express maximum catalytic activity (reviewed in Waldman and Murad, 1987). All nucleotide cyclases require that their purine nucleotide substrates form a chelate with a divalent cation to bind to the catalytic domain and undergo enzymatic cyclization. Mn2+ and Mg2+ are the optimum divalent cation substrate cofactors (reviewed in Waldman and Murad, 1987). Using Mg2+ as the substrate cation cofactor, sGCs and pGCs exhibit basal catalytic activities that are fully sensitive to activation by ATP and ligands. Indeed, Mg2+ likely is the physiological cation supporting guanylyl cyclase activity in vivo. In contrast, using Mn2+as the substrate cation cofactor, guanylyl cyclases exhibit maximum catalytic activity that is insensitive to further activation by nucleotides and ligand (Hardman and Sutherland, 1969; Kimura et al., 1976; Tsai et al., 1978; Garbers, 1979; Levine et al., 1979; Gerzer et al., 1981c; Zwiller et al., 1981). This is a general characteristic of nucleotide cyclases. Adenylyl cyclase also is activated in a ligand-independent fashion using Mn2+ as the substrate cation cofactor. The precise mechanism by which Mn2+activates guanylyl cyclases in a ligand-independent fashion remains unclear. However, it is an intrinsic characteristic of the protein because Mn2+ activated GC-A purified to homogeneity in a ligand-independent fashion (Wong et al., 1995). In addition to serving as a required substrate cofactor, divalent cations also activate guanylyl cyclases in an allosteric fashion (Garbers and Gray, 1974;Waldman and Murad, 1987). Indeed, divalent cation concentrations in excess of nucleotide substrate are required for maximum catalytic activity. The precise molecular mechanisms by which divalent cations allosterically modulate guanylyl cyclases remain unclear.
Calcium also supports guanylyl cyclase catalytic activity as a substrate cofactor, but appears to be a negative allosteric modulator of sGC (Levine et al., 1979). Indeed, Ca2+ and cGMP have antagonistic functions in several physiological systems. In vascular smooth muscle, contraction is mediated by an increase in [Ca2+]i, whereas relaxation occurs as the result of an increase in [cGMP]i. In retinal photoreceptor cells, light induces a decrease in [cGMP]i and a decrease in [Ca2+]i, whereas increases in [cGMP]i and [Ca2+]i are associated with dark recovery after exposure to light. However, it has been difficult to examine the regulation of guanylyl cyclase by Ca2+ because of the presence of calcium-dependent regulatory mechanisms extrinsic to guanylyl cyclases in cells. For example, Ca2+ interaction with GCAPs regulates retinal guanylyl cyclase, and Ca2+ is a required cofactor for NO synthase (NOS), whose product is a potent activator of sGC (Yarfitz and Hurley, 1994; Yu et al., 1999). Recently, Ca2+ regulation of sGC was studied in a heterologous system in which other Ca2+-dependent regulatory mechanisms were absent (Parkinson et al., 1999). Human embryonic kidney cells (HEK 293), which do not express NOS and express low amounts of endogenous sGC activity, were cotransfected with α1- and β1- subunits of rat sGC. Ca2+ inhibited both basal and NO-stimulated crude and immunopurified sGC prepared from those cells. In the physiological range of GTP, Ca2+ inhibition was concentration- and guanine nucleotide-dependent with a K i = 2.6 μM. Inhibition of sGC was mediated by extracellular Ca2+ and by release of [Ca2+]i pools through receptor-mediated mechanisms. Ca2+ decreased bothV max andK m of sGC, consistent with an uncompetitive mechanism of inhibition in which Ca2+ interacts either with the substrate (Mg2+-GTP) or one of the products [cGMP or pyrophosphate (PPi)] at the catalytic site. This mechanism is similar to that of P-site inhibition of adenylyl cyclase, wherein purine nucleotides inhibit enzyme activity by binding to the catalytic apparatus in the presence of the product Mg2+-PPi (Dessauer, 1997). This study suggests sGC may function as a specific sensor of [Ca2+]i that mediates coordinated reciprocal regulation of [Ca2+]i and [cGMP]i.
III. Cyclic GMP and Cell Signaling
A. Introduction
Endogenous and exogenous compounds, including autocoids, hormones, neurotransmitters, and toxins, produce cellular responses through cGMP. The biochemical mechanisms underlying those responses include synthesis (guanylyl cyclase, see above), targeting (various, see below), and degradation (PDEs, see below) of cGMP. The specificity of cellular responses to cGMP is dictated by cGMP-binding motifs in target proteins. Two evolutionarily distinct allosteric sites for binding cGMP are present in eukaryotic cells. One occurs with significant sequence homology in PKGs and cAMP-dependent protein kinase (PKA) and in the cyclic nucleotide-gated (CNG) cation channels, while the other occurs in cGMP-regulated PDEs. In addition, the outcome of increased [cGMP]i is determined by the type and combination of target proteins and substrates, the cGMP-metabolizing enzymes expressed in cells, and their intracellular colocalization and organization into selective compartments and organelles. For example, phospholamban and the IP3 receptor in the sarcoplasmic reticulum are substrates for both PKG and PKA, but the pattern of colocalization of the kinase and its substrates differs according to cell type. PKG colocalizes with these two substrates in SMCs, whereas PKA colocalizes with them in cardiac myocytes (Lincoln et al., 1995). Therefore, the phosphorylation of phospholamban and IP3 receptors in SMCs contributes to the relaxation response and is cGMP-dependent. In contrast, the phosphorylation of the same substrates in cardiac myocytes occurs through the cAMP/PKA pathway and promotes Ca2+sequestration and a shortened cardiac systole. Finally, cGMP may have different effects under physiological and pathophysiological conditions. For example, in activated neutrophils, cGMP/PKG phosphorylates the intermediate filament vimentin after transient colocalization of the enzyme to the filament. In quiescent neutrophils, cGMP analogs do not induce phosphorylation of vimentin (Wyatt et al., 1991).
The following sections provide a brief perspective on how guanylyl cyclases and cGMP are integrated into transmembrane signaling cascades and a broad overview of events downstream from regulation of guanylyl cyclases and [cGMP]i accumulation in placental mammalian systems. For this reason, the physiological role of cGMP in specific cellular systems, such as human blood cells, and in primitive unicellular organisms is not described. For the interested reader, outstanding reviews are available that focus specifically on PKA and PKG (Francis and Corbin, 1999), CNG cation channels (Kaupp, 1995;Zimmerman, 1995; Biel et al., 1999b), and cGMP-regulated PDEs (Beavo, 1995; Juilfs et al., 1999). In addition, a complete compendium of the functions of cGMP is available (Murad, 1994).
B. Protein Kinases
1. Cyclic GMP-Dependent Protein Kinases.
PKG represents the principal intracellular mediator of cGMP signals. Ligand-induced elevation of [cGMP]i induces a binding-dependent activation of PKG leading to the catalytic transfer of the γ-phosphate from ATP to a serine or threonine residue on the target protein. This phosphorylated protein then mediates the translation of the extracellular stimulus into a specific biological function.
Two different genes for PKG have been identified in mammals. One gene is located on human chromosome 10 and codes for the Iα and Iβ isoforms of PKG I, which arise from alternative splicing of the N-terminal region (Tamura et al., 1996a). The other is located on human chromosome 4 and encodes PKG II (Orstavik et al., 1996). PKG I is a cytosolic 76-kDa homodimer widely expressed in mammalian tissues, especially in cerebellum, platelet, and smooth muscle (Lohmann et al., 1997). The difference in the N-terminal domain between the two PKG subtypes confers different binding affinities for cGMP. PKG Iα has high and low affinity binding sites that display positive cooperative behavior. PKG Iβ has two cGMP binding sites characterized by lower affinity and cooperativity (Pfeifer et al., 1999). Moreover, although expression of these two isoforms has been detected in the same human tissues, PKG Iα was detected mainly in the vascular system, kidney, and adrenal gland, whereas only PKG Iβ was detected in the uterus (Tamura et al., 1996a).
PKG II is an 86-kDa membrane-bound homodimer. It is absent from the cardiovascular system, abundant in brain and intestine, and is also expressed in lung, kidney, and bone (Uhler, 1993; Jarchau et al., 1994;Lohmann et al., 1997). The amino acid sequence of PKG II differs from the sequence of PKG I principally at the N terminus; unique sites in this region direct intracellular localization of the enzyme. PKG II contains a myristoylated site that is required for membrane association, whereas PKG I contains an acetylated site (Lohmann et al., 1997). A major difference between the two PKGs is that the cGMP binding sites in PKG II have minimal affinity and cooperativity. Another difference is a significant divergence in their substrate selectivity, which becomes more evident in vivo (Pfeifer et al., 1999). Moreover, the two forms of PKG are expressed in different cells, with the exception of chondrocytes in the growth plate in the tibia of newborn mice (Pfeifer et al., 1996).
All known PKGs are composed of N-terminal, regulatory, and catalytic domains. The N-terminal domain contains five regulatory sites: (1) the subunit dimerization site, consisting of an α-helix with a conserved leucine/isoleucine heptad repeat; (2) autoinhibitory sites, involved in the inhibition of the catalytic domain in the absence of cGMP; (3) autophosphorylation sites, which in the presence of cGMP may increase the basal catalytic activity and the affinity of PKGs for cAMP; (4) a site regulating the affinity and the cooperative behavior of the cGMP binding sites; and (5) the intracellular localization site, which determines the interaction of the enzyme with specific subcellular structures. The regulatory domain contains two cyclic nucleotide binding sites (conventionally termed “A” and “B”) that allow for full activation of the enzyme after specific binding of two molecules of cGMP. Finally, the catalytic domain, located at the C-terminus of PKGs, contains the binding sites for Mg2+-ATP and the target protein (Lincoln et al., 1995; Lohmann et al., 1997; Pfeifer et al., 1999).
A broad range of proteins are phosphorylated by PKGs in vitro, but phosphorylation of only a few has been demonstrated in vivo. This apparent discrepancy may be explained by the fact that, in addition to the recognition sequence of the substrate, under physiological conditions the substrate specificity for PKG is dictated by intracellular spatial localization. This results in a unique macromolecular aggregate comprised of the enzyme-target protein complex.
The current working hypothesis suggests that PKG I acts as a soluble intracellular modulator of [Ca2+]i, while PKG II regulates fluid homeostasis at the cell membrane. Biological substrates for PKG I may be conceptually subdivided into three main groups, “classical,” “new,” and “hypothetical” targets. “Classical” targets are clearly recognized as substrates, in vitro and/or in vivo, are phosphorylated by PKG I, and have well established functions. This group includes: (1) the IP3receptor and phospholamban, which are primarily implicated in SMC relaxation (Raeymaekers et al., 1990; Komalavilas and Lincoln, 1996); (2) the vasodilator-stimulated phosphoprotein and vimentin, which are involved in platelet and neutrophil activation, respectively (Pryzwansky et al., 1995; Aszódi et al., 1999); (3) the G substrate, which is strongly expressed in cerebellar Purkinje cells where it acts as a phosphatase inhibitor (Endo et al., 1999); and (4) the thromboxane A2 receptor, whose activation was found to be inhibited by PKG-mediated phosphorylation in platelets (Wang et al., 1998). “New” target proteins are PKG I substrates that either have been described recently or have conflicting evidence regarding their phosphorylation by PKG I. This group includes: (1) the L-type Ca2+ channel and the Ca2+-activated K+ channel which, upon phosphorylation, contribute to the regulation of vascular smooth muscle tone and cardiac contractility (Jahn et al., 1988; Fukao et al., 1999); (2) the Ca2+-dependent cytosolic phospholipase A2, implicated in intestinal smooth muscle relaxation (Murthy and Makhlouf, 1998); (3) a tyrosine hydroxylase whose activity in intact bovine chromaffin cells was observed to increase after PKG I-mediated phosphorylation (Rodrı́guez-Pascual et al., 1999); and (4) the myosin-binding subunit of myosin light chain phosphatase, which mediates SMC relaxation and vasodilation (Surks et al., 1999). Most, if not all, of these substrates are phosphorylated by the subtype Iα of PKG. “Hypothetical” target proteins have been suggested, but not demonstrated, to be phosphorylated by PKG Iβ. This group contains putative substrates predicted on the basis of experimental demonstrations of cGMP/PKG I mediated processes. For example, cytoskeletal and contractile proteins (i.e., myosin light chain, calponin, desmin, connexins) are thought to be PKG Iβ target molecules in the regulation of vascular remodeling and neoangiogenesis (Lincoln et al., 1998; Eigenthaler et al., 1999). Similarly, synaptic vesicle proteins (i.e., rabphilin-3A) may be phosphorylated by PKG I and mediate synaptic plasticity and neurotransmission (Qian et al., 1996; Gray et al., 1999; Yawo, 1999). The cGMP/PKG I pathway has been implicated in the control of gene expression of various promoter response elements (i.e., serum response element, AP-1 binding site, and cAMP response element) (Gudi et al., 1996, 1997).
In contrast to PKG I, the only recognized “classical” substrate that is phosphorylated by PKG II is the cystic fibrosis transmembrane conductance regulator (CFTR) in intestinal mucosal cells (Vaandrager et al., 1997). Localization of PKG II in the apical membranes of the enterocytes of the small intestine permits it to phosphorylate CFTR in response to GC-C-induced cGMP formation. The phosphorylation of CFTR induces an electrogenic chloride current and subsequent water secretion in the intestine. Colocalization and coregulation of the expression of PKG II and a chloride channel in the inner medulla of rat kidney suggest a similar mechanism may regulate renal function (Gambaryan et al., 1996). PKG II also may control the renin system and endochondral ossification and growth of bone (Pfeifer et al., 1996; Wagner et al., 1998). However, the target molecules in these latter processes are unknown.
2. Cyclic AMP-Dependent Protein Kinases and Cyclic GMP Signaling.
Because PKAs contain specific cyclic nucleotide binding domains with significant homology to PKGs, they may be activated by cGMP, although with a 50-fold lower selectivity than cAMP. Although the nucleotide binding sites of PKAs and PKGs are homologous (Pfeifer et al., 1999), differences between these sites exist, specifically in the substitution of key amino acid residues (Lohmann et al., 1997).
There is some controversy concerning cross-activation of protein kinases by cyclic nucleotides. Interestingly, many of the known physiological substrates for PKGs are also substrates for PKAs. In addition, cGMP appears to act in concert with cAMP in a variety of cellular processes. Thus, PKG and PKA inhibit IP3-dependent release of Ca2+ and induce relaxation in dispersed rabbit and guinea pig gastric muscle cells (Murthy and Makhlouf, 1995). Similarly, isoproterenol or SNP induce the phosphorylation of cytosolic Ca2+-dependent phospholipase A2 by activating PKA or PKG, respectively, in SMCs from the longitudinal muscle layer of rabbit intestine (Murthy and Makhlouf, 1998). The same effect is achieved by simultaneous stimulation of both kinases by VIP and isoproterenol, contributing to the relaxation response of these cells.
In vertebrates, cGMP and cAMP relax vascular smooth muscle, inhibit platelet activation, and regulate chloride and water secretion in the intestine. Cyclic AMP cross-activates PKG in various vascular tissues, including rat aorta and pig coronary artery (Jiang et al., 1992;Eckly-Michel et al., 1997). In vitro, cGMP cross-activated PKA, which mediates secretion of fluid induced by ST in human intestinal cells (Forte et al., 1992; Chao et al., 1994). Functional convergence between cGMP and cAMP also might occur at a downstream level, such as at the level of the kinase target proteins. Based on the identity of substrates phosphorylated by PKA and PKG, there is substantial overlap between the two cyclic nucleotides in their ability to regulate CO2-induced cerebrovasodilatation of adult rat pial arteries (Wang et al., 1999). These kinases may act in a cooperative manner and the phosphorylation activity of one is required for a full effect of the other. Overexpression of NOS, leading to increased levels of NO, results in cross-activation of PKA by a large increase in cGMP (Lincoln et al., 1995). This mechanism mediates inhibition of SMC proliferation and PKG Iα expression in rat aorta and underlies the development of nitrovasodilator tolerance in bovine and rat aortic SMCs continuously exposed to these agents (Cornwell et al., 1994; Soff et al., 1997; Lincoln et al., 1998).
Although the above in vitro observations suggest cGMP could signal via PKA in vivo, other considerations do not support this hypothesis. Intact cells exhibit extensive subcellular compartmentalization that confines second messengers, enzymes, and their substrates to finite “work units” and represents an obstacle to cross-activation. In addition, PKA and PKG must anchor to specific intracellular proteins to perform their physiological functions. Thus, only PKA Iα, but not PKA IIα, is redistributed and colocalized with the T-cell receptor complex during anti-CD3 induced T-lymphocyte activation and capping. PKA Iα thereby mediates the inhibition by cAMP of T-cell proliferation (Skålhegg et al., 1994). Similarly, PKG transiently colocalized with, and phosphorylated, vimentin in human neutrophils activated by A23187, but not in quiescent cells (Pryzwansky et al., 1995). Colocalization of kinases and substrates, mediated by targeting proteins, is now considered a primary determinant of the specificity of their effects (Pfeifer et al., 1999).
Physiologically relevant cross-regulation of protein kinases by cyclic nucleotides has been challenged by studies in mice with homozygous PKG I- or PKG II-null mutations. Cyclic GMP-induced relaxation in aortic rings or gastric fundus muscle strips prepared from PKG I-deficient mice was impaired, whereas cAMP-induced relaxation was not (Pfeifer et al., 1998). These mutant mice were hypertensive and lacked regular intestinal peristalsis, indicating PKG I is the specific mediator of cGMP effects in smooth muscle, in vivo. Furthermore, there was a defective cGMP-mediated inhibition of the activation response in platelets from the mutant mice, whereas cAMP-mediated inhibition was not impaired (Massberg et al., 1999). Indeed, no cross-activation of platelet PKA by cGMP was observed. Finally, mice with a homozygous PKG II null mutation displayed a selective reduction of ST- and 8-bromo-cGMP-induced intestinal secretion (Pfeifer et al., 1996). In contrast, electrogenic anion secretion from the small intestine elicited by cAMP analogs was not affected by the mutation.
These observations suggest that, in vivo, cGMP and cAMP signaling cascades elicit various important physiological effects largely independently, and that neither cGMP activation of PKA nor cAMP activation of PKG can completely compensate for the loss of the other nucleotide. Although direct cross-regulation of protein kinases by cyclic nucleotides may not appear to be a dominant process regulating signaling, there is evidence that cGMP regulates electrogenic chloride secretion in the small and large intestine by inhibiting the type 3 isoform of PDE, increasing intracellular cAMP, and activating PKA (Vaandrager et al., 2000). These data are discussed in more detail in a later section of this review.
C. Cyclic Nucleotide-Gated Channels
CNG channels are a family of voltage-gated cation channels expressed in a variety of cells. Characteristics of CNG channels include: (1) the presence of six transmembrane segments (S1–S6), (2) an ion-conducting pore between the S5 and S6 regions, and (3) two interactive regulatory domains on the cytoplasmic side of the membrane, represented by both the N and C terminus of the channel. In addition, CNG channels responsive to specific cyclic nucleotides contain a cyclic nucleotide-binding domain in the C terminus homologous to that of cyclic nucleotide-dependent protein kinases (Biel et al., 1999c). Their selective sensitivity to cyclic nucleotides resides in the eight β-rolls and three α-helices of the C terminus, as in PKAs and PKGs. Thus, although all CNG channels are activated by both cGMP and cAMP, certain isotypes are more sensitive to cGMP than cAMP, and vice versa.
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are a specific subfamily of CNG channels. These channels are expressed in brain and heart and are modulated by both direct binding of cyclic nucleotides and hyperpolarization of the plasma membrane (Ludwig et al., 1998, 1999). Three distinct HCN channels (HCN 1–3) have been cloned from mouse brain, and a putative fourth member also has been reported (McCoy et al., 1995). These channels possess unique structural characteristics in their pore region and in the S4 transmembrane segment that confer selective ion permeability and unique voltage sensitivity, respectively. Cyclic GMP enhanced the current amplitude and rate of channel activation in membranes from HEK 293 cells transiently expressing HCN2, and cAMP had an even greater effect (Ludwig et al., 1998). HCN2 is permeable to Na+and K+ and demonstrates functional properties similar to the ion current of pacemaker cells in the central nervous system and the heart. Furthermore, two distinct human genes encoding HCN channels cloned from human heart and expressed in HEK 293 cells demonstrated electrophysiological characteristics compatible with a putative function as effectors of cardiac pacemaking in vivo (Ludwig et al., 1999).
The principal family of CNG channels regulates influx of Na+ and Ca2+ into cells and is comprised of tetrameric proteins that are directly “opened” by cyclic nucleotides. In contrast to HCN, CNG proteins display very weak voltage sensitivity and consist of a heterotetrameric assemblage of α- and β-subunits. Only α-subunits form functional monomeric channels when expressed in heterologous systems (Biel et al., 1999c). Three highly homologous α-subunits (CNG 1–3), initially cloned from rods, cones, and olfactory neurons, have been found in pancreas, spleen, testis, ovary, kidney, lung, brain, heart, adrenal gland, and intestine (McCoy et al., 1995). CNG 3-like channels were found in taste buds of rat tongue and may be crucial molecules in sensory organs (Misaka et al., 1997). Only two β-subunits (CNG4 and CNG5) have been identified (Biel et al., 1999c). The CNG 4 gene is differentially spliced, giving rise to several cell-specific isoforms (Biel et al., 1999c). Native CNG channels are thought to be composed of both α- and β-subunits, although homotetramers α4 and β4 may exist. Specific structural features of the pore region of CNG channels confer ion-selective permeability. All channels conduct monovalent cations but are more permeable to Ca2+ than Na+ under physiological conditions (extracellular Ca2+ > other monovalent cations). Furthermore, Ca2+regulates the channel from either the extra- or intracellular compartment in a voltage-dependent manner (Biel et al., 1999c). Ca2+ blocks channel activity directly by interacting with high affinity binding sites and indirectly by activating other proteins, such as calmodulin. The latter effect is an important component of olfactory channel activity.
It has been suggested that β-subunits act as internal regulators of the CNG channel activity, conferring specific properties, such as single channel “flickering” and affinity for cAMP (Biel et al., 1999c). Other modulatory regions of CNG channels include the N-terminal segment and the linker peptide between the S6 transmembrane domain and the cytoplasmic cyclic nucleotide-binding domain. These determine agonist affinity and channel gating and underlie cGMP regulation of visual transduction in rod and cone photoreceptors and cAMP regulation of olfaction (Biel et al., 1999c).
Cyclic GMP mediates phototransduction at the level of rod and cone photoreceptors and regulates neurotransmission within the retina. These processes depend on CNG channels and high [cGMP]i in ganglion, bipolar, and Muller glial cells. Indeed, photoreceptors represent the principal cell types exhibiting cGMP/CNG effects. Deletion of CNG 3 in mice results in selective disruption of cone-mediated photoresponses and is characterized by a progressive degeneration of cone photoreceptors, but not rods or any other retinal cell types (Biel et al., 1999a). Such mice are fertile and develop normally. These data support the hypothesis that a selective mutation in the human CNG 3 gene underlies achromatopsia, a human disease characterized by total color-blindness (Biel et al., 1999a).
D. Cyclic GMP-Regulated Phosphodiesterases
Characterization of biochemical, pharmacological, and structural profiles has identified at least 10 different gene families of PDEs in mammals. Each member contains a conserved catalytic domain of ∼270 amino acids in the C-terminus. This domain cleaves the phosphodiester bond, hydrolyzing the 3′, 5′-cyclic nucleotide to its corresponding nucleotide, 5′ monophosphate (McAllister-Lucas et al., 1995). All PDEs contain heterogeneous regulatory domains and function as dimers, although the functional significance of this latter feature remains undefined.
PDE families 1, 2, 3, and 10 hydrolyze both cGMP and cAMP; PDE families 4, 7, and 8 preferentially cleave cAMP; and PDE families 5, 6, and 9 specifically hydrolyze cGMP. The activity of PDEs is crucial for cellular signaling because metabolism of cyclic nucleotides modulates their intracellular concentrations and affects subsequent cellular and behavioral responses. PDEs regulate cardiac functions, adrenal steroidogenesis, the male erectile response, and phototransduction (Juilfs et al., 1999). Specific PDE targeting sites may localize the enzyme in close proximity to selected proteins, thereby modulating cyclic nucleotide levels in specific compartments.
Cyclic GMP regulates PDEs through three different mechanisms: (1) increasing activity through mass action (PDE5, 6, and 9), (2) altering the rate of hydrolysis of cAMP through competition at the catalytic site (PDE1, 2, and 3), and (3) regulating enzymatic activity through direct binding to specific allosteric sites (PDE2, 5, 6, and 10) (Corbin and Francis, 1999).
The presence of noncatalytic allosteric sites permits cGMP to regulate the activity of PDEs in several ways. The cGMP binding domain in PDE2, 5, 6, and 10 contains two in-tandem homologous sites of about 110 amino acids located at the N terminus. They contain an amino acid sequence different from that of cyclic nucleotide-dependent kinases and CNG channels, representing a separate class of cGMP-regulated proteins (Corbin and Francis, 1999). PDE10 catalyzes the hydrolysis of both cyclic nucleotides, but its physiological function is still unclear. PDE2 is widely distributed, exists as homodimers of 100- to 150-kDa subunits, and catalyzes the hydrolysis of cGMP and cAMP. Cyclic GMP binds to allosteric sites, stimulates PDE2 activity, and increases cGMP hydrolysis, forming a negative-feedback mechanism regulating [cGMP]i. Similarly, cGMP enhances the PDE2-mediated degradation of cAMP, thus cross-regulating its intracellular concentration (McAllister-Lucas et al., 1993).
PDE5, a homodimer of 93-kDa subunits, specifically degrades cGMP. Direct binding of cGMP to allosteric sites promotes phosphorylation of PDE5 by either PKG or PKA, thereby indirectly stimulating enzyme activity. It has been suggested that binding of cGMP to the allosteric sites directly activates PDE5, but this effect has not been demonstrated (Corbin and Francis, 1999). Finally, PDE6 in rod (PDE6-A and -B) and cone (PDE6-C) photoreceptors is comprised of two large catalytically active subunits (α, β in rods; α′2 in cones) associated with various smaller inhibitory γ-subunits and a regulatory δ-subunit. It is believed that cGMP binding to allosteric sites of PDE6 regulates the interaction between catalytic subunits, inhibitory subunits, and transducin, an important step in phototransduction (McAllister-Lucas et al., 1993,1995).
E. Cyclic GMP and Cell Physiology
1. Motility of Vascular Smooth Muscle.
The mechanism underlying contraction of vascular smooth muscle is based on synergistic and antagonistic forces regulating [Ca2+]i. Contractile forces elevate [Ca2+]i and/or sensitize the intracellular environment to Ca2+, whereas dilating forces reduce [Ca2+]i and/or desensitize the intracellular environment to Ca2+. The effect of these opposing processes is phosphorylation of the 20-kDa light chain of myosin (MLC20) on serine 19 by myosin light chain kinase to produce vasoconstriction, and dephosphorylation of MLC20 by myosin light chain phosphatase to produce vasorelaxation, respectively (Bennett and Waldman, 1995).
Exogenous and endogenous compounds (such as nitrovasodilators, endothelium-derived relaxing factor, and natriuretic peptides) produce vasodilatation through increases in [cGMP]i. Cyclic GMP relaxes vascular SMCs by both desensitizing the contractile apparatus to Ca2+ and lowering [Ca2+]i (Fig.4). Glyceryl trinitrate- or SNP-mediated vascular relaxation in the absence of significant reductions of [Ca2+]i has been reported in porcine carotid and coronary arteries (Abe et al., 1990; McDaniel et al., 1992). Cyclic GMP induces a shift to the right of the pCa2+-tension curve in rat mesenteric arteries permeabilized with α-toxin and depleted of stores of [Ca2+]i (Nishimura and van Breemen, 1989). In a similar experiment with de-endothelized smooth muscle strips from rabbit femoral arteries, 8-bromo-cGMP elevated the ED50 of Ca2+ for both contractile force and MLC20 phosphorylation via indirect activation of myosin light chain phosphatase (Lee et al., 1997).

Molecular mechanisms underlying vascular smooth muscle relaxation mediated by cyclic GMP. Cyclic GMP induces smooth muscle relaxation by reducing [Ca2+]i and desensitizing the contractile apparatus to Ca2+. Cyclic GMP reduces [Ca2+]i by (1) inhibiting Ca2+ influx through L-type Ca2+ channels; (2) increasing Ca2+ efflux through activation of (2d) the Ca2+-pumping ATPase and (2b) the Na+/Ca2+ exchanger; also, cGMP may produce membrane hyperpolarization through activation of (2c) the Na+/K+ ATPase and (2a) K+ channels, thereby increasing Ca2+ extrusion by the Na+/Ca2+ exchanger; (3) increasing of Ca2+ sequestration through activation of the sarcoplasmic reticulum Ca2+-pumping ATPase [Ph, phospholamban]; and (4) decreasing of Ca2+ mobilization through inhibition of agonist-induced IP3 formation or inhibition of the IP3 receptor in the sarcoplasmic reticulum. R, receptor; G, G protein; PLC, phospholipase C; IP3R, IP3 receptor. Cyclic GMP desensitizes the contractile apparatus to Ca2+ (5) probably by activating myosin light chain phosphatase, resulting in dephosphorylation of the 20 kDa myosin light chain.
Although PKG I has been proposed as the principal candidate mediating these cGMP effects, other molecular targets cannot be excluded from consideration. Among these, PKA is a possible candidate, based on biochemical and functional convergence between mechanisms underlying cGMP and cAMP-induced vascular relaxation. However, as described above, this hypothetical cyclic nucleotide cross talk does not appear to occur in the cardiovascular system, in vivo, in which cGMP signals via cAMP-independent pathways.
Cyclic GMP mediates vascular smooth muscle relaxation principally by lowering [Ca2+]i (Fig.4). Depending on the tissue, species, and cellular genotype and phenotype, cGMP could affect [Ca2+]i in four different ways: (1) by reducing Ca2+ influx, (2) by increasing Ca2+ efflux, (3) by promoting Ca2+ sequestration in the sarcoplasmic reticulum, and (4) by decreasing Ca2+ mobilization. Thus, cGMP/PKG-dependent mechanisms inhibit voltage-dependent activation of L-type Ca2+ channels by both direct impairment of the channel activity and indirect hyperpolarization of the SMC surface via an increase in the open probability ofK Ca channels (Archer et al., 1994;Carrier et al., 1997; Liu et al., 1997a; Ruiz-Velasco et al., 1998). PKG phosphorylation of putative receptor-operated Ca2+ channels and of the α-subunit of large conductance K Ca channels has been demonstrated, strongly supporting a physiological role for the cGMP-mediated reduction in Ca2+ influx (Blayney et al., 1991; Fukao et al., 1999).
Activation of two different ionic channels, the plasma membrane Ca2+-pumping ATPase and the Na+/Ca2+ exchanger, may mediate an increased efflux of Ca2+ from vascular SMCs. The driving force for extrusion of Ca2+from the cell through the Na+/Ca2+ exchanger, in turn, may be dependent on two other effects mediated by cGMP, depletion of intracellular Na+ via activation of Na+-K+ ATPase and hyperpolarization of the cell membrane via activation of K+ channels. The Ca2+-pumping ATPase and the Na+-K+ ATPase in the plasma membrane are activated by cGMP through PKG (Yoshida et al., 1992;Tamaoki et al., 1997). As described above, cGMP also could indirectly promote Ca2+ transport out of cells through activation of K+ channels and the resultant hyperpolarization. Moreover, 8-bromo cGMP may stimulate the Na+/Ca2+ exchanger independently of any modifications in the membrane potential (Furukawa et al., 1991).
Cyclic GMP induces uptake of Ca2+ into intracellular stores via activation of the sarcoplasmic reticulum Ca2+-pumping ATPase (Andriantsitohaina et al., 1995). The molecular mechanism underlying this effect appears to be PKG phosphorylation of phospholamban, as demonstrated in cultured rat cardiomyocytes (Sabine et al., 1995). Finally, it seems cGMP may inhibit the IP3 signal transduction pathway and thereby lower [Ca2+]i. Indeed, cGMP blocks agonist-induced IP3 formation and induces PKG-mediated phosphorylation of the IP3 receptor in the sarcoplasmic reticulum, subsequently attenuating mobilization of Ca2+(Fujii et al., 1986; Ruth et al., 1993; Komalavilas and Lincoln, 1994,1996).
In summary, there is evidence suggesting a complex role for cGMP in vascular smooth muscle relaxation exerted through the control of [Ca2+]i (Fig. 4). It is likely that, in vivo, different mechanisms operate synergistically to lower levels of [Ca2+]iand induce vascular relaxation utilizing cGMP as a second messenger.
2. Intestinal Fluid and Electrolyte Homeostasis.
ST induces secretory diarrhea by activating guanylyl cyclase and increasing [cGMP]i (Fig. 5). The cellular targets of this pathological effect are the epithelial cells lining the intestine, where the only detected guanylyl cyclase isoform is GC-C, the receptor for ST (Vaandrager and De Jonge, 1994). Disruption of the gene encoding GC-C in mice resulted in resistance to ST-induced diarrhea, demonstrating that GC-C is absolutely required for ST-induced intestinal secretion (Mann et al., 1997; Schulz et al., 1997). In animal models, binding of ST to GC-C stimulated intestinal secretion via increases in cGMP, and the effects of ST on intestinal secretion were mimicked by cell-permeant analogs of cGMP (Field et al., 1978; Hughes et al., 1978; Mezoff et al., 1992). High affinity receptors for ST are localized in brush border membranes of enterocytes from the duodenum to the rectum, with the highest density in the small intestine (Krause et al., 1994b).

Regulation of intestinal secretion by the ST and GC-C. Bacteria, such as E. coli, containing plasmids encoding a member of the homologous peptide family of STs colonize the intestine after the consumption of contaminated food and/or water. Bacterial colonization leads to production of ST in the gut lumen, which specifically recognizes and binds to the extracellular domain of GC-C, expressed in the brush border membranes of intestinal mucosa cells from the duodenum to the rectum. Interaction of ST and the extracellular domain of GC-C is translated across the plasma membrane into activation of the cytoplasmic catalytic domain resulting in the production and accumulation of [cGMP]i. This cyclic nucleotide binds to and activates PKG II, also localized in the intestinal cell brush border membrane. Also, cGMP may activate PKA, either directly or by inhibiting a cAMP-specific PDE and inducing the accumulation of cAMP. The CFTR that is colocalized with GC-C and PKG II in brush border membranes is a substrate for that protein kinase and PKA. CFTR is a chloride channel, and its phosphorylation by PKA or PKG results in a persistent open state, permitting chloride to flow down its concentration gradient from the intracellular to the extracellular compartment. Other ion channels and transporters in the cell maintain the electroneutrality of ST-induced chloride efflux. Vectoral water flux from the basolateral to the apical surface is driven by these ionic conductances, resulting in the accumulation of fluid and electrolytes in the intestinal lumen and secretory diarrhea.
CFTR is a key component mediating the enterotoxigenic effect of ST (Fig. 5). In the absence of functional CFTR, which occurs in patients with cystic fibrosis or in mice with null mutations for CFTR, ST and cGMP analogs fail to induce diarrhea. In addition, the intestine, lung, and pancreas develop severe abnormalities in the regulation of water and salt content (Quinton, 1990). CFTR is recognized as a major mediator of cyclic nucleotide regulation of fluid and electrolyte transport across a variety of epithelia, conducting chloride current across the apical cellular membrane. In addition to CFTR, recent evidence suggests a role for inhibition of brush border membrane electroneutral sodium absorption, possibly mediated by a Na+/H+ exchanger, in mechanisms underlying ST-induced fluid and electrolyte secretion (Vaandrager et al., 2000).
Cyclic GMP activates CFTR and promotes chloride efflux, which presumably drives water transport into the lumen in the intestine (Fig.5). PKG appears to be the principal molecular target of cGMP in the signal sequence leading to CFTR activation. PKG Iα and PKG II phosphorylate CFTR, in vitro, with similar kinetics, suggesting the absence of a specific PKG-mediated function in this process (French et al., 1995). However, PKG II, but not PKG Iα, colocalizes with GC-C in brush borders of enterocytes and activates CFTR in excised membrane patches of various cell lines transfected with CFTR (Lohmann et al., 1997). Cotransfection of rat intestinal cells with CFTR and PKG II results in activation of CFTR, while cotransfection with PKG Iβ does not activate that channel (Vaandrager et al., 1998). Mutation of the PKG II N-terminal myristoylation site reduces localization of the enzyme to the membrane and impairs activation of CFTR. In contrast, a chimeric construct of soluble PKG Iβ and the membrane-directed N-terminal domain of PKG II acquired the ability to activate CFTR. Of significance, agents that increased cGMP in the small intestine of PKG II-deficient mice inhibited the induction of secretion or electrogenic chloride currents, whereas cAMP-induced intestinal secretion was not affected (Pfeifer et al., 1996; Vaandrager et al., 2000). These studies suggest that PKG II is a major physiological mediator of CFTR activation in the small intestine and targeting of this kinase to the membrane is a major determinant for its function in intact cells.
Interestingly, in addition to PKG II, other mechanisms appear to mediate activation of CFTR by cGMP in small intestine and colon (Fig.5). Cyclic GMP can directly activate PKA and stimulate CFTR-mediated chloride currents in human colonic cells that do not express PKG II (Forte et al., 1992; Chao et al., 1994). Also, ST induces electrogenic chloride secretion in the colon and jejunum of PKG II-deficient mice (Vaandrager et al., 2000). The effect of ST on intestinal anion secretion in PKG II-deficient mice was potentiated by addition of amrinone, an inhibitor of the PDE3 isoform that catalyzes the degradation of cAMP but is inhibited by cGMP (Vaandrager et al., 2000). Taken together, these data suggest that increases in [cGMP]i stimulated by ST may induce phosphorylation of CFTR and electrogenic chloride transport by activating PKG II or PKA. The latter protein kinase may be activated directly by cGMP or by local accumulation of [cAMP]i that reflects inhibition of PDE3 by increases in [cGMP]i (Fig. 5).
A key question concerning cGMP-mediated regulation of intestinal fluid and electrolyte transport was the identity of the endogenous ligand that activated GC-C. The paradox that there exists a unique mammalian receptor for bacterial enterotoxins was resolved by the demonstration that extracts of intestine from rats stimulated GC-C and increased cGMP concentrations in T84 human intestinal cells (Currie et al., 1992). The bioactive substance, guanylin, is a 15-residue peptide structurally and functionally homologous to ST. Guanylin is produced by epithelial cells or cells controlling epithelial function in small intestine, colon, adrenal gland, uterus, kidney, pancreas, and oviduct. In T84 human intestinal cells, guanylin stimulates cGMP synthesis with a 10-fold lower potency compared with ST (Forte and Currie, 1995). Uroguanylin, a guanylin-like peptide isolated from the urine of opossum and humans, stimulates GC-C in rat colon, in vitro, and in T84 cells, although with lower potency than ST (Hamra et al., 1993; Kita et al., 1994). Intriguingly, the effect on T84 cells is strongly influenced by the extracellular pH: at low pH (i.e., 5.0–5.5) uroguanylin is 100-fold more potent than guanylin, whereas at a pH of 8.0, guanylin is 4-fold more potent than uroguanylin (Hamra et al., 1997). These latter findings suggest the possibility of a segmental regulation of the intestine by these endogenous peptides because the pH of the intestinal lumen varies considerably from the stomach to the rectum.
The signaling cascade mediated by guanylin/uroguanylin in the intestine is identical with that described for ST (Fig. 5). After binding to GC-C at the apical membrane of enterocytes, the ligands stimulate the intrinsic guanylyl cyclase catalytic activity, initiating a cascade in which there is (1) accumulation of [cGMP]i, (2) stimulation of the membrane-associated PKG II and/or PKA, and (3) phosphorylation of CFTR (Gudi et al., 1996). The electrogenic chloride current, reflecting activation of CFTR, causes net secretion of salt and water into the intestinal lumen. It has been proposed that physiological concentrations of guanylin may act as an intestinal fluid sensor to prevent excessive dehydration and to cleanse the intestinal mucosa (Lohmann et al., 1997). However, at abnormally high concentrations (i.e., in pathological states), guanylin may cause an exaggerated loss of fluid and diarrhea (Forte and Currie, 1995).
The pattern of water and ion movements induced by guanylin, in vivo, was recently studied in closed intestinal loops in rats (Volant et al., 1997). Guanylin (2 μM) stimulated secretion of water, Na+, and Cl− in the duodenum, ileum, and colon through inhibition of Na+ absorption and stimulation of Cl− efflux. This effect was lower than that of ST in all intestinal segments. In contrast, no detectable electrolyte fluxes were observed in the jejunum. However, in a subsequent experiment with closed jejunal loops of anesthetized rats, rat and human guanylin (1 μM) inhibited absorption of fluids and NaCl (Ieda et al., 1999). In the same experiment, guanylin, uroguanylin, and ST induced isotonic fluid movement into the jejunal lumen, but only ST and uroguanylin increased the luminal pH by stimulating bicarbonate secretion.
Although the above discussion suggests an involvement of cGMP in the regulation of intestinal fluid and electrolyte homeostasis, the physiological significance of this mechanism remains unclear. Thus, the presence of GC-C in the intestines of reptiles and birds indicates a clear evolutionary conservation for this isoform of guanylyl cyclase (Krause et al., 1995, 1997). However, mice in which the genes for GC-C or its downstream molecular target, PKG II, have been disrupted have no apparent abnormalities in their intestinal development and function, except for guanylin/uroguanylin/ST unresponsiveness (Schulz et al., 1997; Foster et al., 1999; Pfeifer et al., 1999).
3. Phototransduction.
Phototransduction in the outer segment of the retina represents a vivid example of the importance of cGMP in physiological processes. Indeed, cGMP regulates the recovery phase of visual excitation and adaptation to background light (Ames et al., 1999). Both rod and cone photoreceptors contain unique proteins that act cooperatively to control key second messengers, [cGMP]i and [Ca2+]i. These, in turn, regulate the entire mechanism underlying phototransduction and determine physiological responses to light.
Retinal cells contain two isoforms of membrane-bound guanylyl cyclase, GC-E and -F. These isoforms are expressed only in photoreceptor cells in vertebrates, where they form homodimers that are activated by interaction with specific Ca2+-binding proteins, GCAPs, in the cytoplasmic compartment. In contrast, no putative extracellular ligands have been identified, and these sensory cyclases are considered orphan receptors. GC-E has been detected in both rods and cones, whereas GC-F appears to be present only in rod cells. Disruption of the gene encoding GC-E in mice results in a selective degeneration of cones, but not of rods. Specific mutations of the human GC-E gene are associated with two congenital retinal diseases (Foster et al., 1999).
A similar pathological outcome was observed in CNG 3-deficient mice (Biel et al., 1999a). These mice display a loss of cone cells and an impaired response to light, mimicking achromatopsia (Biel et al., 1999a). The CNG channel, another essential component of the phototransduction mechanisms, is the principal molecular target of cGMP in the plasma membrane of rods and cones. Like the retinal guanylyl cyclases, two distinct, yet homologous, heterotetrameric CNG channels are present in mammalian retina. CNG 3, with high Ca2+ conductance, is in cones, and CNG 1, with low Ca2+ conductance, is in rods. Therefore, in cone photoreceptors, Ca2+ influx during the dark phase is double that in rods (10% of the total ionic current). These inward Ca2+ currents are proportionally counteracted by outward Ca2+ currents carried by Na+/Ca2+K+-exchangers in the plasma membrane of photoreceptors and provide the molecular basis for the difference in timing of photoresponses in cones and rods (Frings, 1997).
The last component of the cGMP-related phototransduction machinery is PDE6, the enzyme that degrades cGMP in photoreceptors. The preferred substrate for this enzyme is cGMP. The enzyme is maximally activated by a cooperative mechanism that involves activated transducin, the γ-subunit of PDE6, and the binding of cGMP to specific allosteric sites in PDE6. In retinal rods, PDE6 exists in a membrane protein complex. In the active state, this complex consists of heterodimers (α- and β-subunits), and in the inactive state it consists of tetramers (α-, β-, and γ2-subunits). A fourth subunit, called δ, serves as a regulatory component of the PDE6 complex and directs translocation of enzymatic activity from membrane to cytoplasm through binding at the prenylated C-terminus of the PDE6β-subunit (Linari et al., 1999). The PDE6 complex in membrane discs of rods is colocalized with rhodopsin and transducin, the two proteins upstream from PDE6 in phototransduction. Rhodopsin contains a chromophore (11-cis-retinal) that confers the ability to respond to photons of light. Transducin is a heterotrimeric GTP-binding protein (αβγ-trimer); it releases an activated complex formed by the α-subunit and a molecule of GTP upon interaction with activated rhodopsin.
Ca2+-binding proteins play important roles in phototransduction (Fig. 6). Recoverin binds Ca2+, and this complex inhibits rhodopsin kinase, which permits activation of rhodopsin by light. Calmodulin activated by Ca2+ binds the β-subunit of retinal CNG channels and lowers their affinity for cGMP. Finally, the family of guanylyl cyclase activating proteins (GCAP 1–3) activate retinal guanylyl cyclase at low [Ca2+]i (Foster et al., 1999). In general, all of these Ca2+-binding proteins work as [Ca2+]isensors to promote an integrated response to light. At low [Ca2+]i they inhibit the hydrolysis of cGMP, stimulate the synthesis of cGMP, and enhance the ability of cGMP to open CNG channels, thereby raising [Ca2+]i. In contrast, at high [Ca2+]i they contribute to decreasing the [cGMP]i and lowering [Ca2+]i.
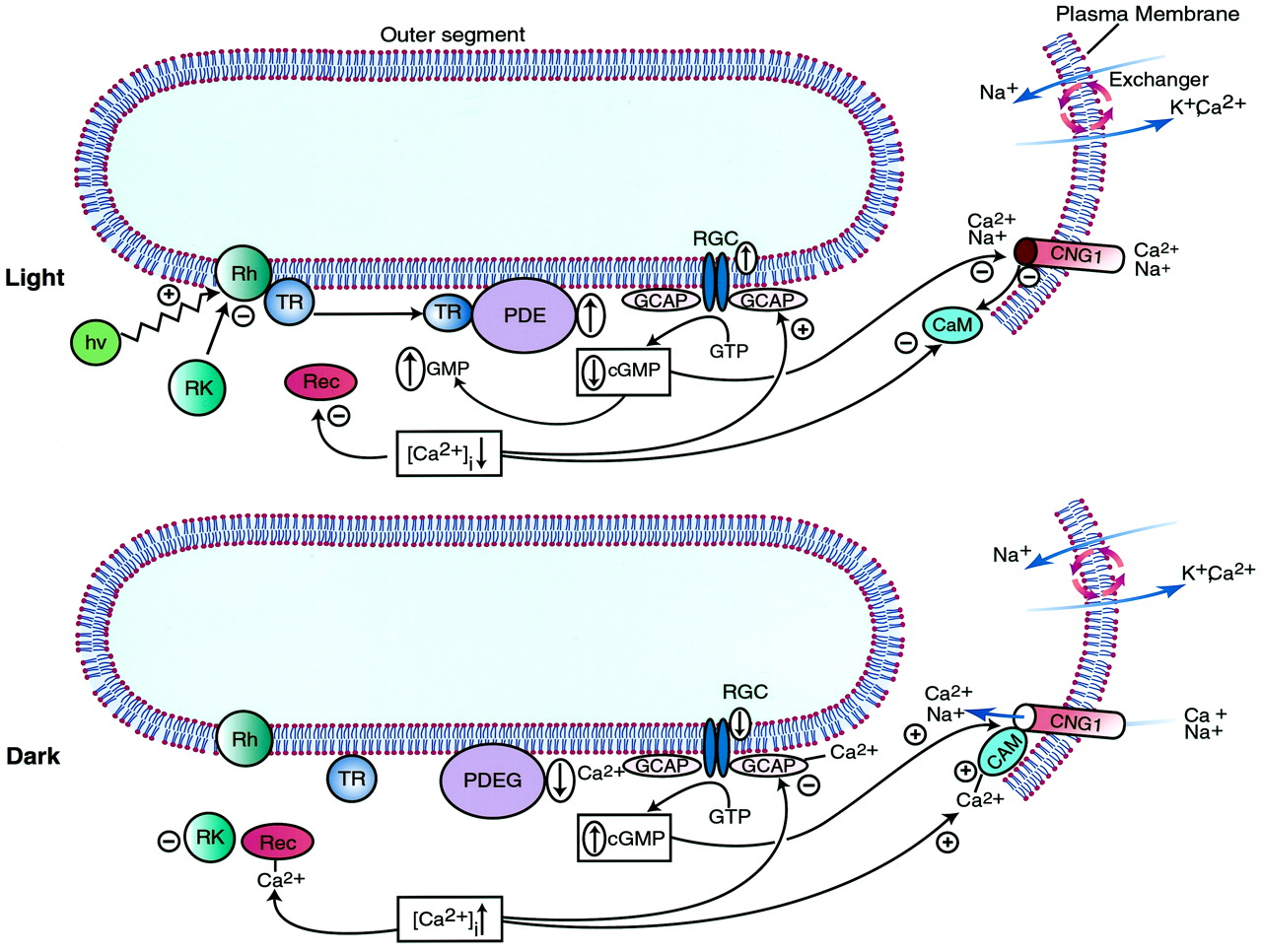
The role of cGMP in phototransduction in vertebrate rods. Light (hv) activates, in sequence, rhodopsin (Rh), transducin (TR), and phosphodiesterase type 6 (PDE), resulting in the hydrolysis of cGMP to GMP. Decreased [cGMP]iresults in the closure of the CNG channel, CNG1, inducing hyperpolarization, a key signal translating photon energy into central nervous system impulses. In addition, closure of CNG1 reduces the influx of Ca2+ while Ca2+ efflux persists through the cation exchanger, reducing [Ca2+]i, which permits modulation of the response to light by decreasing interaction of that cation with (1) GCAPs, resulting in the activation of retinal guanylyl cyclases (RGC) to replenish [cGMP]i; (2) calmodulin (CAM), resulting in its dissociation from CNG1, reducing the potency of cGMP to open that channel; and (3) recoverin (Rec), which inhibits that protein, preventing interaction with rhodopsin kinase (RK), which phosphorylates and inactivates rhodopsin. In the dark state, transducin (TR) and the phosphodiesterase (PDE) are inactive, reflecting the low availability of activated rhodopsin (Rh), and [cGMP]i accumulates, maintaining the cation channel CNG1 in the open conformation and photoreceptors in a depolarized state. In addition, CNG1 in the open state elevates [Ca2+]i, which binds to (1) GCAPs, inhibiting retinal guanylyl cyclase (RGC); (2) calmodulin (CAM), resulting in its association with CNG1, increasing the potency of cGMP to maintain that channel in the open state; and (3) recoverin (Rec), promoting interaction with and inhibition of rhodopsin kinase (RK), potentiating rhodopsin activation. Figure adapted from Current Opinion in Neurobiology, vol. 9, Pugh EN Jr, Nikonov S and Lamb TD. Molecular mechanisms of vertebrate photoreceptor light adaption, 410–418, Copyright 1999, with permission from Elsevier Science.
The molecular components of the mammalian phototransduction machinery combine to create a complex cascade in which cGMP and Ca2+ strictly interact to convert external energy (light in the form of photons) to internal messages (electrical impulses) (Fig. 6). Thus, during the dark state, high levels of cGMP ensure a [Ca2+]i of about 500 nM and maintain the rod outer segment in a depolarized state. Under these conditions, the PDE6 complex is weakly active, the CNG1 channel is open, and the activity of the plasma membrane Na+/Ca2+K+-exchangers limits the rise in [Ca2+]i. Light triggers the sequential activation of rhodopsin, transducin, and the PDE6 complex, leading to hydrolysis of cGMP. The subsequent decrease in [cGMP]i closes the CNG1 channel and disrupts the flux of Ca2+. [Ca2+]i is thereby lowered to about 50 nM, associated with hyperpolarization of the photoreceptor membrane. The light-induced decrease in [Ca2+]i is sensed by the Ca2+-binding proteins, which then inhibit activation of rhodopsin and stimulate guanylyl cyclase, thereby increasing [cGMP]i mediating recovery from photoexcitation.
IV. Conclusions
Over the last decade, with the advent of molecular cloning techniques, our understanding of the family of guanylyl cyclases and their role in physiological and pathophysiological processes has dramatically expanded. However, there are many outstanding questions and exciting challenges for the future. Thus, the precise molecular mechanisms regulating the flow of information between the various domains of guanylyl cyclase to mediate receptor-effector coupling require elucidation. The cellular components regulating guanylyl cyclase activity, including kinases and phosphatases, are important to define. The relationship between guanylyl cyclase activity and cellular metabolism deserves attention, particularly because pGCs are tightly regulated by ATP and alterations in the intracellular concentrations of this nucleotide could have profound consequences on pGC signaling. The integration of guanylyl cyclases into general mechanisms underlying cellular signal transduction and cross talk between the guanylyl cyclase system and other signaling cascades should be examined. Finally, identification and characterization of new downstream receptors of cGMP will further define the role of this important signaling mechanism in cellular physiology.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL59214, CA75123, CA79663). K.A.L. was supported by National Institutes of Health training Grant 5T32 CA09137. I.R.-S. was supported by an NIH minority supplement. J.P. was supported by NIH training Grant T32 DK07705. S.A.W is the Samuel M.V. Hamilton Professor of Medicine, Jefferson Medical College, Thomas Jefferson University, Philadelphia, PA.
Footnotes
-
↵1 Address for correspondence: Scott A. Waldman, MD, PhD, FCP, Division of Clinical Pharmacology, Thomas Jefferson University, 132 South 10th St., 1170 Main, Philadelphia, PA 19107. E-mail: scott.waldman{at}mail.tju.edu
Abbreviations
- NO
- nitric oxide
- [Ca2+]i
- intracellular calcium
- AMPS
- adenosine-5′-O-(3-thiomonophosphate)
- ANP
- atrial natriuretic peptide
- ANPCR
- ANP clearance receptor
- ATPγS
- adenosine-5′-O-(3-thiotriphosphate)
- BNP
- brain natriuretic peptide
- CFTR
- cystic fibrosis transmembrane conductance regulator
- [cGMP]i
- intracellular cGMP
- CNG channel
- cyclic nucleotide-gated channel
- CNP
- C-type natriuretic peptide
- CO
- carbon monoxide
- EGFR
- epidermal growth factor receptor
- EC50
- concentration of ligand yielding a half-maximum response
- eNOS
- endothelial nitric oxide synthase
- GC
- guanylyl cyclase
- GCAP
- guanylyl cyclase activating protein
- PKA
- cAMP-dependent protein kinase
- PKG
- cGMP-dependent protein kinase
- G protein
- heterotrimeric G protein
- GST
- glutathione S-transferase
- Kd
- concentration of ligand yielding half-maximum binding
- KHD
- kinase homology domain
- Ki
- concentration of ligand yielding half-maximum inhibition
- Km
- concentration of substrate yielding half-maximum velocity
- PCR
- polymerase chain reaction
- PDE
- phosphodiesterase
- pGC
- particulate guanylyl cyclase
- PKC
- protein kinase C
- PMA
- phorbol 12-myristate 13-acetate
- PPIX
- protoporphyrin IX
- PAGE
- polyacrylamide gel electrophoresis
- sGC
- soluble guanylyl cyclase
- SMC
- smooth muscle cell
- SNP
- sodium nitroprusside
- ST
- heat-stable enterotoxin
- Vmax
- maximum enzyme velocity
- NOS
- NO synthase
- HCN
- hyperpolarization-activated cyclic nucleotide-gated
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





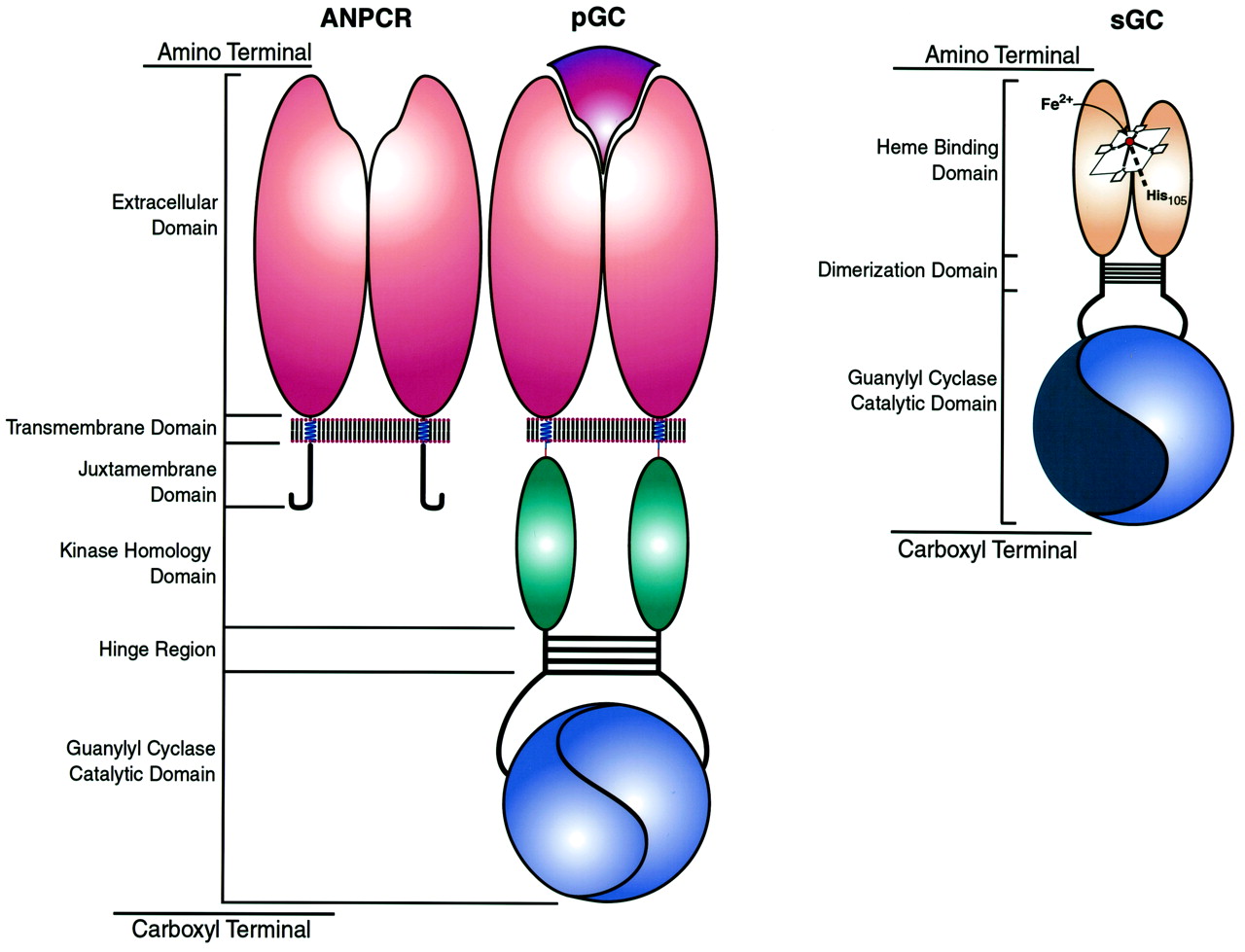{kind=link}
{kind=link}
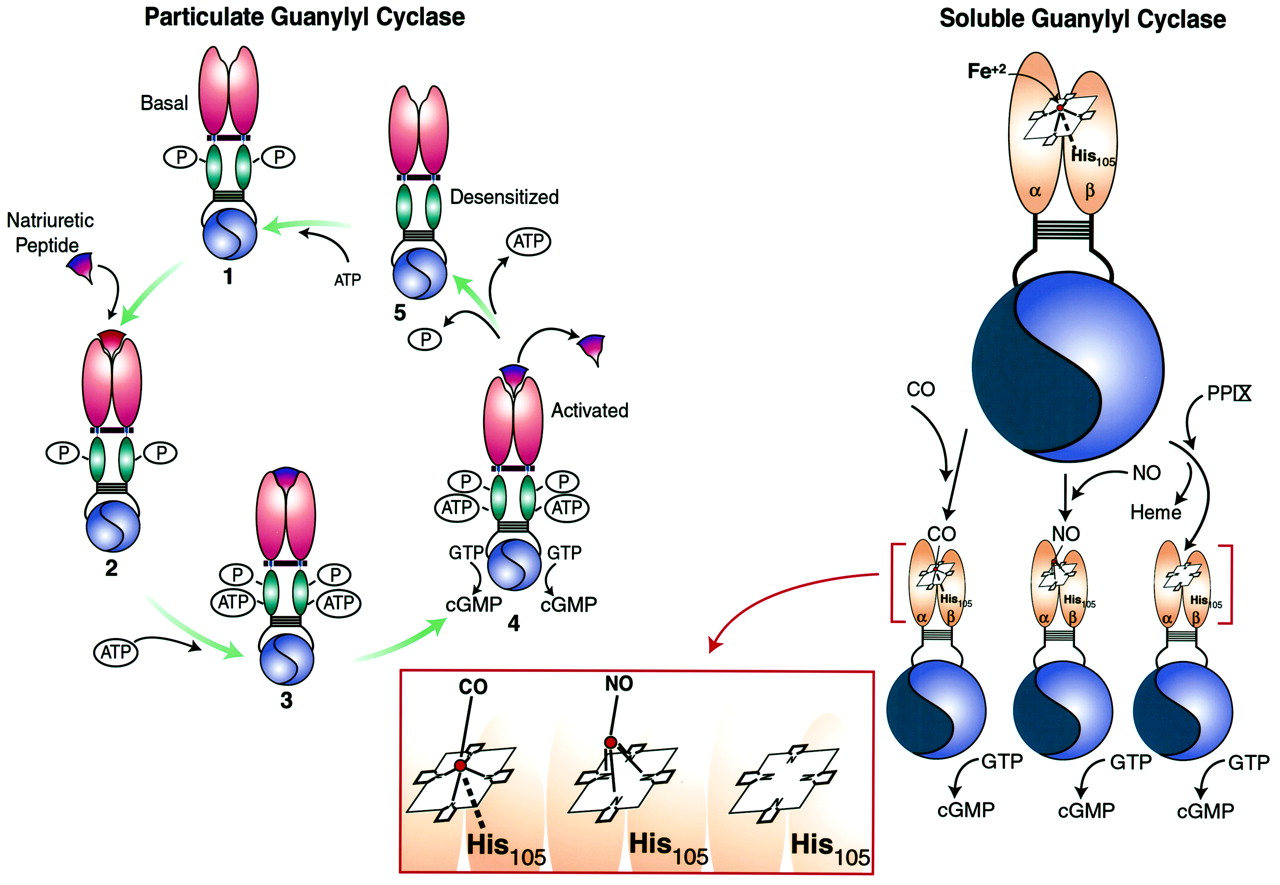{kind=link}
{kind=link}
{kind=link}
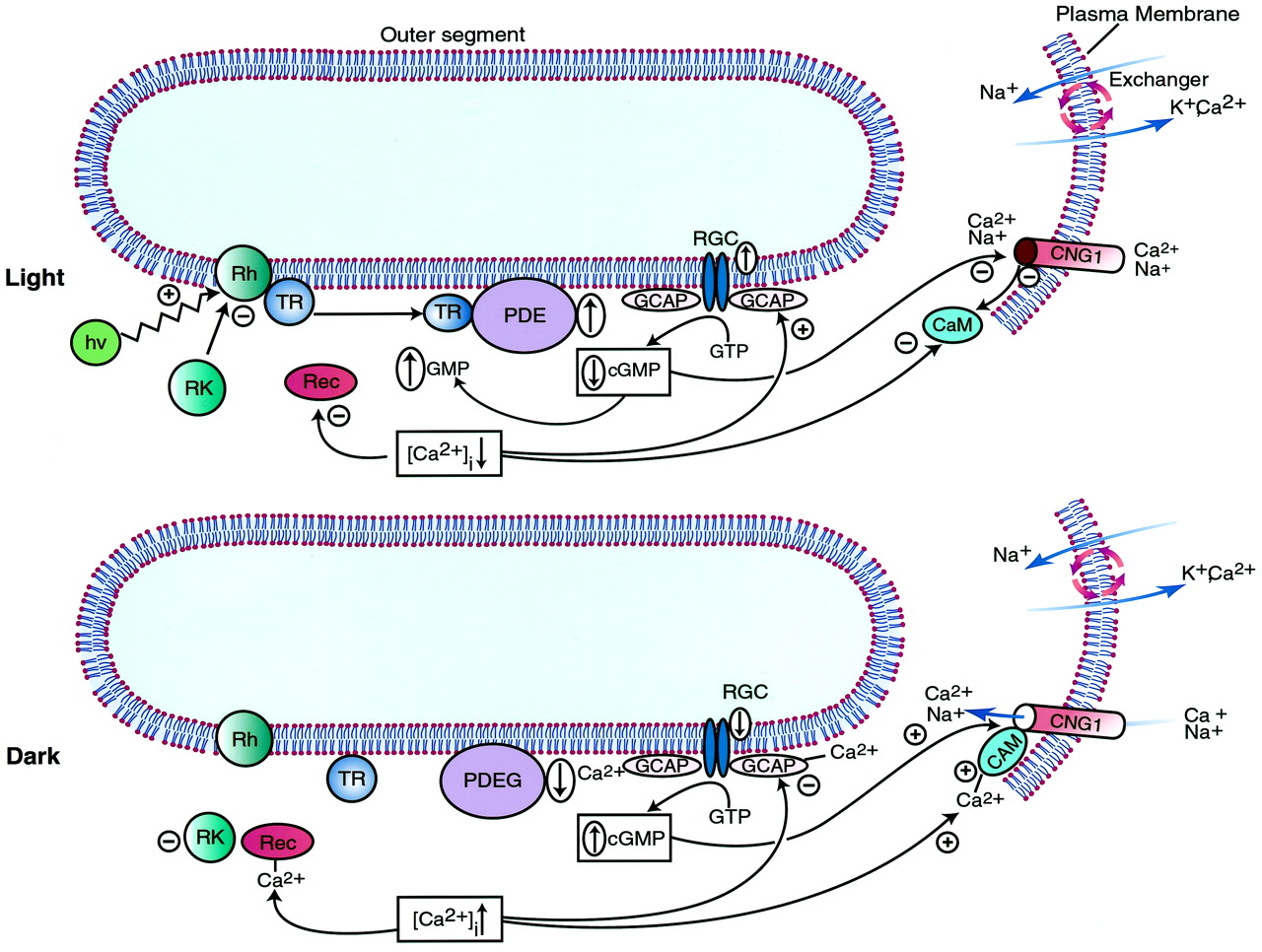{kind=link}