Article Text

Abstract
Background Antitumor vaccines targeting tumor-associated antigens (TAAs) can generate antitumor immune response. A novel vaccine platform using adenovirus 5 (Ad5) vectors [E1–, E2b–] targeting three TAAs—prostate-specific antigen (PSA), brachyury, and MUC-1—has been developed. Both brachyury and the C-terminus of MUC-1 are overexpressed in metastatic castration-resistant prostate cancer (mCRPC) and have been shown to play an important role in resistance to chemotherapy, epithelial–mesenchymal transition, and metastasis. The transgenes for PSA, brachyury, and MUC-1 all contain epitope modifications for the expression of CD8+ T-cell enhancer agonist epitopes. We report here the first-in-human trial of this vaccine platform.
Methods Patients with mCRPC were given concurrently three vaccines targeting PSA, brachyury, and MUC-1 at 5×1011 viral particles (VP) each, subcutaneously every 3 weeks for a maximum of three doses (dose de-escalation cohort), followed by a booster vaccine every 8 weeks for 1 year (dose-expansion cohort only). The primary objective was to determine the safety and the recommended phase II dose. Immune assays and clinical responses were evaluated.
Results Eighteen patients with mCRPC were enrolled between July 2018 and September 2019 and received at least one vaccination. Median PSA was 25.58 ng/mL (range, 0.65–1006 ng/mL). The vaccine was tolerable and safe, and no grade >3 treatment-related adverse events or dose-limiting toxicities (DLTs) were observed. One patient had a partial response, while five patients had confirmed PSA decline and five had stable disease for >6 months. Median progression-free survival was 22 weeks (95% CI: 19.1 to 34). Seventeen (100%) of 17 patients mounted T-cell responses to at least one TAA, whereras 8 (47%) of 17 patients mounted immune responses to all three TAAs. Multifunctional T-cell responses to PSA, MUC-1, and brachyury were also detected after vaccination in the majority of the patients.
Conclusions Ad5 PSA/MUC-1/brachyury vaccine is well tolerated. The primary end points were met and there were no DLTs. The recommended phase II dose is 5×1011 VP. The vaccine demonstrated clinical activity, including one partial response and confirmed PSA responses in five patients. Three patients with prolonged PSA responses received palliative radiation therapy. Further research is needed to evaluate the clinical benefit and immunogenicity of this vaccine in combination with other immuno-oncology agents and/or palliative radiation therapy.
Trial registration number NCT03481816.
- prostatic neoplasms
- immunogenicity
- vaccine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Multiple studies have demonstrated that therapeutic cancer vaccines are safe and can induce specific antitumor immune responses; however, the therapeutic effects observed in clinical trials to date have been modest. Anticancer immunization is a complex process and requires an ideal combination of delivery vectors, tumor-associated antigens (TAAs), and routes of administration that can alter the tumor’s immunosuppressive mechanisms. The first therapeutic cancer vaccine approved by the US Food and Drug Administration in 2010 was sipuleucel-T (Provenge; Dendreon Corporation, Seattle, Washington, USA) for the treatment of minimally symptomatic or asymptomatic metastatic castration-resistant prostate cancer (mCRPC). This paved the way for the development of other therapeutic cancer vaccines for prostate cancer and other solid tumors. Prostate cancer is an ideal model for cancer vaccine immunotherapy because (a) it is slow-growing, (b) prostate-specific antigen (PSA) is a serum marker that can be easily followed, and (c) the prostate is a non-essential organ with several TAAs that can be easily targeted without off-target toxicity.1–3 Another prostate cancer vaccine, PROSTVAC, uses genetically altered poxviruses that deliver the transgenes for PSA to antigen-presenting cells through cellular infection, resulting in the activation of PSA-specific cytolytic T lymphocytes. Despite initial promising results observed in a randomized phase II trial, the placebo-controlled, randomized phase III trial did not show an overall survival (OS) benefit in patients with mCRPC.4 Currently, PROSTVAC is being evaluated in combination with other treatment modalities in several ongoing phase II studies.
Adenoviruses (Ads) are a family of DNA viruses with a linear double-stranded genome contained by an icosahedral, non-enveloped capsid.5 6 Ads have emerged as leading candidate vectors for cancer vaccines. Adenovirus 5 (Ad5) vectors do not integrate (ie, their genomes remain episomal), and risk for germline transmission and/or insertional mutagenesis is extremely low. A new and advanced generation of Ad5 vectors has been developed that, in addition to deletions in the E1 and E3 gene regions, have deletions in the early 2b (E2b) gene regions [E1–, E2b–].7–9 These non-replicating adenoviral vectors may be able to circumvent or decrease neutralizing antiviral immune responses and enable sustained boosting to maximize immune responses. In preclinical studies, Ad5 [E1–, E2b–] vector-based vaccines were used in multiple homologous immunization regimens where they induced immune responses despite the presence of pre-existing Ad5 immunity.10 In in vitro studies of human dendritic cells and in murine vaccination studies, a multitargeted vaccine based on the Ad5 [E1–, E2b–] platform and containing the TAAs CEA, MUC-1, and brachyury induced immune responses directed against all three target antigens with minimal to no antigenic competition.11
We recently published a phase I study of a TriAdeno vaccine regimen composed of the combination Ad5 vaccines containing the TAAs CEA, MUC-1, and brachyury in patients with advanced cancer. We found that concurrent administration of three Ad5 vaccines is safe and can induce CD4+ and/or CD8+ T‐cell responses to at least one TAA encoded by the vaccine with no evidence of antigenic competition.12
The phase I study reported here evaluated the safety and tolerability of an admix of three Ad-based vaccines. ETBX-071 is a PSA-targeting vaccine that employs the Ad5 [E1–, E2b–] vector containing a PSA gene insert. ETBX-061 is a MUC-1-targeting vaccine that employs the Ad5 [E1–, E2b–] vector containing a modified MUC-1 (MUC-1C) gene insert. The investigational product ETBX-051 is a brachyury-targeting vaccine that employs the Ad5 [E1–, E2b–] vector containing a modified brachyury gene insert. ETBX-051, ETBX-61, and ETBX-71 were administered to patients at the same time. The overall goal of this study was to expand immunotherapeutic options for the treatment of mCRPC by employing a multitargeted regimen designed to induce broad antitumor immune responses directed against tumors that overexpress PSA, MUC-1, and/or brachyury. This is the first study to evaluate the concurrent use of the Ad5 PSA/MUC-1/brachyury vaccine in patients with mCRPC.
Patients and methods
Eligibility
Eligible patients had to have incurable mCRPC with radiographic disease progression, defined as any new or enlarging bone lesions or growing lymph node disease consistent with prostate cancer or PSA progression. PSA progression was defined as a sequence of rising values separated by >1 week (two separate increasing values over a minimum of 2 ng/mL as per Prostate Cancer Working Group 2 PSA eligibility criteria). Any number of previous treatment options for mCRPC was allowed, with a withdrawal period of 4–6 weeks following discontinuation of bicalutamide, flutamide, or nilutamide. Patients were required to be ≥18 years of age, no other malignancies within 36 months, have an Eastern Cooperative Oncology Group performance status of ≤1, no autoimmune diseases or significant medical illnesses, and acceptable organ function and hematologic parameters. No systemic or local steroids were permitted, except for physiologic replacement doses that were allowed. Patients were excluded if they had previous treatment with Ad-based immunotherapy, chronic hepatitis B or C infection, HIV, untreated central nervous system metastatic disease, or local treatment of brain metastases within the previous 6 months. Use of herbal products that may decrease PSA levels (such as saw palmetto) was not allowed. Palliative external beam radiation therapy (EBRT) was allowed at the investigator’s discretion. The study was approved by the National Cancer Institute’s Institutional Review Board and was registered on ClinicalTrials.gov.
Assessment of toxicities
Toxicity was evaluated according to the National Cancer Institute Common Terminology Criteria for Adverse Events V.5.0. Toxicities were identified by review of laboratory studies, medical history, and physical examination. The evaluation period for dose-limiting toxicity (DLT) was 28 days from the start of vaccine administration. A DLT was defined as (a) any grade ≥3 toxicity with the exception of transient (≤24 hours) grade 3 flu-like symptoms or fever which could be controlled with medical management; (b) transient (≤24 hours) grade 3 fatigue, local skin reactions, or rash lasting >7 days and associated with desquamation, nausea, headache, or emesis that resolved to grade ≤1; (c) laboratory abnormalities not associated with organ pathology; (d) any grade ≥2 autoimmune reaction (except endocrine-related immune toxicity); or (e) immediate hypersensitivity reaction possibly, probably, or definitely related to vaccine treatment. Any grade 3 autoimmune endocrine-related toxicity that had not resolved clinically within 7 days of initiating therapy was also defined as a DLT.
Study design
This was a single-institution, open-label, phase I clinical study with the goal of determining the safety of concurrent administration of three therapeutic vaccines (ETBX‐071=PSA, ETBX‐061=MUC-1, and ETBX‐051=brachyury). The vaccine was developed as part of a Cooperative Research and Development Agreement between the National Cancer Institute and NantBioScience/ImmunityBio. All three vaccines used the same modified Ad5 vector platform and were administered at a single dose level of 5×1011 viral particles (VP) per vector. In a prior phase I trial of a similar vaccine, Ad5 [E1−, E2b−]-CEA(6D) (ETBX-011) at the dose level of 5×1011 VP per vector was found to be well tolerated, with no DLTs or related severe adverse events (SAEs), and to be the optimal dose for induction of immune responses (NCT01147965).13
All three vaccines were given every 3 weeks for three doses (dose de-escalation cohort in first 6 patients only) and then every 8 weeks for up to 1 year (12 patients in the dose-expansion cohort). Clinical responses were evaluated by CT of the chest/abdomen/pelvis and technetium bone scan at baseline, at week 14, and every 8 weeks thereafter. Response to treatment was measured by RECIST V.1.1. In addition to a baseline CT and bone scan, confirmatory scans were obtained up to 6 (not less than 4) weeks following initial documentation of objective response. Treatment lasted 6 weeks in the dose de-escalation cohort (first six patients) and continued until disease progression, unacceptable adverse events (AEs), withdrawal from study, or week 54 in the dose-expansion cohort. The safety and efficacy of the study drugs were assessed until the end of treatment or for a maximum of 54 weeks.
Immune assays
Blood samples for immune assessments were collected at baseline, week 6, week 14, week 30, and at the end of treatment; in select cases, research blood was also collected at 3 weeks. For peripheral blood mononuclear cell (PBMC) analyses, blood was collected in sodium heparin tubes and PBMCs were separated by Ficoll-Hypaque density gradient separation. The resulting cells were cryopreserved in 90% heat-inactivated human AB serum and 10% dimethyl sulfoxide (DMSO) at a concentration of 1×107 cells/mL. For serum assays, blood was collected in serum separator tubes, spun down, and stored at –80°C before analysis.
Serum assays
The presence of anti-PSA antibodies in the serum of patients before and after vaccination was determined by ELISA using methods previously described with slight modifications.14 15 Polyvinyl chloride 96-well microtiter plates were coated in 2 µg/mL of a purified, recombinant PSA protein (Fitzgerald Industries, Concord, Massachusetts, USA) or bovine serum albumin (BSA) as a negative control. Serum was diluted at 1:50, 1:250, 1:1250, and 1:6250. Purified mouse anti-PSA immunoglobulin G1 (IgG1) antibody (Fitzgerald Industries) was used as a positive control, and an isotype-matched IgG1 antibody (MOPC-21; Sigma-Aldrich, St. Louis, Missouri, USA) served as a negative control. Titer was defined as the maximum dilution where the absorbance at 450 nm was twice that obtained from the same sample on the control BSA plate. Anti-Ad5 neutralizing antibodies were measured as previously described10 16–18 in serum obtained from patients before and after vaccination.
Serum levels of cytokine/soluble factors were determined before and after vaccination using commercially available kits per the manufacturer’s instruction. Levels of serum analytes were also determined in healthy donors obtained from the National Institutes of Health Clinical Center Blood Bank (NCT00001846). Interleukin (IL)-8 was measured by AlphaLISA (PerkinElmer, Waltham, Massachusetts, USA), sCD27 and sCD40L were measured using Instant ELISA kits (Life Technologies, Carlsbad, California, USA), and sPD-1 and sPD-L1 were measured with ELISA kits (Abcam, Cambridge, UK).
PBMC assay to measure antigen-specific T cells
TAA-specific T cells were analyzed using the methods previously described19 in cryopreserved PBMCs isolated from patients before and after vaccination. PBMCs were stimulated in vitro with overlapping 15-mer peptide pools encoding PSA, MUC-1, and brachyury and analyzed by intracellular cytokine staining. Peptide pools encoding the human leukocyte antigen (HLA) and CEFT (a mixture of peptides of cytomegalovirus, Epstein-Barr virus, influenza, and tetanus toxin) served as negative and positive controls, respectively. The absolute number of viable CD4+ or CD8+ T lymphocytes producing cytokine (interferon-γ (IFN-γ), tumor necrosis factor-α (TNFα), IL-2) or positive for a degranulation marker (CD107a) at the end of expansion was calculated per 1×106 cells plated at the start of the stimulation assay. This calculation takes into account not only the percentage but also the total number of viable antigen-specific T cells expanded in the stimulation assay. The background signal (obtained with the HLA peptide pool) and any value obtained prior to vaccination were subtracted from those obtained after vaccination ([post-TAA–post-HLA]–[pre-TAA–pre-HLA]). A patient was scored as developing a TAA-specific T-cell response during therapy if the patient had more than 250 CD4+ or CD8+ T cells that produced IFN-γ, TNFα, or IL-2 or were positive for CD107a at the end of the stimulation assay per 1×106 cells that were plated at the start of the assay. Patients were scored as having pre-existing TAA-specific T cells if pre-TAA–pre-HLA >250/1×106 cells and pre-TAA/pre-HLA >2. Multifunctional TAA responses, defined as CD4+ or CD8+ T cells expressing two or more of IFN-γ, TNFα, IL-2, or CD107a, were also quantified before and after vaccination; the frequency of patients developing a >3-fold, >10-fold, or >100-fold increase in multifunctional TAAs post-vaccination versus pre-vaccination was determined.
Statistical analysis
Summary statistics were used to describe demographic data and baseline performance status characteristics. Descriptive statistics were examined for indications of dose-related toxicity. Disease control rate (DCR) was defined as the percentage of subjects who experience partial response (PR), complete response (CR), or stable disease (SD) lasting for at least 6 months. Progression-free survival (PFS) was defined as the time from the date of first treatment to the date of disease progression or death (any cause) whichever occurs first. OS was evaluated using Kaplan-Meier methods and was defined as the time from the date of first treatment to the date of death from any cause. Immunological parameters were mainly analyzed descriptively and displayed in graphic format using GraphPad Prism V.8 (GraphPad Software, La Jolla, California, USA). In some cases, statistical analysis was performed in RStudio (Boston, Massachusetts, USA); p values for unpaired data were calculated using the Mann-Whitney test, and p values for paired data were calculated using the Wilcoxon matched-pairs signed-rank test.
Results
Patient population
Eighteen patients enrolled between July 2018 and September 2019 received at least one vaccination at the National Cancer Institute in Bethesda, Maryland. Six patients were enrolled in a dose de-escalation cohort, and 12 patients were enrolled in a dose-expansion cohort. Patients were predominantly white (83.3%) with a median age of 71.6 years (range, 54–88 years). Eight patients had received ≥3 prior lines of therapy for mCRPC. The median number of prior therapies was 3 (range, 0–7). Ten patients received prior immunotherapy regimens, seven of whom received therapeutic cancer vaccines and six of whom were treated with checkpoint inhibitors. Baseline characteristics are summarized in table 1.
Patient demographics
Safety
Safety and efficacy were assessed until the end of treatment or for a maximum of 54 weeks. A total of 62 vaccinations were given. Treatment was well tolerated and no DLTs were observed. No SAEs related to treatment were observed. The most common AEs were grade 1 and 2 injection-site reactions (94.4%), flu-like symptoms (58.8%), fatigue (38.9 %), and back pain (22.3 %). All injection-site reactions resolved without intervention. All AEs are listed in table 2 (the cut-off date was September 1, 2020). Grade 3 SAEs included anemia, dehydration and hypotension (one patient in the dose de-escalation cohort), and lung infection (one patient in the dose-expansion cohort). One patient in the dose de-escalation cohort decided to discontinue therapy due to expected side effects (fever/chills) after the first dose of vaccine. The recommended phase II dose was determined to be 5×1011 VP.
Adverse events
Response to therapy
Evaluation of clinical benefits was a secondary objective of this study. Best response (per RECIST V.1.1) was a PR lasting 16 weeks observed in one patient (8.33%) in the dose-expansion cohort. No CRs were observed, but five patients had SD lasting >6 months (figure 1). The DCR (the percentage of patients who experience a PR, CR, or SD lasting at least 6 months) was 60%. Median PFS was 22 weeks (95% CI: 19.1 to 34). Median PSA was 25.58 ng/mL (range, 0.62–1006 ng/mL). As of September 1, 2020, four patients in the dose de-escalation cohort had died due to progressive disease (PD). Five of six patients in the dose de-escalation cohort completed all three planned doses of vaccine. Median OS was not reached, and 12-month OS probability for all patients was 83.3% (95% CI: 56.8% to 94.3%).
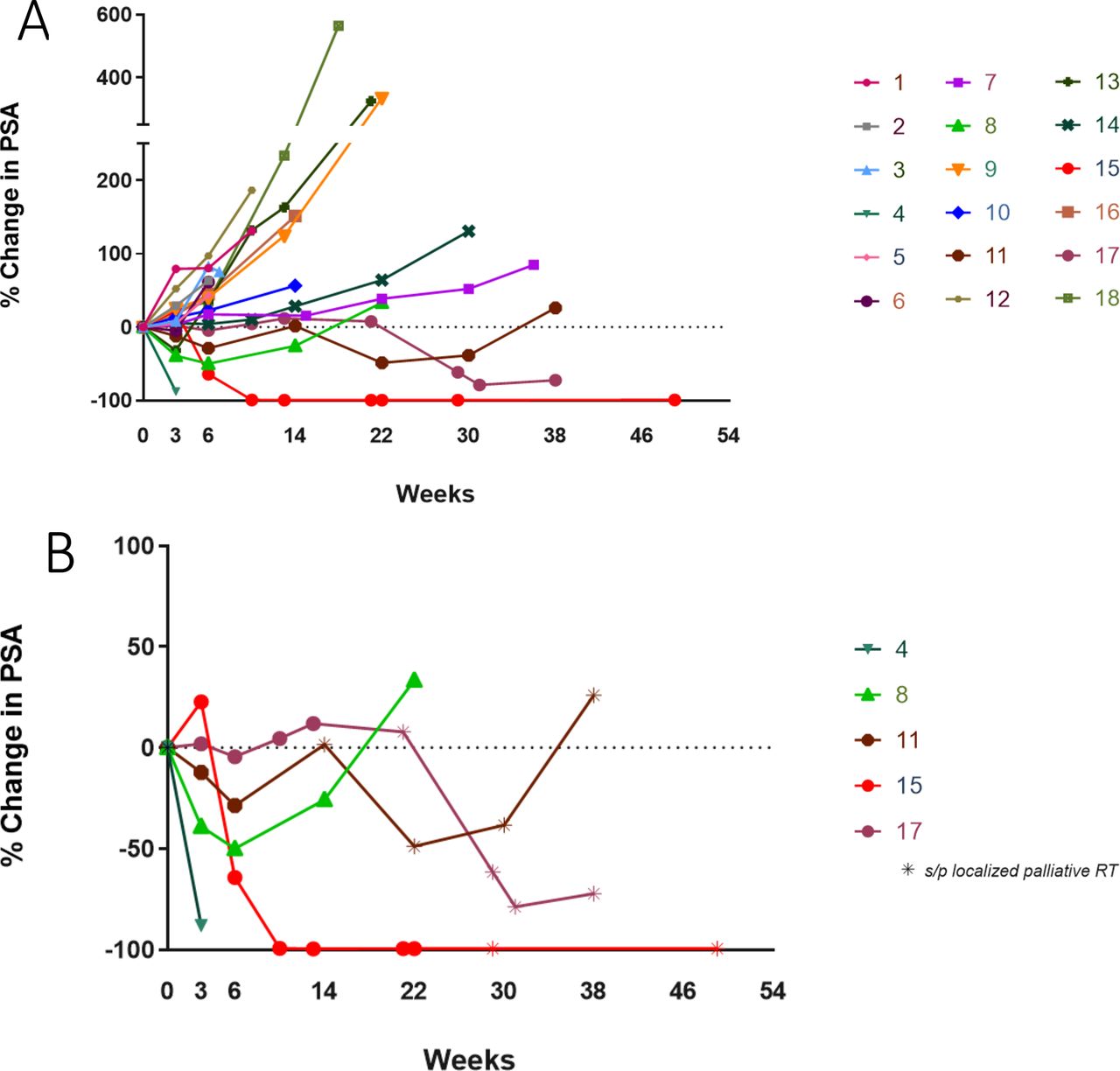
PSA changes. This spider plot of PSA measurement data shows percentage change over time: (A) all patients and (B) PSA responders only. PSA, prostate-specific antigen.
None of the patients in the dose-expansion cohort completed the planned 54 weeks of treatment: five came off study due to PD, two came off study due to preference for another line of therapy, and five came off study per primary investigator discretion due to clinical material availability.
PSA responders
Five patients had confirmed serum PSA declines (table 3, figures 1B and 2). Two patients received palliative EBRT once to a single painful skeletal area while undergoing vaccine treatment (patient #15 to left humerus; patient #17 to left sacrum); patient #11 received EBRT twice to his lumbar spine. All three patients had prolonged PSA declines lasting >6 months. In addition, patient #11 had a confirmed PR by RECIST V.1.1, whereas patient #15 has had a sustained PSA response lasting >54 weeks (figure 1B).

{kind=link}
{kind=link}
Waterfall plot of best PSA response. PSA response was confirmed in five patients. PSA, prostate-specific antigen.
PSA responders
Immune assays
Anti-PSA antibodies were measured in serum collected from patients before and after vaccination. All patients were negative for anti-PSA antibodies at all time points analyzed, including patients with PSA responses following treatment. Neutralizing antibodies to Ad5 were also measured in all patients before and after vaccination. At baseline, 3 of 18 patients had detectable neutralizing Ad5 antibodies. High titers of anti-Ad5 antibodies were induced after multiple vaccinations in all patients, including those who developed PSA responses (online supplemental table 1). Serum cytokine levels of IL-8 and the soluble factors sCD27, sCD40L, sPD-1, and sPD-L1 were also measured in patients before and after vaccination. Prior to therapy, IL-8 and sPD-1 were statistically higher in patients with prostate cancer enrolled in this study than in healthy donors (online supplemental figure 1A and B). Early increases in specific soluble factors, which may be indicative of increased general immune activation, were detected after vaccination. sCD27 and sPD-1 were statistically significantly elevated after 6 weeks of therapy with the Ad5 PSA/MUC-1/brachyury vaccine compared with pretherapy levels (online supplemental figure 1C, D). sCD27 is preferentially derived from activated CD4+ T cells, and studies have shown that immunotherapy can increase this factor.20 There were some trends noted in the change of specific serum analytes between those patients who did versus those who did not develop PSA responses. Levels of sCD40L were decreased in 4 (80%) of 5 patients with PSA responses compared with 1 (8%) of 13 patients who did not develop a PSA response (online supplemental table 2). sCD40L, a functional trimer that is shed from activated T lymphocytes and platelets, has been reported to have an immunosuppressive effect.21 22 Conversely, IL-8, a proinflammatory cytokine/chemokine involved in the recruitment of leukocytes to sites of injury and inflammation, increased at an early time point following vaccination in 5 (100%) of 5 patients who developed PSA responses compared with 5 (38%) of 13 who did not develop PSA responses (online supplemental table 2).
Supplemental material
Based on PBMC availability and quality, antigen-specific T cells could be evaluated before and after vaccination in 17 of 18 patients. Using the criteria described in the Methods section, pre-existing T cells targeting PSA, MUC-1, or brachyury were detected in 47%, 58%, and 69% of patients, respectively. Vaccination with the Ads5 PSA/MUC-1/brachyury vaccine increased CD4+ and/or CD8+ T‐cell responses to at least one TAA encoded by the vaccine in all patients; PSA-specific T cells were developed in 11 (65%) of 17 patients, MUC-1-specific T cells in 17 (100%) of 17 patients, and brachyury-specific T cells in 14 (88%) of 16 patients (table 4). Sixteen (94%) of 17 patients developed T-cell responses to >1 antigen encoded by the vaccine; 8 patients developed T-cell responses to all three TAAs in the vaccine. Polyfunctional TAA‐specific responses, defined as CD4+ or CD8+ T cells that express ≥2 of the markers IFN-γ, TNFα, IL‐2, or CD107a, were also measured before and after vaccination. Using the criteria of a >10‐fold increase post-vaccination versus pre-vaccination, or the presence of >1000 polyfunctional cells post-vaccination per 1×106 PBMCs (if negative at pre-vaccination), polyfunctional T cells targeting at least one of the antigens tested increased in 13 (76%) of 17 patients after vaccination (table 4). Multifunctional T cells targeting PSA, MUC-1, or brachyury were developed after vaccination in 35%, 47%, and 25% of patients, respectively. All patients with PSA responses developed a >10-fold increase in multifunctional T cells targeting at least one of the three antigens evaluated. The generation of long‐lasting polyfunctional T cells, which can persist for years after initial vaccination, has been associated with improved OS.23
Development of TAA-specific T cells during therapy
Discussion
We report here the first-in-human trial of the Ad5 PSA/MUC-1/brachyury vaccine platform in patients with mCRPC. The primary objective of this trial was to evaluate the safety and to determine the recommended phase II dose of the Ad5 PSA/MUC-1/brachyury vaccine. As expected, this vaccine was well tolerated and no DLTs were observed, nor were any SAEs attributed to the vaccine. The only grade 3 toxicity attributed to the vaccine was decreased lymphocyte count in two patients. The only grade 2 toxicities attributed to the vaccine were injection-site reactions (n=7) and decreased lymphocyte count (n=1). SAEs unrelated to the vaccine included anemia (n=1), lung infection (n=1), dehydration (n=1) and hypotension (n=1). One patient in the dose de-escalation cohort discontinued therapy due to expected mild side effects (grade 1 fever and grade 1 chills) after the first dose of vaccine. No biopsies were required in this phase I study. These safety data are consistent with findings from prior clinical trials showing that Ad5 vaccines are safe and well tolerated.12 13 16
There are several limitations to this study, including a small sample size, which requires cautious interpretation of the findings. Although this trial was not powered to evaluate PFS, OS, or clinical benefit, interesting findings were observed. Although no CRs were observed, one patient had a PR lasting 16 weeks and five patients had PFS for at least 6 months. Median PFS was 22 weeks (95% CI: 19.1 to 34). Median OS was not reached, and the 12-month OS probability for all patients was 83.3% (95% CI: 56.8% to 94.3%), indicating that some patients may have derived clinical benefit. The most interesting finding from this study is that of the five patients with a PSA response, two were heavily pretreated (patients #11 and #17) and underwent palliative EBRT due to symptomatic metastases, yet they had prolonged and marked PSA declines lasting from 38 weeks up to 54+weeks, indicating possible synergy between EBRT and the Ad5 PSA/MUC-1/brachyury vaccine (table 3).
We were not able to complete all planned vaccinations in the dose-expansion cohort, and five patients including patients #15 and #17 discontinued treatment early due to clinical material availability. Despite having to discontinue treatment early, patients #15 and #17 had PSA responses lasting 11+ months and 9 months, respectively, after their last dose of vaccine. Recently, it became clear that the effect of EBRT goes beyond direct tumor killing to promoting systemic anticancer immunity.24 Preclinical studies have demonstrated that the combination of EBRT and vaccine can modulate the immunogenicity of the tumor24–26 by increasing its susceptibility to antigen-specific cytotoxic T lymphocytes by changing the tumor surface phenotype.27 In addition, low-dose radiation upregulates major histocompatibility complex (MHC) class I, cellular expression of death receptor Fas/CD95, and intercellular adhesion molecule-1/CD54, promoting tumor cell death via proapoptotic mechanisms and killing by antigen-specific immune cells.28–31 Therefore, radiotherapy that promotes the release of tumor neoantigens together with strategies to overcome dominant immunosuppressive pathways, such as therapeutic vaccines, may lead to effective immune-mediated tumor killing, a phenomenon known as the abscopal effect. Although it is uncommon, the abscopal effect is still evaluated in clinical trials.32
The three TAAs (PSA, MUC-1, and brachyury) encoded in this adenoviral vaccine platform are attractive targets in prostate cancer treatment. Both MUC-1 and brachyury are expressed on several types of human adenocarcinomas, with high expression seen in colorectal cancer, breast cancer, non‐small cell lung cancer, and mCRPC.33–35 Multiple studies have demonstrated that brachyury and/or MUC-1 overexpression are markers of poor prognosis, treatment resistance to chemotherapy and radiation, and tumor aggressiveness, making targeting those antigens a logical strategy.36–40 Preclinical studies have also demonstrated a positive correlation between androgen receptor and MUC-1, including MUC-1’s contribution to the generation of neuroendocrine prostate cancer, making it a viable target for prostate cancer immunotherapy.41
Multiple therapeutic cancer vaccine trials have been conducted using a combination of TAAs, but this is the first trial to use this triad of TAAs (PSA, MUC-1, and brachyury). Immune correlative studies have revealed interesting results. The Ad5 PSA/MUC-1/brachyury vaccine generated CD4+ and/or CD8+ T‐cell responses to at least one TAA encoded by the vaccine in all patients. Notably, all but one patient developed T cells to more than one antigen encoded by the vaccine, and nearly half of the patients (47%) developed responses after vaccination to all three TAAs encoded by the vaccine. Furthermore, polyfunctional TAA‐specific responses, defined as CD4+ or CD8+ T cells that express ≥2 of the markers IFN-γ, TNFα, IL‐2, or CD107a, were increased in the majority of patients after vaccination, and all patients with PSA declines had a >10-fold increase in multifunctional T cells with therapy. These increases in antigen-specific T cells were observed despite all patients developing anti-Ad5 neutralizing antibodies after multiple vaccinations. This study adds to the existing evidence12 15 19 42–45 that antitumor vaccines directed against PSA, MUC-1, and brachyury are well tolerated and can generate antitumor immune responses.
Conclusions
This is the first trial to investigate the effects of Ad5 PSA/MUC-1/brachyury in patients with mCRPC. The primary end points were met and there were no DLTs. The Ad5 PSA/MUC-1/brachyury vaccine platform is well tolerated with acceptable safety and produced PSA decreases and SD lasting >6 months in some patients, especially in those who received concurrent palliative EBRT. Its potential for combination with different immuno-oncology agents makes it a promising agent for future studies. Subsequent trials will evaluate the use of this vaccine in combination with checkpoint inhibitors and/or other immune modulators, including palliative EBRT.
Acknowledgments
The authors would like to thank the patients and research staff who made this trial possible. Also, the authors would like to thank Bonnie L. Casey and Debra Weingarten for editorial assistance in the preparation of this manuscript, and Angie Schwab and Keanan Wright for technical assistance with immune assays. JS and CP have patents involving agonist epitopes for brachyury, MUC-1, and PSA.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @mbilusic, @chfloudas, @HsaterMD, @gulleyj1
Contributors MB, JS, and JG designed the study. MB, SM. FK, RM, CJ, JST, JR, HAS, and JG treated patients and acquired clinical data. MB and JM analyzed the clinical data. SR and PSS developed the vaccine. RD, YT, CP, and JS analyzed and interpreted immune assay data. MB wrote the manuscript. MB, SM, RM, FK, JST, JS, JR, HAS, and JG contributed to overall data interpretation and editing of the manuscript. All authors critically reviewed the manuscript. All authors read and approved the final manuscript.
Funding This research was supported by the Intramural Research Program, Center for Cancer Research, National Cancer Institute, National Institutes of Health, and via a Cooperative Research and Development Agreement (CRADA) between the National Cancer Institute and NantBioScience/ImmunityBio.
Competing interests ImmunityBio authors are employees of ImmunityBio, Inc.
Patient consent for publication Not required.
Ethics approval All patients gave written informed consent for participation. This study was approved by the National Cancer Institute’s Institutional Review Board. The trial registration number is NCT03481816.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.