Article Text

Abstract
Background CD40 agonist immunotherapy can potentially license antigen-presenting cells to promote antitumor T-cell activation and re-educate macrophages to destroy tumor stroma. Systemic administration of CD40 agonists has historically been associated with considerable toxicity, providing the rationale for development of tumor-targeted immunomodulators to improve clinical safety and efficacy. This phase I study assessed the safety, tolerability, preliminary antitumor activity, and preliminary biomarkers of ABBV-428, a first-in-class, mesothelin-targeted, bispecific antibody designed for tumor microenvironment-dependent CD40 activation with limited systemic toxicity.
Methods ABBV-428 was administered intravenously every 2 weeks to patients with advanced solid tumors. An accelerated titration (starting at a 0.01 mg/kg dose) and a 3+3 dose escalation scheme were used, followed by recommended phase II dose cohort expansions in ovarian cancer and mesothelioma, tumor types associated with high mesothelin expression.
Results Fifty-nine patients were treated at doses between 0.01 and 3.6 mg/kg. The maximum tolerated dose was not reached, and 3.6 mg/kg was selected as the recommended phase II dose. Seven patients (12%) reported infusion-related reactions. Treatment-related grade ≥3 treatment-emergent adverse events were pericardial effusion, colitis, infusion-related reaction, and pleural effusion (n=1 each, 2%), with no cytokine release syndrome reported. The pharmacokinetic profile demonstrated roughly dose-proportional increases in exposure from 0.4 to 3.6 mg/kg. Best response was stable disease in 9/25 patients (36%) treated at the recommended phase II dose. CD40 receptor occupancy >90% was observed on peripheral B-cells starting from 0.8 mg/kg; however, no consistent changes from baseline in intratumoral CD8+ T-cells, programmed death ligand-1 (PD-L1+) cells, or immune-related gene expression were detected post-ABBV-428 treatment (cycle 2, day 1). Mesothelin membrane staining showed greater correlation with progression-free survival in ovarian cancer and mesothelioma than in the broader dose escalation population.
Conclusions ABBV-428 monotherapy exhibited dose-proportional pharmacokinetics and an acceptable safety profile, particularly for toxicities characteristic of CD40 agonism, illustrating that utilization of a tumor-targeted, bispecific antibody can improve the safety of CD40 agonism as a therapeutic approach. ABBV-428 monotherapy had minimal clinical activity in dose escalation and in a small expansion cohort of patients with advanced mesothelioma or ovarian cancer.
Trial registration number NCT02955251.
- tumor microenvironment
- therapies
- investigational
- immunomodulation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
CD40 is a key member of the tumor necrosis factor (TNF) receptor superfamily that can act as a bridge for the innate and adaptive immune systems via ligation on antigen-presenting cells (APCs), such as B-cells, dendritic cells, monocytes, and macrophages; this ligation can lead to the upregulation of costimulatory pathways essential for T-cell activation and proliferation. In concert with B-cell receptor signaling, CD40 can directly stimulate the generation of antibody-producing cells, the humoral effector arm for adaptive immunity in an immune response.1 2 In addition, CD40 can re-educate macrophages to destroy tumor stroma.3 4
Various approaches to activating CD40 have been attempted in patients with cancer. For example, subcutaneous administration of multimeric recombinant human CD40 ligand (rhuCD40L) has been associated with objective clinical responses.5 In this study, the maximum tolerated dose (MTD) of rhuCD40L was based on transient grades 3/4 elevations in serum liver transaminases,5 and liver function test abnormalities have since been identified as a class effect of CD40 agonists.6 CD40 agonist monoclonal antibodies have also been tested clinically, including selicrelumab (formerly known as CP-870,893).3 7–13 Although some clinical efficacy has been demonstrated, systemic delivery of these agents has a narrow therapeutic window due to toxicities, such as cytokine release syndrome and hepatotoxicity. Systemic toxicity following systemic delivery of CD40 agonists may be attributed to CD40 receptor expression on numerous cell types (eg, platelets, endothelial cells, fibroblasts, and various immune cell subsets).1 Consequently, alternate delivery methods of CD40 agonists have been employed, including subcutaneous and intratumoral administration of anti-CD40 antibodies or adenoviral vectors expressing CD40 ligand.14–18
The historic toxicity associated with systemic administration of agonists for receptors like CD40 led to the development of an emerging class of tumor-targeted immunomodulators to further improve clinical efficacy and safety.19 20 Tumor-targeted immunomodulators are bispecific antibodies that target both a tumor-associated antigen, such as human epidermal growth factor receptor 2, fibroblast activation protein, or mesothelin, and an immunomodulatory receptor expressed by intratumoral immune cells, such as CD40 or 4-1BB.19–22 ABBV-428 is a first-in-class tumor-targeted immunomodulator designed to achieve CD40 activation in the tumor microenvironment, directed by interaction with the tumor antigen mesothelin, for a localized, non-systemic immune activation.20 Mesothelin is a cell-surface molecule expressed on mesothelial cells lining the pleura, peritoneum, and pericardium, and on the surface epithelium of ovaries, tonsils, and fallopian tubes.23–25 In cancers, mesothelin is highly expressed on epithelial mesotheliomas and carcinomas of the pancreas, lung, and ovary.25 In addition to APC activation, immunomodulation via ABBV-428 may also help counter the regulatory T-cell and M2-like macrophage-rich tumor microenvironment associated with some cancers that highly express mesothelin, such as pancreatic ductal adenocarcinoma and mesothelioma.20 26 27
ABBV-428 is a bispecific antibody that targets mesothelin via a C-terminal single-chain variable fragment flanking Fc-modified human IgG1 and CD40 via an N-terminal single-chain variable fragment. The simultaneous binding to cell-surface mesothelin and CD40 is required to provide cross-linking for activating CD40 multimerization and initiating downstream signaling (online supplemental figure S1). ABBV-428 inhibited PC3 tumor cell growth in cell lines and murine models via enhancement of APC and T-cell activation on binding of the molecule to cell-surface mesothelin. Although mesothelin expression is necessary for CD40 activation by ABBV-428, the resulting enhancement of antigen presentation may potentially lead to broader antitumor activity against both mesothelin cell-surface expressing and non-expressing tumor cells.20
Supplemental material
Our clinical hypothesis was that ABBV-428, despite systemic administration, provides conditional and tumor-localized agonistic CD40 effects (eg, facilitation of T-cell infiltration, augmentation of B-cell activity, and repolarization of macrophage subsets) on binding to mesothelin-expressing tumor cells. Further, tumor microenvironment-localized activation and reduced systemic CD40 activation were hypothesized to limit systemic toxicity. To explore these hypotheses, we performed a first-in-human, phase I study to determine the recommended phase II dose (RP2D), MTD, safety, and pharmacokinetic and pharmacodynamic profiles of ABBV-428 monotherapy in patients with advanced solid tumors.
Patients and methods
Study design
This was an open-label, first-in-human phase I study of ABBV-428 administered as monotherapy to patients with advanced solid tumors. The primary endpoints of the study were to evaluate the safety/tolerability, pharmacokinetics, and MTD/RP2D of ABBV-428 monotherapy.
In dose escalation, an accelerated titration and a standard 3+3 dose escalation scheme were used. ABBV-428 was administered intravenously once every 2 weeks in 28-day cycles. ABBV-428 was administered at nine dose levels (0.01, 0.03, 0.1, 0.2, 0.4, 0.8, 1.6, 2.4, and 3.6 mg/kg every 2 weeks) in patients with advanced solid tumors who had progression on or intolerance to standard therapies. For the first three dose levels, one patient was enrolled in each successive cohort until the occurrence of any treatment-related grade ≥2 adverse event (AE) during cycle 1 (28 days after the first dose). Two additional patients were enrolled at that dose level if any treatment-related grade ≥2 AE occurred. Dose escalation switched to a 3+3 dose escalation scheme starting at the dose level where the treatment-related grade ≥2 AE occurred. If no treatment-related grade ≥2 AEs occurred during accelerated titration at the first three dose levels, then the study was to transition to a 3+3 dose escalation scheme at the 0.2 mg/kg dose level. Each dose level at 0.2 mg/kg and above enrolled ≥3 evaluable patients. The MTD was defined as the highest dose level at which fewer than one-third of evaluable patients experienced a dose-limiting toxicity (DLT). In dose expansion, 24 patients each for platinum-resistant ovarian cancer and epithelioid or biphasic mesothelioma were planned to receive ABBV-428 at the RP2D.
Patients
Eligible patients were aged ≥18 years, with an advanced solid tumor, who had progression on or intolerance to standard therapies known to provide clinical benefit; had an Eastern Cooperative Oncology Group performance status of 0–2; had adequate bone marrow (absolute neutrophil count ≥1500/mm3, platelets ≥100×109/L, hemoglobin ≥9.0 g/dL), renal (creatinine clearance ≥50 mL/min), hepatic (total bilirubin ≤1.5×upper limit of normal (ULN), aspartate aminotransferase and alanine aminotransferase ≤2.5×ULN), and coagulation function; and had measurable disease per Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST V.1.1)28 in the dose expansion arm. Patients were excluded from the study if they had active or prior documented autoimmune disease within the past 2 years; current or prior use of immunosuppressive medication (with certain steroidal exceptions) ≤14 days before the first dose of the study drug; history of primary immunodeficiency, bone marrow transplantation, chronic lymphocytic leukemia, solid organ transplantation, or previous clinical diagnosis of tuberculosis; coagulopathy or a platelet disorder; HIV positive; chronic or active hepatitis B or C; prior grade ≥3 immune-mediated neurotoxicity or pneumonitis while receiving immunotherapy; or known uncontrolled metastases to the central nervous system.
Safety
The severity of each AE was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events V.4.03. DLTs were defined as any grade ≥3 treatment-related AE (TRAE) that occurred during the DLT observation period (days 1–28 after the first dose of study drug). The following conditions were not defined as DLTs: grade 3 infusion-related reactions resolving within 24 hours; grade 3 elevations in aspartate aminotransferase, alanine aminotransferase, or blood bilirubin resolving within 7 days; grade 3 hematological toxicity of any duration; grade 4 lymphopenia lasting ≤8 days; alopecia or vitiligo of any grade; grades 3/4 laboratory tests deemed not clinically significant by the investigator; grade 3 fatigue lasting ≤7 days; and manageable grade 3 nausea and vomiting.
Pharmacokinetics and immunogenicity
Pharmacokinetic parameters of ABBV-428, including the maximum observed serum concentration (Cmax), the time to Cmax (Tmax), the area under the serum concentration-time curve (AUC), and terminal half-life (t1/2), were determined in cycles 1 and 3 using non-compartmental methods. Antidrug antibodies were assessed throughout the study.
Antitumor activity
Responses were assessed every two treatment cycles via CT or MRI until disease progression (based on immune-related RECIST)29 30; retreatment with ABBV-428 after disease progression was not permitted. For all tumor types, response classifications were based on RECIST V.1.1.28
Biomarkers
Longitudinal blood samples were collected for testing of CD40 receptor occupancy on B-cells by real-time flow cytometry. This proprietary receptor occupancy assay used a labeled version of anti-CD40 antibody to detect free receptors and another non-competing antibody to detect the total CD40 receptors for calculation of percentage of CD40 receptors occupied by ABBV-428 treatment. Cytokine secretions were tested from longitudinal serum samples, including interleukin 1 (IL-1), IL-2, IL-6, IL-8, IL-10, IL-12p70, interferon (IFN)-γ, and TNF-α, by using the MSD V-PLEX Proinflammatory Panel 1 Human Kit (MSD, Rockville, Maryland, USA).
For determination of baseline mesothelin tumor expression levels, a fully automated immunohistochemistry (IHC) assay was developed using the mesothelin mouse clone 5B2 antibody (Leica Biosystems, Buffalo Grove, Illinois, USA). Staining was evaluated by a pathologist using a scoring method developed by AbbVie. Evaluation of immunoreactivity involved a combination of the following: staining intensity, subcellular localization, and percentage of cells staining in the primary component of the tissue type of interest. Evaluation of mesothelin was performed in the membrane. The mesothelin assay was evaluated on a semiquantitative scale, and the percentage of cells staining at each of the following four levels was recorded: 0 (unstained), 1+ (weak staining), 2+ (moderate staining), and 3+ (strong staining). An H-score was calculated based on the summation of the product of percentage of cells stained at each intensity, using the following equation: (3×% cells staining at 3+)+(2×% cells staining at 2+)+(1×% cells staining at 1+).
For characterization of changes in the tumor immune microenvironment after ABBV-428 treatment, paired (pretreatment and post-treatment) tumor biopsies were collected during dose expansion (pretreatment: cycle 1, day 1; post-treatment: cycle 2, day 1). The timing of the post-treatment biopsy (cycle 2, day 1) was selected based on evidence in the literature that peak changes in T-cell activation and the T-cell repertoire may occur around 2–4 weeks after the first dose of treatment with existing checkpoint inhibitors.31 32 CD8 and programmed death ligand-1 (PD-L1) dual IHC assays were applied to formalin-fixed paraffin-embedded tissues, and the percentages of CD8+ T-cells and PD-L1+ cells were reported at baseline and for changes from baseline. Changes in immune gene expression resulting from ABBV-428 treatment were analyzed in samples from paired tumor biopsies using the nCounter PanCancer Immune Profiling Panel (NanoString Technologies), per manufacturer’s instructions.
Statistical analysis
For the 3+3 dose escalation phase, up to six patients per dose group were enrolled before establishment of the MTD and/or RP2D of ABBV-428. For the expansion phase, approximately 25 patients were deemed sufficient to provide an acceptably narrow 80% CI for the objective response rate. Continuous data were analyzed using descriptive statistics, including number of observations, mean, SD, median, minimum, and maximum. Categorical data were analyzed using frequency counts and percentages. The two-sided 80% exact binomial confidence intervals of the objective response rate were estimated using the Clopper-Pearson method. Progression-free survival (PFS) was estimated using the Kaplan-Meier method.
Results
Patient population
From November 2016 through June 2018, a total of 59 patients were enrolled and received at least one dose of ABBV-428. For dose escalation, patients were treated at one of nine ABBV-428 dose levels: 0.01 mg/kg (n=2), 0.03 mg/kg (n=2), 0.1 mg/kg (n=6), 0.2 mg/kg (n=6), 0.4 mg/kg (n=5), 0.8 mg/kg (n=4), 1.6 mg/kg (n=3), 2.4 mg/kg (n=3), and 3.6 mg/kg (n=6). The dose escalation cohort included a wide range of tumor types, including pancreatic, sarcoma, cholangiocarcinoma, non-small cell lung cancer, cervical, colorectal, head and neck squamous, small intestine, peritoneal, mesothelioma, ovarian, and unknown primary. Twenty-two patients were enrolled at 3.6 mg/kg for dose expansion, including 10 with ovarian cancer and 12 with mesothelioma. Further enrollment was deferred due to lack of preliminary clinical activity. Patients had received a median of three prior treatments (range, 1–11). Baseline patient and disease characteristics are summarized in table 1.
Baseline patient characteristics
Study drug exposure
The time on treatment for each patient is shown in online supplemental figure S2. Across all dose cohorts, the median number of days on treatment was 43 (range, 1–666). At 3.6 mg/kg (RP2D), patients received treatment for a median of 43 days (range, 1–324). Across dose cohorts, reasons for treatment discontinuation were progressive disease (69%), AEs (10%), physician decision (8%), patient withdrawal (2%), and other (10%), including disease progression, lack of clinical benefit, physician choice, and patient preference.
Safety
Overall, 56 patients (95%) reported treatment-emergent AEs (TEAEs), the most common of which were fatigue (41%), decreased appetite (36%), asthenia (29%), constipation (25%), and dyspnea (25%; online supplemental table S1). A total of 36 patients (61%) reported grade ≥3 TEAEs, of which malignant neoplasm progression (19%), anemia (8%), abdominal pain (7%), fatigue (7%), gamma-glutamyl transferase increased (7%), pleural effusion (7%), and pneumonia (7%) were the most frequently reported (online supplemental table S2). Fourteen patients (24%) died due to TEAEs, including 11 patients (19%) from malignant neoplasm progression, 1 (2%) from sepsis, 1 (2%) from cardiac arrest, and 1 (2%) from general physical health deterioration. Seven patients (12%) reported infusion-related reactions. Increased levels of alanine aminotransferase, aspartate aminotransferase, and blood bilirubin occurred in 3% (grade ≥3, 2%), 5% (2%), and 5% (5%) of patients, respectively. There were no reports of cytokine release syndrome. In the total patient population, 39 patients (66%) experienced a TRAE (table 2). Grade ≥3 TRAEs occurred in three patients (5%), all of whom were treated in the 3.6 mg/kg dose cohort; grade ≥3 TRAEs were pericardial effusion, colitis, infusion-related reaction, and pleural effusion (n=1 each, 2%). Treatment-related fever (any grade) occurred in 8% of patients, and treatment-related nausea (any grade) occurred in 5%.
Incidence of all-grade treatment-related adverse events occurring in ≥5% of patients by dose cohort
The MTD was not reached during this study, and no DLTs were observed. Based on these safety data as well as pharmacokinetic and pharmacodynamic considerations (see subsequent sections later), the maximum administered dose of 3.6 mg/kg was chosen as the recommended expansion dose.
Pharmacokinetics and immunogenicity
The exposure of ABBV-428 increased in an approximate dose-proportional manner from 0.40 to 3.6 mg/kg (figure 1 and online supplemental figure S3). For the 3.6 mg/kg RP2D of ABBV-428 in cycle 1, the median Tmax was 0.25 hours after end of infusion, and the geometric mean Cmax and AUClast were 48.5 µg/mL and 717 µg hour/mL, respectively, and the estimated t1/2 was 13.9 hours. No accumulation of ABBV-428 was observed between cycle 1 and cycle 3.

ABBV-428 serum concentration (mean+SD) versus time profiles in escalation and expansion cohorts after the first dose. Serum concentrations were below the lower limit of quantitation (200 ng/mL) for patients treated at 0.01 and 0.03 mg/kg dose levels.
Antidrug antibody was identified in 63% of treated patients, with antidrug antibody impact on pharmacokinetic parameters observed at dose levels ≤0.80 mg/kg. No apparent impact of antidrug antibody on ABBV-428 pharmacokinetic parameters was observed at the 3.6 mg/kg dose level. No meaningful impact of antidrug antibody on infusion-related reactions was observed; however, one patient with a grade four infusion reaction had very high antidrug antibody titers.
Antitumor activity
Of 53 patients who were evaluable for response across all dose cohorts, none had an objective response (confirmed complete or partial response). Twenty-five patients (47%) had stable disease and 28 (53%) had progressive disease as a best response. Among 35 response-evaluable patients treated at doses where >90% CD40 receptor occupancy was achieved (≥0.8 mg/kg (see the Biomarker section)), 14 patients (40%) had stable disease and 21 (60%) had progressive disease as a best response. Of 25 response-evaluable patients treated with 3.6 mg/kg (RP2D), best responses were stable disease for 9 patients (36%) and progressive disease for 16 patients (64%). The percentage change in tumor size from baseline values for individual tumor types is shown in figures 2 and 3.
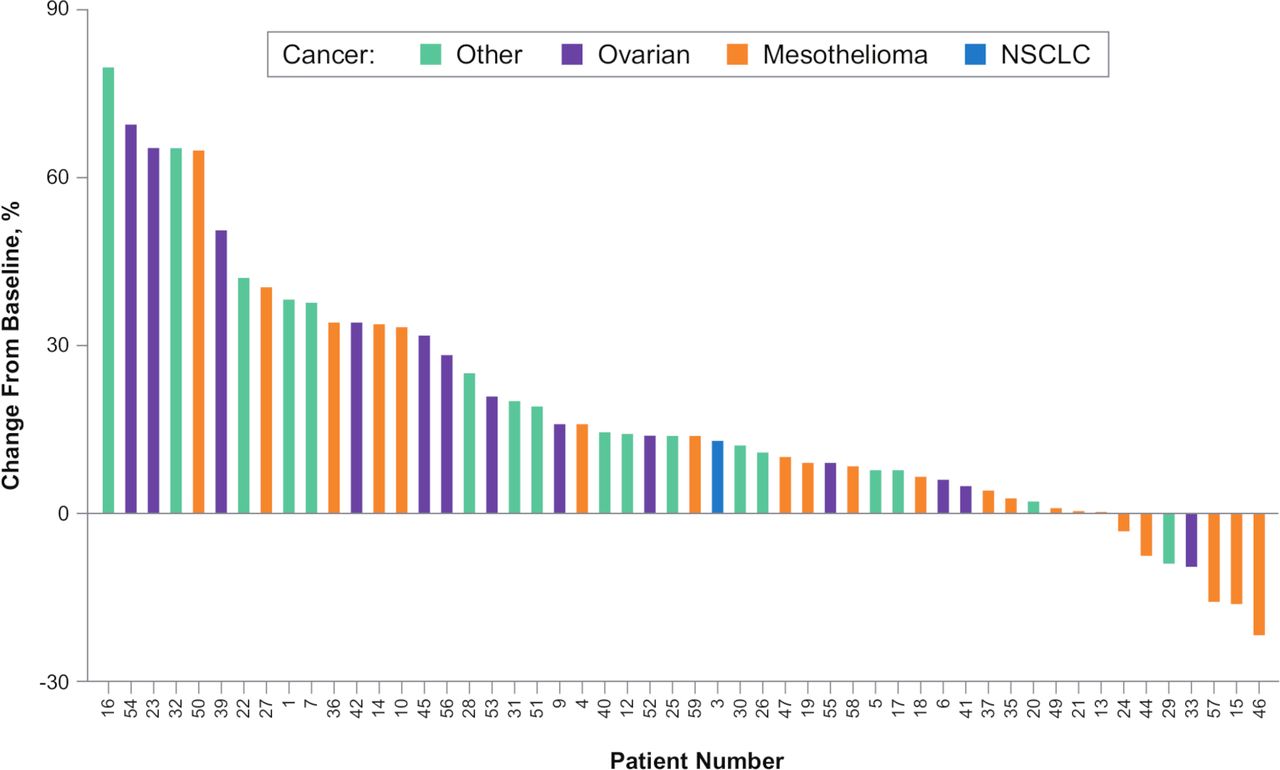
Best percent change in tumor size from baseline. n=52 response-evaluable patients. NSCLC, non-small cell lung cancer.
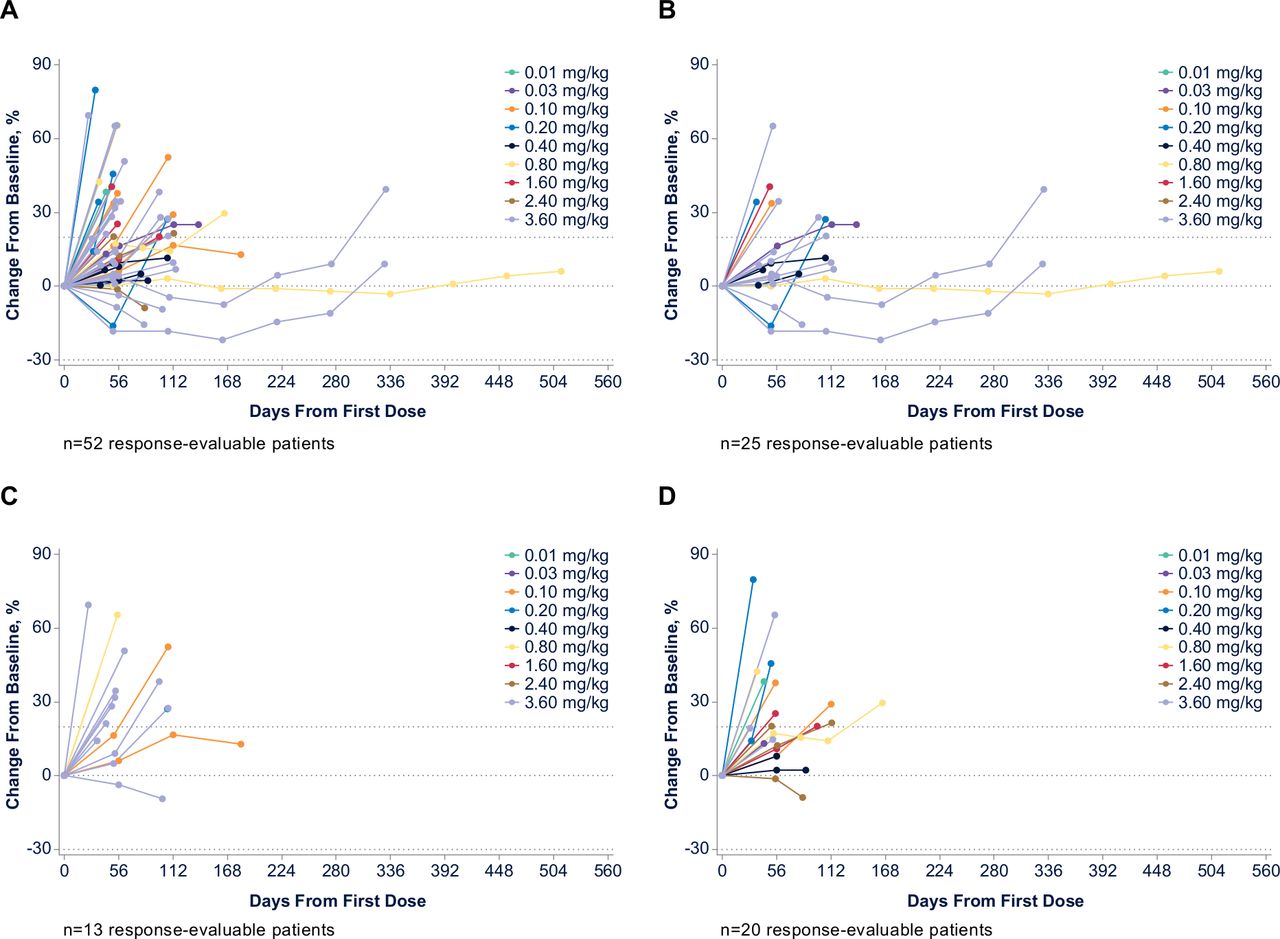
Clinical activity in (A) all response-evaluable patients, (B) mesothelioma, (C) ovarian cancer, and (D) non-small cell lung cancer/other.
Three patients with mesothelioma had stable disease on treatment for >6 months (online supplemental figure S2). Two of these patients were heavily pretreated prior to study enrollment and achieved stable disease (1 with a 20% reduction in target lesion) with ABBV-428 for approximately 1 year, with either no or only low-grade TEAEs (grade 1 neuropathy). After disease progression on ABBV-428, both patients later responded to treatment with a programmed death-1 (PD-1) inhibitor. The third patient had germline-associated bicavitary (pleural and peritoneal) epithelioid mesothelioma and experienced progressive disease following treatment with pemetrexed/carboplatin, pemetrexed maintenance, and tremelimumab versus placebo. This patient received 24 cycles of ABBV-428, with prolonged stable disease and minimal toxicity.
Biomarkers
Blood-based pharmacodynamic biomarkers
We hypothesized that CD40 receptor occupancy would increase in a dose-dependent manner following ABBV-428 administration. Evaluation of CD40 receptor occupancy on peripheral B-cells demonstrated maximal CD40 receptor occupancy between 15 min and 2 hours post-treatment. Greater than 90% CD40 receptor occupancy was achieved starting at a dose of 0.8 mg/kg (measured at cycle 1, day 1, 2 hours post-ABBV-428 dosing; online supplemental figure S4), thereby demonstrating target engagement in peripheral blood.
Consistent with our hypothesized mechanism of action and preclinical data, minimal changes were observed in IFN-γ, TNF-α, and IL-6 levels following administration of 0.1–2.4 mg/kg ABBV-428 (online supplemental figure S4), consistent with the lack of cytokine release syndrome events observed in our study across all dose cohorts (including the RP2D of 3.6 mg/kg). Other tested cytokines (IL-1β, IL-2, IL-8, IL-10, and IL-12p70) also showed minimal changes following treatment with ABBV-428 (data not shown).
Tumor pharmacodynamic biomarkers
Given the role of ABBV-428 as an immunomodulator, we also hypothesized that ABBV-428 treatment would alter the immune tumor microenvironment. However, no consistent changes in tumor-infiltrating CD8+ T-cells or PD-L1+ cells following ABBV-428 treatment were observed in paired tumor biopsy specimens collected during dose expansion (3.6 mg/kg; online supplemental figure S5). Moreover, no statistically significant changes in the tumor immune microenvironment were detected via NanoString gene expression analysis of paired tumor biopsy specimens collected during dose expansion (3.6 mg/kg; online supplemental figure S6).
Baseline mesothelin expression level
Further considering the mechanism of action of ABBV-428, we hypothesized that baseline tumor mesothelin expression might be predictive of clinical outcomes. Among all patients, there was little correlation (Spearman correlation, 0.284; p=0.098) between baseline mesothelin tumor expression and PFS (figure 4). Compared with all patients, dose expansion cohorts (consisting of patients with ovarian cancer and mesothelioma) showed a stronger correlation between baseline mesothelin levels and PFS (Spearman correlation, 0.606; p=0.021).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Baseline mesothelin tissue expression and clinical parameters in (A) all patients, and (B) the expansion cohort. Thirty-six patients were evaluated in all cohorts (A); 14 patients (n=7 with ovarian cancer and n=7 with mesothelioma) were evaluated in the expansion cohort (B). aH-score=1×[% cells 1+]+2×[% cells 2+]+3×[% cells 3+]. bCox proportional hazards progression model was applied. MSLN, mesothelin.
Discussion
Results from this open-label, first-in-human, phase I study demonstrate that ABBV-428 has an acceptable safety profile as a monotherapy in patients with advanced solid tumors. During dose escalation, no DLTs were observed and the MTD was not reached. Pharmacokinetic data suggest non-linearity at lower doses, with target saturation likely around 0.4 mg/kg. Thereafter, exposure to ABBV-428 increased in a dose-proportional manner from 0.4 to 3.6 mg/kg. At the RP2D dose of 3.6 mg/kg ABBV-428, Cmax was 48.5 µg/mL and AUClast was 717 µg hour/mL, which were much higher than the Cmax and AUClast reported for the MTD of selicrelumab, a conventional CD40 agonist monoclonal antibody.9 Furthermore, the impact of ABBV-428 on CD40 receptor occupancy, a classical CD40 agonist pharmacodynamic marker, was well saturated at the RP2D, with achievement of >90% CD40 receptor occupancy on peripheral B-cells starting at a dose of 0.8 mg/kg. It was assumed that sufficient intratumoral drug exposure was achieved at a dose of 3.6 mg/kg because a predicted AUClast of 107 µg hour/mL at the tumor site (based on a conservative estimate of 15% intratumoral drug penetration) would approximate the exposure observed systemically at dose levels of 0.4–0.8 mg/kg, where near maximal pharmacodynamic effects were achieved in the circulation (>90% CD40 receptor occupancy on peripheral B-cells). Although antidrug antibodies were prevalent across all dose levels, they had no observed impact on drug exposure at the RP2D of 3.6 mg/kg. Reflecting these pharmacokinetic, pharmacodynamic, and safety data, the maximum administered dose of 3.6 mg/kg was chosen as the RP2D.
Across dose levels, the most common TRAEs were fatigue, asthenia, infusion-related reactions, decreased appetite, and fever; grade ≥3 TRAEs were pericardial effusion, colitis, infusion-related reaction, and pleural effusion. Although one patient had a grade 4 infusion reaction with a very high antidrug antibody titer, clinically significant infusion-related reactions (observed in 12% of patients) were generally manageable with treatment modification guidelines in place for grade ≥2 occurrences. Systemic administration of immunostimulatory CD40 agonists has historically been associated with frequent hepatotoxicity and cytokine release syndrome.3 5 7–13 However, few liver function test elevations and no instances of cytokine release syndrome were observed with ABBV-428. Furthermore, serum cytokine analysis (including assessment of IL-6, TNF-α, and IFN-γ) demonstrated minimal changes in inflammatory cytokines post-ABBV-428 treatment, corroborating the lack of cytokine release syndrome observed in patients treated with ABBV-428.
No treatment responses by RECIST were observed in the escalation or expansion cohorts. Three patients with mesothelioma had stable disease for >6 months, the best response achieved in this study. The discrepancy in antitumor activity observed in this clinical study versus the prior preclinical study of ABBV-42820 might partly derive from the difficulty in developing preclinical models that recreate the complex tumor biology seen in patients. For example, forced expression of human mesothelin on tumor cells might have increased the immunogenicity of murine syngeneic tumor models, thereby augmenting the antitumor activity of ABBV-428 in the preclinical setting. Therefore, analyses of predictive and pharmacodynamic biomarkers were performed to further explore ABBV-428 treatment outcomes in the clinical setting. Baseline mesothelin levels correlated modestly with PFS (Spearman correlation, 0.606; p=0.021) among RP2D-treated patients with mesothelioma and ovarian cancer, supportive of a tumor-directed mechanism. However, pharmacodynamic biomarker data showed no consistent changes in intratumoral CD8+ T-cells, PD-L1+ cells, or immune-related gene expression following ABBV-428 treatment, which may explain the lack of clinical responses observed in this study.
These data raise several considerations that may guide future development of ABBV-428 or other tumor-targeted immunomodulators. The overall lack of DLTs and systemic cytokine elevations in this study suggest that higher doses of ABBV-428 may need to be evaluated. Similarly, peripheral B-cell CD40 receptor occupancy was used as a pharmacodynamic readout in this study; however, the lack of changes in cytokine secretion and intratumoral pharmacodynamic biomarkers (eg, CD8+ T-cells, PD-L1+ cells, and immune-related gene expression) indicates that ABBV-428 did not induce an effective antitumor immune response at the doses tested in this study. Therefore, subsequent studies may need to focus on pharmacokinetic and pharmacodynamic assessment of intratumoral drug exposure and binding for dose optimization, as higher doses of ABBV-428 may be needed to maximize its intratumoral drug penetration, and consequently, its immunomodulatory effects and antitumor activity. Additionally, spatial distribution and relative frequency of target cell populations need to be evaluated in the indications of interest. In this study, tumor types in expansion cohorts (mesothelioma and ovarian cancer) were selected based on previously described high mesothelin levels; however, study patients were not selected for inclusion based on mesothelin expression. Given the association in our study between baseline mesothelin levels and PFS in cohort expansion in ovarian cancer and mesothelioma, utilization of a patient selection approach based on mesothelin expression might be needed for similar monotherapy or combination studies in the future.
In summary, in this first-in-human, first-in-class phase I study of ABBV-428 monotherapy, we successfully uncoupled the relationship between systemic exposure and systemic toxicity, which has been the hallmark of standard CD40 agonist monoclonal antibodies. ABBV-428 safely provided greater than 12-fold exposure compared with CP-870,893.8 Greater than 90% CD40 receptor occupancy on peripheral B-cells was achieved, consistent with some level of immune cell engagement. However, minimal clinical activity was observed in patients with mesothelioma or ovarian cancer who had progression on or intolerance to prior standard therapies, and intratumoral pharmacodynamic biomarker data showed little evidence of an antitumor immune response. Therefore, administration of higher doses or a patient selection approach based on mesothelin expression may be required for ABBV-428 to achieve effective antitumor immunity as a monotherapy or as part of a combination regimen with chemotherapy or other therapies.
Acknowledgments
AbbVie and authors thank all the trial investigators and the patients who participated in this clinical trial. AbbVie and authors thank the AbbVie Oncology Discovery team for designing ABBV-428, Anita Reddy for biomarker assay strategies, Claire Guo and Chun Zhang for biomarker statistical analysis, Andrew Woolley for IHC data analysis, Tolga Turan for gene expression analyses, Kinjal Hew and Betty Wang for coordinating biomarker samples for analyses, and Catherine Tribouley and Daniel Afar for supporting clinical biomarker research. Medical writing support was provided by Diana Avery, PhD, of Bio Connections, LLC, and funded by AbbVie.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @jasonlukemd
Presented at Presented in part at the European Society of Medical Oncology Conference, Barcelona, Spain, September 27–October 01, 2019, and at the American Association for Cancer Research Virtual Annual Meeting, April 27–28, 2020.
Contributors Conceptualization (ideas; formulation or evolution of overarching research goals and aims): KC, AWT, VS, SY, DTC. Methodology (development or design of methodology; creation of models): JJL, AWT, SY, DTC, WRH, LF. Software (programming; software development; designing computer programs; implementation of the computer code and supporting algorithms; testing of existing code components): WRH. Validation (verification of the overall replication/reproducibility of results/experiments and other research outputs): PAC, SY. Formal analysis (application of statistical, mathematical, computational, or other formal techniques to analyze or synthesize study data): VS, MP, WRH, LF. Investigation (conducting a research and investigation process, performing experiments, or data/evidence collection): JJL, FB, KC, AWT, KK, AH, CLT, VS, FT, SK, PAC, MK, HF, FF, KA, MP, SY, MAM, LF. Resources (provision of study materials, reagents, patients, laboratory samples, animals, instrumentation, computing resources, or other tools): FB, KC, AWT, KK, CLT, VS, SK, PAC, MK, HLK, SY. Data curation (management activities to annotate (produce metadata), scrub data and maintain research data (including software code, where it is necessary for interpreting the data itself) for initial use and later reuse): KC, FF. Writing-original draft (preparation, creation and/or presentation of the published work, specifically writing the initial draft (including substantive translation)): all authors. Writing-review and editing (preparation, creation and/or presentation of the published work by those from the original research group, specifically critical review, commentary or revision—including prepublication or postpublication stages): all authors. Visualization (preparation, creation, and/or presentation of the published work, specifically visualization/data presentation): FB, KC, HF, MP. Supervision (oversight and leadership for research activity planning and execution, including mentorship to the core team): KC, AWT, VS, HF, MP, JSH, MAM. Project administration (management and coordination for research activity planning and execution): KC, AWT, VS. Funding acquisition (acquisition of financial support): KC.
Funding AbbVie funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship.
Competing interests JJL: Data and Safety Monitoring Board for TTC Oncology; consultancy/advisory role for 7 Hills, Abbvie, Actym, Akrevia, Algios, Alphamab Oncology, Array, Astellas, Bayer, Bristol-Myers Squibb, Eisai, EMD Serono, Ideaya, Incyte, Janssen, Kanaph, Mavu (now part of AbbVie), Merck, Mersana, Novartis, Onc.AI, Pyxis, PTx, RefleXion, Regeneron, Silicon, Springbank, Tempest, Tesaro, and Vividion; research support (all to institution for clinical trials unless noted) from AbbVie, Agios (IIT), Array (IIT), Astellas, Bristol-Myers Squibb, CheckMate (SRA), Compugen, Corvus, EMD Serono, Evelo (SRA), Five Prime, FLX Bio, Genentech, Immatics, Immunocore, Incyte, Leap, MedImmune, Macrogenics, Necktar, Novartis, Palleon (SRA), Merck, Springbank, Tesaro, Tizona, and Xencor; patents (both provisional) with the University of Chicago: Serial #15/612,657 (Cancer Immunotherapy) and PCT/US18/36052 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof); travel support from Akrevia, Bayer, Bristol-Myers Squibb, EMD Serono, Incyte, Janssen, Merck, Mersana, Novartis, Pyxis, and RefleXion. FB: consulting/advisory role for AstraZeneca, Bayer, Bristol-Myers Squibb, Boehringer–Ingelheim, Eli Lilly Oncology, F. Hoffmann–La Roche Ltd, Novartis, Merck, MSD, Pierre Fabre, Pfizer, and Takeda. KC: no conflicts to disclose. AWT: Independent Data Monitoring Committee for Mirati and Genentech/Roche; consulting/advisory role (fees paid to NEXT Oncology) for AbbVie, AbGenomics, Adagene, ADC Therapeutics, Agenus, Aro Biotherapeutics, Ascentage Pharma, BioInvent, AxImmune, Bayer, BioInvent, Birdie, Cello Health, Eleven Bio, Ellipses, Elucida, EMD Serono, Five Prime, Forbius (Formation Biologics), Gilde Healthcare Partners, HBM Partners, Immunome, Immunomet, Jazz, Karma Oncology, Mekanistic, Menarini, Mersana, Nanobiotix, NBE Therapeutics, Nuvalent, Pelican, PSI Pharma Support America, Oncology International, OSI, Partner Therapeutics, Pfizer, Pieris, Pierre Fabre, Ridgeway, Ryvu Therapeutics, Scitemex, Seattle Genetics, Symphogen, Syneos, TFS Trial Form Support International, and Zymeworks. KK: consultancy/advisory role for AstraZeneca, Bristol-Myers Squibb, EMD Serono, Eli Lilly Oncology, Genentech/Roche, Inviata, Merck, Novartis, Regeneron, and Symphogen; research funding from Astellas, AbbVie, Bristol-Myers Squibb, EMD Serono, Five Prime Therapeutics, Genentech, Regeneron, Tizona, and Transgene. AH: honoraria from Amgen, Eisai, and Servier; consulting/advisory role for AstraZeneca, Incyte, and Debiopharm; travel support from Amgen, Servier, Lilly, AstraZeneca, and MedImmune; research funding from Incyte. CLT: consultancy/advisory role for MSD, Bristol-Myers Squibb, Merck Serono, Roche, AstraZeneca, GlaxoSmithKline, Rakuten, Seattle Genetics, and Nanobiotix. VS: one-time educational honoraria from Medscape; consultancy/advisory role for Helsinn, Loxo Oncology/Eli Lilly, R-Pharma US, Incyte, QED Pharma, MedImmune, and Novartis; travel support from Novartis, Pharmamar, ASCO, ESMO, Helsinn, and Incyte; research funding from Roche/Genentech, Novartis, Bayer, GlaxoSmithKline, Nanocarrier, Vegenics, Celgene, Northwest Biotherapeutics, Berghealth, Incyte, Fujifilm, Pharmamar, D3, Pfizer, Multivir, Amgen, AbbVie, Alfasigma, Agensys, Boston Biomedical, Idera Pharma, Inhibrx, Exelixis, Blueprint Medicines, Loxo Oncology, MedImmune, Altum, Dragonfly Therapeutics, Takeda, National Comprehensive Cancer Network, NCI-CTEP, The University of Texas MD Anderson Cancer Center, Turning Point Therapeutics, and Boston Pharmaceuticals. FT: stock or other ownership in Salarius Pharmaceuticals; consultancy/advisory role for Tempus Lab; patents, royalties, or other intellectual property for clinical trial software. SK: honoraria from AstraZeneca, Bristol-Myers Squibb, Merck Sharp & Dohme, and Roche; consultancy/advisory role for AstraZeneca, Boehringer, Merck Sharp & Dohme, and Pfizer; travel support from AstraZeneca, Boehringer, Bristol-Myers Squibb, and Roche. PAC: honoraria from Novartis, Roche/Genentech, Blueprint Medicines, Amgen, and Merck Serono; research funding from Novartis, Roche/Genentech, Lilly, Blueprint Medicines, Bayer, AstraZeneca, Celgene, Plexxikon, AbbVie, Bristol-Myers Squibb, Merck Serono, and Merck Sharp & Dohme. MK: consultancy/advisory role for AbbVie, Ipsen, Pfizer, Roche, and Jackson Laboratory for Genomic Medicine; research funding from AbbVie, Bristol-Myers Squibb, and Specialized Therapeutics. HLK: personal fees from Aldeyra Therapeutics, AstraZeneca, Bayer, Boehringer-Ingelheim, Bristol Myers Squibb, Deciphera, Inhibrx, Inventiva, Kyowa, Merck, and Paredox Therapeutics; non-financial support from AstraZeneca, Boehringer Ingelheim, Inventiva, Merck, and Paredox Therapeutics; research funding to institution from Aduro, AstraZeneca, Bayer, Bristol Myers Squibb, Deciphera, GlaxoSmithKline, Lilly, Merck, Polaris, Verastem, Blueprint, Tesaro, and Inhibrx. FF: no conflicts to disclose. KA: no conflicts to disclose. HF, MP, SY, DTC, WRH, JSH, and MAM: employment with AbbVie and may hold stock or other options. LF: research funding from AbbVie, Bavarian Nordic, Bristol-Myers Squibb, Dendreon, Janssen, Merck, and Roche/Genentech. No honoraria or payments were made for authorship.
Patient consent for publication Not required.
Ethics approval Study was conducted at 16 institutions in the USA, Australia, France, and Taiwan in accordance with International Conference on Harmonisation Good Clinical Practice guidelines and the Declaration of Helsinki. Written informed consent was obtained from all patients before enrollment.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (eg, protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.