Article Text

Abstract
With increasing numbers of bispecific antibodies (BsAbs) and multispecific products entering the clinic, recent data highlight immunogenicity as an emerging challenge in the development of such novel biologics. This review focuses on the immunogenicity risk assessment (IgRA) of BsAb-based immunotherapies for cancer, highlighting several risk factors that need to be considered. These include the novel scaffolds consisting of bioengineered sequences, the potentially synergistic immunomodulating mechanisms of action (MOAs) from different domains of the BsAb, as well as several other product-related and patient-related factors. In addition, the clinical relevance of anti-drug antibodies (ADAs) against selected BsAbs developed as anticancer agents is reviewed and the advances in our knowledge of tools and strategies for immunogenicity prediction, monitoring, and mitigation are discussed. It is critical to implement a drug-specific IgRA during the early development stage to guide ADA monitoring and risk management strategies. This IgRA may include a combination of several assessment tools to identify drug-specific risks as well as a proactive risk mitigation approach for candidate or format selection during the preclinical stage. The IgRA is an on-going process throughout clinical development. IgRA during the clinical stage may bridge the gap between preclinical immunogenicity prediction and clinical immunogenicity, and retrospectively guide optimization efforts for next-generation BsAbs. This iterative process throughout development may improve the reliability of the IgRA and enable the implementation of effective risk mitigation strategies, laying the foundation for improved clinical success.
- anti-drug antibodies
- immunogenicity
- bispecific antibody
- T-cell engager
- immunotherapy
- oncology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
A wide variety of novel biotherapeutics with multi-functional domains are currently in development, particularly in the field of cancer immunotherapy, where multiple targets may be required to achieve long-term responses and overcome tumor resistance. Despite a myriad of bispecific and multi-specific products currently under clinical evaluation, there are only four bispecific antibodies (BsAbs) that have been approved for the treatment of cancer as of March 2022: Amgen’s Blincyto® (blinatumomab), Janssen’s Rybrevant® (amivantamab-vmjw), Immunocore’s Kimmtrak® (tebentafusp-tebn), and Fresenius/Trion’s Removab® (catumaxomab). Removab® was approved by the European Medicines Agency in 2009 for the treatment of malignant ascites, but the drug was voluntarily withdrawn from the market in 2017 for commercial reasons.1 2
Recent clinical data highlight immunogenicity as one of the key challenges in the development of these novel biologics with multi-functional domains. Unlike standard human IgG antibodies, which are monospecific with two binding regions directed against the same epitope, BsAbs are genetically engineered antibodies that consist of two distinct binding domains capable of binding to two different antigens or to two different epitopes on the same antigen.3 In contrast to other multi-target therapies (e.g., combination therapies or co-formulated products), BsAbs combine different pharmacology-optimized functional domains into a single drug, which (1) allows for bridging of two target cells through simultaneously cross-linking and co-localizing cell receptors, such as immune cell engagers that deliver an immune cell to a specific target cell for target-dependent immune activation and target cell lysis, or (2) provides synergistic efficacy through engaging dual targets.3 These dual targets may be soluble cytokines, signal pathways, immune checkpoints, or tumor-associated antigens (TAAs), which serve to improve tumor selectivity and/or overcome tumor heterogeneity for more efficient anti-cancer treatment. However, the linkage of different functional domains into one drug can create novel scaffolds with highly engineered sequences that do not resemble a classical IgG antibody.4–6 These atypical formats may reveal cryptic epitopes and/or create neoantigens that trigger immunogenicity. Furthermore, the involvement of multiple mechanisms of action (MOAs) from different domains of the BsAb could also drive clinically impactful immunogenicity, such as the synergistic immunomodulatory MOAs that may induce a break in peripheral tolerance, resulting in an unwanted immune response to the drug. Additionally, several other product-related and patient-related factors, as well as the complexity of the clinical trial settings for anti-cancer BsAbs, could also contribute to immunogenicity. Taken together, the multitude of unique risk factors for BsAbs in the oncology space warrants the need for an early and comprehensive assessment of their immunogenic risk to enhance clinical success.
Clinical relevance of anti-drug antibodies (ADAs) to selected BsAbs in oncology
To date, immunogenicity has been evaluated in large numbers of patients for approved monoclonal antibodies (mAbs) with varying clinical impact,7 but reports for BsAbs are limited. This could be due to the short clinical development history of these molecules. T-cell engagers are a group of BsAbs that have been developed most actively across a broad range of hematological malignancies and solid tumors. Although B cell-depleting agents (e.g., CD19-directed Bispecific T-Cell Engager (BiTE®) molecule Blincyto®, CD20-directed T-cell engager Glofitamab, and BCMA-directed T-cell engagers Elranatamab and Teclistamab) have shown limited ADA formation,8–11 likely due to the elimination of antibody-producing B cells, a wide range of clinical ADA incidence has been observed for T-cell engagers with non-B cell tumor targets. These include case reports of relatively high ADA incidence with clinical impact from early trials of BsAbs, such as carcinoembryonic antigen-directed BiTE® molecule AMG 211 and prostate-specific membrane antigen (PSMA)-directed T-cell engagers (Pasotuxizumab, APVO-414, and JNJ-08112–15), and a recently Food and Drug Administration (FDA)-approved bispecific gp100 peptide-HLA-A*02:01-directed T-cell receptor (TCR) CD3 T-cell engager, Kimmtrak®.16 The majority of non-T cell-targeted engagers (e.g., targeting CD16 for natural killer cells and CD47 for macrophages), immune cell enhancers (e.g., targeting 4-1BB), dual immune checkpoint inhibitors (e.g., targeting PD-1, CTLA-4, and LAG-3), and dual TAA binders (e.g., targeting HER2, EGFR, DLL4, and ANG-2) are currently in the preclinical or early stages of clinical development. Despite scarce data to fully evaluate the clinical relevance of these ADAs at this early stage, emerging data suggest diverse outcomes. ADA-related information from selected BsAbs representing the aforementioned categories is listed in table 1, showing a wide range of ADA incidences with diverse clinical impact across both approved and investigational BsAbs in oncology.
Clinical relevance of ADAs to selected BsAbs in oncology
A high ADA incidence does not always correlate with clinical impact, but a low ADA incidence may sometimes be surprisingly consequential. For example, Removab® is highly immunogenic due to the foreign nature of the rat/murine chimeric antibody origin to the human immune system. Data derived from the pivotal study showed clinical benefit can be obtained, despite the high human anti-murine antibody incidence of 94%.1 17 18 Navicixizumab is a humanized BsAb. Although only 4 out of 25 (16%) patients were ADA-positive in a phase Ib study (NCT03030287), there was an increase in drug clearance and an associated infusion reaction in three patients.19 This diversity in clinical ADA outcomes demonstrates the unpredictability of clinical immunogenicity to BsAbs and its potentially outsized impact on clinical trials. Accordingly, a comprehensive immunogenicity risk assessment (IgRA) becomes an imperative for all BsAbs headed into the clinic.
Factors to consider for IgRA of BsAbs
A complex matrix of drug-related and patient-related factors contribute to the immunogenicity risk for BsAbs. The incidence and clinical consequences of ADAs may differ among different BsAb formats and products for different disease indications. Therefore, an IgRA should outline a risk-based strategy on a case-by-case basis that is aligned with regulatory guidance.20 Risk is defined as the product of probability and consequences (risk=probability×consequence). The risk-based assessment considers the likelihood and the potential consequence of immunogenicity, including the multiple factors contributing to immunogenicity potential and the severity of clinical consequence.20–22 Some of the factors that should be specifically considered for anti-cancer BsAbs are discussed further.
Product-related factors
Sequence-based risk and T-cell epitopes
BsAbs are generally designed to have novel structural formats with bioengineered sequences. This may give rise to neoantigens or expose cryptic epitopes that may potentially become antigenic or elicit immune responses. If the molecule includes a non-mAb functional domain with sequence or structural similarity to its endogenous counterpart (e.g., cytokines, hormones, TCRs, or growth factors), there is potential for generating neutralizing antibodies (Nabs) that cross-react with the endogenous protein, leading to a deficiency in that protein or its function with clinical consequences. Currently, in silico algorithms and in vitro T cell-based assays are the main tools used to assess sequence-based, T cell-dependent immunogenicity risk of therapeutic protein molecules during preclinical development.
In silico algorithms assess the ability of the primary amino acid sequence of the therapeutic protein to bind major histocompatibility complex (MHC) class II. The assessment is based exclusively on the primary amino acid sequence of the molecule without considering structural epitopes and multiple other factors that may contribute to immunogenicity. This method tends to be over-predictive. Therefore, a direct correlation between in silico results and clinical immunogenicity is not expected, and in silico analyses are best used for stack-ranking candidate molecules where other drug attributes such as size and target are equivalent.
The in vitro T cell-based assay evaluates sequence-based risk by assessing the rate and magnitude of T-cell responses to a therapeutic protein using a set of human leukocyte antigen (HLA)-typed donors representing the global frequency of HLA alleles. A standard approach is a peripheral blood mononuclear cell (PBMC):T-cell assay format, in which the test molecule is incubated with PBMCs from individual donors followed by T-cell proliferation or cytokine measurements. For therapeutic protein molecules with immunomodulatory activity or with targets expressed on PBMCs, an alternative assay format such as the dendritic cell (DC):CD4+ T-cell proliferation assay should be considered. In this assay, the test molecule is first incubated with DCs, then co-cultured with autologous CD4+ T cells, followed by T cell-proliferation measurements.
Additional in vitro tools to assess sequence-based risk include DC internalization assays,23 24 liquid chromatography/mass spectrometry-based MHC-associated peptide proteomics (MAPPs) assay,25 26 peptide/HLA-binding assay,27 and peptide T-cell activation/restimulation assay.28–30 These assays may complement the PBMC and DC:CD4+ T assays in settings where the target of the BsAb directly interferes with the T cell-proliferation readout. However, it is important to understand that each assay has its limitations. For example, while the MAPPs assay can narrow down the potential T-cell epitopes within a large therapeutic protein molecule and identify those that are naturally processed and presented by antigen-presenting cells (APCs), it generally only captures the strongest HLA class II binders and assesses peptides bound to HLA-DR (but not HLA-DP and DQ). The challenge of using in vitro assays stems from the difficulty of recapitulating the full breadth of the ADA response which inherently involves multiple cell types in vivo. These assays often evaluate a limited number of steps of the multi-step T cell-dependent ADA formation process. There are emerging technologies such as lymph node systems31 and humanized mouse models32 that take into account a wider spectrum of immune system components and thus may be more amenable to immunogenicity evaluation. However, these tools are often costly, time-consuming, unlikely to be high-throughput, and would most likely require significant expertise to generate high-quality data.
For BsAb-based immunotherapy in oncology, given their diverse formats and targets, each of the standard assays applied on its own may be limited in its ability to inform on sequence-based immunogenicity risk. Therefore, a combination of two or more assays covering multiple aspects of a potential ADA response may help to assign risk based on an integrated approach.33
Pre-existing reactivity and B-cell epitopes
Pre-existing reactivity generally has not been an immunogenicity risk factor for drug development in the past. However, there have been increasing concerns in recent years with rapidly emerging BsAbs and novel therapeutic protein products entering the clinic.34 35 Pre-existing reactivity is not fully understood but could be due to the presence of soluble target, matrix effect, or pre-existing antibodies.36–38 A high prevalence of pre-existing antibodies has been reported against gene therapy vectors (e.g., adeno-associated virus), pegylated proteins, glycan epitopes, selected biotherapeutics (e.g., recombinant cytokines, growth factors, or engineered antibody scaffolds), as well as among patients with rheumatoid arthritis (RA) and other autoimmune diseases.39 The magnitude and epitope specificity of pre-existing antibodies may or may not affect treatment-emergent ADA (TE-ADA), pharmacokinetics (PK), pharmacodynamics (PD), efficacy, or safety of the drug in the clinic.40 A notable case report from Bivi and colleagues34 described a BsAb of an IgG-single-chain variable fragment (scFv) format, which had high pre-existing reactivity with an overall incidence of TE-ADA of 94% in a phase I trial. The TE-ADAs affected drug exposure, and the scFv domain was identified as the target for both the pre-existing reactivity and TE-ADA. In another phase I clinical study (NCT03752177) for LY3415244, a BsAb against TIM-3 and programmed death ligand 1 (PD-L1), most patients had low levels of pre-existing reactivity at baseline, but all (n=12) developed TE-ADAs that were associated with loss of soluble TIM-3 target engagement. Two patients with high ADA titers exhibited hypersensitivity reactions consistent with anaphylaxis, which led to an early trial termination. Notably, the observed pre-existing reactivity and early TE-ADAs mainly targeted the TIM-3-binding domain, which contained amino acid changes for the purpose of engineering the BsAb format.35 41 These results suggest that the specific amino acid changes or chemical modifications introduced to engineer novel BsAb formats may create novel B-cell epitopes or expose cryptic epitopes, which may act as main drivers for pre-existing reactivity and a potentially boosted TE-ADA response. Similar findings were observed for the previously identified pre-existing human anti-hinge antibodies reactive to neoepitopes in the hinge region of therapeutic human monoclonal Fab or (Fab’)2 fragments.42–44 High pre-existing ADAs (36%–52%) were observed for brolucizumab, a recombinant humanized scFv antibody fragment inhibiting human vascular endothelial growth factor.45 46 Therefore, implementation of pre-existing reactivity screening and characterization during the preclinical phase of drug development should be considered for BsAbs. Based on the data, a proactive approach to mitigate the potential risk by selecting the candidate or molecule format with the least pre-existing reactivity should be considered. Some in vitro tools to assess pre-existing reactivity and B-cell epitopes include binding ADA assays, domain characterization, and in vitro B cell-based assays.47 A structure-based engineering approach has been used to abrogate pre-existing ADAs binding to the neoepitope of antibody fragments by small modifications, while maintaining favorable developability characteristics.48
MOA-based risk
Drug MOA-based or pharmacology-based risk is emerging as an important immunogenicity risk factor. In oncology, many BsAbs are designed to potently stimulate the immune system to induce a strong anti-tumor response, a potential corollary of which may be the generation of an unwanted ADA response. In addition, immunotherapy has advanced into front-line settings and become standard of care in select tumor types; BsAbs are now being investigated in patients that may have received, or are concurrently receiving, immune checkpoint inhibitors. We discuss as follows potential mechanisms such as (1) ADA-mediated non-specific immune activation, (2) immune complex (IC) formation, and (3) synergistic immune activation, which may contribute to an increased immunogenicity risk for anti-cancer BsAbs.
ADA-mediated non-specific immune activation for BsAbs targeting cell surface receptors
Some cell surface receptors require dimerization or higher-order crosslinking to trigger activation, and this has informed BsAb design to mitigate unintentional immune activation. Rybrevant®, for instance, uses monovalent engagement of its mesenchymal–epithelial transition tyrosine kinase receptor (C-MET) target to avoid the proliferative signals induced by C-MET dimerization.49 50 There is potential, however, for agonistic products that are designed to activate cell surface receptors to act as a ‘super-agonist’ when cross-linked by ADAs through ADA–drug–target interactions, leading to ADA-mediated immune activation.51 Similarly, for antagonistic products designed to inhibit the activation of cell surface receptors, cross-linking of the drug-receptor complex by ADAs may also result in receptor activation and cytokine release.52 figure 1A is an example of a BsAb agonist drug (IgG-scFv format) which bridges two target cells. The drug is designed to induce target B-dependent target A clustering and immune activation. However, ADAs may potentially cross-link target A via binding to the drug and cause unexpected non-specific immune activation and systemic toxicity. Although there are currently limited clinical examples in oncology, the risk of ADA-mediated cross-linking and activation of cell surface receptors theoretically exists and should be considered when designing BsAbs targeting cell surface receptors.

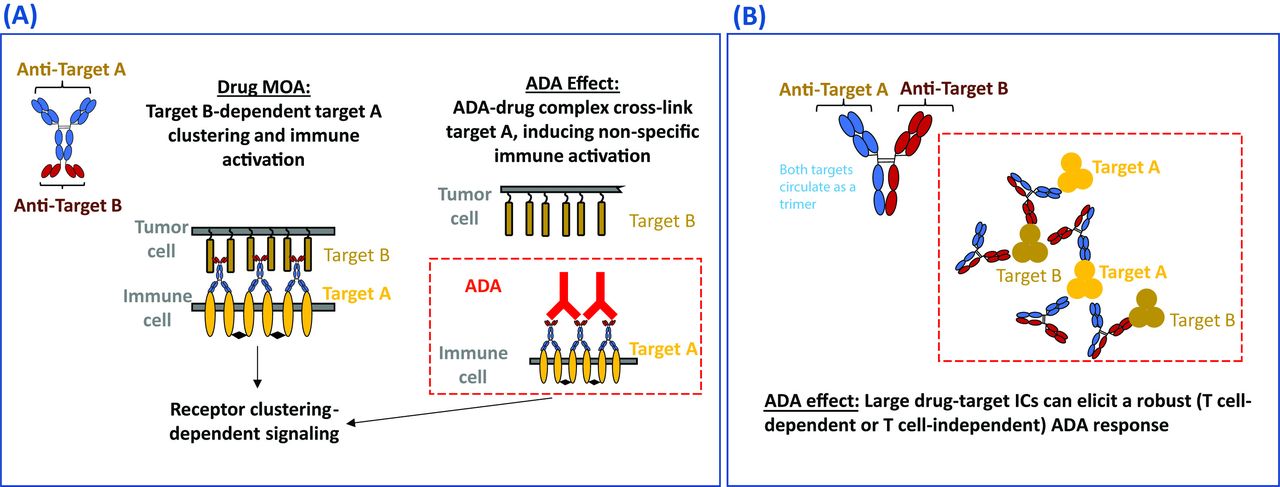{kind=link}
Drug MOA-based immunogenic risk. (A) Diagram of ADA-mediated non-specific activation of a hypothesized BsAb with bridging agonist activity. The drug was designed to induce target B-dependent target A clustering and immune activation. However, ADAs may potentially cross-link target A and cause unexpected non-specific immune activation and systemic toxicity. (B) Diagram of IC formation between BsAbs and multimeric soluble targets for a hypothesized BsAb with dual soluble targets and synergistic MOA. The drug may potentially form large ICs with multimeric soluble targets and induce a robust T cell-dependent or T cell-independent ADA response. ADA, anti-drug antibody; BsAb, bispecific antibody; IC, immune complex; MOA, mechanism of action.
IC formation between BsAbs and multimeric soluble targets
An example of a BsAb drug (hetero-IgG format) targeting two soluble cytokines or ligands resulting in IC-mediated ADA is presented in figure 1B. The drug may potentially form large ICs with both trimeric target A and target B, which may elicit a robust T cell-dependent or T cell-independent ADA response. While there are no examples yet of this phenomenon in the oncology space, there are many examples of associations between IC formation and immunogenicity for anti-inflammatory drugs.53–57 The formation of large ICs between the antibody drug and multimeric soluble targets may result in increased immunogenicity owing to enhanced uptake of the therapeutic agent by APCs. Large ICs can also directly cross-link B-cell receptors, leading to T cell-independent B-cell activation and antibody formation.58 59 Since the drug-to-target stoichiometric ratios can affect IC formation and subsequent ADA development in the clinic, complex formation can be assessed by standard methods such as size- exclusion chromatography when designing BsAbs candidates targeting ligands which could exist as soluble multimers.54
Immunogenicity risk due to synergistic immunostimulatory drug MOA
Immune checkpoint inhibitors
Drugs with immunostimulatory activity may have a greater likelihood of inducing immunogenicity compared with drugs with known immunosuppressive MOA. Despite this possibility, monotherapies of multiple immune checkpoint inhibitors (e.g., anti-PD-1 mAbs and anti-CTLA-4 mAbs) have shown low ADA incidence.7 A variety of BsAbs were recently developed to have synergistic immunostimulatory activities, for example, through dual checkpoint inhibition, immune cell activation, or direct cytokine stimulation.4 Such synergistic immune activation may contribute to an increased immunogenicity risk. While many BsAbs are currently still in early development stages with limited ADA data reported, increased immunogenicity was observed from selected combination therapies with immune checkpoint inhibitors. Most notably, the incidence of anti-nivolumab antibodies increased from 11.2%–12.7% to 23.8%–37.8% when nivolumab (anti-PD-1 mAb) was dosed in combination with ipilimumab (anti-CTLA-4 mAb), a striking two-fold to three-fold increase over nivolumab monotherapy.60 Despite the increased ADA incidence, nivolumab exposure was maintained over time as many of these ADA responses declined within the first 6 months on the combination therapy.61 This was consistent with a subgroup analysis of ADA-negative versus ADA-positive subjects which revealed comparable overall response rates and overall survival between the two groups. Similarly, a phase I first-in-human (FIH) study of FS118, a tetravalent BsAb targeting LAG-3 and PD-L1, reported low-titer ADAs in 42% of treated patients. These anti-FS118 antibodies were transient at higher dose levels and did not affect drug exposure.62 63 Taken together, while ADA incidence and clinical impact have not been fully evaluated, synergistic immune activation via targeting dual checkpoints can theoretically increase immunogenicity risk and warrants consideration.
T-cell engagers
T-cell engagers with B cell-depleting MOAs generally have low immunogenicity risk. Blincyto®, the first BsAb approved worldwide in oncology for the treatment of B-cell precursor acute lymphoblastic leukemia, is an excellent case example. As one of the first-generation BiTE® molecules, the drug is produced from two distinct parental murine mAbs binding to CD19 expressed on malignant B cells and CD3 on T cells. Although the murine sequences in Blincyto® posed a potential immunogenic risk, less than 2% of patients were positive for binding ADAs across hundreds of treated patients in phase III trials.8 The MOA of Blincyto® effectively ablates all B cells expressing CD19, thereby limiting ADA formation. In contrast, non-B cell-depleting T-cell engagers may run a higher risk of inducing clinical immunogenicity if a T-cell epitope is present in the drug, as the MOA of T-cell engagers involve repeated rounds of polyclonal T-cell activation.
4-1BB T-cell enhancers
The T-cell costimulatory receptor 4-1BB is another attractive target for anti-cancer drug development. Thus far, 4-1BB agonists have been proven to be immunogenic in the clinic. In a phase I study (NCT01471210) among patients with r/r B-cell lymphoma, 16%–30% of patients developed ADAs after urelumab monotherapy.64 In another phase I study (NCT01307267) among patients with advanced cancer, 41.8% of patients developed treatment-induced ADA after utomilumab monotherapy.65 The high ADA incidence to 4-1BB agonists may be in part explained by the unique biology of the 4-1BB protein, which is expressed on both APCs and T cells. The primary role of 4-1BB is to provide a potent co-stimulatory signal during the T-cell priming and activation stage of the immunity cycle. Cross-linking of 4-1BB on the APC surface by an anti-4-1BB agonist may accelerate its internalization into the APC. Anti-4-1BB agonism on T cells also provides further co-stimulation and prevents activation-induced cell death, prolonging survival and potentiating CD4+ T-cell help. Together, these mechanisms may contribute to the high ADA incidence of anti-4-1BB agonists. In phase Ib studies of utomilumab in combination with anti-PD-1 pembrolizumab (NCT02179918),66 or in combination with anti-CCR4 mogamulizumab (NCT02444793),67 54.2%–65.2% of patients developed treatment-induced ADA, half of which were neutralizing. There was no substantial impact of ADA/Nab on PK and safety. Therefore, extant data demonstrate that 4-1BB agonists, either as monotherapy, or in combination therapy with other immuno-oncology (IO) agents, induce a relatively high ADA incidence, suggesting that BsAbs incorporating a 4-1BB-targeted functional domain may encounter similar immunogenicity risk. Indeed, preliminary data from a phase I FIH study (NCT03330561) of cinrebafusp alfa (PRS-343), a BsAb targeting HER2 and 4-1BB, showed ADAs were elicited in 27.8% of patients with HER2-positive malignancies at doses of ≥2.5 mg/kg.62 68
Taken together, emerging data from early trials suggest a relatively high ADA incidence for BsAbs with immunostimulatory activity. Whether the ADAs will have clinical impact remains to be fully evaluated. Therefore, the MOA-based immunogenicity risk should be evaluated in the context of multiple other risk factors as well, which may require additional assays specific to the drug MOA for the risk assessment.
Quality attribute/excipient-related risk
The complexity of different BsAb formats creates many manufacturing challenges such as drug stability, increased tendency to form aggregates, product-related impurities, and mispaired species. All these factors may influence immunogenicity. Therefore, it is critical to minimize these drug attributes in the final drug product and to be sure the drug manufacturing process follows the regulatory guidance. Some in vitro assays including T cell-based assays could be used to assess immunogenicity risk related to drug quality attributes.69
Patient-related factors
Patient’s previous treatments
BsAbs and multi-specific therapeutic protein products in oncology often target common TAAs, immune checkpoint inhibitors, or immune cell receptors. ADAs generated to a previous product may cross-react to a related product, resulting in pre-existing antibodies at baseline against the new drug. The antibody response could be boosted to a high titer with early onset due to a rapid memory recall response. Therefore, pre-medication history of the patient including ADA status and specificity (if available) and screening for pre-existing antibodies (if possible) should be considered before switching the patient to a new therapy of the same class. In addition, patients’ prior lines of therapy may affect their immune status at baseline, making them more or less susceptible to developing TE-ADAs during the BsAb therapy.
Dosage and dosing regimens
The drug dosage, dose regimen and route of administration may contribute to immunogenicity. FIH oncology studies investigating immunostimulatory BsAbs often incorporate conservatively low doses (e.g., ng to µg doses for BiTE® molecules, compared with mg doses for mAb drugs) during dose escalation. Unlike traditional mAbs that are often dosed much higher than necessary to execute their MOA and therefore have a broad therapeutic window, T-cell engagers are often met with a toxicity ceiling due to cytokine release syndrome (CRS) and other immune-related adverse events (AEs) that prohibit further dose escalation. Not surprisingly, drug exposure at these low doses tends to be more susceptible to an ADA-mediated effect on drug clearance.
Immunosuppressive premedication such as the glucocorticoid dexamethasone may also be introduced to dampen global immune responses prior to BsAb administration. The use of co-medications such as methotrexate with mAb drugs like adalimumab and infliximab has been shown to be effective in reducing immunogenicity with improved drug efficacy in the treatment of RA and other inflammatory conditions.70 Whether such broad-acting immunosuppressive approaches may be effective for mitigating ADA formation in the context of BsAb immunotherapy among cancer populations remains to be seen.
The route and site of administration of a therapeutic protein can affect the biodistribution, residence time, and likelihood of encounter with APCs, factors that altogether could potentially impact immunogenicity. In certain circumstances, a lower dose administered intermittently, including dose interruptions (e.g., due to neurological toxicity or CRS), or re-exposure after a long treatment-free interval, may be associated with enhanced immunogenicity, compared with a larger dose administered continuously without interruption.20 71 However, higher doses do not uniformly overcome ADA responses. Several studies have shown that intravenous administration may have a lower immunogenicity risk than subcutaneous administration, although there are examples of no apparent differences in the ADA incidence between the subcutaneous and intravenous routes.72 73 BsAbs are often first administered via intravenous routes, but many are being dosed via subcutaneous routes (e.g., BCMA-directed T-cell engagers) in parallel, to balance efficacy and safety with convenient drug access. Pasotuxizumab (AMG 212/BAY2010112), a PSMA-directed BiTE® molecule, is a compelling example of a T-cell engager tested in patients via both subcutaneous and continuous intravenous (cIV) routes with divergent ADA incidences. In an FIH study (NCT01723475) conducted in patients with advanced metastatic castration-resistant prostate cancer,13 30 of 31 evaluable subjects (97%) developed ADAs following subcutaneous administration. While these ADAs were not associated with AEs, the majority of these ADAs were neutralizing and/or associated with decreased drug exposure. Neither application of topical glucocorticoids to the administration site nor prophylactic administration with dexamethasone mitigated the development of ADAs. Due to the high rate of ADA development and the lack of response to ADA mitigation measures, further evaluation of pasotuxizumab via the subcutaneous route was discontinued. Pasotuxizumab was subsequently tested in patients via a cIV dosing regimen. While the maximum tolerated dose was not reached during cIV dose escalation at the time of study discontinuation, ADAs were not detected in any of the 16 patients dosed in the cIV cohorts. Similar results were observed with JNJ-081, a PSMA-directed T-cell engager in the DuoBody® format. The ADA incidence to JNJ-081 was 16.7% (weekly intravenous, 0.1–3 µg/kg) and 63.0% (subcutaneous, 3–60 µg/kg) in the FIH study (NCT03926013). However, the ADA incidence from the intravenous setting was calculated from subjects who had lower doses of JNJ-081 compared with the subcutaneous setting. The high-titer ADAs from subcutaneous cohorts corresponded with a decrease in serum concentration of JNJ-081.15 The high rates of ADA and minimal evidence of therapeutic activity resulted in the premature closure of the JNJ-081 FIH trial. APVO-414 (MOR209/ES414), another T-cell engager targeting PSMA, but formatted in the ADAPTIRTM platform, provides a case example where the dose regimen was modified from a weekly intravenous (0.2–2 µg/kg) dose to cIV (25–300 µg/day) infusion in the initial phase I dose escalation study (NCT02262910). The switch to cIV dosing resulted in a modest reduction from 58% to 50% developing ADA incidence but dramatically decreased titers from 1:250 000 to 1:160–1:320. None of the patients had any adverse reactions due to the ADA, but patients with high ADA titers cleared the drug from their blood to undetectable levels.14 62 74 Despite the decreased titers in the cIV cohorts, sufficient therapeutic benefit was not observed, and the study was discontinued. The learnings from these examples lend caution to the assumption that a switch to a cIV dosing regimen alone would be enough to mitigate ADA formation if a strong T-cell epitope is present in the molecule.
Immunogenicity may be influenced by different dosing strategies investigated during early clinical trials. Particularly for BsAbs in oncology, the ADA incidence and clinical impact data obtained from the early dose escalation studies may not always represent the ADA profile under registrational settings. Thus, it is important to fully evaluate the ADA profile at the predicted efficacious exposure or the recommended registrational dose regimens (if possible).
Ongoing IgRA and IgRA-driven immunogenicity monitoring strategy in the clinical stage
A combination of several in silico, in vitro and ex vivo tools at the preclinical stage to identify and characterize both sequence-based T-cell epitopes and conformational pre-existing B-cell epitopes has improved the reliability of clinical candidate selection. Most preclinical IgRAs consider immunogenicity risks due to treatment-related and patient-related factors, but the drug MOA risk is under appreciated and requires more attention prior to clinical development.27 The IgRA is an ongoing process and must be re-evaluated as clinical data become available. Recent FDA guidance recommends that IgRA aligns with a sample collection and immunogenicity testing plan for inclusion in the original investigational new drug application.75 The immunogenicity testing strategy supporting clinical studies is driven by a drug-specific IgRA. For BsAbs entering early phase trials, a multi-tiered testing approach is recommended.76 77 Samples testing positive in a binding ADA assay may be further characterized, such as for domain specificity or neutralizing activity, according to the IgRA.
The complexity of the structure and MOA of the BsAbs pose increasing challenges for the development of the most appropriate assay format for ADA detection. Multiple assay formats need to be evaluated for both binding ADA78–81 and Nab detection,82 83 as well as the ability to overcome target and matrix interference under multiple study and disease settings.84 85
Immunogenicity management
Development of a reliable IgRA that guides drug candidate and format selection, as well as a proactive approach for de-risking candidates during preclinical development, are critical to mitigate potential risk in the clinic.
However, if an antibody response does occur during the clinical stage, the gold standard for ADA mitigation involves identifying and removing the offending B-cell and T-cell epitopes in the biologic via re-engineering efforts. Apart from ADA monitoring, domain characterization, and ADA impact analyses, a clinical memory recall assay and HLA class II subtyping may be considered during clinical development.
In a clinical memory recall assay, PBMCs from ADA-positive (antigen-experienced) subjects containing a pool of drug-specific memory CD4+ T-cell clones can be ‘recalled upon’ in an ELISpot assay when pulsed with putative immunogenic peptides ex vivo, which may confirm that the selected peptides induce immunoreactivity.86 In the absence of ADA+ subjects’ PBMCshowever, an extended in vitro DC:CD4+ T-cell assay using healthy donors and multiple rounds of stimulation with putative peptides could be considered in order to recapitulate an antigen-experienced memory response.28 29 The clinical memory recall assay identifies specific immunoreactive sequences that can be modified in reverse translation efforts. However, this process of reverse translation to de-immunize the biologic is time-consuming, which presents a unique challenge particularly in the context of an intensely competitive IO landscape.
Several publications have attempted to make associations between specific HLA class II allele subtypes and the development of ADAs.87–89 However, these studies have thus far been conducted on mostly anti-cytokine mAbs and in rheumatic diseases, not in oncology. While the association of ADA formation with specific HLA class II alleles has not been established for anti-cancer biologics, there may be consideration to collect HLA class II allele subtype information from oncology studies to determine any potential HLA class II allele association with immunogenicity risk. Ideally, such associations would be conducted on a ‘discovery’ cohort and confirmed with a ‘validation’ cohort, with large sample sizes being required to provide enough proof of confidence in these associations, if any.
If returning to the drawing board to re-engineer a new version is not an option, one may begin to consider different approaches for potential ADA mitigation. Such approaches must prioritize scientific evidence, safety,logistics with the current dosing regimen of the biologic and commercial implications. Some of these strategies may include using currently approved, broad-acting immunosuppressive agents90 and other immune tolerance induction approaches.91 However, for BsAbs in oncology, ADA mitigation strategies implemented in the clinic should be weighed carefully and balanced with the patient’s benefit and risk during development. Importantly, we should focus on a more comprehensive IgRA with a proactive de-risking approach during the preclinical stage, as well as reverse translation efforts that inform on the platform and scaffold changes to be pursued.
Conclusions
In this review, we describe the importance of a thorough preclinical assessment of immunogenic risk during BsAb drug development. The multi-functional features such as complex structures, highly engineered sequences, and a synergistic, immunostimulatory drug MOA for cancer immunotherapy may all contribute to the increased immunogenicity risk and its potential clinical impact. Consequently, this class of therapeutic candidates requires more preclinical assessments, careful clinical monitoring, and a timely re-evaluation of the IgRA relative to mAb therapeutics in order to improve chances of success during clinical development.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Footnotes
Contributors YZ wrote the first draft based on the outline aligned with all authors. All authors provided critical revision of the manuscript for important intellectual content and approved the final version.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests All authors are employees and shareholders of Amgen Inc.
Provenance and peer review Not commissioned; externally peer reviewed.