Article Text

Abstract
Background Various tumors are insensitive to immune checkpoint blockade (ICB) therapy. Toll-like receptors (TLRs) establish the link between innate and adaptive immunity, which can assist T-cell activation and serve as promising targets for combination to enhance ICB therapy. Here, we aimed to improve efficacy for anti-programmed death ligand 1 (PD-L1) therapy by developing a PD-L1/TLR7 dual-targeting nanobody-drug conjugate (NDC), based on the PD-L1 nanobodies and TLR7 agonist we developed.
Methods PD-L1 nanobodies were obtained by phage display screening and identified through T-cell activation bioassay, in vivo imaging and quantitative biodistribution study. Immune activation and PD-L1-inducing of TLR7 agonists were evaluated in diverse innate cell models. We constructed PD-L1/TLR7 dual-targeting NDCs by chemically coupling PD-L1 nanobodies and TLR7 agonists. The antitumor effect was evaluated via several murine or humanized solid tumor models. Immunophenotyping, immune cell depletion, tumor rechallenge, RNA sequencing and PD-L1-deficient models were combined to determine the mechanism for NDCs function. The dynamics of the in vivo behaviors of NDCs were assessed based on multiorgan changes in PD-L1 levels.
Results The screened PD-L1 nanobodies were characterized as tumor-targeting and alleviated T-cell immunosuppression. The TLR7 agonists induced broad innate immune responses and intratumoral PD-L1 expression on antigen-presenting cells (APCs), and its antitumor effect was dependent on intratumoral delivery. The combination of TLR7 agonists and PD-L1 nanobodies activated both innate and adaptive immunity and upregulated PD-L1-related signaling pathways. After coupling to form dual-targeting NDCs, TLR7 agonists and PD-L1 nanobodies exerted synergistic antitumor effects and safety in either ‘hot’ or ‘cold’ tumor and early or advanced tumor models, reshaped the tumor immune microenvironment and induced antitumor immune memory. CD8+ T cells and natural killer cells were the main effector cells for NDCs to function. NDCs can promote PD-L1 expression on intratumoral APCs and tumor cells, and subsequently achieve targeted enrichment in tumors. Moreover, the efficacy of NDCs is biased toward dependence on host expression of PD-L1.
Conclusions The novel PD-L1/TLR7 dual-targeting NDC exhibited potent efficacy against heterogeneous tumors through orchestrating innate and adaptive immunity, which could act as a promising strategy to improve ICB therapy and shows prospects for clinical development.
- PD-L1
- TLR7
- nanobody-drug-conjugate
- cancer immunotherapy
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Many patients are insensitive to programmed cell death protein 1/programmed death ligand 1 (PD-L1) immune checkpoint blockade therapy (ICB).
Toll-like receptor (TLR)7 agonists can improve the response rates of ICB therapy.
TLR agonists show toxicity associated with extensive immune activation after systemic administration, and TLR agonist tumor-targeted delivery is essential.
Nanobodies have good tumor targeting and are suitable for drug delivery.
WHAT THIS STUDY ADDS
TLR7 agonist activated innate immunity and increased PD-L1 expression in tumors, which boosts anti-PD-L1 nanobody tumor targeting.
The PD-L1/TLR7 dual-targeting nanobody-drug conjugate (NDC) was developed and exerted potent antitumor effects in multiple tumor models by combining innate and adaptive immunity.
The efficacy of PD-L1/TLR7 dual-targeting NDC was biased toward dependence on host expression of PD-L1.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The PD-L1/TLR7 dual-targeting NDC is a novel immunotherapy that exhibits potent efficacy against heterogeneous tumors through orchestrating innate and adaptive immunity and shows prospects for clinical development.
Background
In the tumor microenvironment (TME), programmed death ligand 1 (PD-L1) molecules, which are mainly expressed on tumor cells, macrophages and dendritic cells (DCs), interact with programmed cell death protein 1 (PD-1) on T cells, leading to T-cell exhaustion and antitumor immunity inhibition.1–3 PD-1/PD-L1 immune checkpoint blockade (ICB) therapy is effective against many cancer types,4–7 but only a small fraction of patients respond to treatment.8 Determining the tumor resistance mechanisms for PD-1/PD-L1 antibody therapy and improving therapeutic efficacy are urgent areas to be addressed.
PD-1/PD-L1 antibody therapy alone is more efficacious for highly immunogenic ‘hot’ tumors with adequate T-cell infiltration or high PD-L1 expression, such as melanoma and non-small cell lung cancer, and less efficacious for poorly immunogenic ‘cold’ tumors with inadequate T-cell infiltration or low PD-L1 expression, such as breast cancer and pancreatic cancer.9 10 Converting ‘cold’ tumors into ‘hot’ tumors by combining with drugs that promote tumor immunogenicity and PD-L1 levels, PD-1/PD-L1 antibodies can achieve better efficacy.11
Toll-like receptor 7 (TLR7), a target for innate immune enhancement, is predominantly expressed in macrophages, DCs, natural killer (NK) cells and B cells.12 13 Our studies and others have shown that TLR7 agonists inhibited tumor growth in a wide range of tumor models.14–17 By stimulating TLR7, antigen-presenting cells (APCs) can be activated, and subsequently, their antigen-presenting function is enhanced to facilitate T-cell activation.18 19 In addition, the increased intratumoral infiltration of NK cells, cytotoxic T cells and antigen-specific interferon (IFN)-secreting effector cells is one of the antitumor mechanisms of TLR7 agonists.20–22 TLR7 agonists are a potential option to improve the response rates of ICB therapy.23
In addition to improving the immunogenicity of tumors, favorable targeting of drug molecules is also crucial. Solid tumors have a dense extracellular matrix, which makes it difficult for monoclonal antibodies to penetrate deeply into the tumor interior.24 Nanobodies have good tumor targeting and specificity, making them suitable for targeted therapy and drug delivery.25 Based on nanobodies, nanobody-drug conjugates (NDCs) are new forms of antibody-drug conjugates (ADCs).26 Many NDCs that couple diverse immunotoxins with nanobodies have been developed and have shown beneficial antitumor effects.27
Here, we describe an immunotherapeutic strategy that boosts tumor immunogenicity and breaks immune tolerance by combining TLR7 agonists and PD-L1 nanobodies. We developed a novel PD-L1/TLR7 dual-targeting NDC, which allows TLR7 agonists and PD-L1 nanobodies to exert synergistic antitumor effects. We elucidated the role of cellular localization of PD-L1 on the efficacy of NDC. The antitumor activity, safety and mechanism of action of NDCs were evaluated in multiple tumor models in this work.
Methods
Details about mice, cell lines, plasmids, construction of stable transfected cell lines, quantitative biodistribution study, The Cancer Genome Atlas (TCGA) cohorts analysis, in vitro analysis of immune responses, in vivo treatment study and antibodies used in immunotyping are provided in online supplemental methods.
Supplemental material
Acquisition and expression of murine/human PD-L1 and nanobodies
The nanobodies were obtained as described in our previous study,28 including camel immunization, nanobody library construction and phage display screening. Genes encoding nanobodies and murine/human PD-L1 (amino acid residues 19–239) were cloned into the pFUSE-mIgG2b-Fc or pFUSE-hIgG1-Fc2 vector for expression using HEK293F cells.
Antibody conjugation and characterization
The synthesis of SZU-101 was performed as described in our previous study.16 SZU-107 was prepared by PEG-ylation of SZU-101. SZU-107-N-hydroxysuccinimide was used for conjugation to antibodies. The NDC was denatured and identified by a Xevo G2-XS QTOF mass spectrometer (Waters). The drug-to-antibody ratio (DAR) value was calculated based on the increase in molecular weight of NDC over the uncoupled antibody. Details are provided in online supplemental methods.
Bioactivity evaluation of nanobodies and NDC
The binding activity of nanobodies and NDC was detected with mPD-L1-Fc-based ELISA and 293T/mPD-L1 cell-based flow cytometry. The detection antibodies were anti-mFc-horseradish peroxidase (Thermo Fisher) or goat anti-human IgG-Fc (FITC, Abcam). To assess the blocking capacity, candidates with serial dilutions and 20 μg/mL mouse PD-1-biotin were incubated with 293T/mPD-L1 cells, which were then stained with goat anti-streptavidin (PE, Abcam) solution and detected by flow cytometry.
In vivo treatment study
To evaluate the antitumor activity of candidates, CT26, B16-F10, CT26/hPD-L1 and PD-L1-deficient B16-F10 syngeneic tumor models and LLC orthotopic lung tumors, and B16F10 lung metastasis tumors were used. For more specific methods, see the online supplemental materials.
In vivo imaging
CT26 tumor-bearing mice were injected intraperitoneally with fluorescein Cy7-labeled Nb16 or 10F.9G2. Fluorescent antibody distribution in vivo was monitored using an IVIS imaging system (Caliper PerkinElmer) at predetermined time points (0 hour, 1 hour, 2 hours, 4 hours and 7 hours). At the end of the experiment, mice were sacrificed, and tumors were obtained and imaged in vitro.
Immune cell depletion study
BALB/c mice were randomly divided into six groups and injected with CT26 cells (1×106) to the shaved right flank subcutaneously. CD4+ T cells, CD8+ T cells or NK cells were deleted by intraperitoneal injection of antibody drugs (anti-CD4 (Biolegend), anti-CD8 (Biolegend), anti-NK1.1 (Biolegend)) on day 3 (200 μg/day), day 4 (100 μg/day) and day 5 (100 μg/day), respectively. Macrophages were deleted by intraperitoneal injection of chlorophosphate-liposomes (200 μL/day, Yeasen) on day 3. Deletion effect was identified by flow cytometry.
Immunophenotype analysis
Tumor tissues were digested and filtered into single cell suspensions and subjected to cell extraction by lymphocyte separation medium. The cells were blocked with anti-CD16/CD32 antibody (BD Biosciences) and stained with indicated surface antibodies. Intracellular or intranuclear antibodies were stained after permeabilized with Cytofix/Cytoperm buffer or TF Fix/Perm buffer (BD Biosciences). Antibody staining was performed following the antibody manufacturer’s recommendations. Flow cytometry analysis was performed using a BD FACSCalibur or ACEA NovoCyte, and data processing was performed using FlowJo software or NovoExpress software.
RNA sequencing
RNA was isolated from fresh tumor tissues. Transcriptome libraries were constructed with the VAHTS mRNA-seq V3 Library Prep Kit for Illumina (Vazyme). Transcriptome library sequencing was performed by Oebiotech. Differential expression was evaluated with DESeq. A fold change of 1.5:1 or greater and a false discovery rate-corrected p value of 0.05 or less were set as a threshold for differential genes. Immune signature scores are defined as the mean log2(fold change) among all genes in each gene signature. T-cell infiltration within tumor tissues was performed by ImmuCellAI.
NDC safety analysis
Clinical observations were conducted daily from the day next to the randomization to experimental completion. Plasma chemistry was measured by a biochemical analyzer (Cobas C501, Roche). Hematology was measured by blood counters (Hemavet 950 FS). Tissues and organs were embedded and stained with H&E and then examined microscopically.
Statistical analyses
Statistical analysis was performed using GraphPad Prism V.8 Software. Statistical differences were analyzed by Student’s t-test or one-way analysis of variance followed by Tukey’s multiple comparisons test. Animal survival analysis was performed by the Kaplan-Meier method. Detailed statistical methods and sample sizes in the experiments are described in each figure legend. All statistical tests were two sided and p values <0.05 were considered to be significant.
Results
Antitumor activity and tumor targeting of anti-PD-L1 nanobody Nb16
Compared with monoclonal antibodies, nanobodies have many advantages, such as small size, high stability and low immunogenicity. Based on phage display technology, we generated nanobodies that target the extracellular domain of mouse PD-L1. The optimized candidate (termed Nb16) was selected and linked to Fc fragments to form fusion proteins. Nb16 bound to PD-L1 and blocked the interaction between PD-1 and PD-L1, with a 50% effective concentration (EC50) value of 0.04336 μg/mL or 0.516 nM (figure 1A) and a 50% inhibitory concentration (IC50) value of 0.5691 μg/mL or 6.78 nM (figure 1B). The binding and blocking activity of Nb16 was verified in HEK293T cells stably expressing PD-L1 (figure 1C,D). Besides, Nb16 blocked the inhibitory signal from CT26 cells (induced by IFN-γ to express high levels of PD-L1) to Jurkat-NFAT-mPD-1-luciferase cells we constructed (figure 1E), suggesting it could effectively activate T cells. Notably, Nb16 suppressed tumor growth with an inhibition rate of 39.1% (figure 1F) in the IFN-γ-induced CT26 model (online supplemental figure S1). To compare the tumor-targeting ability of nanobody and monoclonal antibody, Nb16 or anti-PD-L1 monoclonal antibody 10F.9G2 were labeled with fluorochrome Cy7 and then injected into mice to detect the tumor accumulation from 0 to 7 hours. The fluorescence intensity in the tumor gradually increased and peaked from 4 to 7 hours, and Nb16 was significantly higher than 10F.9G2 (figure 1G,H). The ex vivo imaging of dissected tumor tissue also showed Nb16 had a 2.7-fold higher tumor accumulation than 10F.9G2 (figure 1I,J). Similarly, ELISA-based quantitative biodistribution indicated that Nb16 rapidly targeted to tumor tissues within 2 hours and accumulated gradually within 8 hours (figure 1K). These demonstrated that the anti-PD-L1 nanobody had enhanced solid tumor targeting compared with anti-PD-L1 monoclonal antibodies. Nevertheless, Nb16 had moderate in vivo activity and combination strategies were required to improve the antitumor efficacy.

PD-L1 nanobody inhibited tumor growth and have better solid tumor targeting. (A) Binding affinity of Nb16 to PD-L1 was assessed via ELISA. (B) PD-1/PD-L1 blocking activity of Nb16 via ELISA. (C) Flow cytometry histograms showing the binding of Nb16 to PD-L1 on HEK293T cells stably expressing PD-L1. (D) Flow cytometry histograms showing that Nb16 blocked the PD-1/PD-L1 interaction in HEK293T cells stably expressing PD-L1. (E) Effect of Nb16 on blocking the inhibitory signal from CT26 cells (induced by IFN-γ) to Jurkat-NFAT-mPD-1-luciferase cells. (F) BALB/c mice bearing CT26 tumors were treated with 200 µg of Nb16 or mFc every 3 days. The tumor growth in mice is shown (n=6). (G and H) In vivo imaging showed the enhanced tumor accumulation of Nb16. (I and J) Ex vivo imaging of the tumors and radiant efficiency of the tumors. (K) ELISA-based quantitative biodistribution study was performed for Nb16 in CT26 tumor-bearing mice. The data are presented as the mean±SEM. **P<0.01; ***p<0.001; ns as determined by unpaired t-test. IFN, interferon; ns, not significant; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; RLU, relative luciferase units.
TLR7 agonist SZU-101 activated innate immunity and increased PD-L1 expression in tumors, and intratumoral administration was required
TLR7 is an important innate immune receptor that is expressed low in various clinical malignancies, and TLR7 expression level was negatively correlated with overall survival of patients in TCGA datasets (online supplemental figure S2A–D). Also, TLR7 showed a positive correlation with innate immune cell infiltration (macrophages, DCs and neutrophils) and negative correlation with myeloid-derived suppressor cells infiltration in tumors (figure 2A–C). Notably, the expression levels of TLR7 with tumor necrosis factor-α (TNF-α), IFN-γ, major histocompatibility complex class II (MHC II), PD-L1 and PD-L1-related regulatory molecules in tumors showed a strong positive correlation (figure 2D and online supplemental figure S2E–J). These data and previous studies23 29–33 suggested that the combination of TLR7 agonists with anti-PD-L1 antibodies may be a rational strategy for cancer therapy.

TLR7 expression correlated with immune infiltration and PD-L1 expression in TCGA cohorts. (A–C) The correlation of TLR7 expression with the immune infiltration level in different tumor types as indicated in the TCGA database was determined by TIMER2.0. Heatmap showing the correlation between TLR7 expression levels and immune cells (DC, macrophage, MDSC, neutrophil, CD4+ T cell and CD8+ T cell) infiltration levels in different tumor types (A). Scatter plots showing the correlation between TLR7 expression levels and immune cells (DCs, macrophages and MDSCs) in COAD (B) and KIRC (C). (D) Correlation analysis of TLR7 and PD-L1-related genes (TNF, IFNG, CD274, HLA-DRB1, HLA-DRA, JAK1, STAT1, STAT3, NFKB1, HIF1A and IRF1) in different tumor types in the TCGA database as determined by TIMER2.0. COAD, colon adenocarcinoma; DC, dendritic cell; IFN, interferon; KIRC, kidney renal clear cell carcinoma; MDSC, myeloid-derived suppressor cell; PD-L1, programmed death ligand 1; TCGA, The Cancer Genome Atlas; TIMER2.0, Tumor Immune Estimation Resource; TLR, toll-like receptor; TNF-α, tumor necrosis factor-α.
TLR7 agonist SZU-10116 we developed could activate mouse bone marrow-derived DCs and stimulate the expression of their maturation markers CD80, CD86 and MHC II higher than three times (figure 3A–C and online supplemental figure S3A–D). Furthermore, in bone marrow-derived macrophages models, SZU-101 repolarized macrophages through upregulating the M1 macrophages by 16.7% and reducing the M2 macrophages by 29.1%, and SZU-101 simultaneously stimulated macrophages to express the IFN-γ (figure 3D–F and online supplemental figure S3E–H), which were then confirmed on the peritoneal macrophage model (online supplemental figure S4). In addition, SZU-101 stimulated NK cells to highly express the activation markers CD69 and CD25 (online supplemental figure S5). These results suggested that SZU-101 can activate innate immunity and has potential antitumor activity.
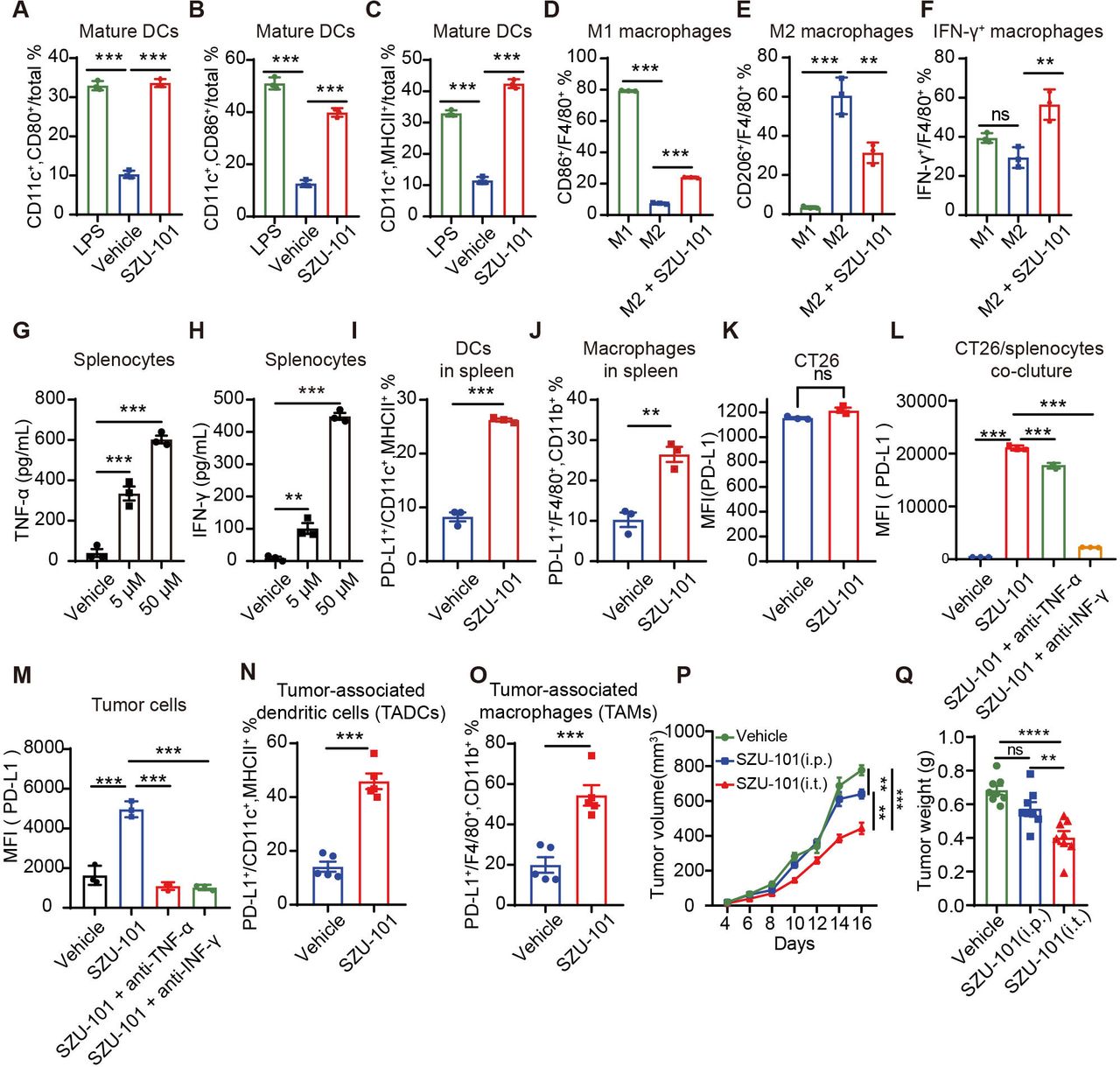
TLR7 agonists activated innate immune cells and induced the upregulation of intratumoral PD-L1 levels. (A–C) BMDCs from BALB/c mice were treated with complete medium, LPS or SZU-101 for 24 hours (n=3). CD80 (A), CD86 (B) and MHC II (C) on DCs were detected by flow cytometry. (D–F) BMDMs from BALB/c mice induced to differentiate into M2 macrophages were stimulated with SZU-101 for 24 hours (n=3). CD86 (D), CD206 (E) and IFN-γ (F) expression in macrophages was determined by flow cytometry. (G and H) Spleen cells from BALB/c mice were treated with PBS or SZU-101 (n=3). TNF-α (G) and IFN-γ (H) were detected by ELISAs. (I and J) Spleen cells from BALB/c mice were treated with SZU-101 for 24 hours (n=3). PD-L1 expression on DCs (I) and macrophages (J) was determined by flow cytometry. (K) Flow cytometry histograms showing the PD-L1 levels on CT26 cells induced by SZU-101 or vehicle. (L) CT26 cells were cocultured with spleen cells (n=3). After treatment with SZU-101, anti-IFN-γ antibody or anti-TNF-α antibody for 24 hours, PD-L1 expression on CT26 cells was determined by flow cytometry. (M) PD-L1 expression on tumor cells in CT26 tumor tissues treated with SZU-101, anti-IFN-γ antibody or anti-TNF-α antibody was determined by flow cytometry (n=3). (N and O) PD-L1 expression on DCs (N) and macrophages (O) in CT26 tumor tissues treated with SZU-101 or vehicle was determined by flow cytometry (n=5). (P and Q) Mice bearing CT26 tumors were treated with SZU-101 intratumorally (i.t.) and intraperitoneally (i.p.) or with a vehicle. Tumor volume and weight are shown (n=8). The data are presented as the mean±SEM. **P<0.01; ***p<0.001; ns by one-way analysis of variance followed by Tukey’s multiple comparisons test or unpaired t-test. BMDC, bone marrow-derived DC; BMDM, bone marrow-derived macrophage; DC, dendritic cell; IFN, interferon; LPS, lipopolysaccharide; MFI, median fluorescence intensity; MHC, major histocompatibility complex; ns, not significant; PBS, phosphate-buffered saline; PD-L1, programmed death ligand 1; TLR, toll-like receptor; TNF-α, tumor necrosis factor-α.
In addition, SZU-101 promoted the secretion of TNF-α and IFN-γ from splenocytes in a dose-dependent manner in vitro, which is consistent with the analysis of clinical TCGA data (figure 3G,H). Considering that TNF-α and IFN-γ are potent PD-L1 inducers, we investigated the effect of SZU-101 on cellular PD-L1 expression levels in vitro. We found that SZU-101 significantly increased PD-L1 expression on splenic DCs and macrophages by more than two times (figure 3I,J). However, SZU-101 did not directly promote PD-L1 expression in CT26 cells cultured alone (figure 3K), and only in the coculture system of CT26 cells/splenocytes, SZU-101 elevated PD-L1 expression in CT26 tumor cells (figure 3L). Moreover, PD-L1 upregulation in tumor cells can be largely overridden by anti-IFN-γ neutralizing antibody and slightly influenced by anti-TNF-α neutralizing antibody (figure 3L), which indicated that PD-L1 expression promoted by SZU-101 in tumors dependent on TNF-α and IFN-γ. Similarly, tumor cells and APCs (DCs and macrophages) were also induced to overexpress PD-L1 by SZU-101 in vivo (figure 3M–O), and either anti-TNF-α or anti-IFN-γ neutralizing antibodies completely abolished PD-L1 expression in tumor cells (figure 3M), which was somewhat different from cytokine neutralization assay in vitro and possibly due to the complex TME in vivo. Besides, SZU-101 significantly inhibited CT26 tumor growth, whereas systemic administration of SZU-101 did not induce antitumor activity to the extent induced by local intratumoral administration (figure 3P,Q), which suggested that precise delivery of SZU-101 to tumors could achieve better antitumor effects. These results suggested that combined anti-PD-L1 antibody and TLR7 agonist therapy may achieve better antitumor efficacy.
The combination of anti-PD-L1 nanobody and TLR7 agonist for tumor inhibition
To evaluate the combination strategy, we tested the effect of SZU-101 and Nb16 combination treatment in vivo (figure 4A). The growth of CT26 tumors treated with a combination of Nb16 and SZU-101 was significantly inhibited with an inhibition rate of 60.8% compared with single-agent treatment (figure 4B,C). We also compared the antitumor effects of Nb16 and 10F.9G2 and their combination with SZU-101. Although there was no significant difference in tumor volume between the Nb16 combination and the 10F.9G2 combination, Nb16 combination possessed better survival (online supplemental figure S6).

The combination of PD-L1 nanobody and TLR7 small molecule agonist exerted potent antitumor effects. (A) BALB/c mice bearing CT26 tumors were treated with 200 μg of mFc, SZU-101 (3 mg/kg), 200 μg of Nb16 or a combination of these compounds. (B and C) Tumor growth and tumor weight are shown (n=9). (D) Immune effector cells in above tumors were quantified by flow cytometry (n=3). Heatmap shows the proportion of cells in a specific cell population. (E–J) RNA-sequencing analysis of tumor tissues from BALB/c mice (n=3) treated as described above. T-cell infiltration analysis by ImmuCellAI (E). Heatmap showing that immune pathway analysis (F). Heatmap showing that cytotoxic cell effect-associated genes (G) and antigen presentation process-associated genes (H) changed after treatment (defined as the log2(fold change)). The CD274 (I) expression and related genes (J) expression (defined as the log2(fold change) of FPKM) are shown as scatter plots. The data are presented as the means±SEM. *P<0.05; **p<0.01; ***p<0.001; ns as determined by one-way analysis of variance followed by Tukey’s multiple comparisons test. IFN, interferon; MHC, major histocompatibility complex; ns, not significant; PD-L1, programmed death ligand 1; Th, T helper; TLR, toll-like receptor.
Next, we used flow cytometry to quantify immune effector cells in mFc-treated, Nb16-treated, SZU-101-treated and combination-treated tumors (online supplemental figure S7). SZU-101 treatment and combination treatment significantly increased DC infiltration and activation in the TME to the same extent (figure 4D, online supplemental figure S8A-B and S9A-B), indicating that the changes in DCs in the combination group were mainly caused by SZU-101. Combination therapy further increased the infiltration of M1 macrophages by 14.1% (figure 4D, supplemental figure S8C and S9C) and decreased the infiltration of M2 macrophages by 25.16% (figure 4D, online supplemental figure S8D and S9D). Only the combination treatment significantly downregulated transforming growth factor-β (TGF-β) expression in macrophages by 1.5-fold (figure 4D, online supplemental figure S8E and S9E). Analysis of tumor-infiltrating lymphocyte alterations showed that both the number of regulatory T cells and TGF-β-associated immune cells were significantly decreased after treatment with combination therapy, whereas neither was affected by administration of Nb16 or SZU-101 (figure 4D, online supplemental figure S8J-K and S9J). Further analysis of the activation of adaptive immunity showed that Nb16 and combination treatment increased the infiltration of intratumoral CD8+ T cells equally, while SZU-101 did not, suggesting that the changes in CD8+ T cells in the combination group were mainly due to Nb16 (figure 4D, online supplemental figure S8F and S9F). Notably, only the combination treatment promoted the expression of granzyme B (GB) in CD8+ T cells by 28.9% and IFN-γ in both CD4+ T and CD8+ T cells, showing elevated T-cell antitumor activity (figure 4D, online supplemental figure S8G-I and S9G-I). In brief, Nb16 combined with SZU-101 could better suppress tumor growth by reshaping the TME, relieving tumor immunosuppression and activating antitumor immune responses.
The combination of anti-PD-L1 nanobody and TLR7 agonist promoted the expression of immune cell signature genes and PD-L1-associated genes
To examine the gene expression changes induced by the combined Nb16 and SZU-101 treatment, we analyzed CT26 tumor tissues by RNA sequencing (RNA-seq, online supplemental figure S10A). Compared with isotype control treatment, combination treatment resulted in differential gene expression, which was mainly enriched in immune system-related pathways, particularly antigen presentation and cytotoxic cell function pathways (online supplemental figure S10B).
To assess the effects of combination treatment on immune infiltration and immune processes, we analyzed the expression of gene signatures associated with distinct immune cell types and processes. The combination treatment resulted in overall improvement of tumor immune pathways (figure 4F) and increased the signature scores of T cells, NK cells, DCs, the IFN-α response and the IFN-γ response (online supplemental figure S10C-H). Since T cells have functionally complex subpopulations, we analyzed T-cell infiltration within tumor tissues and found that the combination therapy caused a reduction in the percentage of intratumoral naive T cells and T helper (Th)17 cells (figure 4E) and an increase in intratumoral Th1 cells (figure 4E). In particular, the combination treatment significantly increased the intratumoral infiltration of cytotoxic cells (NK cells and CD8+ T cells, figure 4E).
We further analyzed the expression levels of genes characterizing the cytotoxic cell effector molecules and antigen presentation molecules. The combination therapy significantly increased the activity of intratumoral antigen-presenting machinery and promoted the expression of effector molecules of cytotoxic cells, such as IFN-γ, TNF-α, GB, perforin and FASL (figure 4G,H). In addition, the expression of PD-L1 and related regulatory genes was significantly upregulated in combination group (figure 4I,J). In summary, the RNA-seq results revealed that the combination of the anti-PD-L1 nanobody and TLR7 agonist promoted antitumor cell infiltration, antigen presentation, cytotoxic effects and PD-L1 expression, which was consistent with our aforementioned findings.
Preparation and identification of the PD-L1 and TLR7 dual-targeting nanobody-drug conjugate
Based on the efficacy of combination therapy, considering that the anti-PD-L1 nanobody possessed effective tumor-targeting properties and that SZU-101 promoted intratumoral PD-L1 expression, coupling SZU-101 with Nb16 to prepare a dual-targeting NDC was thought to achieve better tumor delivery and tumor distribution of both compounds, and then achieve strong tumor suppression.
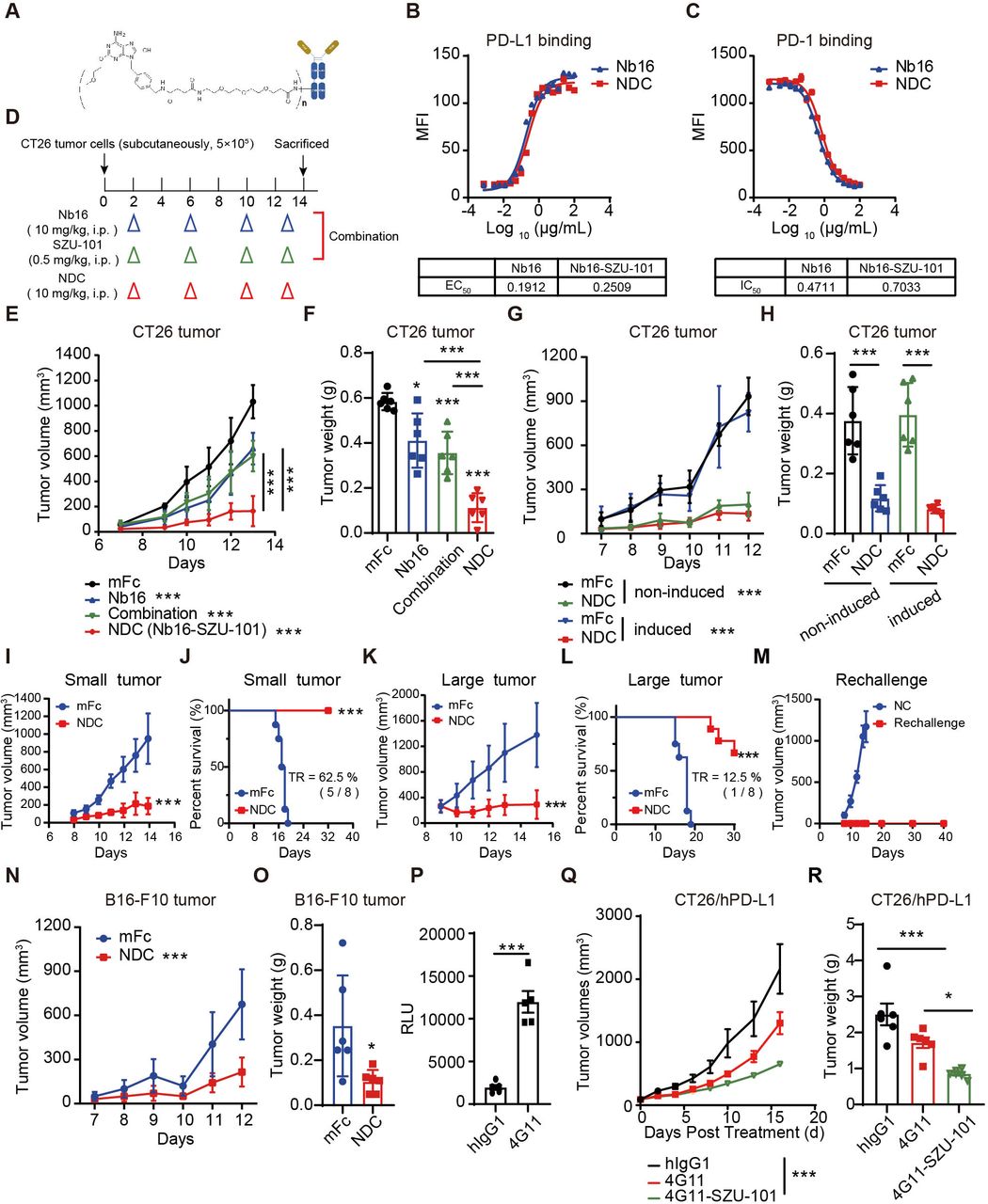
To prepare the dual-targeting NDC, SZU-101 was connected to PEG to yield the intermediate compound SZU-107 (online supplemental figure S11A). The innate immune cell activation effect of SZU-101 and SZU-107 showed no significant difference, which indicated PEGylation did not affect the biological activity of SZU-101 (online supplemental figure S11B-H). The novel dual-targeting NDC Nb16-SZU-101 was formed by coupling carboxyl group-containing SZU-107 and primary amine group-containing Nb16 via the condensation agent EDC (figure 5A). The DAR of the NDC were identified by mass spectrometry before and after coupling and determined to be 4.5 according to the change in the molecular weight of the antibody (online supplemental figure S11I, J). Besides, we measured the EC50 and IC50 values of the nanobodies and found no significant difference before and after coupling, indicating that NDC retained the good bioactivity activities of the original nanobody (figure 5B,C). In addition, we compared the different batches of NDC and the DAR values were around 3–6 (online supplemental figure S11K-M). And the DAR value of the NDC (online supplemental figure S11N), which was placed at 4℃ for 1 week, barely changed compared with the original conjugates (online supplemental figure S11K). As a result, we obtained a novel PD-L1/TLR7 dual-targeting NDC.
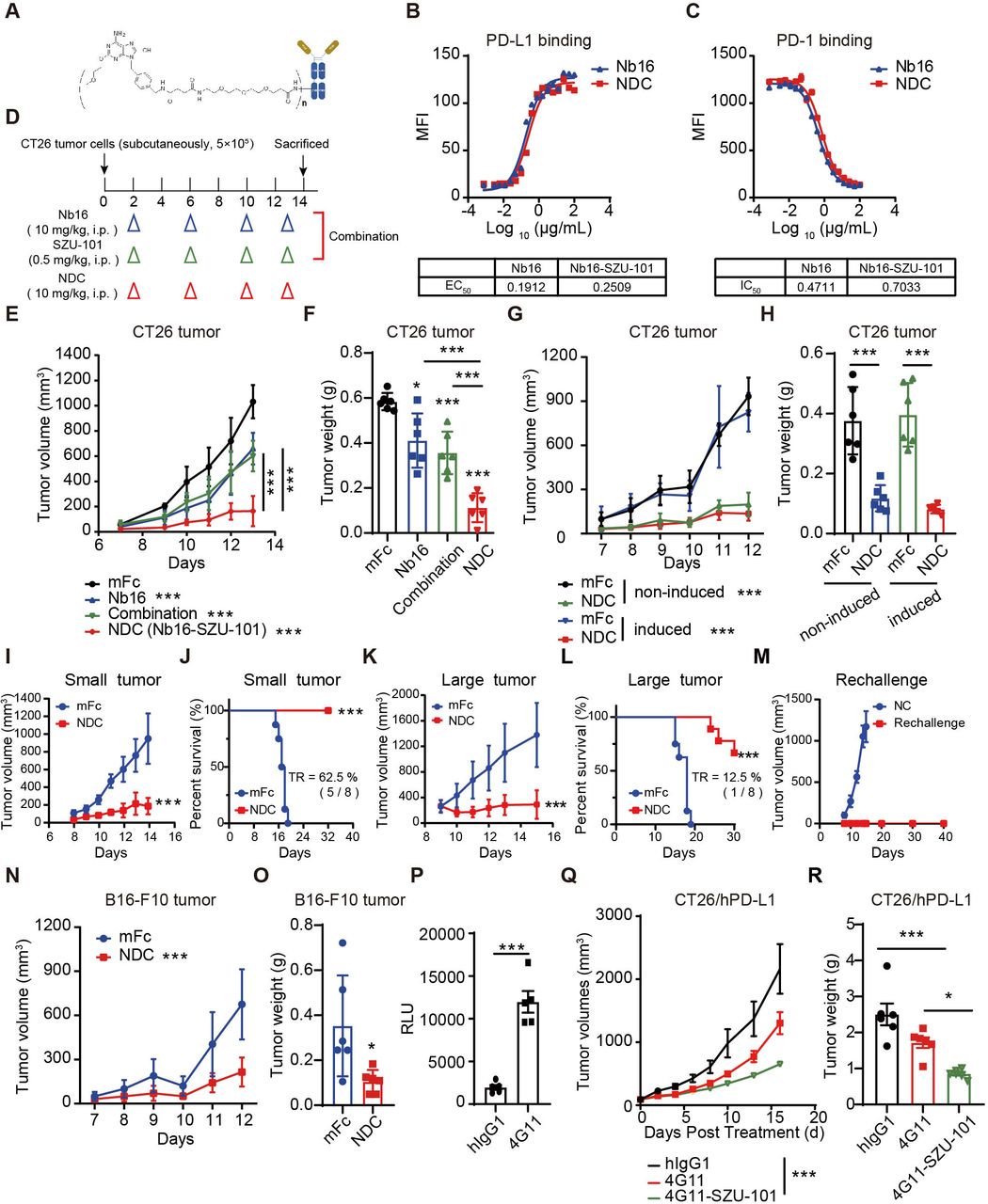
PD-L1/TLR7 dual-targeting NDC exerted a potent antitumor effect. (A) The structure schematic of Nb16-SZU-101. (B) Binding affinity of Nb16-SZU-101 for PD-L1. (C) PD-1/PD-L1 blocking activity of Nb16-SZU-101. (D–F) BALB/c mice bearing CT26 tumors were administered intraperitoneally with mFc (200 μg), Nb16 (200 μg), NDC (200 μg) or SZU-101 (0.5 mg/kg/day, according to the DAR of NDC) (D). Tumor growth in mice (E) and tumor weight (F) are shown (n=6). (G and H) BALB/c mice bearing CT26 tumors (induced or not) were treated with mFc or NDC. Tumor growth (G) and tumor weight (H) are shown (n=6). (I–L) BALB/c mice bearing CT26 tumors were treated with mFc or NDC when tumor volume was <50 mm3 (small tumor, I and J) or >200 mm3 (large tumor, K and L). Tumor growth and Kaplan-Meier survival curves are shown (n=8). (M) Tumor growth of rechallenge mice is shown. (N and O) C57BL/6 mice bearing B16-F10 tumors were treated with mFc or NDC. Tumor growth (N) and tumor weight (O) are shown (n=6). (P) Effect of 4G11 to block the inhibitory signal from RKO cells (induced by IFN-γ) to Jurkat-NFAT-luciferase cells. (Q and R) PD-1/PD-L1 dual-humanized BALB/c mice bearing CT26/hPD-L1 tumors were treated with hIgG1, 4G11 or 4G11-SZU-101. Tumor growth (Q) in mice and tumor weight (R) are shown (n=6). The data are presented as the mean±SEM. *P<0.05; ***p<0.001; ns as determined by one-way analysis of variance followed by Tukey’s multiple comparisons test or unpaired t-test. DAR, drug-to-antibody ratio; IFN, interferon; MFI, median fluorescence intensity; NDC, nanobody-drug conjugate; ns, not significant; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; TR, tumor regression.
Evaluation of antitumor activity of the PD-L1/TLR7 dual-targeting NDC in multiple tumor models
Due to SZU-101 enhanced PD-L1 expression, we assumed that the PD-L1/TLR7 dual-targeting NDC may have good inhibitory effects on tumors with different PD-L1 expression and both ‘hot’ and ‘cold’ tumors. When the doses of Nb16 and SZU-101 administered were consistent across Nb16-SZU-101 and combination treatments (figure 5D), NDC was significantly more effective than the combination treatment (figure 5E,F). In CT26 tumor models (with or without IFN-γ induction), Nb16-SZU-101 significantly inhibited tumor growth, with 78.9% inhibition in the uninduced CT26 model and 83.6% in the postinduced CT26 model (figure 5G,H). In addition, the antitumor effects of nanobody-based conjugate Nb16-SZU-101 and monoclonal antibody-based conjugate 10F.9G2-SZU-101 were compared, and Nb16-SZU-101 had a better therapeutic effect in prolonging the survival of the mice (online supplemental figure S12).
Also, the NDC showed effective tumor control with 80.3% inhibition in small CT26 tumors (<50 mm3) and with 78.9% inhibition in large CT26 tumors (>200 mm3) when the tumors became more established (figure 5I–L and online supplemental figure S13). Moreover, the administration of NDC in early or advanced tumor models both prolonged the survival of mice (figure 5J,L). Impressively, NDC treatment caused tumor regression in 62.5% of mice for the small tumor model and 12.5% of mice for the large tumor model (online supplemental figure S13). One week after tumor regression in mice, the mice were rechallenged with CT26 tumors. All mice showed no further tumor growth for more than 40 days, indicating that NDC treatment induced an effective antitumor immune memory (figure 5M). In addition, NDC also exerted potent antitumor effects with 68.2% inhibition in a B16-F10 tumor model with low PD-L1 expression levels (figure 5N,O and online supplemental figure S14A, B). Surprisedly, NDC still performed antitumor effect in LLC orthotopic lung tumors (online supplemental figure S14C–E) and B16F10 lung metastasis tumors (online supplemental figure S14F, G).
The excellent antitumor efficacy of Nb16-SZU-101 confirms our proof-of-concept success. To make this novel drug molecule ready for clinical application, we obtained the nanobody 4G11, which targets human PD-L1 with high blocking activity (figure 5P). Based on the PD-L1 nanobody 4G11, we developed another PD-L1/TLR7 dual-targeting NDC named 4G11-SZU-101 using the same method as described above. Considering that both host-expressed PD-L1 and tumor cell-expressed PD-L1 may contribute to the efficacy of PD-L1 nanobodies, we chose PD-1/PD-L1 dual-humanized mice to evaluate the in vivo antitumor activity of 4G11-SZU-101. 4G11-SZU-101 showed potent antitumor effects in a CT26/hPD-L1 tumor model established with PD-1/PD-L1 dual-humanized BALB/c mice, showing a tumor inhibition rate of 68.7% (figure 5Q,R and online supplemental figure S15). 4G11-SZU-101 demonstrated superior antitumor activity compared with single agents.
Moreover, systematic safety evaluations were performed in BALB/c mice after five doses of NDC, including 20 hematology indexes, 18 plasma chemistry indexes, 45 pathological indexes and clinical observation (online supplemental figure S16–18). Compared with the hIgG1 group, no pathological damage of concern to organs and no abnormalities in hematology or plasma chemistry parameters were observed after 4G11-SZU-101 treatment, demonstrating the good safety of this NDC (online supplemental figure S16–18). All the results demonstrated the clinical development potential of this novel drug molecule.
PD-L1/TLR7 dual-targeting NDC exerted antitumor effects by combining innate and adaptive immunity
To further illustrate the effect of NDC, immunophenotyping was performed. In CT26 tumor-bearing mice, NDC promoted the maturation of DCs, repolarized tumor-associated macrophages, reduced TGF-β+ macrophages and increased PD-L1 expression in macrophages compared with the isotype control (figure 6A–F and online supplemental figure S19A–F). Consistent with the results of RNA-seq analysis, NDC significantly increased the function of tumor-infiltrating cytotoxic cells, promoting their expression of GB and IFN-γ (figure 6G–I and online supplemental figure S19G–I). NK cell infiltration, activation and function were likewise significantly enhanced after administration (figure 6K–M and online supplemental figure S19K–M). IFN-γ+ CD4+ T-cell infiltration was also significantly increased in the NDC treatment group (figure 6J and online supplemental figure S19J). Thus, the PD-L1/TLR7 dual-targeting NDC played an antitumor effect by combining innate and adaptive immunity.
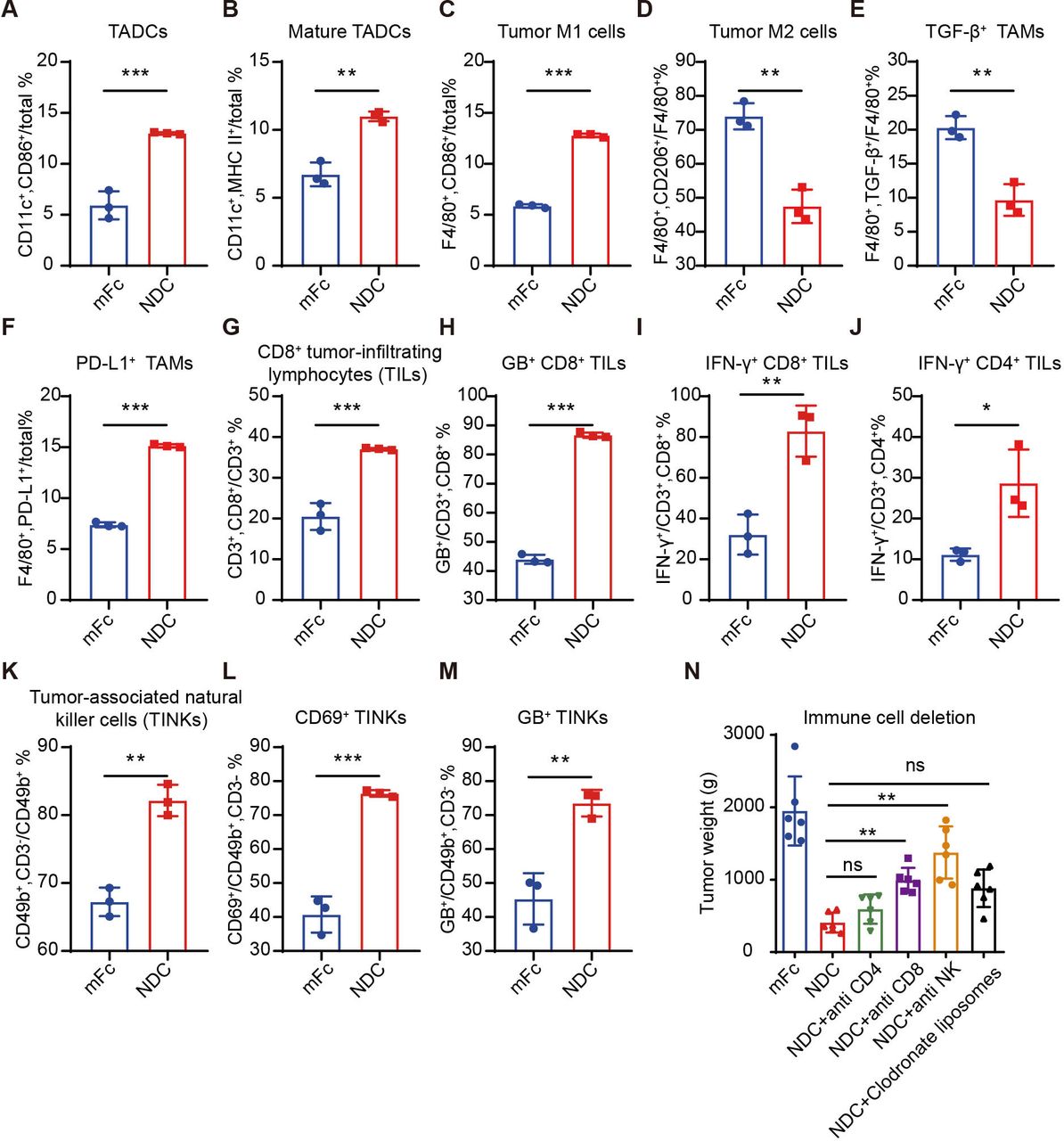
PD-L1/TLR7 dual-targeting NDCs orchestrated innate and adaptive immune responses. (A–M) Immune effector cells in CT26 tumors from mice treated with 200 μg of mFc or NDC were quantified by flow cytometry (n=3). (N) Mice bearing CT26 tumors were treated with 200 μg mFc or Nb16-SZU-101 dosed with antimurine CD4, antimurine CD8, antimurine NK1.1 or clodronate liposomes. Tumor weight is shown (n=6), and the group was compared with the NDC group. The data are presented as the mean±SEM. *P<0.05; **p<0.01; ***p<0.001 as determined by one-way analysis of variance followed by Tukey’s multiple comparisons test or unpaired t-test. IFN, interferon; NDC, nanobody-drug conjugate; PD-L1, programmed death ligand 1; TLR, toll-like receptor.
Using a series of antibody-based immune cell depletion studies in CT26 tumor-bearing mice (online supplemental figure S20A–D), the antitumor efficacy of NDC was abrogated by the depletion of CD8+ T cells and NK cells, while the depletion of CD4+ T cells and macrophages did not markedly hinder the antitumor efficacy of NDC (figure 6N and online supplemental figure S20E,F). Besides, immunohistochemistry results displayed that NDC boosted the infiltration of CD8+ T cells and NKs and secretion of GB (online supplemental figure S20G). Therefore, the antitumor effects NDC exerted relied mainly on CD8+ T cells and NK cells, which is consistent with our existing results. The deletion of all macrophages and CD4+ T cells was not sufficient to account for their role in the efficacy of NDC, due to their subtypes in the TME are complex.
The efficacy of PD-L1/TLR7 dual-targeting NDC was biased toward dependence on host expression of PD-L1
To analyze the changes in NDC action in vivo during continuous dosing to better understand the efficacy and safety mechanisms of NDC, we administered NDC every 2 days in CT26 tumor-bearing mice for a total of five doses and collected peripheral blood, spleen and tumor tissues after the first and fifth doses to determine PD-L1 levels. First, analyzing mFc-administered mice, we found that tumor tissues had 8.7-fold higher PD-L1 expression than peripheral blood and 6.0-fold higher PD-L1 expression than the spleen, suggesting that tumors are one of the organs with significant PD-L1 expression (figure 7A). This may be why PD-L1 nanobodies can be rapidly enriched in tumors in <8 hours. At 48 hours after the first dose, PD-L1 was significantly elevated in blood cells and there was a slight elevation of PD-L1 in splenocytes, which may be an effect of NDC entering the circulatory system after dosing (figure 7B,C). Although PD-L1 was slightly elevated in total intratumoral leukocytes, PD-L1 expressed by tumor cells and intratumoral macrophages and DC cells was decreased after NDC for reasons unknown (figure 7D). At 48 hours after the fifth dose, PD-L1 expression in both peripheral blood cells and spleen cells was downregulated compared with the first dose (figure 7B,C). Interestingly, the expression of PD-L1 was significantly increased in total intratumoral leukocytes, and PD-L1 expression was upregulated by 2.90-fold, 2.91-fold and 1.75-fold in tumor cells, macrophages and DC cells, respectively (figure 7D). In addition, the results showed that macrophages had the highest level of PD-L1 in tumors. According to the above results, it can be inferred that NDC gradually promoted PD-L1 expression in tumors as the dosing progresses, thus increasing the targeting of the tumor while decreasing the effect in other tissues, which may be one of the reasons for the efficacy and safety of NDC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The efficacy of PD-L1/TLR7 dual-targeting NDC was biased toward dependence on host expression of PD-L1. (A–D) BALB/c mice bearing CT26 tumors were treated with mFc or NDC every 2 days for total five doses. PD-L1 levels in total cells from blood, spleen and tumor tissues were assayed prior to drug administration (A). Peripheral blood (B), spleen (C) and tumor tissues (D) were collected after the first and fifth doses to determine PD-L1 levels by flow cytometry. (E) C57BL/6 mice bearing B16-F10 (Cas9 control or PD-L1 KO) tumors were treated with mFc or NDC. Tumor weight is shown (n=6). (F and G) PD-L1 expression on macrophages (F) and DCs (G) in B16-F10 (Cas9 control or PD-L1 KO) tumor tissues from tumor-bearing mice treated with mFc or NDC was determined by flow cytometry (n=3). (H) PD-L1-deficient C57BL/6 mice bearing B16-F10 tumors were treated with mFc or NDC. Tumor weight is shown (n=6). The data are presented as the mean±SEM. *P<0.05; **p<0.01; ***p<0.001; ns by one-way analysis of variance followed by Tukey’s multiple comparisons test or unpaired t-test. DC, dendritic cell; IFN, interferon; MFI, median fluorescence intensity; NDC, nanobody-drug conjugate; ns, not significant; PD-L1, programmed death ligand 1; TADC, tumor-associated dendritic cell; TAM, tumor-associated macrophage; TLR, toll-like receptor.
Furthermore, we wanted to investigate the necessity of tumor-expressed PD-L1 and host-expressed PD-L1 for the efficacy of NDC. NDC administration inhibited the growth of Cas9 control B16-F10 tumors at a 65% inhibition rate (figure 7E). PD-L1 knockout inhibited the growth of B16-F10 cells in vivo, but NDC administration still inhibited the growth of PD-L1-deficient B16-F10 tumors at a 38% inhibition rate (figure 7E). Immunophenotyping was used to show that NDC administration promoted PD-L1 expression in intratumoral macrophages and DCs in both Cas9 control and PD-L1-deficient B16-F10 tumors (figure 7F,G), indicating the deficiency of PD-L1 in tumor cells did not affect the ability of NDC to target and function in tumors. Next, we evaluated the effect of NDC on B16-F10 tumor growth in PD-L1-deficient C57BL/6 mice and found that NDC exhibited only a weak and non-significant tumor suppressive effect (figure 7H). Therefore, we concluded that the host expression of PD-L1 has a more vital role in the efficacy of NDC.
Discussion
As a new type of ADC, NDCs have drawn increased attention. For cancer treatment, due to the strengths in terms of tumor targeting and speed of clearance, nanobodies can better carry drugs to kill tumor cells while reducing toxicity to normal tissues. Existing therapeutic NDCs are commonly nanobodies conjugated with toxins, such as a glypican-3-targeting nanobody conjugated with Pseudomonas exotoxin against liver cancer.34 Distinguishingly, the small molecule conjugated in our NDC is a functional targeting drug rather than a toxin. To the best of our knowledge, we developed the first PD-L1/TLR7 dual-targeting NDC reported to date. In this study, PD-L1 nanobodies were used as promising components for developing of NDCs to deliver drugs, addressing the problem that systemic administration of TLR7 agonists did not achieve good solid tumor targeting. This study, our previous work and others’ work all suggest that PD-L1 is a potential drug delivery target but may be limited in tumors with low PD-L1 expression.28 35 Interestingly, TLR7 agonists could promote intratumoral PD-L1 expression, thus enabling the application of PD-L1 nanobodies for enrichment in tumors with low PD-L1 expression.
The potent efficacy of our NDC supports the concept of combining PD-L1 antibody and TLR7 agonist to promote tumor immunogenicity and attenuate tumor immune tolerance. The absence of a sufficient innate immune response and tumor immunogenicity may be limiting factors that restrict the development of an effective adaptive antitumor immune response. In addition to the TLR7 agonists applied in this study, drug targeting of other innate immune receptors, such as stimulator of interferon genes, cyclic GMP-AMP synthase and other TLRs also holds promise for improving the response rate to ICB therapy. Other strategies to promote tumor immunogenicity have also been tentatively investigated. IFN-α could promote anti-PD-L1 therapeutic effects by facilitating the cross-presentation of DCs.35 36 PCSK9 antibodies can promote the expression of MHC I in tumor cells.37 These studies all indicate that promoting intratumoral antigen presentation and increasing tumor immunogenicity can improve the therapeutic efficacy of ICB therapy. Notably, the crosstalk between the two target signaling pathways needs to be carefully evaluated, and the appropriate immunogenicity-enhancing targets for combination may vary depending on the immune checkpoint molecule.
PD-L1 levels in tumor tissues are considered to be one of the major predictive markers for PD-L1 antibody therapeutic efficacy.38 In studies of atezolizumab in patients with non-small cell lung cancer, clinical responses were associated with the infiltrating of PD-L1-positive immune cells.39 Similar findings were found in the clinical treatment of metastatic bladder cancer, where the response rate of PD-L1 antibodies was 52% and 11% in PD-L1-positive and PD-L1-negative groups, respectively.40 Presumably, induction of PD-L1 expression may be necessary to improve PD-L1 antibody therapeutic efficacy and tumor targeting. The TLR7 agonist SZU-101 played this role. Interestingly, in vivo, SZU-101 and NDC induced higher PD-L1 expression in APCs than in tumor cells, suggesting that the NDCs we developed prefer to target host APCs for action. Currently, tumor proportion score, immune positive score and combined positive score are used to assess the expected response to ICB therapy based on tumor cells or immune cells expressing PD-L1 levels. There is an ongoing controversy about whether PD-L1 from tumor cells or PD-L1 from immune cells can be used to predict the effect of ICB therapy. Herbst et al even concluded that there was no correlation between PD-L1 levels in tumor cells and the efficacy of atezolizumab.39 Our NDC still showed good antitumor activity in PD-L1-deficient tumor models and weak effects in host PD-L1-deficient animal models, suggesting that the effectiveness of NDC was not entirely dependent on the tumor cells themselves, or even biased to depend on the host PD-L1 expression. This is consistent with some reports suggesting that host expression of PD-L1 determines the efficacy of PD-L1 pathway blockers.41 42
Of course, our study has many limitations, and further research is needed. First, the NDC we developed currently functions through a non-deterministic coupling technique, and it is necessary to subsequently adopt a deterministic coupling technique and optimize the DAR value of the NDC. It is expected that by optimizing the appropriate DAR values, PD-L1/TLR7 dual-targeting NDCs will achieve better antitumor effects. Second, we have evaluated the antitumor activity of NDC only on PD-1/PD-L1 dual-humanized BALB/c mice thus far, and therefore, the inhibitory effect of NDCs needs to be tested on human-derived tumors growing in the immune system-humanized mice. In addition, the role of NDC in tumor growth needs to be further investigated to identify targeted drugs that can be combined with NDC to achieve better antitumor efficacy.
In conclusion, the combination of the PD-L1 nanobody and TLR7 agonist to form PD-L1/TLR7 dual-targeting NDC shows a notable rationale, which represents the strategy that boosts tumor immunogenicity and breaks immune tolerance. First, PD-L1 nanobody have the desired tumor-targeting ability such that TLR7 agonists can be precisely delivered to tumor tissues. Second, TLR7 agonists can induce high expression of PD-L1 within tumors; therefore, PD-L1 nanobodies can achieve better antitumor effects and responses to tumors with low levels of PD-L1. Third, the lack of an effective innate immunity activating makes it difficult for PD-L1 nanobodies to fully restore T-cell antitumor immune responses. TLR7 agonists can enhance tumor immunogenicity and convert ‘cold’ tumors into ‘hot’ tumors by activating innate immunity and promoting the activity of antigen-presenting machinery within the tumors. Accordingly, we have developed a type of PD-L1/TLR7 dual-targeting NDC with potent and broadly responsive antitumor activity and with potential for clinical development.
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
We thank H Wang, Y Hang, M Zhang and the staff of GemPharmatech for their help with mouse husbandry and experiments. We thank Professor Y Huang for scientific discussions. We thank the staff of the Center for Drug Safety Evaluation and Research (CDSER), Shanghai Institute of Materia Medica (SIMM), for the safety evaluation. We thank Conjugenix Pharma-Tech (Shenzhen, China) for assisting molecule synthesis. We also thank Professor Y Geng for kindly providing the nanobody 4G11.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
XY and YL contributed equally.
Contributors LG, GJ, JL, XY and YL designed the experiments and analyzed the data. XY and YL performed the experiments and prepared the manuscript. BC, YT, MS and PX assisted in performing the experiments and preparing the manuscript. CH, ML and RC assisted in performing the animal experiments. XJ expressed and purified the nanobody 4G11. WH and FT performed the identification of NDC by mass spectrometry. JZ performed the synthesis of SZU-101 and SZU-101-NHS. YW, JR and JS assisted in data interpretation. HL performed pathological analysis. LG acted as the guarantor for this work.
Funding This work was supported by the National New Drug Creation Program of China (No. 2019ZX09732002-013 and 2018ZX09201017-004) and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA 12050305).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.