Article Text

Abstract
Background The use of immunotherapeutic vaccination in prostate cancer is a promising approach that likely requires the induction of functional, cytotoxic T cells . The experimental approach described here uses a well-studied adenovirus-poxvirus heterologous prime-boost regimen, in which the vectors encode a combination of prostate cancer antigens, with the booster dose delivered by either the intravenous or intramuscular (IM) route. This prime-boost regimen was investigated for antigen-specific CD8+ T cell induction.
Methods The coding sequences for four antigens expressed in prostate cancer, 5T4, PSA, PAP, and STEAP1, were inserted into replication-incompetent chimpanzee adenovirus Oxford 1 (ChAdOx1) and into replication-deficient modified vaccinia Ankara (MVA). In four strains of mice, ChAdOx1 prime was delivered intramuscularly, with an MVA boost delivered by either IM or intravenous routes. Immune responses were measured in splenocytes using ELISpot, multiparameter flow cytometry, and a targeted in vivo killing assay.
Results The prime-boost regimen was highly immunogenic, with intravenous administration of the boost resulting in a sixfold increase in the magnitude of antigen-specific T cells induced and increased in vivo killing relative to the intramuscular boosting route. Prostate-specific antigen (PSA)-specific responses were dominant in all mouse strains studied (C57BL/6, BALBc, CD-1 and HLA-A2 transgenic).
Conclusion This quadrivalent immunotherapeutic approach using four antigens expressed in prostate cancer induced high magnitude, functional CD8+ T cells in murine models. The data suggest that comparing the intravenous versus intramuscular boosting routes is worthy of investigation in humans.
- Immunotherapy
- Adaptive Immunity
- CD8-Positive T-Lymphocytes
- Immunity, Cellular
- Immunogenicity, Vaccine
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Effective immunotherapeutic vaccination for cancer is likely to require induction of high magnitude, functional and durable antigen-specific CD8+ T cells. We have previously shown that a heterologous prime boost approach using a simian adenovirus vector encoding the tumor antigen 5T4 followed by a modified poxvirus (modified vaccinia Ankara (MVA)) encoding the same protein results in high level CD8+ T cells in mice and promising initial clinical results in prostate cancer in humans.
WHAT THIS STUDY ADDS
Here we show that this candidate can be expanded to a prostate-specific four-antigen cassette and that the administration of the MVA intravenously in the prime/boost setting results in higher magnitude and increase functionality compared with delivery of the boost by the intramuscular route.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Comparing the intravenous versus intramuscular boosting routes is worthy of investigation in humans.
Introduction
Prostate cancer (PCa) is the second most frequent cancer diagnosis made in men and the fifth leading cause of cancer death worldwide.1 There is an increasing body of evidence supporting the role of immune responses in the control and eradication of particular cancers.2 3 T cell responses can be directed toward antigens expressed in tumors, either tumor-associated antigens or neoantigens.4 5
To date, only one therapeutic cancer vaccine, sipuleucel-T, has been approved by the Food and Drug Administration (FDA) and is indicated for asymptomatic or minimally symptomatic metastatic, castration-resistant, PCa.6–8 This immunotherapy targets one of the prostate-associated antigens: prostatic acid phosphatase (PAP). The vaccine has shown statistically significant, though modest, efficacy in clinical trials; increasing overall survival in treated patients by 4 months. However, no effect on time to tumor progression compared with placebo group has been observed, and the T cell responses induced appeared modest.9
Previous preclinical and clinical studies have been conducted using a heterologous prime-boost strategy in which a replication-incompetent chimpanzee adenoviral vector vaccine10 encoding the oncofetal antigen 5T4 was administered intramuscularly and then followed 1–4 weeks later by a poxvirus (modified vaccinia Ankara (MVA)) also encoding 5T4.11–14 In early stage PCa patients, the vaccine induced ex vivo 5T4-specific T cell responses in over 60% of patients, and when combined with the CI, nivolumab, in metastatic castration-resistant PCa, 22% of patients achieved a 50% decrease in PSA levels compared with baseline (A Hill, personal communication, 2021).
To advance and improve on this finding, two changes to this approach were investigated. The first broadened the range of target antigens to increase response breadth while simultaneously decreasing the possibility of immune evasion by antigen loss. Evaluation of the literature and preclinical data led to the selection of PSA, PAP, STEAP1, along with the original 5T4 antigen as promising candidates for this approach.15–17
The second was the administration of MVA boosts by the intravenous route of administration, and the effectiveness of this approach was measured through monitoring of an important subset of memory CD8+ T cells, characterized by the expression of T cell-specific transcription factor 1 (TCF1), in the context of other markers of early differentiation (eg, KLGR), which were previously shown to be preferentially induced through intravenous vaccination.18–21 As both MVA and chimpanzee adenoviral vectors have been delivered previously to humans by the intravenous route,22 23 we set out to design a new immunization constructs and assess their immunogenicity in a variety of animal models to compare the previously studied intramuscular (IM) boosting route to intravenous administration.
Methods
Construct design and confirmation of antigen expression
PCa immunogen design was carried out using the full-length coding sequences (with N-terminal signal sequences removed) of PSA, PAP, STEAP1 and 5T4. The resulting immunogen sequences were codon optimized for human codon usage and cloned into the ChAdOx1 E1 locus under the control of the immediate early cytomegalovirus promoter and the bovine growth hormone polyA sequence.10 The same immunogen sequences were also codon optimized for vaccinia virus codon usage and cloned into the MVA F11 gene locus under the control of the native F11 promoter, as previously described.24
Antigen expression from the ChAdOx1 vector was confirmed by western blot according to standard methods. Briefly, HEK293 cells were transduced with ChAdOx1 expressing prostate immunogens or ChAdOx1 expressing GFP (negative control) at a multiplicity of infection (MOI) of three infectious units per cell, and cells were harvested 24 hours later. Cell lysates were denatured, separated on a 4%–20% precast polyacrylamide gel and blotted onto a nitrocellulose membrane. Membranes were probed with primary antibodies against hPSA (ab76113, abcam), hPAP (ab109004, abcam), hSTEAP1 (ab3679, abcam) or h5T4 (ab129058, abcam) and appropriate secondary antibodies. Bands were visualized using the Pierce ECL western blotting substrate kit (ThermoFisher) and a Chemidoc (BioRad). Positive controls were as follows: recombinant human PSA (ab151676, abcam), recombinant human PAP (ab219224, abcam), recombinant human STEAP1 (16178432, Fisher Scientific), recombinant human TPBG/5T4 (19845-H08H, SinoBiological).
Mice
Six to 8 week old female CD-1, C57BL/6 or male HLA-A2 transgenic mice were purchased from Charles River UK Limited or Taconic Biosciences (Denmark). Animal experiments were conducted at Evotec, Toulouse, France (studies 1 and 2) and Charles River Laboratories (CRL), Portishead, UK (studies 3–6). Mice were housed in groups of four to five per cage and had access to food and water ad libitum. Mice were maintained in a temperature (20°C–24°C) and humidity (45%–65%) controlled environment on a 12 hours light/dark cycle. Animals were allowed 1 week to acclimatize to the facility prior to commencing the study. On conclusion of the study, animals were humanely terminated following cervical dislocation. For studies conducted at CRL, spleen samples were shipped to Vaccitech plc, Oxford, UK, for immunogenicity analyses.
Immunizations
Mice were injected intramuscularly in the hind leg or intravenously in the tail vein with 50 µL of sterile PBS containing 108 IU ChAdOx1 constructs (administered as prime) or 106 PFU MVA constructs (administered as boost). Regimens across different experiments were either prime alone, prime-boost or prime-boost-boost. Animals were terminated 14 or 57 days after the final immunization, and spleens were collected. Experimental designs are summarized in table 1.
Summary of mouse studies
Preparation of splenocytes
Isolated spleens were prepared into a single cell suspension by passing through a 70 µM cell strainer with 15 mL RPMI using a 5 mL syringe plunger. Cells were centrifuged (+4°C, 1200 rcf, 5 min), and red blood cells were lysed with 1 mL ACK lysis buffer for 3 min. Cells were washed and resuspended in 3 mL Complete Media (RPMI+Glutamax-1, 10% heat inactivated foetal bovine serum, 10 µg/mL gentamicin). For flow cytometry experiments, cells were counted and resuspended at 1×107 cells/mL in Complete Media. For ELISpots cell suspensions were diluted to 2×106 or 1×106 cells/mL in Complete Media (all reagents sourced from Fisher Scientific UK Ltd).
IFNγ ELISpot
Ninety-six well PVDF plates (Millipore) were coated overnight at 4°C with 4 µg/mL antimouse IFNγ capture antibody (BD Biosciences). Plates were washed and blocked with Complete Media for 2 hours, then loaded with 1 µg/peptide/mL peptide pools: PSA, PAP, 5T4, STEAP1 (Mimotopes) or PSA nonamer (HCIRNKSVI, ProImmune Ltd), 0.4% DMSO as a negative control and 2 µg/mL Concanavalin A as a positive control, prepared as 4X solutions in Complete Media. Splenocytes were seeded at 1×105 or 2×105 viable cells/well and plates were incubated for 18 hours±30 min at 37°C and 5% CO2. Splenocytes were removed, and plates were incubated with 1.6 µg/mL antimouse IFNγ detection antibody (BD Biosciences) for 2 hours, followed by horseradish peroxidase-conjugated streptavidin (1:100) (BD Biosciences) for 1 hour. Plates were developed for ~10 min using AEC Substrate Set (BD Biosciences), and spots were counted using an AID (iSpot) Spectrum Reader.
Flow cytometry
Splenocytes were assessed by flow cytometry using an ex vivo Dextramer/T cell marker/transcription factor panel (panel 1, online supplemental figure 1) or a T cell marker/intracellular cytokine panel (panel 2, online supplemental figure 2) following an 18-hour restimulation with PCAQ peptide pools.
Supplemental material
Supplemental material
For ex vivo staining (panel 1), 1×106 splenocytes/well were incubated in 50 nM Dasatinib in PBS (Universal Biologicals) to prevent TCR downregulation prior to Dextramer staining. Cells were stained with: (1) Dextramer reagent containing H-2Db molecules presenting an immunodominant PSA antigen (HCIRNKSVI, Immmudex), (2) Fc Block (BD Biosciences) + Viability Dye (Biolegend) and (3) surface antibody cocktail. Cells were fixed and permeabilized using a FoxP3 transcription buffer set (eBiosciences) then stained intracellularly for Ki67 and TCF1.
For intracellular cytokine staining (panel 2), 1×106 splenocytes/well were stimulated for 18 hours with 2× PCAQ peptide pools: PSA+5T4 and PAP+STEAP1 (1 µg/peptide/mL) at 37°C, 5% CO2. DMSO alone wells were set up as negative controls. For the last 3 hours of culture, phorbol myristate acetate and ionomycin (Biolegend, 1:1000) were added to positive control, set-up and unstained control wells; Brefeldin A (Biolegend, 1:1000) was added to all wells to inhibit secretion of intracellular cytokines. Cells were stained with Fc Block (BD Biosciences) + Viability Dye (Biolegend) followed by staining with surface antibodies. Cells were fixed and permeabilized using Cytofix/Cytoperm buffer (BD Biosciences) then stained for intracellular cytokines.
In both panels, fluorescence minus one (FMOs) and reference controls were generated from one sample and applied to the data analysis. Cells were analyzed using a Cytek Northern Lights flow cytometer, and data analysis was performed on FlowJo Software (V.10.8.1). For details of dyes and antibodies used in panels 1 and 2, refer to online online supplemental table 1.
Supplemental material
In vivo killing assay
The in vivo killing assay measured the functionality of antigen-specific T cells elicited in PCAQ-immunized C57BL/6 mice through assessment of their cytotoxic function. Target cells were prepared using splenocytes from syngeneic, naïve C57BL/6 mice (n=4). Spleens were prepared into single splenocyte suspensions and divided into two equal populations by volume. One population was mock-pulsed with PBS, and the second was pulsed with 1 µg/mL PSA nonamer comprising a well-recognized immunodominant epitope in C57BL/6 mice (HCIRNKSVI, ProImmune Ltd) for 1 hour at 37°C. Cells were washed and resuspended in PBS. Equal volumes of 0.4 µM or 4 µM Cell Proliferation Dye (CPD) eFluor 450 (eBioscience) were added to the mock-pulsed and PSA nonamer-pulsed cell populations, respectively, labeling cells with final concentrations of 0.2 µM or 2 µM CPD. Pulsed and labeled cells were washed, counted and resuspended at 2×107 cells per mL. The two populations were combined in a 1:1 ratio to create the final target cell preparation. One hundred microliter of target cell preparation, containing 1×106 each of mock-pulsed and PSA nonamer-pulsed cells, were injected into tail veins of mice on day 84 of the experiment #4. Naïve mice (n=3) were also injected with target cell preparation as a negative control group. Eighteen hours after target cell infusion, mice were terminated, and spleens were harvested and prepared into single splenocyte suspensions for analysis by flow cytometry using a Cytek Northern Lights cytometer. The percentages of target cells in unpulsed (CPDLow) and pulsed peaks (CPDHigh) were measured for each animal and the % specific target cell lysis was calculated as [1 – (% PSA-pulsed ÷ % unpulsed)] × 100.
Results
Antigen expression and confirmation of immunogenicity in outbred mice
We first cloned the prostate immunogens into the recombinant adenoviral vector ChAdOx1 and assessed their expression in vitro by western blot. Expression of the full-length immunogen proteins was confirmed by probing vector-transduced cell lysates with antibodies against each immunogen (figure 1).

Confirmation of antigen expression by western blot. Cells were transduced with ChAdOx1 (lane 1) or ChAdOx1 (negative control, lane 2) expressing prostate immunogens, and cell lysates were probed with antibodies against PSA, PAP, STEAP1 and 5T4 as shown. The respective commercially available recombinant proteins were included as positive controls (lane 3). No signal was detectable for the 5T4 positive control protein.
We next assessed whether the ChAdOx1 vector expressing prostate immunogens could stimulate a T cell immune response against the encoded immunogens. Outbred (CD-1) mice were immunized with ChAdOx1, and spleens were harvested for IFNγ ELISpot assays 2 weeks later. We observed robust T cell responses against PSA and PAP antigens, with subdominant responses toward STEAP1 and 5T4 (figure 2A). To further boost responses, we used MVA expressing the same prostate immunogens. A prime-boost regimen of ChAdOx1 followed by MVA in CD-1 mice showed increased responses to all four antigens (figure 2B).
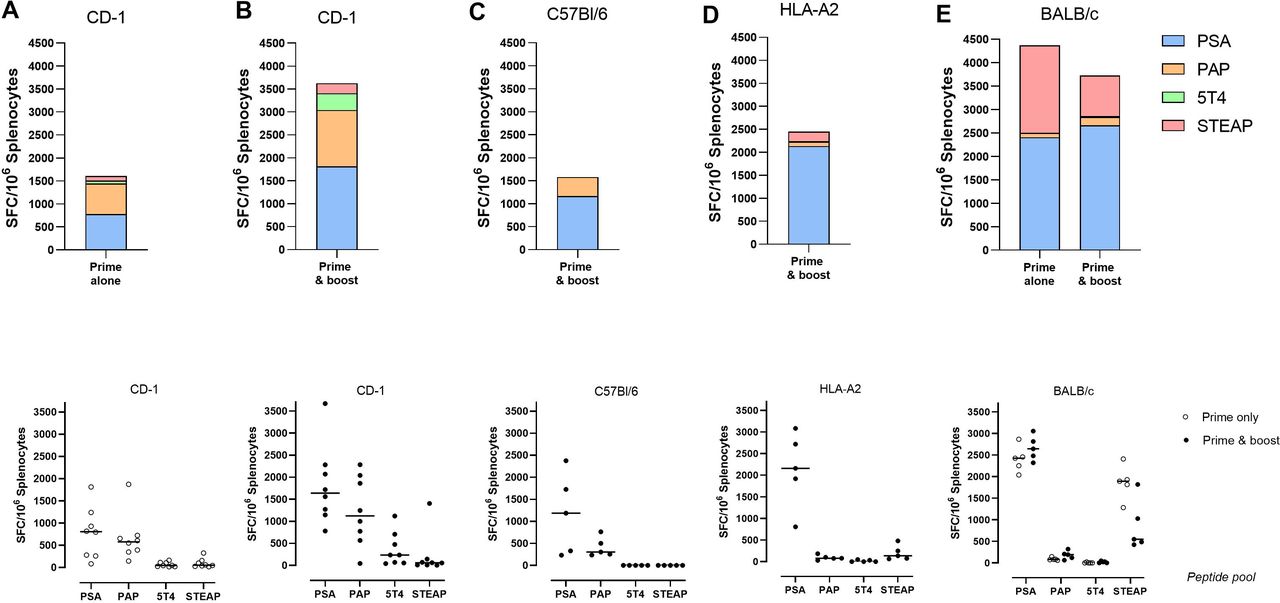
Immunogenicity of ChAdOx1/MVA in different mouse strains. Mice were immunized intramuscularly either prime alone with ChAdOx1 or prime-boost with ChAdOx1/MVA, following different regimens outlined in table 1. Data for (A) CD-1 prime alone, (B) CD-1 prime and boost, (C) C57BL/6, (D) HLA-A2 transgenic and (E) BALB/c were generated in studies 1, 2, 3, 6 and 5, respectively. At the end of each study, mice were terminated, and spleens were harvested. Splenocytes in single cell suspension were applied to IFNγ ELISpot at 200 000 per well and restimulated with 1 µg/peptide/mL of PSA, PAP, 5T4 or STEAP1 overlapping 15-mer peptide pools for 18 hours. 0.4% DMSO and ConA were used as assay negative and positive controls. Top row: stacked bar graphs show average SFC/106 splenocytes generated in response to each peptide pool; bottom row: scatter graphs show responses produced by individual animals to each peptide, line indicates mean. Data are displayed following subtraction of DMSO background control and application of assay positivity threshold (max of 2×SD DMSO control or 25 spot-forming cells (SFC)/106 splenocytes). MVA, modified vaccinia Ankara.
Different response profiles to prostate immunogens are elicited in different mouse strains, confirming immunogenicity of ChAdOx1/MVA platform
We next evaluated immunogenicity of ChAdOx1 and MVA expressing prostate immunogens using prime alone or prime–boost regimens in different inbred mouse strains (C57BL/6 and BALB/c) and an HLA-A2 transgenic strain. T cell responses measured by IFNγ ELISpot were mainly dominated by PSA and PAP in C57BL/6 mice (1169±818 and 412±200 mean±SD SFU/106 splenocytes, respectively), PSA and STEAP1 in HLA-A2 transgenic mice (2136±782 and 201±154 mean±SD SFU/106 splenocytes) and to PSA and STEAP1 in BALB/c mice (prime and boost – 2663±256 and 861±524 mean±SD SFU/106 splenocytes) (figure 2).
These results indicate that ChAdOx1 and MVA can elicit T cell responses to each of the prostate immunogens in either outbred, inbred or HLA-A2 transgenic mice, demonstrating the immunogenic capacity of the ChAdOx1/MVA prime–boost immunotherapeutic platform.
Intravenous administration of MVA boost augments T cell immunogenicity compared with MVA boost administered IM
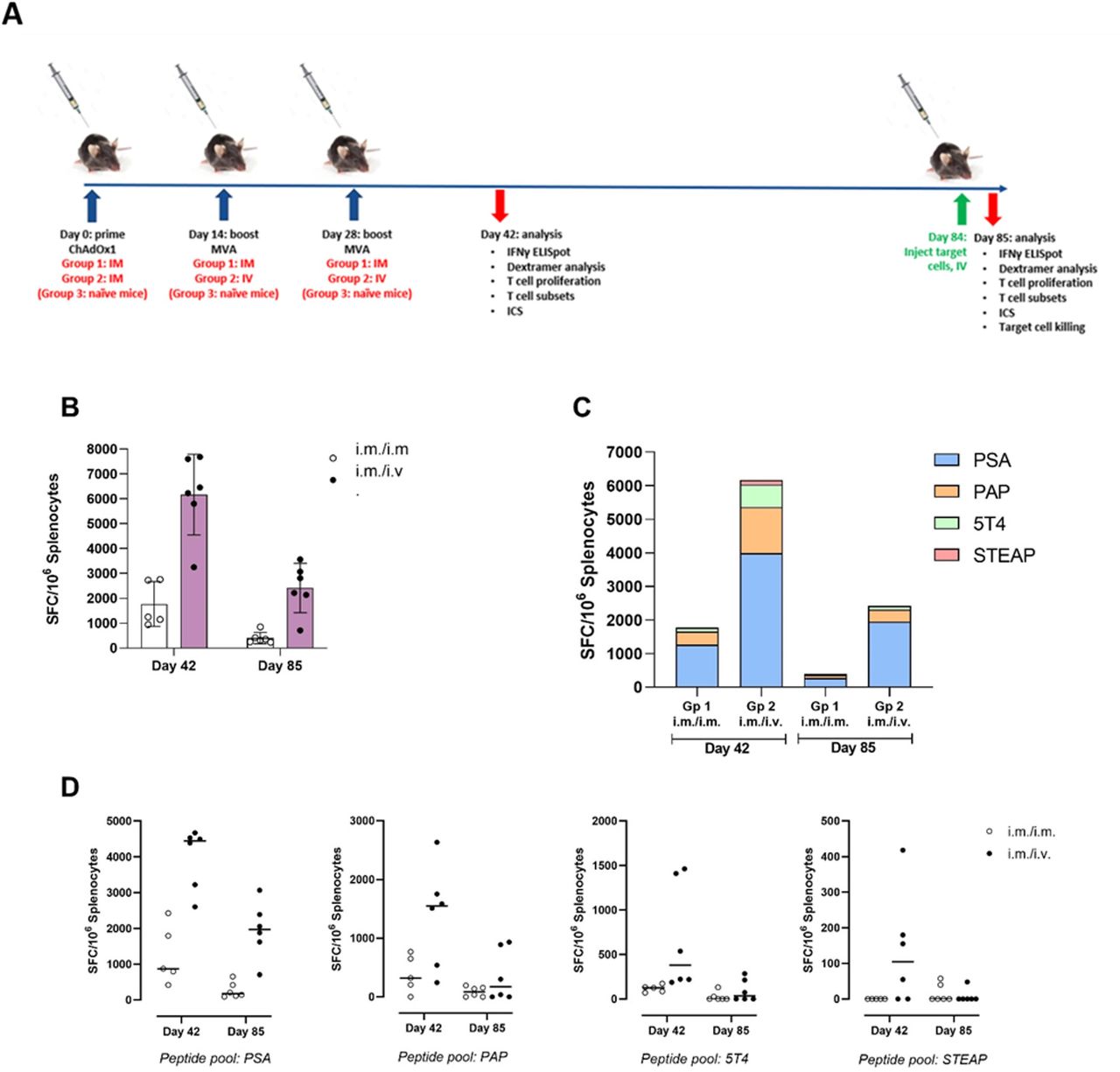
We sought to determine whether the route of MVA boost administration (intravenous vs intramuscular) influenced antigen-specific T cell responses primed by intramuscular administration of ChAdOx1. An initial pilot study was performed (table 1, experiment #3), in which a single MVA boost was administered on day 14 and immunogenicity was assessed on day 28. This was followed by a larger study in which animals were boosted twice with MVA (days 14 and 28) then assessed at days 42 and 85 (table 1, experiment #4). The total magnitude of T cell response measured by IFNγ ELISpot was 3.5-fold and 6-fold higher at days 42 and 85, respectively, when MVA boost immunizations were administered intravenous compared with IM (figure 3B, day 42: 6171 ± 1477 intravenous versus 1767 ± 805 IM mean±SD SFU/106 splenocytes; day 85: 2417±907 intravenous vs 402±210 IM mean±SD SFU/106 splenocytes). Intravenous administration also increased the breadth of responses to the prostate immunogens, notably at day 42, where responses to 5T4 and STEAP1, which were negligible following IM administration, were detected following IV administration, while at day 85 responses to PSA, PAP and 5T4 were elicited with intravenous but not IM (figure 3C,D). Similar response profiles and fold-changes were observed in the initial pilot study (data not shown).

Comparing T cell immunogenicity of ChAdOx1 prime administered IM followed by 2× MVA boost administered IM or intravenous: IFNγ ELISpot. Study design (A): C57BL/6 mice were primed with ChAdOx1 intramuscularly and boosted on days 14 and 28 on with MVA either intramuscularly (group 1) or intravenously (group 2). Mice were sacrificed on day 42 or day 85 (n=6 per timepoint, per group), and spleens were harvested and prepared into single cell suspensions for immunogenicity analysis. (B) Splenocytes were applied to IFNγ ELISpot at 200 000 per well and restimulated with 1 µg/peptide/mL of PSA, PAP, 5T4 or STEAP1 overlapping 15-mer peptide pools for 18 hours. 0.4% DMSO and ConA were used as assay negative and positive controls. Bar graph shows average±SD of the total magnitude of response for groups 1 and 2 at day 42 and day 85, calculated as the sum of SFC/106 splenocytes generated in response to 4× prostate immunogen antigen peptide pools; dots represent individual mice. (C) stacked bar graphs show average SFC/106 splenocytes generated in response to individual peptide pools. (D) Scatter graphs show responses produced by individual animals to each peptide pool; line indicates mean. Data are displayed following subtraction of DMSO background control and application of assay positivity threshold (max of 2xSD DMSO control or 25 SFC/106 splenocytes). IM, intramuscular; MVA, modified vaccinia Ankara.
The same splenocyte samples were evaluated by flow cytometry to measure CD8+ T cell proliferation (using the marker Ki67) and antigen-specific recognition of an immunodominant PSA epitope: HCIRNKSVI (using a PE-conjugated Dextramer where HCIRNKSVI is presented in the context of H-2Db).
Intravenous administration of MVA generated 2.2-fold and 2.0-fold increases in CD8+ T cell proliferation relative to intramuscular administration at day 42 (73±3% IM/IV vs 33±17% IM/IM) and day 85 (58±5% IM/IV vs 29±2% IM/IM), respectively (figure 4A). The proportion of dextramer-specific CD8+ T cells increased 3.7-fold at day 42 (5.48±1.55% IM/IV vs 1.47±0.94% IM/IM) and 6.0-fold at day 85 (3.53±1.67% IM/IV vs 0.59±0.15% IM/IM) with intravenous versus intramuscular administration (figure 4B), closely mirroring the relative fold-increases observed with IFNγ ELISpot (figure 3B). Together, these data demonstrate that intravenous administration of the MVA boost enhances the magnitude of CD8+ T cell responses to antigens encoded by the ChAdOx1 and MVA constructs, and the corresponding ability of antigen-specific T cells to proliferate.
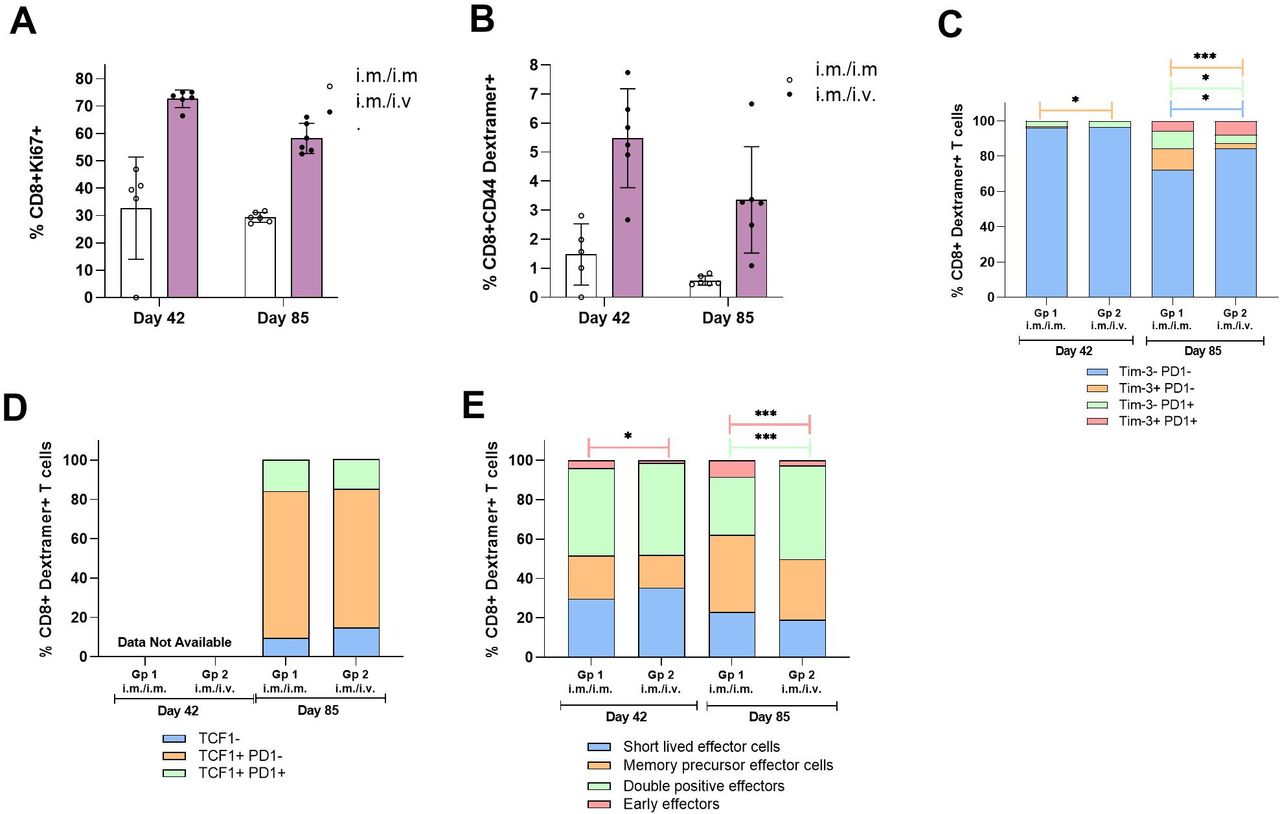
Comparing CD8+ T cell immunogenicity and phenotype after ChAdOx1 prime administered IM followed by 2× MVA boost administered IM or intravenous. Study design: refer to figure 3A. Spleens samples were collected and prepared into single cell suspensions for application into flow cytometry assays. Splenocytes (1×106 cells per well) were stained ex vivo with Dextramer, Viability Dye, surface markers and Ki67/TCF1 following a fixation/permeabilization step (panel 1: refer to online supplemental table 1 and figure 1). (A) Bar graph shows mean±SD% of CD8+ T cells which express proliferation marker Ki67; (B) bar graph shows mean±SD% of CD8+ CD44+ T cells, which are PSA antigen-specific through detection using Dextramer. Stacked bar graphs show proportions of CD8+ Dextramer+ T cells with different expression profiles of (C) Tim3 and PD1; (D) TCF1 and PD1; (E) KLRG1 and CD44 to characterize MPECs and SLECs and (F) CD62L and CD44 to characterize effector and central memory subsets. Unpaired T tests were performed to compare proportions of subsets in IM and intravenous groups: n.s. p>0.05, *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001. IM, intramuscular; MPECs, memory precursor effector cells; MVA, modified vaccinia Ankara; SLECs, short-lived effector cells.
PSA antigen-specific CD8+ T cells detected by Dextramer analysis were further characterized for expression of stem-like T cell marker TCF1 transcription factor, exhaustion markers (PD-1, Tim-3) and memory subset markers: KLRG1 and CD127 to characterize short-lived effector cells (SLECs) and memory precursor effector cells (MPECs), and CD44 and CD62L for effector and central memory cells.
TCF1 is a transcription factor that maintains a pool of precursor cells within the overall exhausted tumor CD8+ T cell population.19 CD8+ T cells coexpressing PD1 and TCF1 are characterized as ‘stem-like’: they are responsive to CIs, can proliferate and self-renew. At day 85, we did not observe any difference in the percentages of this stem-like CD8+ population in Dextramer-positive CD8+ T cells with intravenous versus IM MVA boost administration: mean±SD frequencies were 15.6±1.91% for intravenous and 15.3±5.2% for IM, respectively (figure 4C; data not available for Day 42). This trend was consistent with the initial pilot study (table 1, experiment #3; data not shown).
The frequencies of Dextramer-positive CD8+ T cells characterized by their coexpression of PD1 and Tim3 were negligible at day 42 (0.1±0.1% IM and 0.03±0.02% intravenous) but increased at day 85 (5.71±1.92% IM and 7.9±2.34% intravenous). No statistically significant differences in percentages of this population were seen between the routes of administration (figure 4D).
The percentages of Dextramer-positive CD8+ T cells characterized as MPECs and SLECs did not differ between the routes of administration at either time point. At day 85 relative to day 42, there was an increase in MPECs (39.1±7.3% IM and 30.8±11.5% intravenous at day 85; 21.8±6.0 IM and 16.5±3.7% intravenous at day 42) and corresponding decrease in SLECs (23.1±7.0% IM and 19.0±7.4% intravenous at day 85; 29.8±3.4 IM and 35.4±7.6% intravenous at day 42). Of note, there was a significant increase in differentiated KLRG1+CD44+ double positive effector cells with intavenous boost administration compared with IM at day 85 (47.6±4.7% intavenous vs 29.5±6.4% IM; unpaired t-test p=0.005). There was also a statistically significant decrease with intravenous boost administration compared with IM in KLRG1-CD44- double negative early effectors both at day 42 (1.4±0.5% IV vs 29.5±6.4% IM; unpaired t-test p=0.005) and day 85 (47.6±4.7% intravenous vs 29.5±6.4% IM; unpaired t-test p=0.005) (figure 4E).
The majority (>92%) of Dextramer-positive CD8+ T cells induced across groups were characterized as CD44+CD62L− effector memory cells at both day 42 and day 85, except for the IM boost administration group at day 85. Overall, these data suggest that the route of MVA boost administration does influence somewhat the differentiation of antigen-specific CD8+ effector T cells.
Splenocytes were also assessed for intracellular cytokine production following an overnight restimulation with peptide pools covering the selected prostate immunogens, grouped into pairs: PSA+5T4 and PAP+STEAP1.
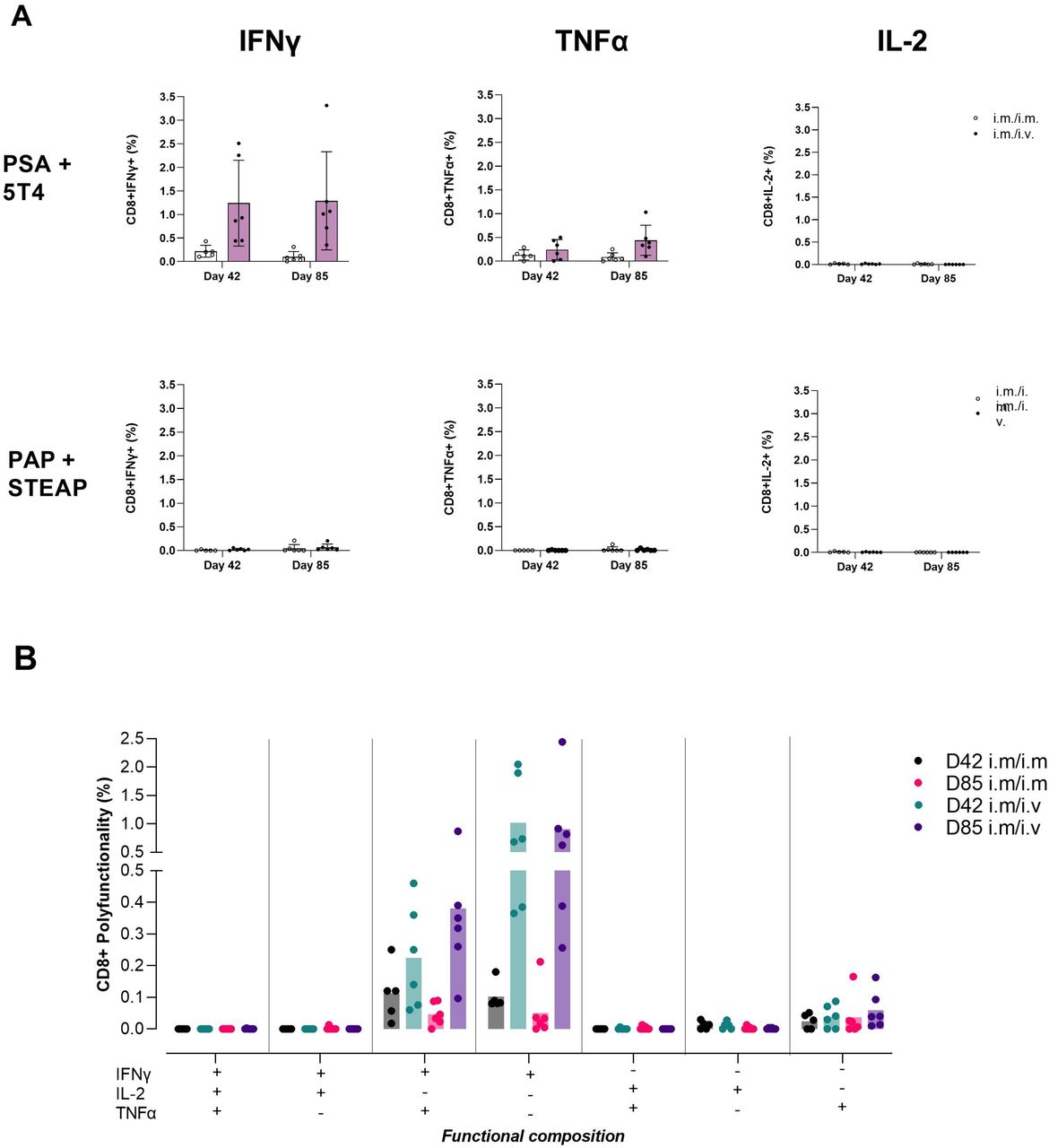
We observed production of IFNγ and TNFα in CD8+ T cells in response to PSA+5 T4 peptide restimulation. The IFNγ response was 5.7-fold and 13.1-fold higher with intravenous MVA boost administration at days 42 and 85, respectively (1.24±0.83% intravenous vs 0.22±0.11% IM at day 42, p=0.357; 1.29±0.95% intravenous vs 0.1±0.1% at day 85, p=0.0194), the corresponding TNFα response was 1.8-fold and 5.1-fold higher, respectively (0.24±0.19% intravenous vs 0.13±0.09% IM at day 42, not significant; 0.44±0.29% intravenous vs 0.09±0.08% at day 85, p=0.0254). We did not detect CD8+ IL-2 responses to PSA+5T4 or production of any cytokine in response to PAP+STEAP1 peptide pool (figure 5A).
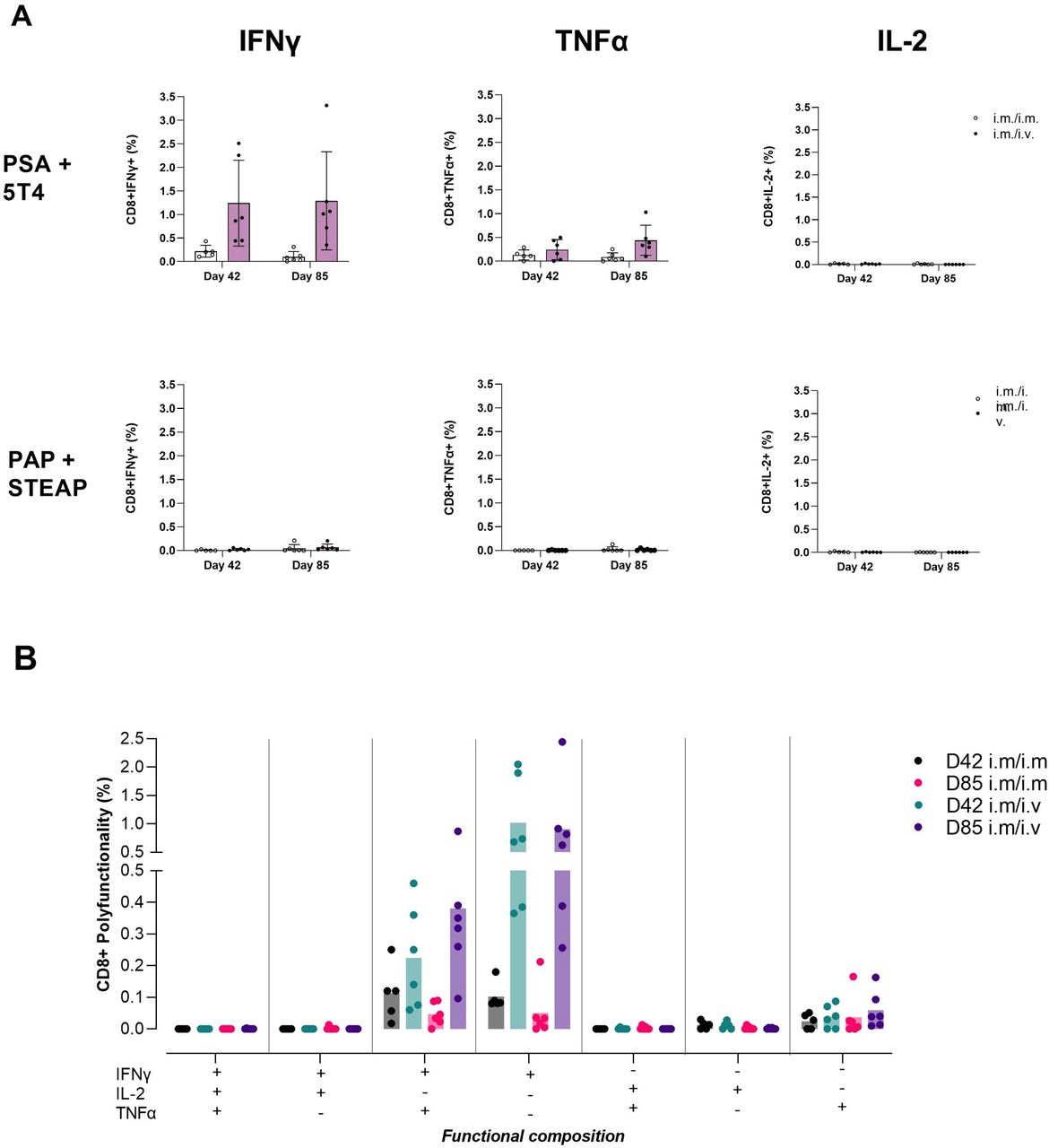
Comparing T cell immunogenicity of ChAdOx1 prime administered IM followed by 2× MVA boost administered IM or intravenouos: intracellular cytokines. Study design: refer to figure 3A. Spleens samples were collected and prepared into single cell suspensions for application into flow cytometry assays. Splenocytes (1×106 cells per well) were stained for surface markers, fixed and permeabilized then stained for intracellular markers following an 18-hour stimulation with 2× prostate immunogen peptide pools; PSA+5T4 and PAP+STEAP1, 1 µg/peptide/mL. (A) Bar graph panel shows mean±SD monofunctional CD8+ T cell responses to PSA+5T4 and PAP+STEAP1 peptide pools at days 42 and 85, with IM/IM and IM/intravenous prime–boost regimens. (B) Bar graph panel shows mean polyfunctional CD8+ T cell responses to PSA+5 T4 peptide pool at days 42 and 85, in IM/IM and IM/intravenous prime–boost regimens. dots represent individual mice. IM, intramuscular.
The functional composition of CD8+ T cell responses to PSA+5T4 was evaluated to measure polyfunctionality of responding cells. The profile of TNFα-secreting cells indicated the majority were IFNγ+TNFα+ double-positive. IFNγ-secreting cells were split between having a mono-functional IFNγ+ and polyfunctional IFNγ+TNFα+ profile (figure 5B).
Intravenous administration of MVA boost augments CD8+ T cell killing of PSA-pulsed target cells in vivo compared with MVA boost administered IM
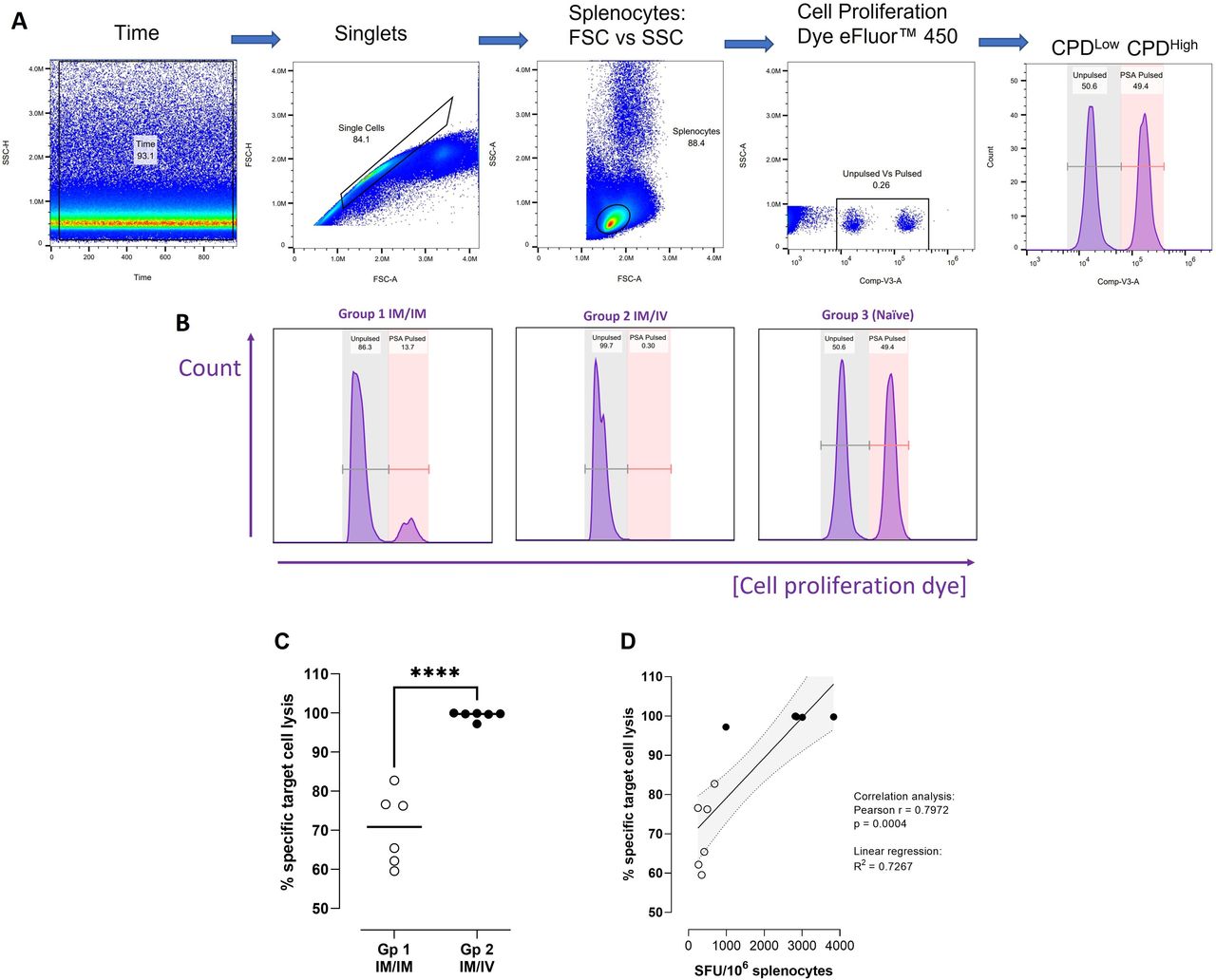
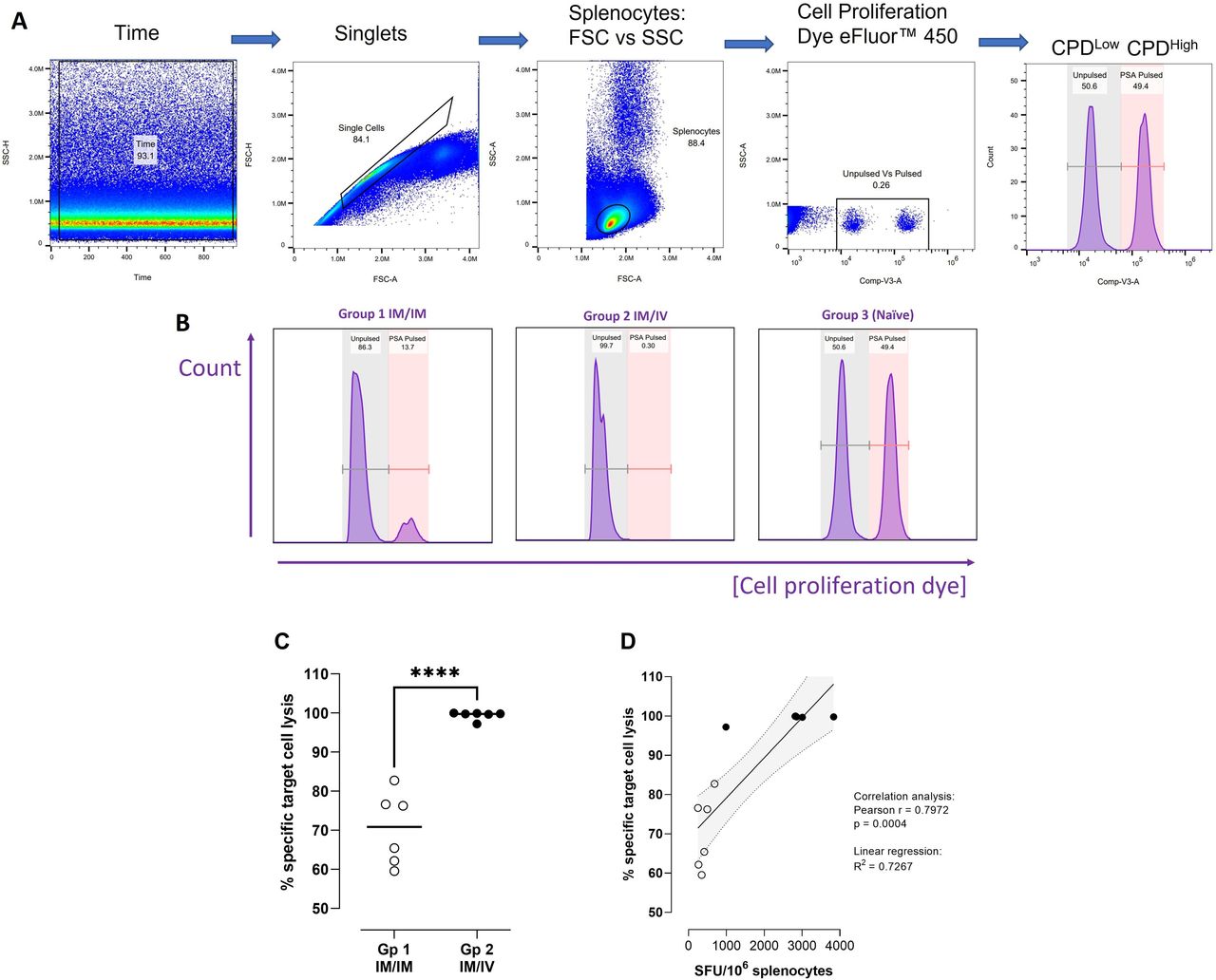
We investigated the functional cytolytic capacity of PSA antigen-specific CD8+T cells elicited through either IM or intravenous administration of a MVA boost (following ChAdOx1 prime) using an in vivo killing assay. Syngeneic naïve mouse splenocytes pulsed with a PSA epitope restricted to H-2Db with known immunodominance in C57BL/6 mice (HCIRNKSVI, the same epitope comprising the peptide used in the Dextramer construct), were used as target cells. PSA-pulsed CPDHigh target cells combined in a 1:1 ratio with unpulsed CPDLow cells were injected intravenously into previously vaccinated mice. Percentage specific target cell lysis was determined after 18 hours (day 85), where changes in the proportion of PSA-pulsed relative to not pulsed target cells indicated the degree of systemic antigen-specific T cell killing of target cells (figure 6A,B). The results demonstrated that while IM administration of a MVA boost generated a strong degree of antigen-specific T cell killing (70.5±8.5%), intravenous administration resulted in almost total elimination of PSA-pulsed target cells (99.4±1.0%); (figure 6C). In naïve mice, the proportion of PSA pulsed to unpulsed cells remained unchanged from the target cell preparation measured prior to injection, confirming that no antigen-specific T cell killing occurred in unvaccinated animals.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparing functionality of CD8+ T cells elicited following 2× MVA boost administered IM or intravenous. Study design: C57BL/6 mice were primed with ChAdOx1 intramuscularly and boosted on days 14 and 28 on with MVA either intramuscularly (group 1) or intravenously (group 2). On day 84, target cells were prepared by pulsing splenocytes from naïve syngeneic mice with a known immunogenic epitope from PSA antigen (‘PSA-pulsed’) or PBS alone (‘Unpulsed’), staining the populations with CPDHigh or CPDLow and combining in a 1:1 ratio. Target cells were injected intravenously into vaccinated (n=6 per group) and naïve control mice (n=3). Splenocytes prepared on day 85 from injected mice for immunogenicity analyses were assessed for target cell killing by flow cytometry. (A) Gating strategy for identification of unpulsed and PSA-pulsed populations. (B) Representative histograms from groups 1 (IM/IM), 2 (IM/intravenous) and 3 (naïve control), peaks on left represent unpulsed target cells, peaks on right represent PSA-pulsed target cells. (C) Graph shows % specific target cell lysis calculated for groups 1 and 2. An unpaired t-test was performed; ****p≤0.0001. (D) Splenocytes from groups 1 and 2 mice were restimulated with PSA antigen used to pulse target cells in an 18-hour IFNγ ELISpot assay. Graph shows correlation between % specific target cell lysis and SFU/106 splenocytes generated in response to PSA antigen for individual mice. Empty circles: group 1; black circles: group 2. IM, intramuscular; MVA, modified vaccinia Ankara.
The same splenocyte samples were applied to an 18-hour IFNγ ELISpot assay and restimulated with PSA peptide HCIRNKSVI to quantify CD8+ T cells elicited in vaccinated animals that specifically recognize this epitope and would be responsible for killing corresponding peptide-pulsed target cells. The percentage specific target cell lysis was correlated with SFU/106 splenocytes on a per-animal basis for groups 1 and 2 (Pearson correlation: r=0.8525; p=0.0004).
Discussion
Previous studies conducted using a heterologous prime–boost approach with ChAdOx1 and MVA encoding the tumor antigen 5T4 showed the induction of T cell responses in early-stage PCa patients and of potential clinical benefit in late stage, metastatic disease.11 25 Building on the work done with this single antigen candidate, ChAdOx1 and MVA encoding 5T4, PSA, PAP and STEAP1 were constructed and shown to be highly immunogenic, especially when given intravenously.
Several studies have shown differential effects of intravenous delivery of immunotherapeutics on both T cell responses and functional outcome in murine cancer models.26–28 TCF1 has been shown to identify a population of stem-like cells in the tumor-associated exhausted CD8+ population and to drive commitment to further differentiation.19–21 29–31 Clinical studies that have identified such self-renewing cells in tumors have associated their induction with improved outcomes.32–35 Baharom et al18 demonstrated that the use of an intravenous peptide-based nanoparticle that contained a TLR7/8 agonist induced higher levels of stem-like T cells that expressed TCF1 and that these CD8+ T cell populations correlated with positive outcomes in mouse tumor models. In the prime–boost setting, Bridle et al28 hypothesized that the protective environment of antigen presentation in the spleen may counteract any pre-existing CD8+ responses to the vaccine construct induced by priming.
While we did not observe a percentage increase in TCF1 positive T cells, the sixfold increase in antigen-specific CD8+ T cells suggests that the total absolute number of these stem-like T cells were also increased. Use of other surface markers revealed that the majority of the CD8+ T cells induced by intravenous delivery of the boost in our prime–boost regimen exhibited markers consistent with early effectors and were characterized by the production of either IFNγ alone or IFNγ and TNFα. However, single or homologous immunizations to investigate increased TCF1 percentages induced by the intravenous route were not performed as a part of this study, as was the case in Baharom et al,18 and the timing of immune response assessments and use of covalently linked TLR adjuvants also differed.
We did not investigate antivector immune responses in this study; however, there are examples in the literature that support use of multiple MVA vaccinations and demonstrate how the magnitude of antigen-specific T cell responses is not impacted. In a phase III therapeutic cancer vaccine trial using MVA-5T4, up to 12 MVA vaccinations were given 3 or 4 weeks apart. Anti-MVA antibodies were detected at 7 and 10 weeks postbaseline, but these did not prevent the generation of anti-5T4 antibodies, which were associated with longer survival.36 In a phase I HIV vaccine trial, up to three MVA vaccinations were given 8 weeks apart.37 It is unlikely that anti-MVA neutralizing antibodies blunt the impact of the second or subsequent MVA administration(s). The T cells generated by intravenous boost delivery that recognized a single specific peptide reflected the overall response in magnitude and memory when compared with the intramuscular route and were also shown to increase in vivo killing potential. Essentially, all the peptide-labeled target cells infused into mice previously vaccinated and boosted intravenously were quickly eliminated from the circulation, showing biological cytotoxic activity.
Immunodominance of the PSA antigen was observed in most strains of mice, although the pattern of response varied. PSA was immunodominant in outbred mice as well; however, PAP responses increased in CD-1 and STEAP1 in BALB/c. Whether this reflects some degree of cognate self-antigen status of these human ‘host’ antigens was not assessed, for example, by measurement of prevaccine antigen-specific precursor frequency. This restricted T cell response is due to the limited class I heterogeneity in inbred mice, and it will be critical to assess dominance patterns in higher outbred mammals. In initial toxicology studies in rabbits, the responses have been far more balanced with PAP T cell induction greater than PSA, which was of similar magnitude to STEAP1 and 5T4 (Anderson, personal communication, 2022).
An assessment of homology between human and mouse PAP, STEAP and 5T4 proteins indicated identity of 81%, 82% and 82% and similarity of 88%, 90% and 89% at the amino acid sequence level, respectively. As these protein sequences are highly homologous between species, it is expected that a similar magnitude of functional T cells would be elicited in humans. There is no homolog of PSA in mice, which may account for immunodominance of this antigen across mouse strains with the recognition as a foreign antigen.
In HLA-A2 transgenic mice, either no or very low reactivity to non-PSA antigens were detected. The mice are transgenic for a single human HLA allele so this result should not be overinterpreted. In addition, these HLA-A2 transgenic mice do not express a PSA homolog (as is the case for mice in general). An immunodominant response to PSA was therefore to be expected, since PSA epitopes were recognized as foreign, whereas PAP, 5T4 and STEAP would function as self-like antigens. In addition to HLA-A*02, the transgenic mice express wild-type MHC from C57BL.6 (H2Kb and H2Db) and BALBc (H2Kd and H2Dd), as the parental mouse was backcrossed onto a wild type C57BL/6 background, then bred with a wildtype BALB/c mouse to yield the F1 generation. In the prime–boost–boost study in C57BL/6 mice, the antitumor immunity to non-PSA antigens largely wanes by D85. Boosting with MVA at later timepoints has been shown to restore initial magnitudes of CD8+ T cell responses,36 37 which suggests the boosting regimen we adopted in our study could be investigated further with potential to improve memory T cell responses. A review of over 100 completed clinical trials with MVA on ClinTrials indicate that up to half used MVA boost vaccinations from two to five times, which further supports this strategy.
How the magnitude of the peripheral CD8+ T cell response correlates with antitumor activity in patients is not determined in this study. The only cancer immunotherapy approved by the US FDA to date is Sipuleucel-T, in which induced T cell responses are weak.9 It is assumed that higher T cell magnitudes will result in greater antitumor efficacy, although measurement of T cell magnitude in peripheral blood may not translate to intratumoral T cell activity. ChAdOx-MVA prime–boost approaches have been shown to elicit high antigen-specific CD8+ T cell magnitudes with the capacity to proliferate and maintain the memory pool over time.25 38
In summary, vaccination using a ChAdOx1/MVA prime–boost regimen induces cellular responses to the prostate immunogens studied here, and intravenous boosting results in responses superior in both magnitude and functionality to those of intramuscular boosting. The increased breadth of the immune response induced by the selected prostate immunogens should help to prevent tumor escape by downregulation of antigen expression but would not overcome beta-2-microglobulin loss. The magnitude, functionality, and breadth of response observed in these studies suggest that investigation of this approach in humans may be valuable, and human studies will also allow further comparison of the intramuscular to intravenous boosting routes.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Animal experiments were conducted at Evotec, Toulouse, France (studies 1 and 2) and Charles River Laboratories, Portishead, UK (studies 3–6). All in vivo work was performed in accordance with the Animals (Scientific Procedures) Act 1986 and EU directive, following a local Ethical Review Process (ERP), approved by the UK Home Office. All protocols involving mice were also approved by the Institutional Animal Care and Use Committee (IACUC) and the Association for Assessment and Accreditation of Laboratory Animal Care, (AAALAC), under license number 001814 2021.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SS, TE, GS, BT and KP designed and prepared vaccine constructs. SS, TE, MM and VW conceived and designed animal studies. Charles River Laboratories, Portishead UK conducted animal studies and isolated samples. AV, CD and KA designed and performed immunological assays. AV, KA, CD and IM analysed data and prepared figures. KA, TE, AV and SS wrote manuscript. TE is the guarantor, responsible for the overall content.
Funding All studies were funded by Vaccitech PLC.
Competing interests All authors are employees of Vaccitech, a public company engaged in biomedical research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.