Article Text

Abstract
Background The antitumor effects of external beam radiation therapy (EBRT) are mediated, in part, by an immune response. We have reported that a single fraction of 12 Gy EBRT combined with intratumoral anti-GD2 hu14.18-IL2 immunocytokine (IC) generates an effective in situ vaccine (ISV) against GD2-positive murine tumors. This ISV is effective in eradicating single tumors with sustained immune memory; however, it does not generate an adequate abscopal response against macroscopic distant tumors. Given the immune-stimulatory capacity of radiation therapy (RT), we hypothesized that delivering RT to all sites of disease would augment systemic antitumor responses to ISV.
Methods We used a syngeneic B78 murine melanoma model consisting of a ‘primary’ flank tumor and a contralateral smaller ‘secondary’ flank tumor, treated with 12 Gy EBRT and intratumoral IC immunotherapy to the primary and additional EBRT to the secondary tumor. As a means of delivering RT to all sites of disease, both known and occult, we also used a novel alkylphosphocholine analog, NM600, conjugated to 90Y as a targeted radionuclide therapy (TRT). Tumor growth, overall survival, and cause of death were measured. Flow cytometry was used to evaluate immune population changes in both tumors.
Results Abscopal effects of local ISV were amplified by delivering as little as 2–6 Gy of EBRT to the secondary tumor. When the primary tumor ISV regimen was delivered in mice receiving 12 Gy EBRT to the secondary tumor, we observed improved overall survival and more disease-free mice with immune memory compared with either ISV or 12 Gy EBRT alone. Similarly, TRT combined with ISV resulted in improved overall survival and a trend towards reduced tumor growth rates when compared with either treatment alone. Using flow cytometry, we identified an influx of CD8+ T cells with a less exhausted phenotype in both the ISV-targeted primary and the distant secondary tumor following the combination of secondary tumor EBRT or TRT with primary tumor ISV.
Conclusions We report a novel use for low-dose RT, not as a direct antitumor modality but as an immunomodulator capable of driving and expanding antitumor immunity against metastatic tumor sites following ISV.
- Immunotherapy
- Tumor Microenvironment
- Radiotherapy
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Radiation therapy (RT), including targeted radionuclide therapy (TRT), is capable of altering the tumor immune microenvironment and in preclinical studies this improves responses to systemically administered immunotherapies like immune checkpoint inhibition. Whether and how RT may improve systemic propagation of antitumor immunity following a locally directed (eg, intratumoral) immunotherapy in the setting of metastatic disease is unknown.
WHAT THIS STUDY ADDS
External beam radiation doses as low as 2 Gy delivered to distant macroscopic tumors and TRT doses as low as 4–6 Gy enhance the propagation of antitumor immunity when combined with an in situ vaccination regimen that targets a single-tumor site in murine models of metastatic melanoma. This results in improved survival and enhanced effector immune cell infiltrate at distant tumors not directly targeted by in situ vaccination.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study supports growing evidence that tumor sites in settings of metastatic disease have the capacity to influence the immune response at other tumor sites and demonstrates that delivering low-dose radiation to this collective tumor immune microenvironment is an effective strategy for enhancing the propagation of antitumor effects that arise following a locally directed in situ vaccine regimen at one tumor site.
Introduction
In situ vaccination (ISV) is a therapeutic strategy that aims to turn a tumor into a focus for stimulation of an adaptive antitumor immune response, turning that patient’s own tumor into a personalized anticancer vaccine.1 Radiation therapy (RT), including external beam radiation therapy (EBRT), has been shown to drive an ISV effect,2–4 and this may be augmented when combined with additional immunotherapies.5–9 This may be accomplished through elimination of suppressive immune cell lineages, upregulating major histocompatibility complex type 1 and natural killer (NK) group 2D ligand expression, eliciting type I interferon (IFN) responses through cyclic GMP-AMP synthase (cGAS) stimulator of interferon genes (STING) pathway activation, and stimulating local cytokine release and endothelial cell changes that enhance tumor infiltration by immune cells.3 10–12 These mechanisms have been shown in preclinical and clinical studies to result in enhanced neoantigen recognition and T-cell diversification, consistent with an ISV effect.3 EBRT can enhance efficacy of a variety of immunotherapies including immune checkpoint blockade, toll-like receptor agonists, oncolytic viruses, and cytokine therapies.13–17 Immunostimulatory EBRT in doses of 8–12 Gy may be particularly beneficial in poorly immunogenic tumors characterized by low tumor mutational burden, low T-cell infiltrate, and limited response to standard immunotherapies.10
Clinical data indicate that the abscopal effects of RT alone are weak and rarely manifest as systemic antitumor responses.18 Abscopal effects may be observed more frequently when EBRT is combined with immune checkpoint blockade, but clinical results have been mixed.19 Most patients require additional intervention beyond EBRT to one site alone to mount an effective systemic immune-mediated antitumor response.19 20 Our group has developed an enhanced RT-based ISV approach consisting of local EBRT combined with an intratumoral injection of the hu14.18-IL2 immunocytokine (IC).9 This IC consists of the hu14.18 antibody targeting the GD2 disialoganglioside (expressed on neuroectoderm-derived tumors such as melanoma, neuroblastoma, and small cell lung cancer) fused at the FC domain with interleukin-2.21 Though intratumoral immunocytokine (IT-IC) was shown to be more effective than intravenous IC in clearing small (<50 mm3) tumors,22 the combination of 12 Gy EBRT followed by IT-IC (RT/IC) improved survival in mice bearing larger, poorly immunogenic, syngeneic B78 melanoma tumors and rendered ~70% of mice disease-free with immune memory.9 However, when delivered to one tumor in a mouse bearing two macroscopically detectable B78 tumors, RT/IC ISV was only capable of clearing ~10% of the distant tumors via a suppressive mechanism at least partially mediated by T regulatory (Treg) cells.23 These results suggest that while EBRT does augment IT-IC in a mouse with a single tumor, these effects do not propagate sufficiently into a systemic response against distant macroscopic tumor sites. This may be due to insufficient generation of effector antitumor immune cells, immune editing for resistant clones, or recruitment of immune suppressors that can circulate systemically from an immunosuppressive tumor-immune microenvironment (TME).24 25 RT/IC ISV clearly alters the targeted tumor, but the degree to which other established TMEs must be altered to enable propagation of effective antitumor immunity and whether this can be achieved by RT is unknown.
We hypothesized that it might be necessary to irradiate all tumor sites, altering the collective TME to effectively propagate systemic antitumor responses resulting from local ISV in models of established metastatic disease. Clinically, oligometastatic cancer is increasingly treated with EBRT to all sites of disease based on demonstrated survival benefit.26 In some patients receiving combination EBRT and immunotherapy, low-dose radiation to other tumor sites is associated with improved radiographic response.27 Herrera and colleagues have also demonstrated that low-dose (2 Gy) EBRT can synergize with a combination of immune checkpoint blockade, CD40 agonism, and cyclophosphamide in metastatic murine models in an IFN-dependent manner.28 In patients with widely metastatic disease, ablative EBRT could be associated with substantial toxicity including lymphopenia and immune suppression. In this setting, intravenously delivered targeted radionuclide therapies (TRTs) with selective uptake and retention within tumor tissue may provide an attractive solution to deliver RT to all sites of metastases.
Our group has been exploring the use of alkylphosphocholine analogs as a vector for targeting the delivery of radionuclides to tumors. We have previously demonstrated selective uptake and prolonged retention of NM600, an alkylphosphocholine analog, in nearly all tumor types and locations, including in human, canine, and murine tumors.29 30 Using 90Y-NM600, we have demonstrated that selective delivery of low-dose radiation to all tumor sites in murine models of metastatic disease results in favorable immunomodulatory effects on the TME and enhanced response to immune checkpoint blockade.31 32 Here, we report findings testing our hypothesis that intravenous injection of 90Y-NM600, delivered prior to local RT/IC ISV, would permit greater systemic control of established distant tumors compared with RT/IC ISV alone. To our knowledge, this is the first time that low-dose TRT has been tested for its capacity to enhance the propagation of antitumor immunity following local ISV (or any vaccine-based) immunotherapy strategy.
Methods
Syngeneic tumor cell line
The B78-D14 (B78) murine melanoma cell line, derived from B16-F10 melanoma,33 34 was provided by Ralph Reisfeld (Scripps Research Institute). Panc02 murine pancreatic adenocarcinoma cells were obtained from the NCI. B78 and Panc02 cells were cultured in RPMI 1640 (Mediatech, Manassas, Virginia, USA) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 µg/mL streptomycin, and 2 mM L-glutamine.
Animals and tumor models
Animals were housed and cared for using an approved protocol (M005670) reviewed by the University of Wisconsin-Madison Institutional Animal Care and Use Committee. Tumors were engrafted in female C57BL/6 mice 6–8 weeks old (Taconic Biosciences, Rensselaer, New York, USA) by injecting 2×106 cells (either B78 or Panc02) in 100 µL intradermally in the right flank. Secondary tumors were injected similarly 10–14 days after the primary tumor in the contralateral (left) flank. Mobile status of the tumor was used to confirm intradermal tumor placement.35 For all studies, mice without palpable secondary tumors were excluded. Mice were evaluated two times per week using calipers to measure tumor volume estimated as (width2×length)/2. Survival endpoints were tumor dimension of >20 mm or veterinary recommendation of euthanasia. Disease-free mice were rechallenged by injecting 2×106 B78 cells intradermally to the left shoulder 30 days after confirmation of disease-free status. Animals rejecting B78 rechallenge were injected with 2×106 Panc02 murine pancreatic adenocarcinoma cells intradermally in the right shoulder.
Tumor radiation treatments
EBRT was delivered using an X-RAD 320 system (Precision X-Ray, North Branford, Connecticut, USA). Mice were immobilized using custom lead jigs that expose both flanks and were irradiated with additional lead shields to protect the secondary tumor as previously described.9 23 To deliver EBRT to the secondary tumor on the left flank, flank shields were removed during irradiation. Once the scheduled dose (2, 6, or 12 Gy) was delivered to both tumors, the left flank was reshielded so that the remaining dose could be delivered to the primary tumor.
The alkylphosphocholine molecule used for molecular targeted radionuclide therapy (TRT), 2-(trimethylammonio)ethyl(18-(4-(2-(4,7,10-tris(carboxymethyl)−1,4,7,10-tetraazacyclododecan-1-yl)acetamido)phenyl)octadecyl) phosphate (NM600), was kindly provided by Archeus Technologies (Madison, Wisconsin, USA). 90YCl3 was purchased from PerkinElmer (Waltham, Massachusetts, USA). The radiolabeling and characterization of 90Y-NM600, as well as its PET-detectible counterpart 86Y-NM600, have been described elsewhere.36 37 Mice receiving RT were radiated on treatment day 1, and mice receiving TRT were injected with 100 µCi of 90Y-NM600 preparation by tail vein injection on treatment day 1. An injected activity of 100mCi, which corresponds to a dose of approximately 4 Gy at the secondary tumor, was chosen as it was previously demonstrated not to limit (via immunosuppression) efficacy of RT/IC ISV in a one-tumor B78 model (data not shown).
IC treatments
Lyophilized hu14.18-IL2 IC (4 mg/vial) was provided by Apeiron Biologics (Vienna, Austria) and reconstituted with 8 mL of sterile phosphate buffered saline (PBS). For mice treated with RT/IC, 100 µL of 0.5 mg/mL IC solution was injected intratumorally using a 30 G needle daily on treatment days 6–10, for a total dose of 250 µg per mouse.
Flow cytometry
At the time of harvest, mice were euthanized by CO2 asphyxiation. Tumors were harvested and dissociated as previously described.38 Sample dissociate was filtered through a 70 µm cell strainer, washed with 10 mL of cold PBS, and stained according to two antibody panels; the antibodies for each are detailed in online supplemental table 4. After all surface staining, cells were fixed and permeabilized using the eBioscience fix/perm kit (00-5523-00), internally stained, and then cryopreserved at −80°C in a solution of 10% dimethyl sulfoxide (DMSO)/90% heat-inactivated fetal bovine serum.38 Samples were stored at −80°C behind radioactive shielding until 30 days past treatment injection to ensure 90Y decay to background level.
Supplemental material
Samples were acquired on an Attune NxT flow cytometer (Thermo Fisher, Waltham, Massachusetts, USA) with manufacturer-provided acquisition software. All flow cytometry experiments included Fluorescence Minus One controls used for setting gates. Data were analyzed using FCS Express 7 (De Novo Software, Pasadena, California, USA) platform.
Statistical analysis
All experiments were repeated, generally in triplicate, to confirm validity of results. All data presenting replicates include the individual values as well as mean±SEM except where otherwise noted. Differences in tumor growth rates were assessed using linear mixed-effects models. Growth rates were estimated from the model as a ratio of change every 21 days. Models predicted tumor volume based on treatment group, days since treatment started, an interaction between treatment and time, and a random intercept per mouse. The statistics of interest were the interaction between treatment and time, which represented the ratio between one treatment’s estimated 21-day growth rate (expressed as an estimated average fold change in volume every 21 days, applied across the entire growth curve) and the average rate corresponding to another treatment. These estimates, along with 95% CIs and Kenward-Rogers p values, were calculated and are presented in online supplemental table 1–3. Tumor volumes for some analyses were transformed with the natural log function to correct patterns in residuals and to account for the exponential growth of tumors. Zero-valued measurements were imputed as 4 mm3 (the smallest recordable tumor volume using calipers) in order to be defined under the transformation. Statistical analysis was conducted using R V.4.0.2; the lme4 package was used for fitting mixed models and Kenward-Rogers p values were estimated from the lmerTest package. Significance was assessed at the alpha=0.05 level, and no corrections were made to account for inflated type 1 error rate of multiple hypotheses.
Survival analyses were conducted using the Kaplan-Meier method, with comparisons using the log-rank test. Comparisons in immune cell populations measured by flow cytometry were conducted using a one-way analysis of variance, with Tukey’s method for conducting multiple comparisons. Comparisons of proportions were conducted using the χ2 test or Fisher’s exact test if there were zero values.
Results
EBRT delivered to an established secondary tumor combined with ISV to primary tumor improves overall survival
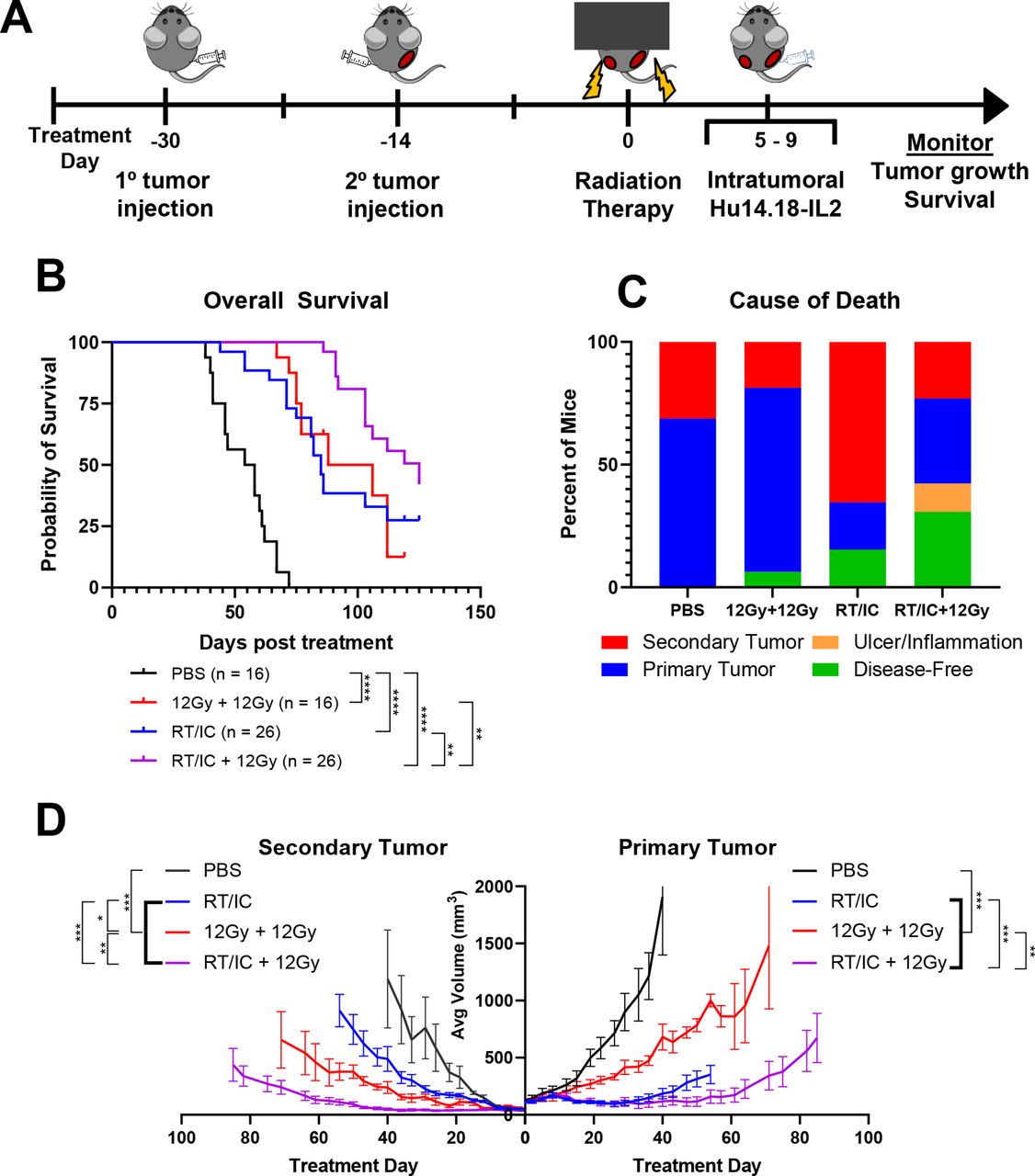
To test the effect of secondary tumor EBRT combined with primary tumor ISV on overall survival and antitumor efficacy, C57BL/6 mice with ~150 mm3 primary and ~50 mm3 secondary B78 melanoma tumors were treated with either PBS, 12 Gy EBRT to both tumors, RT/IC ISV to the primary tumor, or ISV to the primary plus 12 Gy EBRT (RT/IC+12 Gy) to the secondary tumor (figure 1A). Results demonstrated that the RT/IC+12 Gy group had improved survival compared with RT/IC alone (p=0.0025) or to 12 Gy EBRT to both tumors (p=0.0052, figure 1B and online supplemental table 1). This effect is also reflected in slower tumor growth rates as calculated by mixed effects modeling. Comparing these average rates, we estimated that both primary and secondary tumors on average in the RT/IC+12 Gy group grew slower than primary and secondary tumors in mice treated with ISV alone, or 12 Gy EBRT to both tumors (online supplemental table 1). All three interventions (RT only, RT/IC only, or RT/IC+12 Gy EBRT) extended overall survival and slowed tumor growth compared with the PBS control (p<0.001, online supplemental table 1). Importantly, the data suggest a cooperative effect at both the primary and secondary tumors when ISV to the primary is combined with 12 Gy EBRT to the secondary.

12 Gy EBRT to both primary and secondary tumor allows for better tumor control induced by subsequent IT-IC immunotherapy. (A) Schema of mouse multiple tumor model and subsequent in situ vaccination approach. C57BL/6 mice were engrafted intradermally with 2×106 B78 syngeneic melanoma cells. On the right flank, engraftment was performed to initiate primary tumors approximately 4 weeks prior to start of treatment. On the left flank, tumor cells were engrafted 2 weeks later to initiate the secondary tumor, approximately 2 weeks prior to the start of treatment. Mice were treated with EBRT to either the primary only or the primary and secondary tumors on treatment day 0 and given IT-IC immunotherapy on treatment days 5–9. (B) Pooled assessment of overall survival of animals from three separate replicate experiments following the indicated treatment. Animals either died from their tumor burden or reached a predetermined size of tumor to require euthanasia. Statistical comparisons by log-rank test are indicated. (C) Breakdown of the cause of death for all animals treated over the course of the three individual experiments. Mice that were sacrificed due to either primary tumor burden (blue), secondary tumor burden (red), or due to inflammation or dermatitis (yellow) are indicated. Mice that were still alive at the conclusion of the experiment were labeled as disease-free if no palpable tumor was detectable (green). Mice that were still alive with tumors were considered, for this analysis, as dying from either the primary or secondary tumor, depending on which tumor was larger at the conclusion of the experiment. (D) Average ±SEM tumor volume from a representative experiment. primary tumors are on the right side, and secondary tumors are on the left. Average growth curves terminate on the day post treatment where the first member of that treatment cohort died. See online supplemental figure 1 for individual tumor growth curves corresponding to this experiment. See online supplemental table 2 for evaluation of linear mixed-effects modeling of tumor growth rates and survival analyses. Comparisons in (D) represent Kenward-Rogers p values for the ratio of estimated growth rates between the indicated groups. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Groups being compared are indicated by brackets connecting the comparison groups. Heavy bolded brackets indicate pairwise comparisons between the group outside of the bolded brackets and each group within the bolded bracket. Each of the comparisons described by the heavy bolded brackets has a p value consistent with the range indicated by asterisks. For example, the first bolded bracket from the left (D) indicates that the p values for the comparisons of PBS to RT/IC, PBS to 12 Gy+12 Gy, as well as PBS to RT/IC+12 Gy are each <0.001. EBRT, external beam radiation; IC, immunocytokine; IT-IC, intratumoral immunocytokine; RT, radiation therapy.
In addition to survival and tumor growth rate, the cause of death was evaluated for each mouse (figure 1C). Eleven out of 16 mice (68.8%) in the PBS group died from their primary tumor, as did 12/16 mice (75%) treated with 12 Gy EBRT only to both tumors. By contrast, 5/26 (19.2%, p=0.0027 compared with PBS) mice treated with RT/IC ISV died from their primary tumor, and 17/26 (65.4%) died from their secondary tumor. Four out of 26 mice (15.4%, p=0.1429 compared with PBS) treated with RT/IC were disease-free. In mice treated with RT/IC+12 Gy, 11/26 (42.3%, p=0.032 compared with RT/IC) mice were disease-free, 9/26 (34.6%) mice died from primary tumor burden, and 6/26 (23.1%, p=0.002 compared with RT/IC) mice died from secondary tumor burden. Three of the disease-free mice were euthanized due to ulceration/inflammation at the tumor site. These data suggest that the addition of IC to RT at the primary tumor site can control the primary disease, yet untreated secondary tumors continue to grow. Complete control of both sites of disease is improved with RT/IC ISV to one tumor and additional 12 Gy EBRT delivered to the secondary tumor.
In mice rendered disease-free by treatment, tumor-specific memory was tested with a rechallenge of B78 tumor cells. 100% (8/8, p=0.002 compared with control) of disease-free mice in the RT/IC+12 Gy group rejected tumor rechallenge. The three disease-free mice with ulceration/inflammation were euthanized prior to memory testing. In the RT/IC group, 75% (3/4) of disease-free mice rejected tumor rechallenge. However, in the EBRT only group, the one disease-free mouse did not reject rechallenge. Four out of four (100%) age-matched, tumor-naïve mice implanted with B78 at the same time each developed tumors. To confirm this rejection was tumor-specific, the 11 remaining disease-free mice developed palpable Panc02 tumors after they were subsequently challenged with an unrelated Panc02 pancreatic cancer cell line. In total, these data suggest EBRT delivered to all sites of disease improves the antitumor immune response (with tumor-specific memory) not only at the ISV-treated primary but also at the distant secondary tumor, beyond the effect of either ISV or RT alone.
EBRT improves ISV response at primary and secondary tumors in a dose-dependent fashion
To determine the relationship between radiation dose to the secondary tumor and its additive effect with primary tumor ISV, a dose titration experiment was conducted. C57BL/6 mice with~150 mm3 primary and~50 mm3 secondary tumors were established as described in figure 2A. Mice were then randomized to receive either PBS (no treatment control), 12 Gy EBRT to both tumors (12 Gy/12 Gy RT only control), or ISV (RT/IC) at the primary tumor combined with 0, 2, 6, or 12 Gy EBRT to the secondary tumor. We hypothesized that additional RT to the secondary tumor, combined with ISV to the primary tumor, would slow tumor growth beyond either treatment alone in a dose-dependent fashion.

Titration of EBRT dose to secondary tumor combined with RT/IC immunotherapy to primary tumor. Female C57BL/6 mice were implanted intradermally with 2×106 B78 syngeneic melanoma cells approximately 4 weeks prior to treatment at the right flank and approximately 2 weeks prior to treatment on the left flank. Primary tumors were on average ~150 mm3, while secondary tumors were on average ~50 mm3. (A) On day 0 (when EBRT was given), mice were treated with PBS control (orange), 12 Gy EBRT to both primary and secondary (black), and 12 Gy EBRT+IT-IC to the primary with either 0 Gy (blue), 2 Gy (green), 6 Gy (red), or 12 Gy (purple) of EBRT to the secondary tumor. IT-IC was given at the primary tumor on treatment days 5–9 as described in the Methods section. Treatment group nomenclature in the legends is indicated as treatment to primary tumor/treatment to secondary tumor. tumor volumes were measured two times per week. Mice with no palpable secondary tumor on treatment day 0 were excluded from the analysis. (B) Average ±SEM tumor volume for both primary (right side) and secondary (left side) tumors. Statistical comparisons in the legend indicate Kenward-Rogers p values for comparison of estimated growth rate (as determined by linear mixed-effects modeling on log-transformed data) for the primary tumor (right side) and secondary tumor (left side). See online supplemental table 2 for additional statistical comparisons. (C–H) Individual tumor growth curves for each of the mice in the treatment groups as described. Data presented here are representative of two independently conducted experiments. *P<0.05, **P<0.01, ***P<0.001. Groups being compared are indicated by brackets connecting the comparison groups. Heavy bolded brackets indicate pairwise comparisons between the group outside of the bolded brackets and each group within the bolded bracket. Each of the comparisons described by the heavy bolded brackets has a p value consistent with the range indicated by asterisks. For example, the first bolded bracket from the left (B) indicates that the p values for the comparisons of secondary tumors of 12 Gy 1o/0 Gy 2o to 12 Gy+IC 1o/6 Gy 2o, as well as 12 Gy 1o/0 Gy 2o to 12 Gy+IC 1o/12 Gy 2o are each <0.001. EBRT, external beam radiation therapy; IC, immunocytokine; IT-IC, intratumoral immunocytokine; RT, radiation therapy.
Results demonstrated that at the primary tumor, all four treatment groups treated with the ISV (regardless of EBRT dose to the secondary tumor) had slower tumor growth rates compared with PBS or EBRT alone (figure 2 and online supplemental table 2). EBRT alone slightly reduced tumor growth rate compared with PBS alone. Importantly, the primary tumors in mice treated with RT/IC+12 Gy EBRT to the secondary tumor had a trend towards slower growth than mice treated with RT/IC alone (p=0.051). This is consistent with the same ‘reciprocal effect’ observed at the primary tumor described in figure 1.
At the secondary tumor, growth rates were reduced in a dose-dependent fashion proportional to radiation dose at the secondary tumor. At the highest tested dose, 12 Gy EBRT to the secondary combined with ISV to the primary had slower secondary tumor growth rates compared with ISV alone, 12 Gy EBRT alone, and ISV+6 Gy to the secondary (figure 2B and online supplemental table 2). Treatment with 6 Gy to the secondary plus ISV to the primary resulted in slower secondary tumor growth than delivering a greater RT dose (12 Gy) alone to both tumors. This potentiation effect, combined with the aforementioned observations, suggest that RT delivered at doses less than 12 Gy to a secondary tumor can combine with ISV to slow the growth of the secondary tumor.
TRT combined with ISV improves overall survival
To determine if we could apply the same findings from combination ISV and EBRT to combination ISV and TRT, we used the beta particle-emitting TRT agent 90Y-NM600. Dosimetry studies using 86Y-NM600 demonstrate that this TRT agent is selectively taken up and retained in a wide variety of tumor types and locations37 and that absorbed dose over time (as a function of injected activity) is similar between ‘large’ (>200 mm3) and ‘small’ (<70 mm3) B78 tumors, regardless of the number of tumors (online supplemental figure 2).
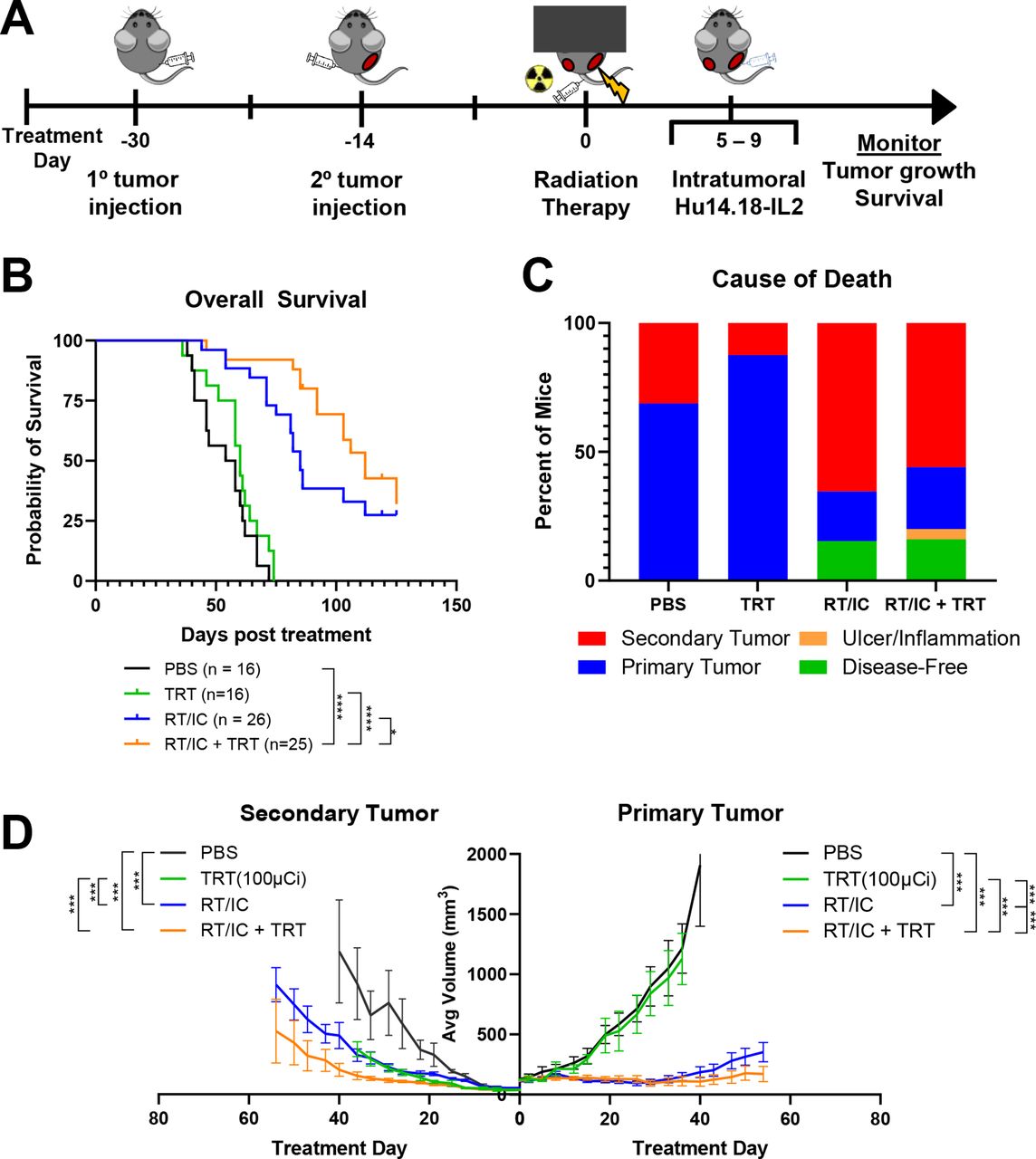
To determine if systemically administered 90Y-NM600 can provide benefit in controlling multiple tumors when combined with local RT/IC ISV, C57BL/6 mice with ~150 mm3 primary and ~50 mm3 secondary B78 tumors were randomized to either PBS control, 100 µCi of TRT only, RT/IC to the primary tumor, or RT/IC+100 µCi TRT (figure 3A). Results demonstrated that the combination of RT/IC+TRT had significantly increased overall survival compared with RT/IC ISV alone (median 112 vs 85 days, p=0.025; figure 3B) or TRT alone (112 days vs 60 days, p<0.0001; online supplemental table 3). In addition, mixed-effects modeling of log-transformed growth curves demonstrated that primary tumors treated with RT/IC+TRT grew slower than those treated with either RT/IC or TRT alone. Secondary tumors in mice treated with RT/CI+TRT grew slower than those in mice receiving TRT alone; however, secondary tumor growth rate in mice receiving RT/IC+TRT was not significantly slower than in mice receiving RT/IC alone (figure 3D and online supplemental figure 3 and table 3).

Systemically delivered 90Y-NM600 combined with local RT/IC in situ vaccination improves overall survival and may allow for better tumor control compared with in situ vaccination alone. C57BL/6 mice were implanted intradermally with primary B78 syngeneic murine melanoma tumors in the right flank, followed by secondary tumors in the left flank 14 days later. Mice were randomized to receive either PBS control, 100 µCi of 90Y-NM600 TRT, RT/IC in situ vaccine to the primary tumor, or the combination of RT/IC+TRT. Of note, the PBS and RT/IC groups were shared with the experiments depicted in figure 2. (A) Schema of the multiple tumor model set-up and subsequent in situ vaccination and radiation approach. (B) Overall survival of animals pooled over three separate experiments. Animals either died from their tumor burden or reached a predetermined size of tumor to require euthanasia. (C) Mice that were sacrificed due to either primary tumor burden, secondary tumor burden, or inflammation or dermatitis are indicated. Mice that were still alive at the conclusion of the experiment were labeled as disease-free if no palpable tumor was detectable. Mice that were still alive with tumors were considered as dying from either the primary or secondary tumor, depending on which tumor was larger at the conclusion of the experiment. (D) Average ±SEM tumor volume from a representative experiment. Primary tumors are on the right side, and secondary tumors are on the left. Average growth curves terminate on the day post treatment, where the first member of that treatment cohort died. See online supplemental figure 3 for individual tumor growth curves corresponding to this experiment. see online supplemental table 3 for evaluation of linear mixed-effects modeling of tumor growth rates and survival analyses. Comparisons (D) represent Kenward-Rogers p values for the ratio of estimated growth rates between the indicated groups. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. IC, immunocytokine; IT-IC, intratumoral immunocytokine; RT, radiation therapy; TRT, targeted radionuclide therapy.
Analysis of cause of death revealed that 14/16 (87.5%) mice in the TRT-only group died from primary tumor burden, compared with 6/25 (24%, p<0.0001 compared with TRT only) mice in the RT/IC+TRT group (figure 3C). Four out of 25 (16%) mice in the RT/IC+TRT group became disease-free. All these four disease-free mice rejected subsequent subcutaneous engraftment with an additional B78 tumor inoculum but developed tumors following similar injection of unrelated, syngeneic Panc02 tumor cells. This demonstration of improved overall survival, and induction of tumor-specific memory suggests that addition of TRT supported propagation of systemic antitumor immune responses following RT/IC ISV, similar to the effect observed with EBRT.
Increased immune cell infiltrate and activation following ISV combined with radiation to all tumor sites
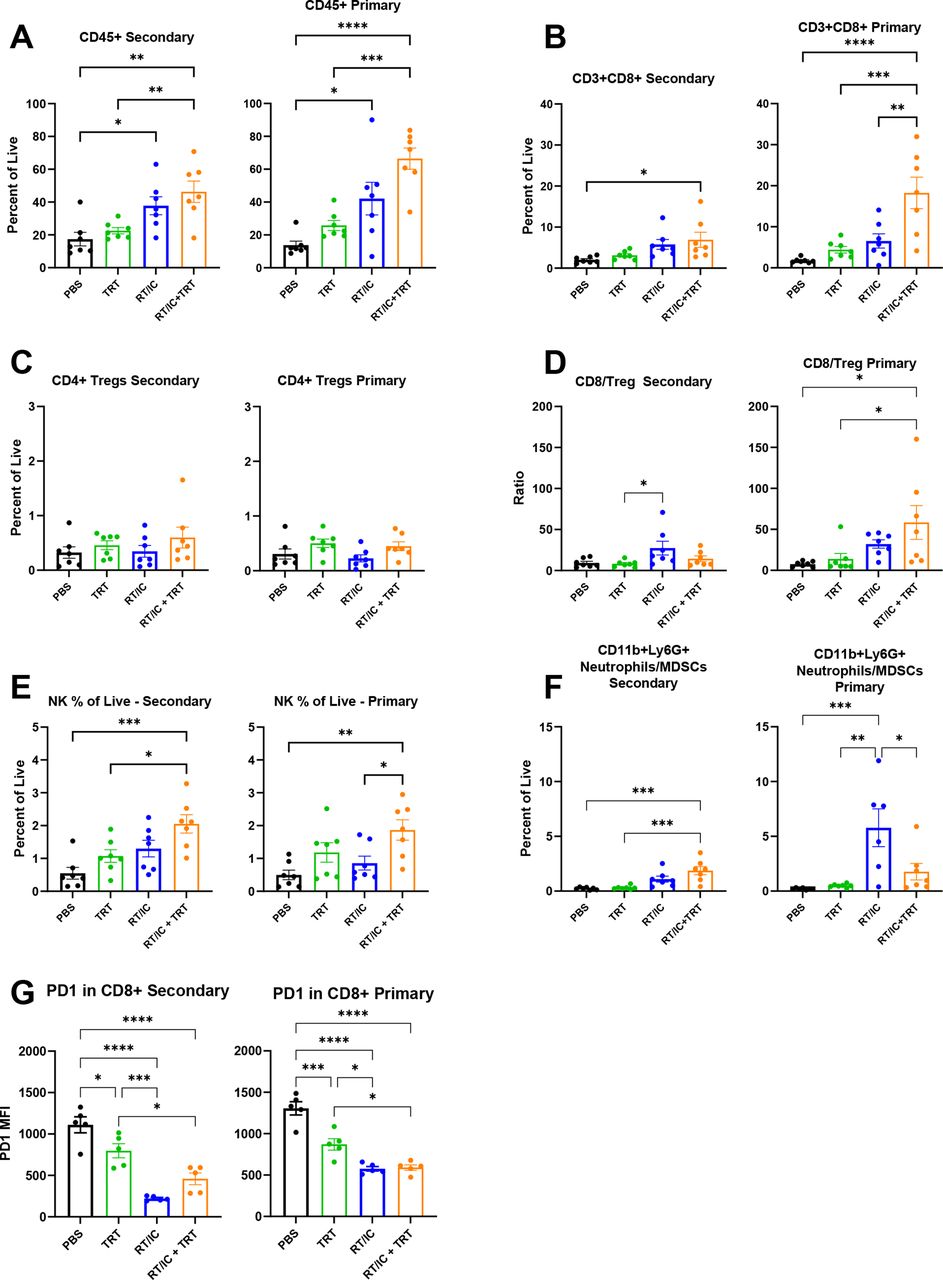
To characterize immune changes in the TME following each of the treatments described in figure 1, we used flow cytometry to evaluate mice bearing two B78 tumors (figure 4 and online supplemental tables 4 and 5). Tumors treated with RT/IC and RT/IC+12 Gy EBRT exhibited an influx of CD45+ immune cells at both the primary and secondary tumors compared with PBS. At the secondary tumor, mice treated with RT/IC+12 Gy had more CD45+ immune cells, CD3+CD8+ T cells, and NK cells compared with either RT/IC or 12 Gy/12 Gy RT alone. In mice receiving EBRT only, a significant increase in CD4+CD25+Foxp3+ Treg cells was noted in both tumors. This increase was not observed in the primary tumors of mice treated with RT/IC or RT/IC+12 Gy but was noted in the secondary tumors of mice treated with RT/IC+12 Gy (figure 4C). This resulted in an increased (compared with PBS-treated mice) CD8:Treg ratio in primary tumors of both RT/IC and RT/IC+12 Gy mice, but this ratio was increased only in secondary tumors of the RT/IC group (not the RT/IC+12 Gy group) (figure 4D). Among the CD8+ T cells, PD1 expression (a marker of activation and exhaustion) was substantially reduced in the primary and secondary tumors of mice treated with both RT/IC and RT/IC+12 Gy, though secondary tumors in the RT/IC+12 Gy group had CD8+ T cells with higher PD1 expression compared with the secondary tumors in the RT/IC alone group (figure 4G). There was also a greater proportion of CD11b+Ly6G+ granulocytes (often considered neutrophils and/or myeloid-derived suppressor cells (MDSCs)) in primary tumors of both RT/IC and RT/IC+12 Gy-treated mice compared with PBS-treated mice, but at the secondary tumor, they were only increased in mice treated with RT/IC alone.

Flow cytometry analysis of primary and secondary tumors in mice treated with in situ vaccine combined with radiation to distant tumors. Female C57BL/6 mice were implanted intradermally with B78 tumors on the right and left flanks simultaneously. After 4 weeks, mice were treated with either PBS (black), 12 Gy EBRT to both primary and secondary tumors (red), 12 Gy EBRT and IT-IC to the primary tumor only (blue), or RT/IC to the primary tumor and 12 Gy EBRT to the secondary tumor (purple). RT was given on treatment day 0, and IC was given on treatment days 5–9 as described in figure 2A. On treatment day 14, tumors were harvested and dissociated as described in the Methods section. Aliquots of tumors were stained using the adaptive and innate antibody panels outlined in online supplemental table 4, fixed, and cryopreserved as described in the Methods section for 30 days. Cells were gated according to the expression of the markers outlined in online supplemental table 5. Immune populations are expressed as a percentage of all live, single cells, except where otherwise noted. In each population pair of graphs depicted (A–G), results corresponding to the right flank primary tumor are on the right, and results corresponding to the left flank secondary tumor are on the left. Representative plots from two independent experiments are presented. Statistical analyses were conducted using one-way analysis of variance, with multiple comparisons using Tukey’s method. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. EBRT, external beam radiation therapy; IC, immunocytokine; IT-IC, intratumoral immunocytokine; ns, not significant; RT, radiation therapy.
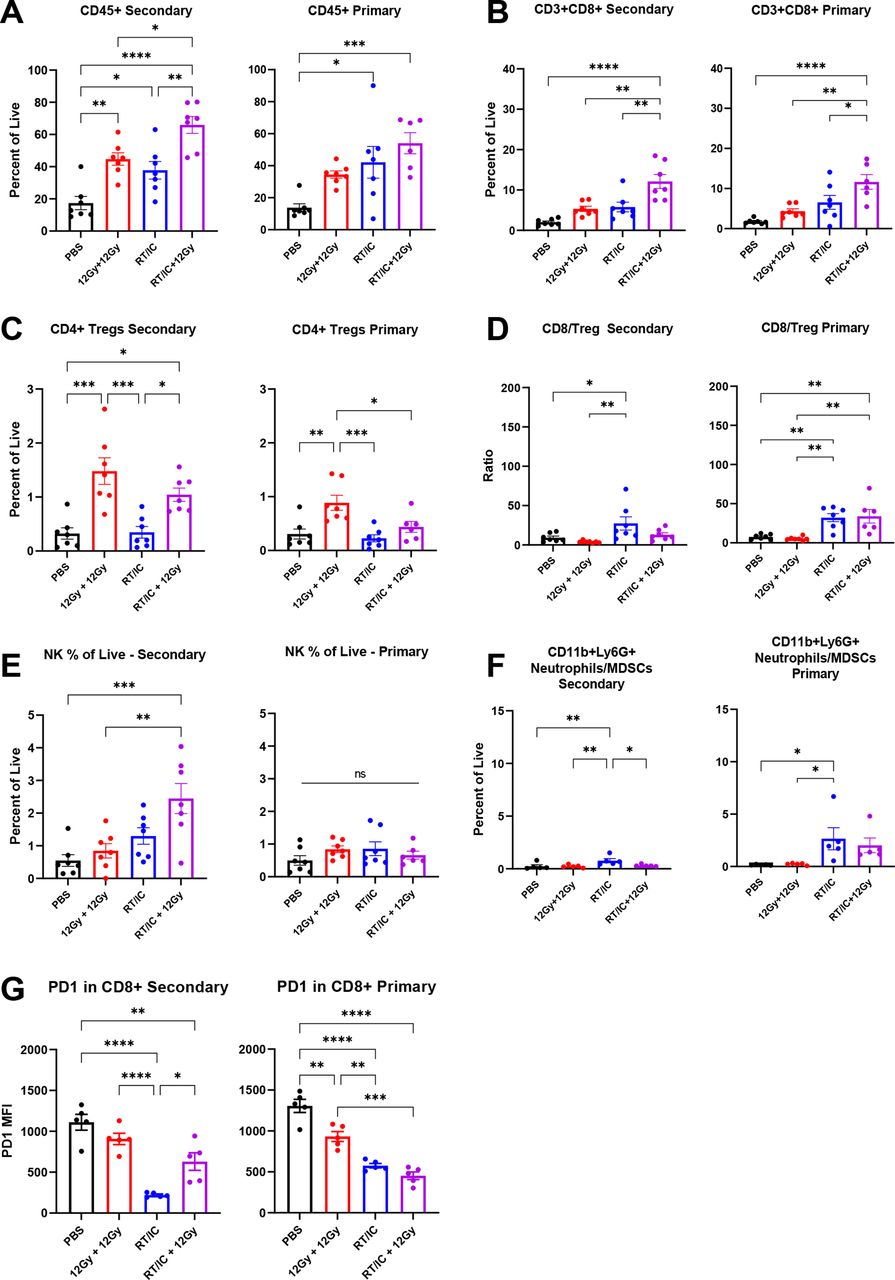
Using a similar methodology, we evaluated changes to the TME in mice following treatment with ISV and TRT corresponding to figure 3 (figure 5 and online supplemental tables 4 and 5). As was the case with EBRT, an influx of CD45+ immune cells was detected in both primary and secondary tumors for mice treated with RT/IC or RT/IC+TRT compared with PBS (figure 5A). A similar increase was seen for CD8+ T cells and NK cells (figure 5B,E) in both primary and secondary tumors of mice treated with RT/IC+TRT compared with PBS treatment. No changes were detected in Treg proportions among all measured tumors. The CD8:Treg ratio was again increased in the primary tumors of mice treated with RT/IC plus TRT compared PBS but was not significantly different at the secondary tumor. Again, PD1 expression on CD8+ cells was reduced in primary and secondary tumors of mice treated with RT/IC or RT/IC+TRT compared with PBS (figure 5G). Under these conditions, an increase (over the PBS control) in CD11b+Ly6G+ neutrophils (or MDSCs) was noted among RT/IC-treated primary tumors, as well as for RT/IC+TRT-treated secondary tumors (figure 5F). In total, these two sets of flow observations (figures 4 and 5) document an influx of effector immune cells and an alteration in the effector-to-suppressor ratio among both primary and distant tumors treated with a combination of primary-tumor directed ISV and RT targeting the distant tumor(s) using either EBRT or TRT.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Flow cytometry analysis of primary and secondary tumors in mice treated with in situ vaccine combined with 90Y-NM600 TRT female C57BL/6 mice implanted intradermally with B78 tumors on the right and left flanks simultaneously. After 4 weeks, mice were treated with either PBS (black), 100 µCi of 90Y-NM600 TRT by tail vein injection (green), 12 Gy EBRT and IT-IC to the primary tumor only (RT/IC, blue), or RT/IC to the primary tumor and 100 µCi of 90Y-NM600 (orange). EBRT and TRT was given on treatment day 0, and IC was given on treatment days 5–9. On treatment day 14, tumors were harvested and dissociated as described in the Methods section. Aliquots of tumors were stained using the adaptive and innate antibody panels outlined in online supplemental table 4, fixed, and cryopreserved as described in the Methods section for 30 days. Cells were gated according to expression of the markers outlined in online supplemental table 5. Immune populations are expressed as a percentage of all live, single cells, except where otherwise noted. In each population pair of graphs depicted, results corresponding to the right flank primary tumor are on the right, and results corresponding to the left flank secondary tumor are on the left. Representative plots from two independent experiments are presented. Statistical analyses were conducted using one-way analysis of variance, with multiple comparisons using Tukey’s method. Note that these data were collected as part of the same experiment outlined in figure 4 and share common PBS and RT/IC groups, which are presented again here. IC, immunocytokine; IT-IC, intratumoral immunocytokine; RT, radiation therapy; TRT, targeted radionuclide therapy.
Discussion
We have previously demonstrated the utility of adding EBRT to IT-IC as a potent antitumor ISV in preclinical poorly immunogenic tumors.9 While this therapy effectively eradicated single tumors, it was much less efficacious in treating animals with a concurrent distant tumor. Therefore, in this study, we sought to test our hypothesis that RT delivered to distant sites of disease would enhance tumor complete response rates to local ISV. We first tested the use of 12 Gy EBRT to a secondary tumor combined with ISV to the primary tumor and found this combination significantly improved survival and reduced average tumor growth rate at both the primary and secondary tumors compared with either EBRT or ISV alone (figure 1). We then demonstrated that even lower doses of EBRT (as low as 2 Gy) could be used at the secondary tumor, combined with RT/IC ISV to the primary, to enable propagation of antitumor immunity from the ISV resulting in reduction of primary and secondary tumor growth rates beyond that achieved with ISV or EBRT alone (figure 2). In substituting for EBRT with approximately 4–6 Gy equivalent of 90Y-NM600 TRT, we again demonstrated improved survival and reduced tumor growth rates when combined with RT/IC ISV (figure 3). Analysis of the immune populations within the TME demonstrated an influx of effector immune cells and favorable alterations of effector-to-suppressor ratio in tumors treated with combination of local ISV and systemic tumor-directed EBRT or TRT (figures 4 and 5). In total, our findings support the hypothesis that low-dose RT delivered to all established sites of disease alters the collective immune TME such that a local in situ vaccine can generate a stronger systemic antitumor immune response.
Generation of an abscopal response, or a systemic antitumor response following local therapy, has been a goal of cancer immunologists and radiation oncologists for several decades. Our data show that although it does not result in complete tumor clearance in the B78 model, RT/IC ISV alone at the primary tumor can result in improved survival and reduced primary and secondary tumor growths, with increased immune infiltrate in both tumors. This suggests that there is a systemic antitumor immune response induced by local ISV, although insufficient to overcome immune suppressive effects at the secondary tumor. Previous studies show that radiation doses as low as 2 Gy can modulate an immune response in part through activation of a cGAS/STING-mediated type I IFN response.10 28 31 39 Our studies not only demonstrate antitumor effects at the secondary tumor by delivering low-dose RT to the secondary tumor combined with ISV to the primary but also show that the antitumor effect at the primary tumor is greater following delivery of RT to the secondary tumor. Even though the primary tumor is treated with the same ISV, RT to distant sites permits a stronger local response at the primary tumor. This finding is similar to previous observations in the setting of two-tumor mice23 and demonstrates a reciprocal abscopal response: treatment of a primary tumor site with an ISV that includes moderate dose RT induces some antitumor response at a secondary tumor, while simultaneous treatment of that secondary tumor site with low-dose RT improves antitumor response at the primary tumor. This benefit of immunomodulatory radiation to distant tumor sites may have immediate clinical applicability. These findings might also suggest that other ISV approaches using IT injection of oncolytic viruses, TLR agonists, or cytokines could be combined with low-dose RT to all sites of metastatic disease to achieve stronger propagation of antitumor immunity and improved clinical efficacy.40
There is growing interest in radiating all known sites of disease in cases of certain oligometastatic (five or fewer metastases) cancers. The results of a randomized phase II trial of stereotactic ablative radiotherapy to all disease sites versus standard of care in 99 patients with oligometastatic disease demonstrated improvement in median survival (41 vs 28 months) among patients undergoing RT to all tumors.26 Yet, delivering ablative RT to all sites of disease in patients with widely metastatic disease would be limited by toxicity and systemic myelosuppression. Moreover, this approach would likely leave undetectable micrometastases unirradiated. An approach which combines immunotherapy with low-to moderate-dose radiation (via EBRT or TRT) targeting all sites of disease for the purpose of immunomodulation may help overcome local immunosuppression in established tumors while also taking advantage of a systemic antitumor immune response from ISV to eradicate sites of metastatic disease. There is also potential to further potentiate the systemic antitumor effect of ISV with other immunotherapies including checkpoint blockade, adoptive cell transfer, and other antibody-directed therapies.41 42 Notably, we reported cooperative therapeutic efficacy for anti-PD-1 and anti-CTLA-4 immune checkpoint blockade and 90-Y-NM600 in murine tumor models.31
Addition of 12 Gy EBRT or 100 µCi TRT to a distant site with primary tumor RT/IC increased CD45+ immune cells and CD8+ cytotoxic T cells in primary and secondary tumors, and increased NK cells in secondary tumors compared with single or no treatment controls (figures 4 and 5). These alterations in the TME are consistent with prior studies conducted in this B78 model.31 38 We also observed significantly decreased MDSCs in the secondary tumors of mice treated with 12 Gy and RT/IC compared with those getting RT/IC alone. This effect was not seen in secondary tumors treated with 100 µCi of TRT and may suggest a dose-rate effect or that higher biologically effective doses of radiation may help in overcoming distant tumor immunosuppression mediated by myeloid cells such as MDSCs or M2 macrophages. Unirradiated secondary tumors in mice treated with RT/IC had significantly lower effector immune cell infiltrate than irradiated secondary tumors in mice treated with RT/IC+12 Gy. However, the secondary tumors in mice treated with RT/IC did demonstrate significantly increased effector cell and Treg populations compared with PBS controls. These findings support our prior observation that local RT/IC does indeed result in a systemic immune response that can be observed at distant sites of disease, which may be augmented and/or unmasked to achieve a clinical effect. Future studies will explore the use of other immunomodulatory agents that synergize with TRT, namely, immune checkpoint blockade antibodies, combined with ISV, to further overcome the suppressive collective TME.31
While we did not directly compare the two modes of radiation (EBRT and TRT), our findings suggested greater secondary tumor response with 12 Gy of RT vs 100 µCi of TRT when combined with RT/IC. This may be due in part to differences in the radiobiological efficacy of absorbed dose, as 100 µCi is equivalent to 4–6 Gy of radiation delivered over 25 days compared with 12 Gy delivered over 2–3 min. Interestingly, we did find that addition of TRT to RT/IC significantly slowed estimated growth rates at the primary tumor, suggesting that a higher dose of radiation to a primary tumor may enhance local control of our RT/IC regimen, or that TRT-mediated systemic immunomodulation improves local ISV response, as is the case observed with 12 Gy of EBRT. Additional studies comparing these RT modalities are currently underway.
Our study does have several limitations. Our findings were demonstrated in a single syngeneic transplantable tumor model, which is expected to exhibit TME differences compared with spontaneously arising heterogeneous human tumors. Additionally, our flow cytometric analyses were performed at a single time point (day 14 after RT) based on previous understanding of peak immune activation kinetics from a robust endogenous immune response, as opposed to a non-specific stimulation from the therapy itself. Nevertheless, the immune TME is dynamic, and additional studies are under way to evaluate the kinetics of immune activation following ISV treatment. While antigen-specific adaptive antitumor immunity is generated by RT+IT IC when combined with radiation to distant tumor sites, the identity of recognized tumor antigens is not known. This limits our ability to determine whether the effects of RT on distant tumor are selective to tumor-specific immune cells. Notably, we previously demonstrated that T cells were required for the ISV effect of RT+IT IC, but NK cells were not.9 In future studies, we will evaluate the role of these and other immune populations in the immunomodulatory effects of RT. Finally, while our findings establish that immunomodulatory RT to distant sites of disease can enhance the systemic ISV effects of local RT/IC, tumor progression and immune escape do occur in some treated mice. Studies examining differences between responding, non-responding, and late progressing mice are under way and will be helpful in making further improvements to this treatment paradigm.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
Acknowledgments
The authors thank the UWCCC Flow Cytometry Core for assistance in data collection, panel construction, and data analysis for the flow cytometry data in this article, especially Dagna Sheerar, Kathryn Fox, and Lauren Nettenstrom. The authors also thank the University of Wisconsin Small Animal Imaging and Radiotherapy Facility, particularly Justin Jeffery and Ashley Weichmann, for their assistance in handling, imaging, and disposing of the radioactive mice; Drs Jacquelyn Hank, Taylor Aiken, and Vladimir Subbotin and Ms Anna Hoefges for their thoughtful discussion of project goals, experimental design, experimental troubleshooting, and collection of mouse tissue; Dr Stephen D Gillies for creating the hu14.18-IL2 immunocytokine and for its provision and that of other reagents, and for years of helpful collaboration and advice in our team’s preclinical and clinical research with this agent.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @PeterCarlsonMD, @PiepsTweeets
Contributors PMC designed the experiments; conducted the primary preparation, mouse handling, and data collection for all studies; analyzed the data; generated figures; and drafted and edited the manuscript. RBP significantly contributed to the design, execution, and analysis of all experiments, and in drafting and editing the manuscript. JB conducted statistical modeling and analysis for tumor growth curves and survival outcomes. MR, CS, AMB, ASF, and AAP assisted in mouse handling, experiment execution, and data collection. AAP also contributed to manuscript review, editing, and formatting. AKE contributed to experiment design, mouse handling, and data collection. IM, JG, RH, and JPW provided the NM-600 TRT agent, chelated with radioactive isotope, collected PET/CT images, and conducted Monte Carlo-based dosimetry calculations. ALR contributed to the experimental design and manuscript review. PMS and ZSM contributed to experimental design, project oversight, analysis of the data, and revision of the manuscript drafts. PMC, PMS, and ZSM share responsibility for the overall content as guarantors.
Funding This work was supported by Midwest Athletes Against Childhood Cancer (N/A), Stand Up 2 Cancer (N/A), the St. Baldrick’s Foundation (N/A); the Crawdaddy Foundation (N/A), and the University of Wisconsin Carbone Cancer Center. This research was also supported in part by public health service grants (TR002373, U54-CA232568, R35-CA197078, 5K08CA241319, P01 CA250972, and U01 CA233102) from the National Cancer Institute; and 1DP5OD024576 from the National Institutes of Health and the Department of Health and Human Services. PMC was supported by National Cancer Institutes of the NIH award (F30CA228315), NIH award (TL1 TR002375), and NIH award (T32 GM140935). Shared resource flow cytometry reagents and equipment are funded by the University of Wisconsin Carbone Cancer Center (support grant P30 CA014520). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests ZSM, JPW, RH, and JG have financial interests in Archeus Technologies. ZSM is a member of the Scientific Advisory Boards for Archeus Technologies and for Seneca Therapeutics. PMS is an unpaid medical advisor for Invenra. JPW is a cofounder, CSO, and director of Archeus Technologies, which holds the licence rights to NM600-related technologies. BPB and JG are cofounders of Voximetry, and BPB is the CSO. The following patents have been applied for or filed by the University of Wisconsin Alumni Research Foundation: US Patent 10,736,949, 'Radiohalogenated agents for in situ immune modulated cancer vaccination', with ZSM, PMS, JPW, and BPB as inventors; US Patent 10,751,430, 'Targeted radiotherapy chelates for in situ immune modulated cancer vaccination' with ZSM, PMS, JPW, BPB, and PMC as inventors; application no. 15/809,427, 'Using targeted radiotherapy to drive anti-tumor immune response to immunotherapies', ZSM, PMS, JPW, PMC, JG, RBP, and RH as inventors; and US 2011/0060602, 'A1 treatment planning system for radiopharmaceuticals', with BPB and JG as inventors.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.