Article Text

Abstract
Background Anti-GD2 monoclonal antibody immunotherapy has significantly improved the overall survival rate for high-risk neuroblastoma patients. However, 40% of patients fail to respond or develop resistance to treatment, and the molecular mechanisms by which this occurs remain poorly understood. Tumor-derived small extracellular vesicles (sEVs) have emerged as critical regulators in modulating the response to immunotherapy. In this study, we investigated the role of neuroblastoma-derived sEVs in promoting resistance to the anti-GD2 monoclonal antibody dinutuximab. Moreover, to determine whether pharmacologic inhibition of sEV secretion sensitizes tumors to dinutuximab treatment, we combined dinutuximab with tipifarnib, a farnesyltransferase inhibitor that inhibits sEV secretion.
Methods We investigated the role of neuroblastoma-derived sEVs in modulating the response to dinutuximab by utilizing the syngeneic 9464D-GD2 mouse model. The effect of neuroblastoma-derived sEVs in modulating the tumor microenvironment (TME) and host immune system were evaluated by RNA-sequencing and flow cytometry. Importantly, we used this mouse model to investigate the efficacy of tipifarnib in sensitizing neuroblastoma tumors to dinutuximab. The effect of tipifarnib on both the TME and host immune system were assessed by flow cytometry.
Results We demonstrated that neuroblastoma-derived sEVs significantly attenuated the efficacy of dinutuximab in vivo and modulated tumor immune cell infiltration upon dinutuximab treatment to create an immunosuppressive TME that contains more tumor-associated macrophages and fewer tumor-infiltrating NK cells. In addition, we demonstrated that neuroblastoma-derived sEVs suppress splenic NK cell maturation in vivo and dinutuximab-induced NK cell-mediated antibody-dependent cellular cytotoxicity in vitro. Importantly, tipifarnib drastically enhanced the efficacy of dinutuximab-mediated inhibition of tumor growth and prevented the immunosuppressive effects of neuroblastoma-derived sEVs in vivo.
Conclusions These preclinical findings uncover a novel mechanism by which neuroblastoma-derived sEVs modulate the immune system to promote resistance to dinutuximab and suggest that tipifarnib-mediated inhibition of sEV secretion may serve as a viable treatment strategy to enhance the antitumor efficacy of anti-GD2 immunotherapy in high-risk neuroblastoma patients.
- Neuroblastoma
- Drug Therapy, Combination
- Immunotherapy
- Tumor Microenvironment
Data availability statement
Data are available in a public, open access repository. The RNA-sequencing data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE189921.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known on this topic
Tumor-derived small extracellular vesicles (sEVs) have immunosuppressive functions and modulate the response to immunotherapy.
What this study adds
This study identifies an immunosuppressive tumor microenvironment as a novel mechanism of resistance to anti-GD2 immunotherapy mediated by neuroblastoma-derived sEVs and identifies tipifarnib as a promising adjunct to dinutuximab, which could be translated to the clinic to improve patient outcomes.
Introduction
Neuroblastoma is the most common extracranial solid tumor in children, accounting for approximately 6% of all pediatric malignancies and more than 10% of childhood cancer-related deaths.1 The standard treatment regimen for patients with high-risk neuroblastoma includes multiagent chemotherapy, surgery, autologous stem cell transplantation, radiotherapy, and maintenance therapy.2 Despite multimodal treatment, the 5-year overall survival rate for patients with high-risk disease is only around 50%.3
The recent incorporation of dinutuximab and immunostimulatory agents (granulocyte-macrophage colony-stimulating factor and interleukin-2) into maintenance therapy for patients with high-risk neuroblastoma has substantially improved patient outcomes.4 Dinutuximab (ch14.18) is a chimeric monoclonal antibody against the disialoganglioside GD2, which is expressed on the outer leaflet of the plasma membrane of peripheral neurons, skin melanocytes, and the central nervous system, and is ubiquitously present on tumors of neuroectodermal origin, including most neuroblastomas.5–7 Tumor-bound anti-GD2 antibodies recruit immune effector cells to trigger Fc-receptor-mediated killing by both complement-mediated cytotoxicity and antibody-dependent cell-mediated cytotoxicity (ADCC).4 Due to the specificity of GD2 for tumors of neuroectodermal origin, recent studies have investigated the use of anti-GD2 CAR-NKT and CAR-T cell therapies for patients with refractory neuroblastoma.8 9 However, despite its relative clinical success, more than 40% of neuroblastoma patients fail to respond or develop resistance to anti-GD2 therapy.10 Moreover, although anti-GD2 immunotherapy is highly effective against minimal residual disease, it has limited efficacy for targeting solid tumors.10 However, the mechanisms underlying therapeutic failure and resistance to anti-GD2 immunotherapy remain unknown.10 11
Small extracellular vesicles (sEVs) have recently emerged as critical regulators of tumor growth, metastasis and cancer progression.12 These 30–150 nm vesicles are secreted by almost all cell types through outward budding of the plasma membrane or direct fusion of multivesicular bodies with the plasma membrane. Notably, sEVs contain biologically active molecules capable of modulating the extracellular environment and immune system.13 Recent studies have found that tumor-derived sEVs play an important role in promoting resistance to immunotherapy by interacting with immune effector cells and suppressing the host immune system.14 15 NK cells, which express the receptor FcgRIIIa (CD16), are the major effector cells for anti-GD2 immunotherapy and use ADCC to target neuroblastoma cells.16 Tumor-derived sEVs attenuate ADCC in vitro by inhibiting the binding of antibodies to tumor cells.17 Moreover, tumor-derived sEVs have been shown to dysregulate NK cell function and induce NK cell exhaustion.18 19 However, whether tumor-derived sEVs regulate resistance to anti-GD2 monoclonal antibody immunotherapy in vivo remains unclear.
In this study, we use a well-characterized preclinical mouse model of neuroblastoma to reveal that neuroblastoma-derived sEVs induce resistance to anti-GD2 immunotherapy. We show that neuroblastoma-derived sEVs modulate the systemic immune response and alter tumor immune cell infiltration following dinutuximab treatment to establish an immunosuppressive TME and promote evasion of dinutuximab-induced cytotoxicity. Importantly, we identify tipifarnib, an FDA-approved farnesyltransferase inhibitor shown to inhibit sEV secretion, as a novel agent that enhances the efficacy of dinutuximab and prevents the development of sEV-induced immunosuppression. Taken together, our preclinical results provide a promising new treatment option that can be rapidly translated to the clinic to improve outcomes for high-risk neuroblastoma patients.
Materials and methods
Cell lines and plasmids
The mouse 9464D neuroblastoma cell line was a gift from Dr Paul Sondel (University of Wisconsin, Madison, Wisconsin, USA). Human IMR32 neuroblastoma (CCL-127), HEK 293T/17 (CRL-11268), and NK92-EGFP-CD16 (PTA-8836) cell lines were purchased from ATCC. Neuroblastoma and HEK 293T/17 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Corning, 10–013-CV) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich F2442) and 1% antibiotic-antimycotic (Corning, 30–004 CI). NK-92-EGFP-CD16 cells were cultured in RPMI 1640 (Corning, 10–040-CV) containing 10% horse serum (Equitech-Bio, SE30-0100), 10% heat-inactivated FBS, and 100 units/mL IL-2 (BioLegend, 589104-BL). Cells were incubated at 37°C in a humidified chamber containing 5% CO2. All cell lines used in this study were thawed from frozen stock and maintained in cell culture for less than 6 months. Cells were periodically authenticated by mycoplasma testing, morphologic inspection, and STR analysis.
The pCDH1-CMV-MCS-SV40-Hygro construct was previously described.20 SFG.GD3 synthase (St8sia1)−2A-GD2 synthase (B4galnt1) was obtained from Martin Pule through Addgene (#75013). pCDH1-CMV-St8sia1-2A-B4galnt1-SV40-Hygro construct was generated by subcloning the PCR amplified (primer set: 5’- ATCCTCTAGACTGCCACCATGAG-3’, 5’- TAAATTCGAATCACTCGGCGGTCATGCACT-3’) St8sia1-2A-B4galnt1 cassette into the XbaI-BstBI site of pCDH1-CMV-MCS-SV40-Hygro. 9464D-GD2 cells were generated by transducing 9464D cells with lentiviral particles harboring pCDH1-CMV-St8sia1-2A-B4galnt1-SV40-Hygro followed by selection with hygromycin (400 µg/mL).
Drugs and antibodies
Dinutuximab (Unituxin®) was a gift from the Penn State Health Pharmacy (Hershey, Pennsylvania, USA). Tipifarnib (AdooQ, MedChemExpress) was dissolved in dimethylsulfoxide (DMSO) to create a 10 mM stock solution for in vitro use or suspended at 4 mg/mL in 20% w/v hydroxypropyl-β-cyclodextrin (Millipore Sigma, 3 32 607–100G) in distilled water, pH 2.5 for in vivo studies. The following antibodies were used for immunoblotting: Alix (Cell Signaling Technology, 3A9, 1:1000), CD63 (Abcam, ab217345, 1:1000), Calnexin (Abcam, ab22595, 1:1000), Tsg101 (GeneTex, 70255, 1:500), Rab27a (Cell Signaling Technology, 69 295S, 1:1000), β-Actin (Sigma, A5441, 1:10,000), and Golgin97 (Thermo Fisher, A-21270, 1:1000). Antibodies used for flow cytometry are included in online supplemental table S1.
Supplemental material
Small extracellular vesicles (sEVs) were isolated from conditioned cell culture medium according to a previously described differential ultracentrifugation method.21 Briefly, FBS was depleted of sEVs by centrifuging heat-inactivated FBS two times at 120,000 relative centrifugal force (RCF) for 12 hours at 4°C (Beckman, SW32Ti) followed by filtration of the supernatant through a 0.2 µm filter. Conditioned cell culture medium was collected from cells cultured for 24 hours in DMEM supplemented with 10% sEV-depleted FBS. The conditioned medium was centrifuged at 500 RCF for 10 min at 4°C (Beckman, SX4750A) to remove cells and large cell debris. The supernatant was filtered through a 0.2 µm syringe filter (VWR 28 145–501) and concentrated using a 100K molecular weight cutoff (MWCO) protein concentrator (Thermo Fisher, 88533). The concentrated supernatant was centrifuged at 10,000 RCF for 20 min at 4°C (Eppendorf, FA-45-30-11) to remove larger microvesicles and apoptotic bodies followed by centrifugation at 120,000 RCF for 4 hours at 4°C (Beckman, SW55Ti). The sEV-containing pellet was washed two times in ice-cold phosphate-buffered saline (PBS) and pelleted by centrifugation at 120,000 RCF at 4°C for 4 hours and 12 hours, respectively (Beckman, SW55Ti). The sEV-containing pellet was resuspended in PBS and stored at −20°C.
Animal experiments
All animal studies were performed according to guidelines established by the Institutional Animal Care and Use Committee (IACUC #PRAM201145989) at the Penn State College of Medicine (Hershey, Pennsylvania, USA). An immunocompetent mouse model of neuroblastoma was generated by subcutaneously injecting 1×106 9464D-GD2 cells in a 50:50 mixture of DMEM and Matrigel (Corning, 354234) into C57BL/6J mice (8–10-week-old, JAX 000664) with male and female mice represented at an equal ratio. One week following tumor cell inoculation, mice were randomized into treatment groups. Where indicated, mice were treated two times per week by tail-vein injection with PBS (100 µL), dinutuximab (25 µg in 100 µL PBS), purified sEVs from 9464D-GD2 cells (20 µg in 100 µL PBS), or the combination of dinutuximab and sEVs. Where indicated, tipifarnib (25 mg/kg) or an equivalent volume of vehicle was administered two times a day by oral gavage. Primary tumor growth was monitored by measuring tumor volume using calipers (volume = π(length*width2/6). At the experimental endpoint, mice were euthanized, and tumor, blood, spleen, and bone marrow (BM) were harvested for ex vivo analysis. Endpoint tumor volume was calculated by measuring tumors using calipers (volume = π(length*width*height)/6).
Flow cytometry
For cell surface staining, approximately 1×106 cells were stained with pre-mixed antibody cocktail panels (online supplemental table S1) in 100 µL fluorescence-activated cell sorting (FACS) buffer (1% FBS, 0.2% NaN3 in PBS) for 30 min on ice. Cells were washed two times with FACS buffer and fixed with 2% paraformaldehyde (Fisher Scientific, 50-980-487) for 15 min. Fixed cells were washed once with PBS and resuspended in 0.5 mL FACS buffer for flow cytometric analysis using an LSR II or Symphony flow cytometer (BD Biosciences). Compensation was performed using UltraComp eBeads™ compensation beads (Invitrogen). FlowJo v10 (FlowJo, LLC) software was used for data analysis.
In vitro NK cell-mediated ADCC assay
The coculture assay was adapted and modified from Barry et al.22 Briefly, neuroblastoma cells were seeded at 2×104 cells per well in a 96-well plate. The following day, the medium was removed, cells were washed in PBS, and 1×104 NK92-CD16-EGFP cells (effector:target ratio of 1:2) were added in RPMI 1640 containing 10% FBS, 100 units/mL IL2, and 0.5 µM YOYO-3 iodide (ThermoFisher, Y3606). Where indicated, NK92-CD16-EGFP cells were preincubated with neuroblastoma-derived sEVs (25 µg/mL) for 2 hours prior to addition, and dinutuximab was added to the co-culture at a concentration of 100 ng/mL. Images were taken at 1-hour intervals using the Incucyte S3 Live Cell Imaging System (Sartorius) and quantified using the Incucyte Cell-by-Cell Analysis Software Module (Sartorius).
Statistical analysis
GraphPad Prism (GraphPad Software) was used for statistical analysis. Two-tailed unpaired student t-tests were used for single comparisons. One-way analysis of variance with Tukey’s/Sidak’s posthoc tests were used for multiple comparisons. Statistical significance was set to p<0.05.
Results
Neuroblastoma-derived sEVs promote resistance to dinutuximab in vivo
The 9464D cell line is derived from a spontaneous neuroblastoma that arose in a TH-MYCN transgenic mouse on the C57BL/6 background, creating a genetically defined transplantable tumor model.23 24 While the complex, acidic ganglioside GD2 is highly expressed on most neuroblastoma, 9464D cells express a lower cell surface level of GD2 compared with other neuroblastoma cell lines.24 25 To establish a syngeneic model of neuroblastoma suitable for investigating mechanisms of resistance to anti-GD2 immunotherapy, murine GD3 synthase (St8sia1), the rate-limiting enzyme for GD2 biosynthesis, and GM2/GD2 synthase (B4galnt1) were stably overexpressed in 9464D cells to upregulate cell surface GD2 expression (9464D-GD2; online supplemental figure S1A). sEVs were isolated from 9464D-GD2 cells using a well-established differential ultracentrifugation protocol21 (online supplemental figure S1B) and were found to demonstrate the characteristic morphology, protein markers, and size distribution of sEVs (figure 1A–C).26 To determine how sEVs regulate tumor growth and resistance to anti-GD2 immunotherapy in vivo, immunocompetent C57BL/6 mice were subcutaneously inoculated with 1×106 9464D-GD2 cells. One-week post-inoculation, tumor-bearing mice began receiving two times per week tail-vein injections of PBS, dinutuximab, and/or sEVs isolated from 9464D-GD2 cells (hereafter denoted sEVs unless otherwise noted) and were monitored for tumor growth (figure 1D). As anticipated, dinutuximab dramatically suppressed tumor growth compared with PBS control, highlighting the efficacy of anti-GD2 immunotherapy against 9464D-GD2 tumors in vivo (figure 1E). Strikingly, while sEVs alone failed to alter tumor growth compared with control, mice treated with the combination of dinutuximab and sEVs displayed nearly complete resistance to the antitumor effect of dinutuximab (figure 1E). Ex vivo quantification of tumor volume and weight at the experimental endpoint confirmed that sEV treatment significantly suppressed the antitumor efficacy of dinutuximab but had no effect on 9464D-GD2 tumor growth when administered alone (figure 1F,G, online supplemental figure S1C).

Neuroblastoma-derived sEVs promote resistance to dinutuximab in vivo. (A) Representative electron microscopy image of sEVs isolated from 9464D-GD2 cells. Scale bar, 200 nm. (B) Immunoblot of positive and negative sEV markers in WCL and purified sEVs isolated from 9464D-GD2 cells. All lanes loaded with equal amount of protein. (C) Representative size distribution and concentration of sEVs isolated from 9464D-GD2 cells quantified by NTA. Mode 95.3 nm, concentration 4.69x108 (D) Experimental design. C57BL/6 mice were subcutaneously inoculated with 1×106 9464D-GD2 cells. One-week postinoculation, tumor-bearing mice began receiving tail-vein injections of PBS, dinutuximab (25 µg/mouse) and/or sEVs derived from indicated cell lines (20 µg) two times per week. On day 30, mice were sacrificed and tumors were harvested for analysis. (E) Quantification of tumor volume at indicated time points post-tumor inoculation. Mean±SEM, PBS, n=11; sEVs, n=10; dinutuximab, n=12; dinutuximab+9464D-GD2 sEVs, n=11. Data represent one experiment. (F) Quantification of tumor weight from indicated treatment groups on day 30. Mean±SEM, n as in (E). Two-tailed unpaired t-test. *p<0.05; **p<0.01. (G) Representative images of tumors from treatment groups as in (E) on day 30. Scale bar, 1 cm. (H) Quantification of tumor volume at indicated time points. Mean±SEM, n=8 per group. (I) Tumor weight from indicated treatment groups on day 30. Mean±SEM, n=8 per group. Two-tailed unpaired t-test. *p<0.05; **p<0.01; ***p<0.001. (J) Representative images of tumors from indicated treatment groups on day 30. Scale bar, 1 cm. sEV, small extracellular vesicle; WCL, whole cell lysate.
While the surface expression of GD2 was significantly upregulated in 9464D-GD2 cells, sEVs isolated from these cells showed a moderate increase in GD2 compared with control (online supplemental figure S1D). To investigate whether sEV-associated GD2 promoted resistance to dinutuximab by acting as an antibody decoy to facilitate tumor immune evasion, similar to the immunosuppressive mechanism exhibited by sEV-associated programmed death-ligand 1 (PD-L1), we repeated the in vivo experiment and included sEVs derived from both 9464D-GD2 and parental 9464D cell lines (figure 1H).14 Notably, mice treated with the combination of dinutuximab and either sEVs derived from 9464D parental or 9464D-GD2 cell lines demonstrated a comparable increase in tumor growth compared with mice treated with dinutuximab alone (figure 1H–J, online supplemental figure S1E), suggesting that in this experimental system, sEV-associated GD2 is not the primary factor involved in mediating resistance to anti-GD2 immunotherapy.
Neuroblastoma-derived sEVs suppress dinutuximab-induced NK cell tumor infiltration and enhance the recruitment of tumor-associated macrophages
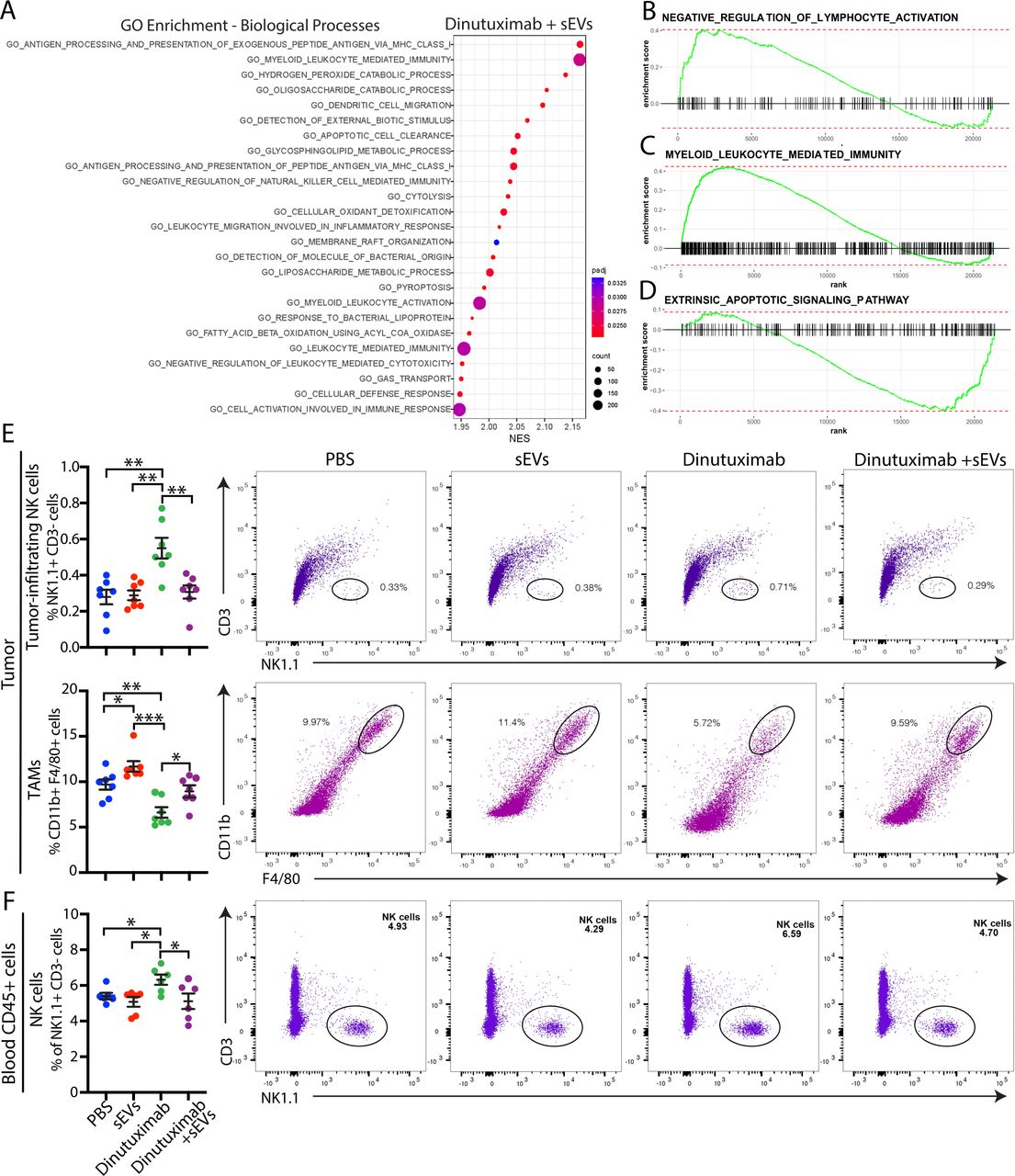
To elucidate the transcriptome changes underlying sEV-mediated resistance to dinutuximab, we performed RNA sequencing analysis of 9464D-GD2 tumors isolated from mice treated with either dinutuximab or dinutuximab plus sEVs (online supplemental figure S2A–C). Tumors derived from mice treated with the combination of dinutuximab and sEVs demonstrated a significant upregulation in genes involved in myeloid leukocyte-mediated immunity and myeloid cell recruitment, including Ccr2,27 Clec10a,28 and Clec1a29 (online supplemental figure S2D). To further investigate how sEVs mediate changes to the tumor TME, we performed gene set enrichment analysis (GSEA). Intriguingly, pathways involved in myeloid leukocyte-mediated immunity, as well as negative regulation of NK cell-mediated immunity and cytotoxicity, were among the top 20 most significantly enriched pathways in tumors derived from mice treated with dinutuximab plus sEVs (figure 2A, online supplemental tables S2 and S3). Using GSEA of gene sets of interest, we confirmed that sEV treatment induced enrichment of genes involved in myeloid leukocyte-mediated immunity as well as negative regulation of lymphocyte activation (figure 2B,C). In contrast, genes involved in extrinsic apoptotic signaling were less enriched in tumors treated with dinutuximab and sEVs compared with dinutuximab alone (figure 2D).
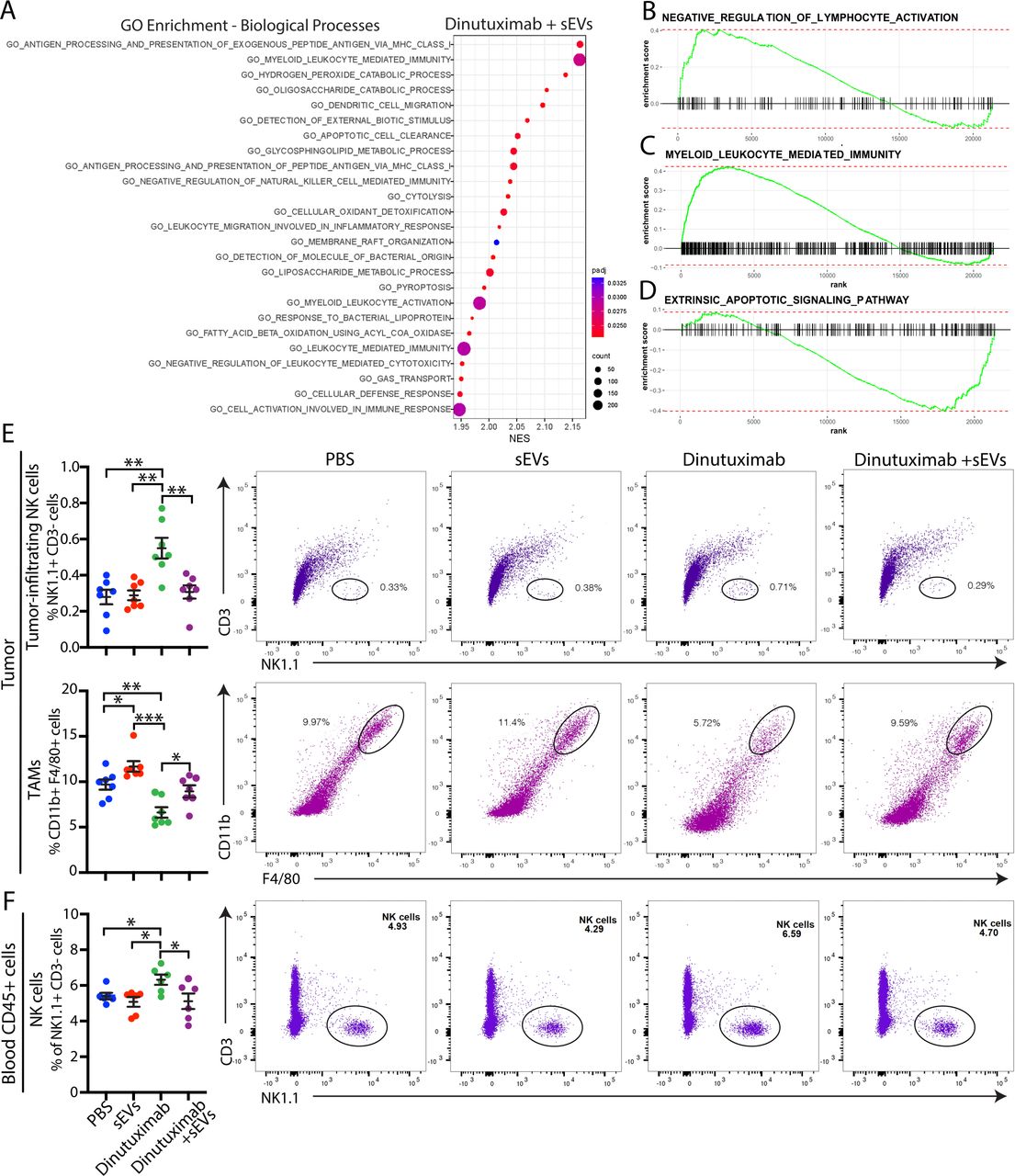
Neuroblastoma-derived sEVs suppress dinutuximab-induced NK cell tumor infiltration and enhance the recruitment of tumor-associated macrophages. (A) Top 20 biological processes identified by GO enrichment analysis of 9464D-GD2 tumors treated with the combination of dinutuximab and sEVs compared with tumors treated with dinutuximab alone, as described in online supplemental figure S2A. (B,C) Select GSEA enrichment plots of clusters enriched in tumors treated with dinutuximab plus sEVs: (B) negative regulation of lymphocyte activation and (C) myeloid leukocyte mediated immunity. (D) GSEA enrichment plot for the extrinsic apoptotic signaling pathway. (E,F) C57BL/6 mice were inoculated with 9464D-GD2 cells and treated as described in figure 1D. (E) Quantification and representative flow cytometry plots of tumor-infiltrating NK cells (NK1.1+CD3-; top panel) and TAMs (CD11b+F4/80+; bottom panel) in 9464D-GD2 tumors isolated from mice in indicated treatment groups on day 30. Mean±SEM, n=7 per group. Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. (F) Quantification and representative flow cytometry plots of NK cells (NK1.1+CD3-) isolated from the blood of tumor-bearing mice on day 30 in indicated treatment groups. Mean±SEM, n=7 per group. Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. GSEA, gene set enrichment analysis; sEV, small extracellular vesicle; TAM, tumor-associated macrophage.
As the composition of tumor-infiltrating immune cells correlates with sensitivity to immunotherapy, we hypothesized that neuroblastoma-derived sEVs alter the tumor immune microenvironment. To test this hypothesis, we performed flow cytometry to analyze immune cell populations in tumors derived from mice treated with PBS, dinutuximab and/or sEVs (as in figure 1). We were specifically interested in how sEVs affected NK cell tumor infiltration, as patients with tumors that express high levels of the NK cell marker CD56 or NK cell activating receptor NKG2D demonstrate an increased event-free 5-year survival probability compared with patients with low expression of these markers (online supplemental figure S2E,F, see online supplemental table S4 for patient characteristics). We found that treatment with dinutuximab significantly increased the tumor-infiltrating NK cell population compared with control or sEVs alone (figure 2E, see online supplemental figure S3 for gating strategy). Additionally, mice treated with dinutuximab displayed a significant increase in the percentage of serum NK cells compared with control or sEV treatment groups (figure 2F). Strikingly, sEVs significantly suppressed dinutuximab-induced NK cell mobilization and tumor infiltration to a level comparable to control (figure 2E–F).
Myeloid cells also play important roles in regulating neuroblastoma response to immunotherapy, as they create an immunosuppressive TME that suppresses T cell and NK cell proliferation and cytotoxicity.3 As our RNA sequencing results implicated myeloid cells in sEV-mediated resistance to dinutuximab, we next analyzed tumors for the presence of tumor-associated macrophages (TAMs). Dinutuximab significantly decreased the TAM population compared with all other treatment conditions (figure 2E). Interestingly, tumors derived from mice treated with sEVs alone demonstrated an upregulation in the percentage of TAMs, while those derived from mice treated with the combination of dinutuximab and sEVs demonstrated a percentage of TAMs comparable to control (figure 2E). Collectively, these results suggest that neuroblastoma-derived sEVs induce resistance to dinutuximab by promoting an immunosuppressive TME characterized by a decrease in tumor-infiltrating NK cells and an increase in TAMs.
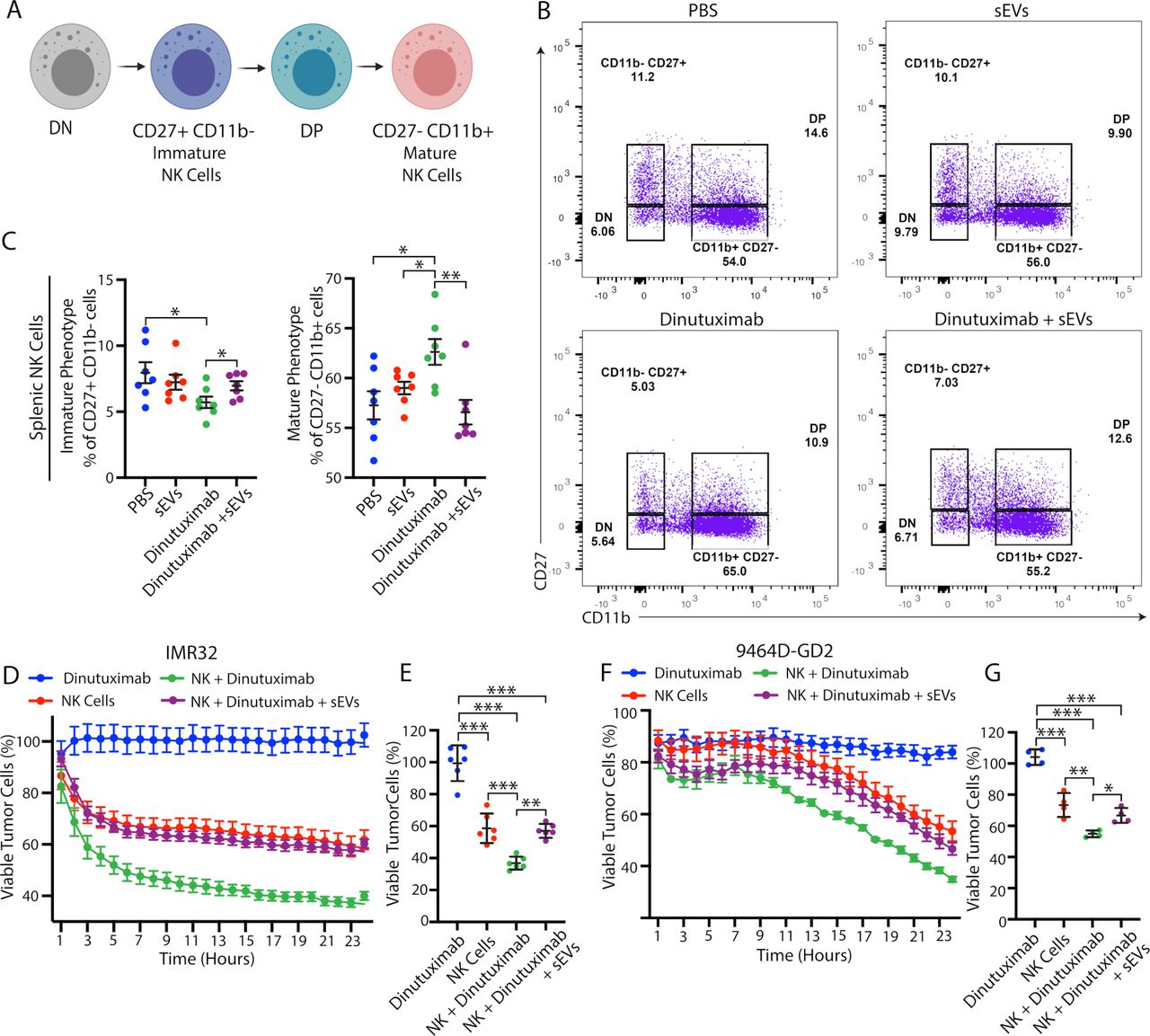
Neuroblastoma-derived sEVs modulate NK cell maturation in vivo and NK cell-mediated ADCC in vitro
To determine how sEVs promote an immunosuppressive TME, we next asked where they are primarily taken up in vivo. We found that Vybrant DiD-labeled sEVs demonstrated significant uptake in the lungs, liver, and spleen within the first 6 hours after tail vein injection (online supplemental figure S4). To determine whether splenic uptake of neuroblastoma-derived sEVs alters immune cell maturation, we performed flow cytometry analysis on spleens isolated from 9464D-GD2 tumor-bearing mice treated with PBS, dinutuximab and/or sEVs, as in figure 1.30 Since NK cells are the main immune effector cells that respond to anti-GD2 immunotherapy, we were particularly interested in how sEVs modulate NK cell subpopulations and maturation.3 We used CD27 and CD11b to subdivide NK cells into four different maturation subsets and focused on both immature CD27+CD11b- cells and mature CD27- CD11b+ cells (figure 3A,B).31 32 Interestingly, dinutuximab significantly decreased the percentage of immature splenic NK cells and increased the percentage of mature splenic NK cells compared with all other treatment groups, suggesting that dinutuximab promotes NK cell maturation in vivo (figure 3C). Notably, spleens isolated from mice treated with the combination of dinutuximab and sEVs demonstrated NK cell percentages comparable to control levels, suggesting that sEVs suppress dinutuximab-induced NK cell maturation in vivo (figure 3C).

Neuroblastoma-derived sEVs modulate NK cell maturation in vivo and NK cell-mediated ADCC in vitro. (A) Schematic of NK cell subpopulations. Created with BioRender.com. (B) Representative flow cytometry plots of splenic NK cell subpopulations isolated from 9464D-GD2 tumor-bearing mice receiving the indicated treatments as described in figure 1D. (C) Quantification of the percentage of immature splenic NK cell (CD27 +CD11b- ; left panel) and mature NK cell (CD27- CD11b+; right panel) subpopulations in 9464D-GD2 tumor-bearing mice treated as described in figure 1D. Mean±SEM, n=7 per group. Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. (D–G) Human (IMR32) or murine (9464D-GD2) neuroblastoma cells treated as indicated in the presence or absence of NK92-CD16-EGFP cells (NK) and monitored for viability utilizing cell impermeant nucleic acid stain YOYO-3 with the IncuCyte S3 Live-Cell Analysis System. The cell-by-cell analysis module was used to quantify viable tumor cells (YOYO3- EGFP-). (D) Kinetic analysis of the IMR32 in vitro NK-cell-mediated ADCC assay. Mean±SEM, n=6. Viable cells calculated as percentage of viable cells in each treatment condition divided by viable cells in untreated control group. (E) Percentage of viable IMR32 cells at 24 hours, n=6. Mean±SD. One-way ANOVA with Tukey’s/Sidak’s post hoc tests. **p<0.01; ***p<0.001. (F) Kinetic analysis of the 9464D-GD2 in vitro NK-cell-mediated ADCC assay. Mean±SEM, n=4. Viable cells calculated as percentage of viable cells in each treatment condition divided by viable cells in untreated control group. (G) Percentage of viable 9464D-GD2 cells at 24 hours, n=4. Mean±SD. One-way ANOVA with Tukey’s/Sidak’s post hoc tests. *p<0.05; **p<0.01; ***p<0.001. ADCC, antibody-dependent cell-mediated cytotoxicity; ANOVA, analysis of variance; DN, double negative; DP, double positive; sEV, small extracellular vesicle.
To determine whether neuroblastoma-derived sEVs directly suppress NK cell-mediated ADCC, we next performed an in vitro co-culture experiment using the human NK92 cell line stably expressing CD16 and EGFP (NK92-CD16-EGFP) with either 9464D-GD2 cells or the GD2-positive human neuroblastoma cell line IMR32 (online supplemental figure S5A,B). NK cell-mediated ADCC was monitored in the presence or absence of dinutuximab and sEVs using the cell impermeant nucleic acid stain YOYO-3 and the IncuCyte S3 live-cell analysis system. As expected, the cytotoxic effects of dinutuximab required the presence of NK92-CD16-EGFP immune effector cells, while preincubation of NK92-CD16-EGFP cells with sEVs suppressed dinutuximab-induced NK cell-mediated ADCC against both human and mouse neuroblastoma cell lines (figure 3D–G; online supplemental figure S5C,D). Taken together, these data suggest that sEVs induce resistance to dinutuximab by both modulating NK cell maturation in vivo and suppressing NK cell-mediated ADCC.
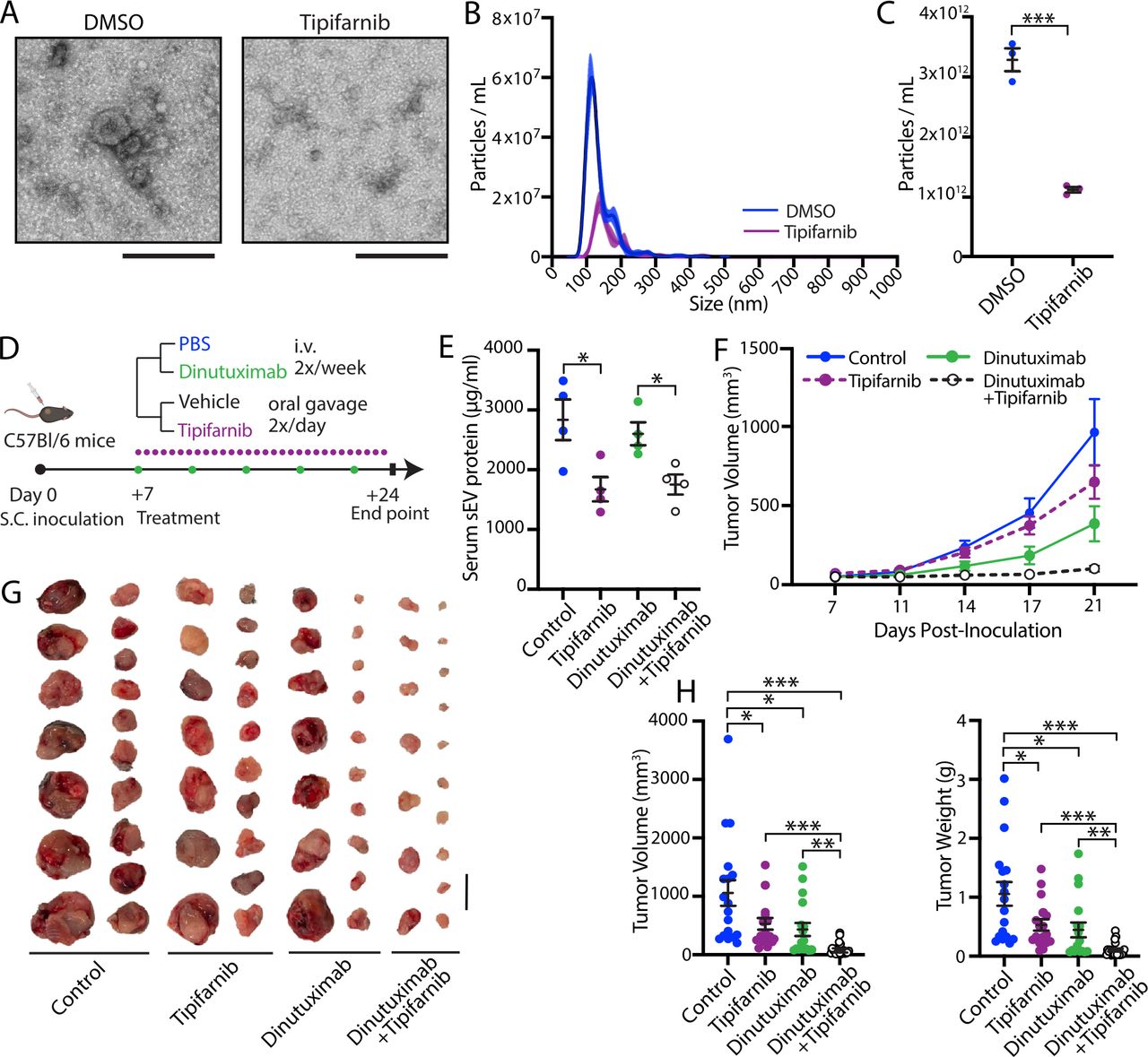
Inhibition of sEV secretion by tipifarnib suppresses neuroblastoma growth by sensitizing 9464D-GD2 tumors to dinutuximab. (A) Representative electron microscopy images of sEVs isolated from 9464D-GD2 cells treated with DMSO or 0.1 µM tipifarnib for 48 hours. Scale bar, 200 nm. (B,C) NTA analysis of sEVs isolated from 9464D-GD2 cells treated with DMSO or 0.1 µM tipifarnib for 48 hours. (B) Representative size distribution and particle concentration. (C) Quantification of particle concentration. Mean±SD, n=4. Student’s t-test. ***p<0.001. (D) Experimental design for E–H. C57BL/6 mice were subcutaneously inoculated with 1×106 9464D-GD2 cells. One-week post-inoculation, tumor-bearing mice began receiving tail-vein injections of PBS or dinutuximab (25 µg/mouse) two times per week combined with oral administration of vehicle or tipifarnib (25 mg/kg/half day) two times per day. On day 24, mice were sacrificed and tumors were harvested for analysis. (E) Quantification of sEV protein content isolated from serum of mice receiving indicated treatments. Mean±SEM, n=4 per group. Student’s t-test. *p<0.05. (F) Quantification of tumor volume at indicated time points. Mean±SEM, n=17 per treatment group. Data represent two independent experiments. (G) Representative images of tumors from indicated treatment groups on day 24. Scale bar, 1 cm. (H) Quantification of tumor volume and weight from indicated treatment groups on day 24. Mean±SEM, n=17 per treatment group. Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. Data represent two independent experiments. sEV, small extracellular vesicle.
Inhibition of sEV secretion by tipifarnib suppresses neuroblastoma growth by sensitizing tumors to dinutuximab
After determining that neuroblastoma-derived sEVs induce resistance to anti-GD2 immunotherapy through modulation of the immune system, we next sought to determine whether inhibition of sEV secretion would resensitize tumors to dinutuximab. We first blocked sEV secretion through genetic depletion of Rab27a, an essential Rab GTPase involved in sEV secretion.26 As expected, loss of Rab27a in 9464D cells dramatically suppressed sEV secretion in vitro (9464D-crRab27a, online supplemental figure S6A,B). However, despite no difference in proliferation in vitro compared with parental cells, loss of Rab27a dramatically suppressed 9464D tumor growth in vivo (online supplemental figure S6C–E), which prevented us from using this genetic model to further dissect the role of sEVs in dinutuximab resistance.
We next sought to use a pharmacologic approach to inhibit sEV secretion through treatment with tipifarnib. Tipifarnib is a potent, selective, and orally bioavailable inhibitor of farnesyltransferase that was recently shown to selectively inhibit sEV secretion from cancer cell lines in vitro.33 As tipifarnib inhibits additional cellular pathways, including RAS signaling, we established that a dose of 0.1 µM significantly suppressed sEV secretion in 9464D-GD2 cells in the absence of cytotoxicity (figure 4A–C, online supplemental figure S7A,B).
To determine whether tipifarnib-mediated inhibition of sEV secretion sensitizes neuroblastoma tumors to dinutuximab in vivo, we subcutaneously inoculated C57BL/6 mice with 1×106 9464D-GD2 cells. One-week post-inoculation, tumor-bearing mice began receiving two times a week tail-vein injections of PBS or dinutuximab in combination with tipifarnib or an equivalent volume of vehicle by oral gavage two times per day (figure 4D). Mice treated with either tipifarnib or the combination of dinutuximab and tipifarnib demonstrated a significant decrease in circulating sEV concentration compared with mice treated with either vehicle control or dinutuximab alone, as determined by measurement of serum sEV protein and NTA analysis, confirming that tipifarnib suppresses sEV secretion in vivo (figure 4E, online supplemental figure S7C,D). Interestingly, mice treated with tipifarnib alone demonstrated a reduction in tumor volume and weight compared with vehicle control, suggesting that tipifarnib suppresses tumor growth in vivo through inhibition of sEV secretion and/or other cellular mechanisms (figure 4F–H). Critically, combination treatment with tipifarnib and dinutuximab significantly enhanced the antitumor efficacy of dinutuximab, resulting in a substantial reduction in tumor growth throughout the duration of the experiment compared with single treatment with either agent (figure 4F). At the experimental endpoint, tumors isolated from mice treated with the combination of dinutuximab and tipifarnib demonstrated a significant reduction in weight and volume compared with tumors from mice in all other treatment groups, suggesting that the combination treatment exerts a synergistic antitumor effect (figure 4G,H).
Combination treatment with tipifarnib and dinutuximab suppresses neuroblastoma growth through remodeling of the TME and inhibition of sEV-induced immune suppression
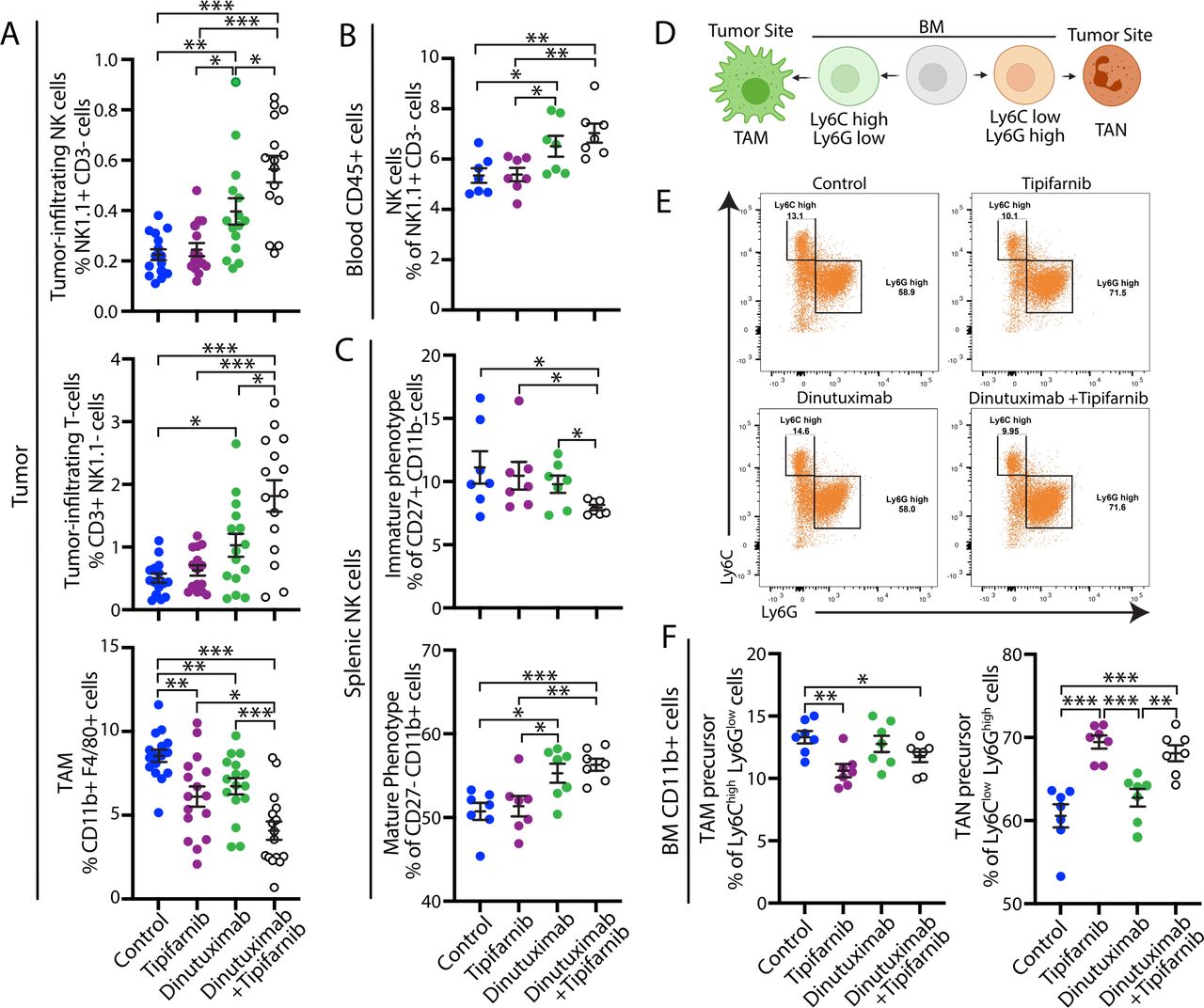
To examine whether tipifarnib sensitizes tumors to dinutuximab by preventing sEV-induced immunosuppression, we analyzed the immune cell composition of 9464D-GD2 tumors and blood isolated from mice treated with vehicle, dinutuximab and/or tipifarnib, as in figure 4D. Consistent with above data, tumors derived from mice treated with dinutuximab demonstrated an immunoreactive TME characterized by enhanced mobilization of NK cells, increased percentages of tumor-infiltrating NK cells and T-cells, and decreased TAMs (figure 5A). Combination treatment with tipifarnib and dinutuximab led to an increase in the percentage of circulating NK cells, as well as the percentage of tumor-infiltrating NK cells and T-cells compared with control or either treatment alone (figure 5A,B), suggesting that tipifarnib and dinutuximab work synergistically to prevent the development of an immunosuppressive TME. Interestingly, while sEV treatment alone upregulated the percentage of TAMs compared with dinutuximab or control (figure 2E), tipifarnib treatment decreased the percentage of TAMs compared with control, and tumors derived from mice treated with the combination of tipifarnib and dinutuximab demonstrated a significant reduction in TAMs compared with all other conditions, suggesting that tumor-derived sEVs promote the recruitment of immunosuppressive macrophages to the TME (figure 5A).
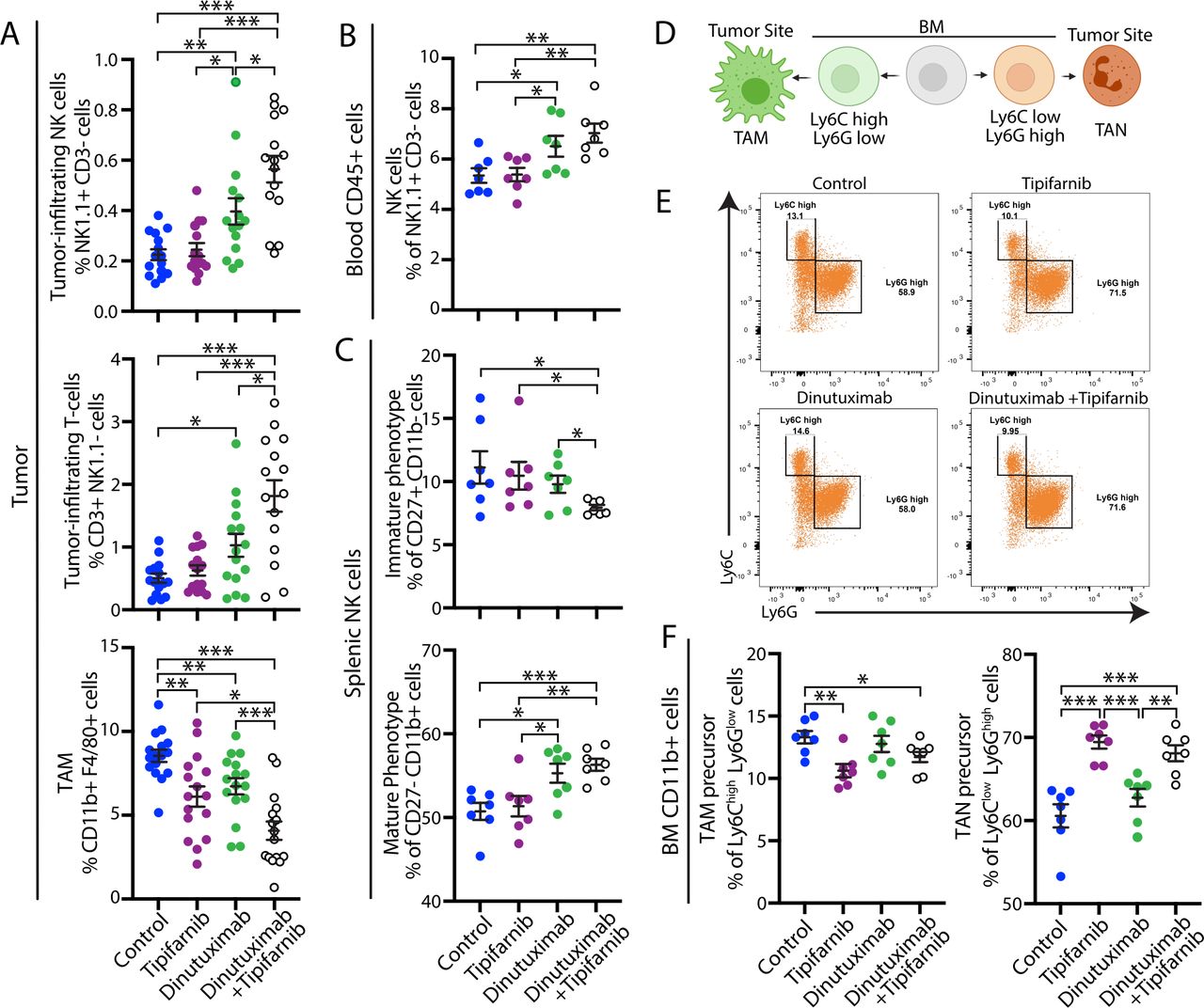
Combination treatment with tipifarnib and dinutuximab suppresses neuroblastoma growth through remodeling of the tumor microenvironment and inhibition of sEV-induced immune suppression. (A) Quantification of tumor-infiltrating NK cells (NK1.1+CD3-; n=15; top panel), tumor-infiltrating T-cells (CD3+NK1.1-; n=15; middle panel) and TAMs (CD11b+F4/80+; n=17; bottom panel) in 9464D-GD2 tumors isolated from mice in the indicated treatment groups on day 24 as described in figure 4D. Mean±SEM. Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. Data represent two independent experiments. (B) Quantification of NK cells (NK1.1+/CD3-) isolated from the blood of tumor-bearing mice in the indicated treatment groups on day 24 as described in figure 4D. Mean±SEM, n=7. Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. Data represent one experiment. (C) Quantification of the percentage of immature NK cell (CD27+CD11b-; top panel) and mature NK cell (CD27- CD11b+; bottom panel) subpopulations in splenic NK cells isolated from tumor-bearing mice receiving the indicated treatments on day 24 as described in figure 4D. Mean±SEM, n=7. Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. (D) Schematic of myeloid cell populations. Image created with BioRender.com. (E) Representative flow cytometry plots of the CD11b+cell subpopulations in BM samples isolated from tumor-bearing mice receiving the indicated treatments as described in figure 4D. (F) Quantification of the percentage of Ly6Chigh Ly6Glow TAM precursor (left panel) and Ly6Clow Ly6Ghigh TAN precursor (right panel) subpopulations in BM CD11b+cells isolated from mice on day 24 as described in figure 4D. Mean±SEM, n=7. Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. BM, bone marrow; sEV, small extracellular vesicle; TAM, tumor-associated macrophage; TAN, tumor-associated neutrophil.
To determine whether tipifarnib prevents sEV-induced systemic immune suppression, we next examined NK cell subsets in the spleen and myeloid cell subpopulations in the BM. We found that both dinutuximab and the combination of dinutuximab and tipifarnib treatment significantly increased the percentage of mature NK cells in the spleen compared with control or tipifarnib alone (figure 5C). Moreover, combination treatment decreased the percentage of immature splenic NK cells compared with other treatment groups (figure 5C). Recent literature revealed that TAMs are primarily derived from BM rather than splenic progenitor cells34 and arise from the CD11b+Ly6Chigh population of circulating mouse monocytes in grafted tumors.35 As tipifarnib decreased the percentage of TAMs, we next investigated whether treatment also altered immature myeloid cell populations in the BM.36 Progenitor cells that differentiate toward TAMs are characterized as CD11b+Ly6ChighLy6Glow, while cells that differentiate toward tumor-associated neutrophils are CD11b+Ly6ClowLy6Ghigh (figure 5D).36 Tipifarnib treatment significantly decreased the percentage of CD11b+Ly6ChighLy6Glow myeloid cells in the BM and increased the percentage of CD11b+Ly6ClowLy6Ghigh myeloid cells in both the presence and absence of dinutuximab (figure 5E,F, gating strategy in online supplemental figure S3D).
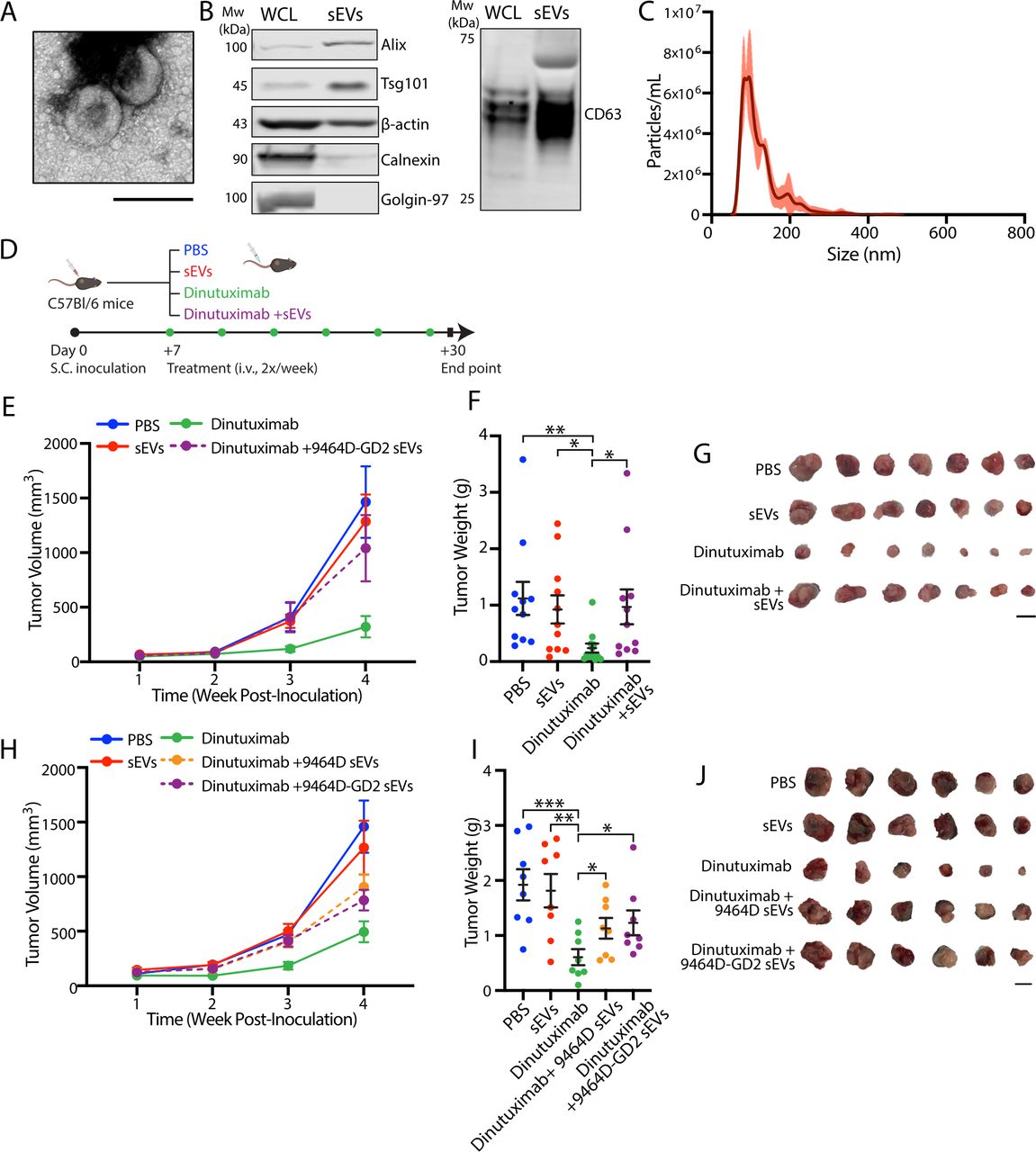
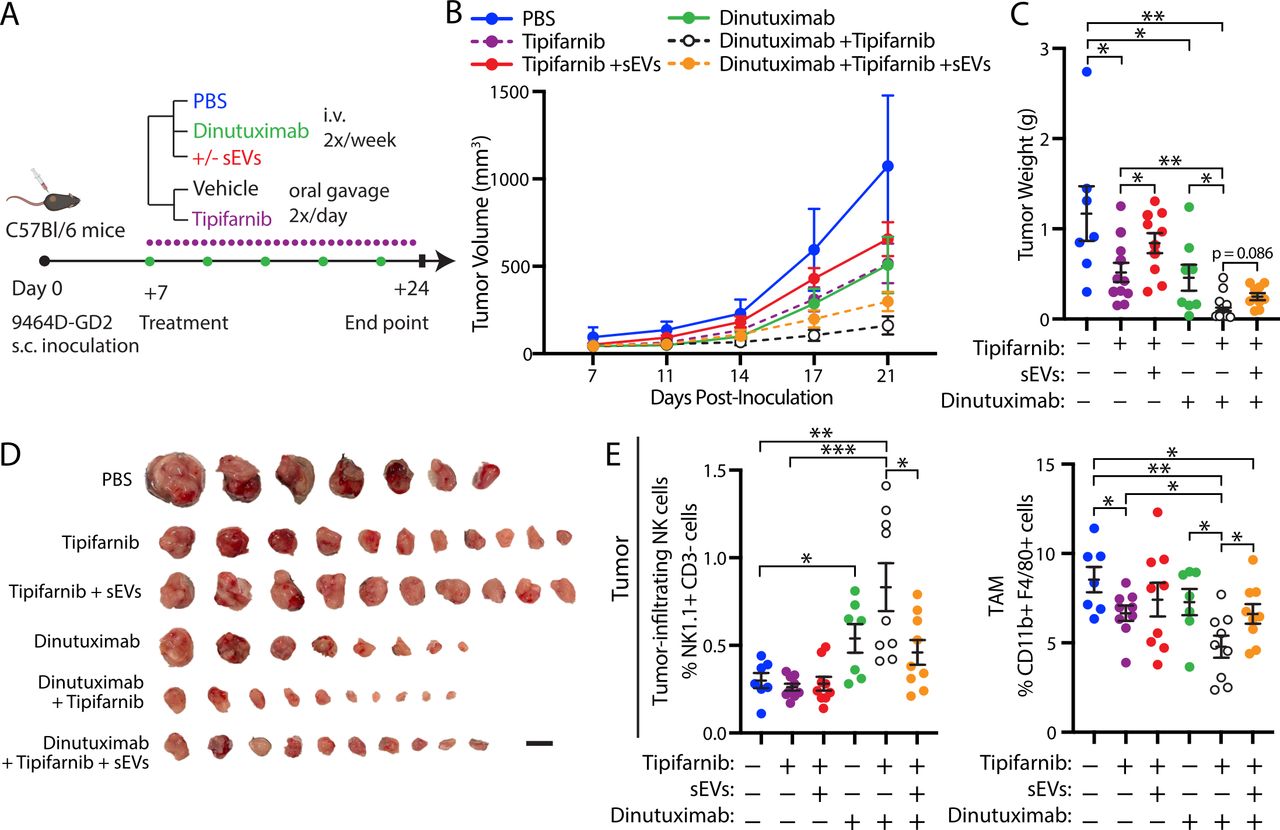
To validate that tipifarnib sensitizes tumors to dinutuximab in vivo through inhibition of sEV secretion, we performed a rescue experiment by treating mice with dinutuximab and/or tipifarnib in the presence or absence of neuroblastoma-derived sEVs (figure 6). While both dinutuximab and tipifarnib demonstrated mild antitumor activity alone, the combination of dinutuximab and tipifarnib significantly decreased tumor growth and weight compared with either agent (figure 6B–D). Notably, the addition of sEVs rescued tumor growth and weight in tipifarnib-treated mice to a level comparable to control, suggesting that inhibition of tumor-derived sEV secretion contributes to the antitumor effects of the drug in vivo (figure 6B–D). Furthermore, sEVs appeared to reverse the antitumor efficacy of dinutuximab plus tipifarnib (figure 6B–D), although the result did not reach statistical significance (p=0.086). Interestingly, we found that the addition of sEVs to combination treatment with dinutuximab and tipifarnib significantly decreased the percentage of tumor-infiltrating NK cells and enhanced the TAM population to a level comparable to dinutuximab treatment alone (figure 6E). Taken together, these results suggest that tipifarnib enhances the efficacy of dinutuximab and modulates the immune system in part through inhibition of sEV secretion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neuroblastoma-derived sEVs rescue tipifarnib-induced attenuation of tumor growth and reverse the effects of tipifarnib and dinutuximab on tumor immune cell infiltration. (A) Experimental design. C57BL/6 mice were subcutaneously inoculated with 1×106 9464D-GD2 cells. One-week postinoculation, tumor-bearing mice began receiving tail-vein injections of PBS control, dinutuximab or dinutuximab plus sEVs two times per week combined with oral administration of vehicle or tipifarnib two times per day. On day 24, tumors were harvested for analysis. (B) Quantification of tumor volume at indicated time points. Mean±SEM. PBS, n=7; tipifarnib, n=11; tipifarnib+sEVs, n=10; dinutuximab, n=8; dinutuximab+tipifarnib, n=10; dinutuximab+tipifarnib+sEVs, n=10. (C) Quantification of tumor weight from indicated treatment groups on day 24. Mean±SEM, n as in (B). Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. (D) Representative images of tumors from indicated treatment groups on day 24. Scale bar, 1 cm. (E) Quantification of tumor-infiltrating NK cells (NK1.1+/CD3-; left panel) and tumor-associated macrophages (TAM) (CD11b+/F4/80+; right panel) in 9464D-GD2 tumors isolated from mice in the indicated treatment groups. Mean±SEM, n=7 for PBS and Dinutuximab groups, n=9 for all other groups. Student’s t-test. *p<0.05; **p<0.01; ***p<0.001. sEV, small extracellular vesicle.
Discussion
In this study, we demonstrate that neuroblastoma-derived sEVs significantly suppress the efficacy of dinutuximab in vivo and identify the FDA-approved drug tipifarnib as a promising novel adjunct to anti-GD2 immunotherapy, which can be rapidly translated to the clinic for high-risk neuroblastoma patients. Interestingly, our data demonstrate that resistance to dinutuximab is independent of sEV-associated GD2 expression in this model, suggesting that unlike the well-established mechanism by which sEV-associated PD-L1 interacts with the programmed death-1 receptor on the surface of T cells to suppress their function and promote tumor growth,14 neuroblastoma-derived sEVs do not serve as a decoy to inhibit the binding of GD2 antibodies and tumor cells. However, this does not preclude the possibility that significant upregulation of sEV-associated GD2 could result in a decoy mechanism to suppress the efficacy of dinutuximab.
Our data reveal that neuroblastoma-derived sEVs modulate immune effector cells both locally and systemically, including NK cells, which are the primary effector cells that mediate dinutuximab-induced killing.16 Systemically, we found that sEVs suppressed dinutuximab-induced NK cell mobilization and maturation in the spleen. Within the TME, sEVs suppressed dinutuximab-induced NK cell infiltration and upregulated the population of TAMs, promoting an immunosuppressive environment that favored tumor growth. In agreement with our observations, high levels of TAMs are associated with poor outcomes in neuroblastoma patients, while intratumoral NK cells predict improved overall survival.37 38 Interestingly, we found that tumors isolated from mice treated with both tipifarnib and dinutuximab demonstrated a higher percentage of CD3+ cells, suggesting that T cells may exert an antitumor effect independent of the role of NK cells in this study. These results warrant further investigation, given the well-established function of T cells in neuroblastoma antitumor immunity and the role of sEVs in modulating T cell response.3 39 Collectively, these data establish that neuroblastoma-derived sEVs modulate the tumor immune cell environment to confer resistance to dinutuximab.
The molecular mechanisms by which neuroblastoma-derived sEVs regulate NK cell function, tumor immune cell recruitment and systemic immune suppression remain unknown. Tumor-derived sEVs have a complex role in modulating the response to immunotherapy.12 40 sEVs carry an array of immunosuppressive cargo, including miRNA, long non-coding RNA, DNA, and proteins that interfere with the host immune system and reprogram immune effector cells.12 For example, tumor-derived sEVs inhibit NK cell function through the transfer of miR-23a, leading to CD107a downregulation.18 Similarly, tumor-derived EVs carry transforming growth factor beta, which inhibits NK cell cytotoxicity by downregulating expression of the activating receptor NKG2.18 19 41 As sEVs contain a large number of bioactive molecules, we hypothesize that multiple cargo likely contribute to the immunosuppressive effects of neuroblastoma-derived sEVs, and future studies will be aimed at identifying the critical cargo(s) responsible for resistance to anti-GD2 immunotherapy. In addition, investigation into the molecular mechanisms of sEV biogenesis in neuroblastoma cells will provide additional insight into their immunosuppressive nature and potentially identify other molecules for therapeutic targeting.
The use of the 9464D subcutaneous model offers an immune TME comparable to human neuroblastoma and provides a practical method to test clinically applicable therapies.42 Future studies into the role of sEVs in anti-GD2 immunotherapy resistance will include the use of orthotopic models, which provide a more representative neuroblastoma TME than subcutaneous tumor models.43 Interestingly, as dinutuximab targets primary tumor cells less effectively than minimal residual cells in the BM,10 it is tempting to speculate that the accumulation of tumor-derived sEVs within solid tumors may contribute to local immunosuppression and promote resistance to dinutuximab. Further investigation into the role of circulating and tumor-associated sEVs as well as intratumoral immune cell infiltrates in patients unresponsive to anti-GD2 immunotherapy will provide further insight into the role of sEVs in drug resistance.
Critically, we demonstrated for the first time that tipifarnib, which was recently identified as a selective inhibitor of sEV secretion from cancer cells, significantly enhanced the antitumor efficacy of dinutuximab.33 44 Mechanistically, tipifarnib inhibits sEV secretion by downregulating several molecules involved in sEV biogenesis and/or secretion, including ALG-2-interacting protein X (Alix), neutral sphingomyelinase 2 and Rab27a.33 44 Tipifarnib is a potent farnesyltransferase inhibitor that has antitumor activity by inhibiting protumorigenic HRAS signaling.45 Our data revealed that when given as a single agent tipifarnib exhibited mild antitumor efficacy in neuroblastoma that could be rescued by sEVs, indicating that inhibition of tumor-derived sEV secretion contributes to the antitumor efficacy of tipifarnib in this model. Likewise, neuroblastoma-derived sEVs partially rescued tumor growth and reversed the effects of tipifarnib and dinutuximab on tumor immune cell infiltration. However, we cannot exclude the possibility that systemic administration of tipifarnib inhibited additional farnesylation-dependent pathways that may have contributed to the enhanced antitumor efficacy observed in combination with dinutuximab. Interestingly, alterations in the anaplastic lymphoma kinase-RAS/MAPK pathway correlate strongly with poor clinical outcomes in all neuroblastoma risk categories46 and are found to be present at a higher frequency in relapsed tumors.47 Therefore, in addition to inhibiting sEV secretion, administration of tipifarnib with dinutuximab may provide an additional therapeutic benefit to patients harboring aberrant RAS/MAPK signaling pathways. Given that tipifarnib recently entered phase II clinical trials in pediatric patients with advanced solid tumors including neuroblastoma, this combination therapy can be rapidly translated to the clinic to improve outcomes for patients with high-risk neuroblastoma.
Data availability statement
Data are available in a public, open access repository. The RNA-sequencing data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE189921.
Ethics statements
Patient consent for publication
Ethics approval
All animal studies were performed according to guidelines established by the Institutional Animal Care and Use Committee (IACUC #PRAM201145989) at the Penn State College of Medicine (Hershey, PA).
Acknowledgments
We would like to acknowledge the Department of Comparative Medicine, Bioluminescent Imaging Core, Transmission Electron Microscopy Core, Flow Cytometry Core, and Genome Sciences Core at the Penn State College of Medicine.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
XL and CAW contributed equally.
Contributors Conceptualization: XL, CAW, H-GW. Data curation: XL, LC, JZ, YZ, MZ. Formal analysis: XL. Validation: XL. Investigation: XL, LC. Visualization: XL. Methodology: XL, CAW, LC, JMS, TS, VSS, H-GW. Writing-original draft: XL, CAW, MMY. Writing-review and editing: XL, MMY, CAW, H-GW. Guarantor: H-GW.
Funding This work was supported in part by the Lois High Berstler Research Endowment Fund and the Four Diamonds Fund of the Penn State College of Medicine.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.