Article Text

Abstract
During oncogenesis, tumor cells present specific carbohydrate chains that are new targets for cancer immunotherapy. Whereas these tumor-associated carbohydrates (TACA) can be targeted with antibodies and vaccination approaches, TACA including sialic acid-containing glycans are able to inhibit anticancer immune responses by engagement of immune receptors on leukocytes. A family of immune-modulating receptors are sialic acid-binding Siglec receptors that have been recently described to inhibit antitumor activity mediated by myeloid cells, natural killer cells and T cells. Other TACA-binding receptors including selectins have been linked to cancer progression. Recent studies have shown that glycan-lectin interactions can be targeted to improve cancer immunotherapy. For example, interactions between the immune checkpoint T cell immunoglobulin and mucin-domain containing-3 and the lectin galectin-9 are targeted in clinical trials. In addition, an antibody against the lectin Siglec-15 is being tested in an early clinical trial. In this review, we summarize the previous and current efforts to target TACA and to inhibit inhibitory immune receptors binding to TACA including the Siglec-sialoglycan axis.
- antigens
- tumor-associated
- carbohydrate
- lymphocytes
- tumor-infiltrating
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Recent advancements in the stimulation of the immune microenvironment and anticancer immune responses with immune checkpoint inhibitors (ICI) has improved the outcome of treatments for patients and has led to impressive long-term remissions in some patients with advanced disease.1–5 However, primary and acquired resistance significantly diminish the success of ICI and only a minority of patients benefit from currently available cancer immunotherapies.6 7 Thus, new strategies are urgently needed in order to induce long-term remissions with cancer immunotherapy in many more of our patients.
Carbohydrates belong to the major biomolecules of living organisms. Carbohydrates can be attached to proteins (glycoproteins), lipids and exist as chains of carbohydrates (glycosaminoglycans). Glycans—carbohydrate-containing macromolecules—are ubiquitous in biological systems and are essential for numerous biological functions.8–10 Cell surfaces and extracellular proteins are significantly glycosylated. In addition, glycosaminoglycans can be found in the extracellular matrix. Glycans are used as storage for energy (glycogen), are structurally important (see later for the stability of programmed cell death protein 1 (PD-1)) and can mediate signals. Whereas proteins undergo substantial post-translational modifications, in particular N-glycosylation and O-glycosylation,8–10 intracellular modification of tyrosine with O-GlcNAc serves for intracellular signaling.11 Changes of glycosylation have a significant impact on cancer biology and cancer progression.12–15 Of note, altered glycan structures represent antigenic targets for cancer immunotherapy. In this review, we summarize how cancer-associated changes in glycosylation can be used to improve cancer immunotherapy.
Cancer-specific changes in glycosylation
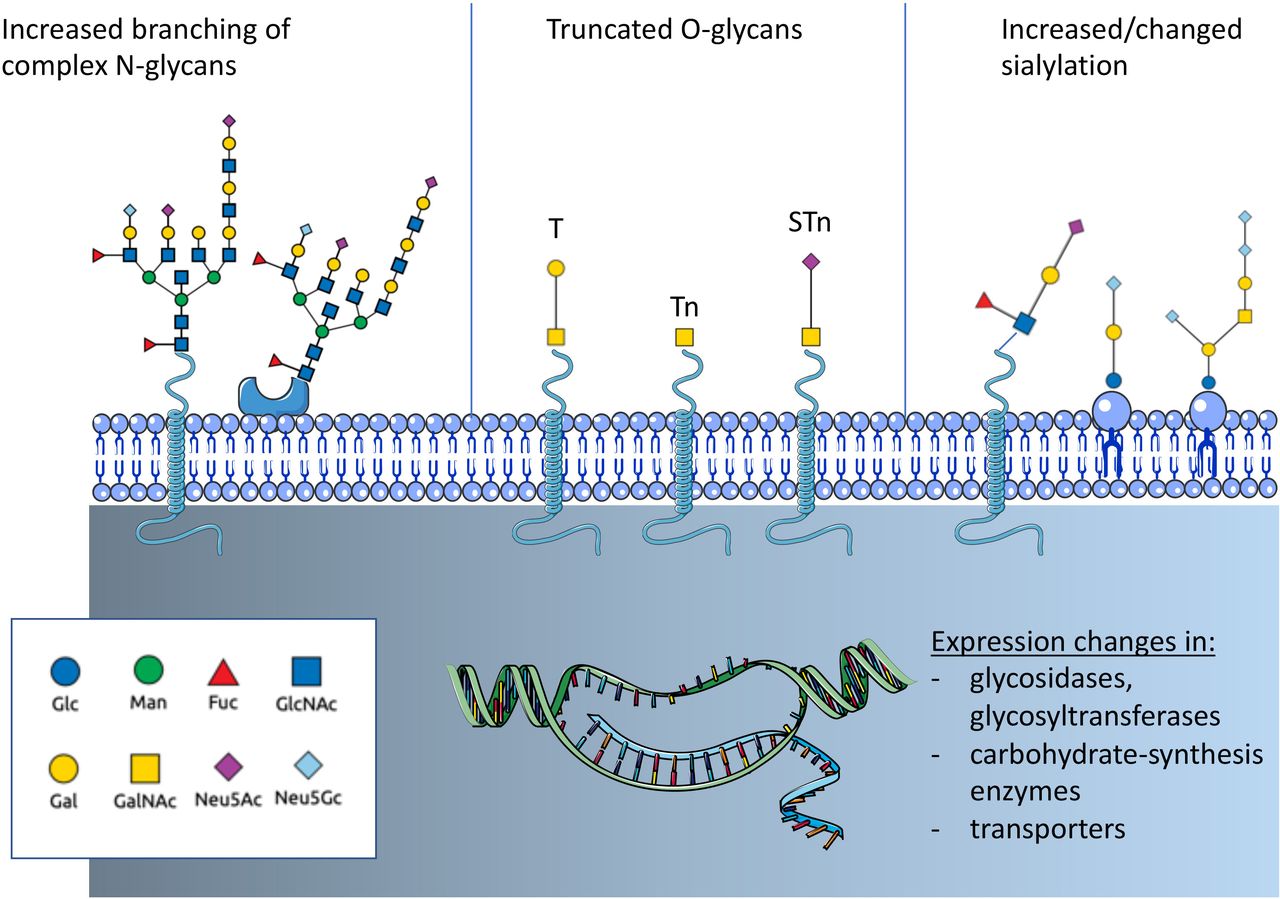
Altered glycosylation is a common feature of tumor cells and leads to the formation of tumor-associated carbohydrates (TACA) (figure 1). Three common changes are often associated with cancer: a) increased expression of truncated or incomplete glycans, b) increased branching of N-glycans and c) augmented or changed presence of sialic acid-containing glycans.15 Tumor-cell-surface glycans are known to promote cancer progression by affecting tumor growth, cell invasiveness and negatively regulate immune responses.15–17 Changes of glycosylation observed in cancer depend on the expression and changes of enzymes involved in glycan biosynthesis and glycan-modifying enzymes including transferases and glycosidases as well as transporter for saccharides and precursors.12 14 18 Expression of these glycan-modifying proteins are altered in cancer due to genetic and epigenetic alterations and differ between cancer types.

Overview on cancer-associated glycosylation. Three main changes can be found in cancer that are regulated by genetic or epigenetic alterations in genes of glycan-modifying enzymes or enzymes involved in carbohydrate biosynthesis. N-glycans show often an increased branching due to increased MGAT5 expression. Another often observed change is the truncation of O-glycans and the exposure of new tumor-associated carbohydrates (TACA) including the T antigen, Tn antigen and the sialyl-Tn antigen (STn). In addition, changes of sialylation of both glycoproteins and glycolipids can be observed. Increased sialylation (hypersialylation) is often observed. The introduction of the non-human sialic acid Neu5Gc can also be observed. Fuc, fucose; GlcNAc, N-acetyl-glucosamine; Gal, galactose; GalNAc, N-acetyl-galactosamine; Glc, glucose; Man, mannose; N-acetyl-neuraminic acid; Neu5Gc, N-glycosyl-neuraminic acid.
Changes in sialylation
Cancer-associated glycans often exhibit an increased amount of sialic acid. Augmented sialylation of tumor cells has been correlated with a metastatic phenotype and poor prognosis in patients with cancer.19 20 Sialic acids are predominantly found at the non-reducing end of N-linked and O-linked glycans attached to proteins or glycolipids.21 Hypersialylation facilitates interactions with sialic acid binding receptors such as selectins and Siglecs with consequences for cancer progression. Moreover, the incorporation of the non-human sialic acid N-glycolyl-neuraminic acid (Neu5Gc) into glycans and the interaction with circulating anti-Neu5Gc antibodies influences cancer progression.22–26 Neu5Gc is biosynthesized from N-acetyl-neuraminic acid (Neu5Ac) via the enzyme CMP-Neu5Ac hydroxylase (CMAH), which is not present in humans.27 However, various studies have found an increased presence of Neu5Gc-containing glycans in cancer, which could be associated with uptake of meat from Neu5Gc producing mammals.28 29
Truncation of O-glycoproteins
O-glycans are ubiquituously found on cells and are particularly secreted into the extracellular matrix or into the lumen of internal organs. For example, mucins, highly O-glycosylated proteins such as CA19-9,30 can serve as a biomarker for some cancer types. One of the most common cancer-associated changes in glycosylation is the truncation of O-linked carbohydrate chains (figure 1).31 Usually, a GalNAc sugar residue is attached to a serine or threonine of the glycoprotein (GalNAcα1-O-Ser/Thr, Tn antigen) and usually elongated by the T-synthase (core 1 β3-galactosyltransferase) in the Golgi apparatus that attaches a galactose residue to Tn antigen. The resulting glycan is called T antigen (sometimes also called the Thomsen-Friedenreich (TF) antigen). The T-synthase requires a chaperone for the correct folding and enzymatic activity.31 The chaperone was termed Core 1 β3-galactosyltransferase Specific Molecular Chaperone (COSMC).32 The X linked COSMC gene is mutated in various cancer types leading to the presence of Tn antigen or its sialylated form, the sialyl-Tn (STn) antigen.31 Interestingly, truncated O-glycosylation is shown to have an immunomodulatory effect. Tn antigen binds to macrophage galactose-type lectin on dendritic cells and macrophages that inhibits the migration of immature antigen-presenting cells (APCs) and increases M2-like tumor associated macrophages.33–35
Truncated O-glycans represent epitopes which may selectively target cancer cells. Various cancer tissues have been analyzed for the expression for the T, Tn and STn antigen.36–38 The human-mucin 1 (MUC1) is overexpressed in many adenocarcinomas, presenting high levels of truncated glycans as STn-MUC1, Tn-MUC1 and T-MUC-1. Yet, these antigens are rarely expressed in normal tissue compared with cancer tissue.39–44 Further studies are needed to determine the specificity of the expression patterns if we consider to target these epitopes with antibodies and chimeric antigen receptors (CARs).
Altered branching of N-glycoproteins
Increased branching of N-glycans, mediated by β1,6-N-acetylglucosaminyltransferase V (MGAT5, figure 1),12 15 can influence cell adhesion, migration and metastasis of tumor cells.45 46 Upregulation of MGAT5 has also been shown to directly influence cytokine signaling and tumor progression47 while the knockdown of Mgat5 led to activation of CD4+ T cells and macrophages in breast cancer.48 Altered N-glycosylation of immune cells could also affect the antitumor immune response. Increased branching of N-glycans can directly inhibit T cell activation by increasing T cell receptor (TCR) clustering.49 50 This effect was attributed to interaction with galectin-3.49 On the other side, TCR signaling also directly influences enzymes modulating N-glycosylation.51
Alterations in glycolipids
Gangliosides are sialylated glycan-containing lipids of the cell membrane show also often changes on cancer cells. The gangliosides GM3, GM2, CD3 and GD2 are present in normal tissue but are often overexpressed in different cancers including lung cancer, melanoma and neurogenic tumors such as neuroblastoma.52–55 Glycolipids can significantly influence cell signaling by mediating the formation of lipid rafts.56 Tumor-associated gangliosides have been investigated for their immunosupressive properties and role in cancer progression. Furthermore, the plasma concentration of gangliosides is often elevated,57 making them potential therapeutic targets and diagnostic tools. GM3 contains sialic acid-residues and several studies have shown that GM3 containing the non-human sialic acid Neu5Gc (Neu5Gc-GM3) is relatively specific for different types of cancer.29
Cancer-associated changes in glycosylation and immune phenotypes
The association of specific glycan changes with the immune state of a cancer is currently being studied. Immune phenotypes such as T cell excluded tumors were associated recently with galectin-1 expression and interactions with glycan-ligands.58 The Cancer Genome Atlas has been used to study the expression of different glycan-modifying enzymes, for example, for sialic acid-modifying enzymes.59 However, further systematic studies including also lectin stainings are needed on tissue sections to correlate immune phenotypes with.
Modalities to target tumor-associated glycans
ADCC and CDC
Antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC) is triggered by the interaction between antibody-bound target cells—for example, infected or tumor cells—and effector immune cells or complement factors, respectively. Several glycan-targeted monoclonal antibodies (mAbs), which are in clinical use or development, are known to elicit ADCC and/or CDC (figure 2). Among these, dinutuximab, which targets ganglioside GD2 on melanoma, and neuroblastoma (table 1),60–62 is approved for the treatment of high-risk neuroblastoma pediatric patients.63 Similarly, KW871, a chimeric mAb which targets the ganglioside GD3, exhibited antitumor activity in combination with with IFNα2b in vitro.64 In patients with metastatic melanoma, the combination of KW871 with interferon was shown to be well tolerated, although not highly efficacious.65 An anti-idiotype antibody was generated to elucidate an immune response against Neu5Gc-GM3.66 Early trials have shown interesting activity in patients with non-small cell lung cancer (NSCLC).66 67 Trials testing the efficacy of this antibody called racotumomab in a larger population are currently recruiting (eg, NCT01460472).

{kind=link}
{kind=link}
Overview on targeting approaches for cancer-associated glycosylation. (A) Tumor-associated carbohydrates (TACA) can serve as tumor-specific antigen and be approached with antibody-dependent cellular cytotoxicity (ADCC)-inducing or complement-dependent cytotoxicity (CDC)-inducing antibodies or with antibodies carrying a payload (chemotherapy or even enzyme). (B) Chimeric antigen receptor (CAR) expressing immune cells could be redirected towards TACA-presenting tumors. Similarly, bispecific antibodies could be used to direct immune cells to TACA-expressing tumors (not shown). (C) TACA such as sialoglycans can engage immune receptors including inhibitory Siglec receptors on T cells and myeloid cells and improve anticancer immunity directly. ADC, antibody-drug conjugates.
Previous and ongoing clinical trials targeting tumor-associated glycans or lectins
Since the 1990s, several other mAbs have been designed that target Lewis antigens (Le), expressed by a broad range of tumor cells, and that are able to elicit ADCC and/or CDC in preclinical and clinical studies. Examples of such mAbs are BR96,68 targeting Ley; hu3S193,69 a humanized anti-Ley mAb which showed low toxicity in an early clinical trial,70 but insufficient efficacy in a subsequent phase II trial.71
As for targeting of tumor-associated MUC1 (TA-MUC1) expressed on several malignancies including ovarian, breast and cervical cancers,72 the mAb PankoMab-GEX (gatipotuzumab) has shown therapeutic efficacy, particularly in heavily pretreated patients with ovarian cancer.73 74 Yet, the subsequent phase II trial did not to show any outcome advantage.75 Another high-affinity mAb named 5E5 targets the aberrant Tn glycoform of mucin MUC1 and has been shown to lyse breast cancer cells via both ADCC and CDC.76
Glycosylation is also under investigation to be used as tool for the enhancement of ADCC and CDC immune response mechanisms. Indeed, manipulation of specific residues of the Fc N-glycan has been shown to modulate antibody-dependent effector functions via modification of the Fc binding affinity to Fc receptors expressed on different immune cells. Particularly, core fucosylation and sialylation of Fc regions results in decreased ADCC, while N-glycans with low or no sialic acid are better suited to trigger ADCC. These approaches are reviewed by Wang and Ravetch77 and Mastrangeli and colleagues. 78
Antibody-drug conjugates
Antibody-drug conjugates (ADCs) unify the properties of cytotoxic chemotherapy and mAbs, in an effort to selectively target and lyse cancer cells. Similarly, antiglycan-directed antibodies can be exploited to deliver selected anticancer payloads specifically to tumor cells (figure 2). First attempts used the previously mentioned anti- Ley BR96 mAb, conjugated to doxorubicin and docetaxel. This ADC was tested in phase II trials for advanced NSCLC (table 1)79 and for metastatic breast cancer.80 Although initial results were encouraging, the mAb was later discontinued, likely due to low efficacy. More recent attempts at exploiting tumor glycan expression for drug delivery include work by Sedlik et al, who used a Tn-directed mAb (Chi-Tn) to deliver cytotoxic drugs; they showed promising antitumor activity of the chiTn ADC in vitro and in vitro, when this was conjugated to saporin or auristatin F.81 These effects were particularly dependent on high Tn expression in tumor cells.81 Similarly, Prendergast et al developed murine mAbs able to target tumor cells expressing STn with high avidity and further exploited a subset of these mAbs as ADCs by conjugating them to monomethyl auristatin E (MMAE).82 These ADCs showed high efficacy in vitro, in the presence of STn-expressing cancer cell lines, as well as tumor inhibition in multiple in vivo model, including breast and colorectal cancer.82 Furthermore, humanized anti-STn antibodies conjugated to MMAE have been shown to determine in vitro cytotoxicity specific to STn-expressing ovarian cancer cell lines as well as efficacy in tumor control in in vivo models for ovarian cancers, including in patient-derived xenografts.83 These humanized aSTn-ADCs were further shown to present a low toxicity profile as they did not cross-react to any tissue of human origin. Most recently, the murine mAb FG129 and corresponding chimeric human variant CH129, were developed to target sialyl-di-Lea-containing glycoproteins and shown to bind to a range of cancer tissues including colorectal, pancreatic, gastric and ovarian tumors.84 Conjugation of CH129 to either MMAE or maytansinoid (DM1 and DM4) resulted in cell death in vitro, as well as in vivo tumor control.84
Bispecific antibodies
Bispecific antibodies (bsAbs) are molecules designed to recognize two distinct antigens or epitopes and have been recently emerging as potential key actors in cancer immunotherapy. In terms of tumor-associated glycans, research on bsAbs has so far focused on targeting GD2 ganglioside and MUC1 combined (table 1).85–88 Recent developments involve also targeting of glycan-binding lectins on tumor cells. bsAbs designed to target lectins including Siglecs are currently under investigation, for example, bsAb against CD22/CD19 on B cells—currently in a phase I–II trial, targeting B cell malignancies89 or bsAb bridging CD33 on AML cells to CD3 on T cells90 91 or to CD16 on natural killer (NK) cells.92
Redirection of immune cells
CARs can redirect immune cells to tumor cells or the tumor microenvironment by targeting tumor-specific antigens.93 94 However, design of CARs directed at solid cancers presents several challenges, due to the immunosuppressive mechanisms within the TME and the difficulty in finding antigens that are solely cancer-specific.94
Targeting CARs to glycans may be advantageous due to the specificity of aberrant glycosylation on tumor cells. Several attempts at designing glycan-targeted CARs have been made and are currently being tested in preclinical and clinical studies (table 1). Tumor-associated glycoprotein 72 (TAG72) antigen is the truncated sialyl-Tn found on many O-glycoproteins and overexpressed by various types of cancer cells, including lung, colorectal and ovarian cancer cells. TAG72-direceted CAR T cells were shown to be effective in vitro on gastrointestinal cancer cell lines.95 A much more recent clinical trial using anti-TAG72 CAR T cells in advanced colorectal cancer patients failed to show effective clinical responses; this was potentially due to the murine origin of the scFv or to lack of co-stimulation96; the second-generation TAG72-targeted CAR was shown to effectively target ovarian cancer cell lines and patient-derived primary ovarian cancer cells in vitro, as well as to reduce tumor growth and improve survival in mice, with sequential intraperitoneal administrations.97 Attempts at designing MUC1-targeted CARs relied on two main antibodies, SM3 and HMFG2.98 More recent Tn-MUC1-targeting CARs have been developed from the 5E5 mAb and shown to present cancer-specificity and weak reactivity against healthy tissues, as well as to effectively target and kill cancer cells in pancreatic and leukemia xenograft models.99 A successful target for CAR T cell therapy is the TACA Ley. A second generation fully humanized CAR construct targeting Ley was shown to be effectively transduced in PBMC-derived T cells and to lyse Ley positive tumor cell lines in vitro.100 Similarly, in vivo, adoptive transfer of Ley− targeted CAR T cells resulted in tumor homing and subsequently inhibiting growth of myeloma and ovarian cancer xenografts.100 101 The same CAR T cells were tested in four patients with relapsed AML, presenting Ley positive blasts, in a phase I clinical trial.102 An ongoing Phase I clinical trial is now testing the safety and tolerability of using these CAR T cells in patients with advanced solid tumors presenting Ley surface expression (NCT03851146).
In high-risk neuroblastoma patients with Epstein-Barr virus (EBV)-associated malignancies, infusion of EBV-specific and GD2 CAR-transduced T cells showed persistence of these cells and tumor necrosis in some patients,103 with three patients showing complete responses and two long-term remission (up to 48 months) in a long-term follow-up study.104 Combination of GD2-targeted CARs with PD-1 blockade is also under investigation, with studies showing that, in vitro, anti-PD-1 can rescue GD2 CARs from activation-induced cell death,105 a phenomenon also observed in vivo in patients with metastatic melanoma treated with GD2-specific CAR T cells in a phase I clinical trial.105 In another phase I clinical trial, combination of a third-generation GD2-targeted CAR with lymphodepletion and PD-1 blockade resulted in improved in vivo expansion of the CAR T cells, which were safely tolerated, but modest clinical responses.106 A recent proof-of-concept study showed that incorporating IL-12 or IL-18 within a GD2-targeting CAR resulted in enhanced effector functions and increased monocyte recruitment in vitro.107 Another phase I clinical trial testing IL15-GD2 CAR NKT cells in pediatric patients with neuroblastoma is currently ongoing (NCT03294954). More recent attempts at using immune cells other than T cells for GD-2 targeted CARs include work by Mitwasi et al, who used a ‘universal’ CAR platform (UniCAR) to target NK cell line NK-92 to GD2 expressing cells.108 UniCARs present an ‘on/off system’, which allows for controlling the activity of CAR expressing cells, depending on the presence of a target module, that is, an antibody-based cell-binding domain specific to GD2.108
Vaccination
Several vaccines have been developed based on different glycans Tn-MUC1,109 MUC16,110 GD2,111 112 GM2112 and Neu5Gc-GM3,113 114 but most vaccines had major issues relating to the poor immunogenicity of glycans and glycopeptides. Glycan-based vaccine research is therefore currently focused on addressing these problems and enhancing immunogenicity of these immunotherapies. A strategy to increase immunogenicity is conjugation to carrier proteins; one such example is Theratope, an STn-based vaccine conjugated to carrier protein keyhole limpet hemocyanin. Though initial evaluations of Theratope in patients with metastatic adenocarcinomas produced encouraging results,115 including increased antibody titres and prolonged survival, this vaccine was later shelved after failing to meet its primary endpoints in a phase III clinical trial.116 Other strategies to enhance glycan immunogenicity include designing polyvalent vaccines which contain multiple glycan structures, such as Globo-H, STn, Tn, TF and Ley 117 or using peptide mimetics of TACAs, such as for SLex, SLea, SLey or Tn, to prevent enzymatic degradation and improve antitumor efficacy.118 119
Glycan modifications can also be used to target shared or neo-antigens to APCs including dendritic cells.120 Dendritic cells express lectins binding to glycan epitopes. For example, dendritic cells express the lectins Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) and Langerin that can be targeted with fucosylated glycans of Lewis-type oligosaccharides.121 In another work, Siglec-1 on macrophages was targeted to induce an immune response to a lipid antigen and robustly activate NK T cells.122
Targeting glycan-receptor interactions to improve cancer immunotherapy
Siglec-sialoglycan interactions
Recent evidence also suggests that cancer-associated glycosylation can directly influence anticancer immunity by binding to glycan binding receptors, which are also called lectins.12–14 123 124 Hypersialylated glycan structures have been identified in various types of cancer.14 22 24 The high density of sialoglycans on some tumor cells can engage inhibitory sialoglycan-binding receptors called Siglecs.25 125 126 Siglecs belong to the family of immunoglobulin (I)-type lectins.125 127 128 Siglecs are single-pass transmembrane proteins and bind sialoglycans through their carbohydrate-binding domain (table 2).125 127 128 In humans, there are 14 functionally active Siglec receptors that can be divided into two groups. On one side, there are evolutionary conserved Siglec receptors including Siglec-1, Siglec-2 (CD22), Siglec-4 and Siglec-15. On the other side, CD33-related Siglecs have rapidly evolved also within mammalian species.22 129 Siglec-3 (CD33), Siglec-5, Siglec-6, Siglec-7, Siglec-8, Siglec-9, Siglec-10, Siglec-11 and Siglec-14 belong to the CD33-related Siglecs and there are for most of them no direct orthologs, for example, in mice and humans.128 Most Siglec receptor have intracellular domains containing immunoreceptor tyrosine-based inhibitory motifs (ITIM) or ITIM-like motifs. Engagement of these inhibitory receptors lead to a phosphorylation of Src homology region 2 domain-containing phosphatase (SHP)-1 and SHP-2, which inhibit cell activation.22 128 Most Siglec receptors are expressed on immune cells and engagement inhibits immune cell activation. Diversification of CD33-related Siglecs is probably due to interactions with pathogens exploiting inhibitory Siglec receptors by covering themselves with sialoglycans.130 Some pathogens as the group B streptococci have even developed proteins to engage inhibitory Siglecs on myeloid cells and thereby evade immune-mediated killing.131
Summary of human Siglec receptors, their expression on the surface of different cells, their intracellular signaling and their binding to sialic acid-containing glycan ligands
The role of Siglec receptors as new targetable immune checkpoints has been recently reviewed in depth.23 25 125 126 132 Expression of inhibitory Siglec receptors on innate immune cells have been linked to inhibition of anticancer immunity.12 13 125 Siglec-7 is expressed on the majority of NK cells and Siglec-9 can be found on some subpopulations of NK cells.128 133 Upregulation of sialoglycans on cancer cells can inhibit NK cell-mediated tumor cell killing by engaging Siglec-7 and in some instances also Siglec-9.133 134 Siglec-9 expression has been shown to skew macrophage polarization to a protumorigenic phenotype and increased programmed death ligand 1 (PD-L1) expression on macrophages.135 Siglec-9 engagement was shown to be dependent in this set of experiments on STn-modified mucins.135 Other experiments in mice showed that myeloid polarization is affected by inhibitory Siglec receptors including Siglec-E, the functional paralog of Siglec-9 in mice.136 Recent evidence demonstrated that Siglec-10 on macrophages can act as a “don’t eat me” signal to inhibit phagocytosis.137
Of note, the expression of Siglecs on adaptive immune cells including CD8+ T cells are inhibiting effective anticancer immunity as shown by us and others. Siglec-9 is upregulated on tumor-infiltrating T cells in various cancers including NSCLC, colorectal cancer, epithelial ovarian cancer and melanoma.138 139 Tumor growth was significantly enhanced in a transgenic mouse model of overexpression of Siglec-9 on T cells.138 Siglec-9 was expressed mainly on tumor-specific T cells shown by a reduced TCR repertoire in tumor infiltrating CD8+ T cells.139 Interestingly, Siglec-9 positive CD8+ T cells represent a less dysfunctional intratumoral T cell subtype and Siglec-9 blockade could reactivate these T cells.138 The conserved Siglec-15 was identified as inhibitor of T cell activation on APCs.140
Approaches to target sialoglycan-Siglec interactions could involve both blocking the Siglec receptor, for example, with an antibody. Siglec-15 was targeted with an antibody in preclinical mouse models140 and is already in an early clinical trial for advanced solid tumors (NCT03665285). On the other side, the ligand could be targeted enzymatically with sialidases or with small molecules interfering with sialoglycan biosynthesis.141 142 The use of sialic acid analogs in vivo by intratumoral injection could block the sialoglycan biosynthesis and led to a strong activation intratumoral T cells,141 however the systemic application of these analogs would be quite toxic. Bacterial sialidase was tested in a system linked to the HER2-targeting antibody trastuzumab and NK cell-mediated tumor cell killing was tested in vitro.142 Recently, HER2-targeted bacterial sialidase was tested in vivo in syngeneic mice and showed efficacy.143 Currently, a humanized version of this tumor-targeted sialidase is in clinical development and the toxicity profile needs to be further tested.
Targeting selectins
Selectins belong to the class of C-type lectins and bind mainly to sialoglycans.144 The three selectins, E-selectin, P-selectin and L-selectin (CD62E, CD62P and CD62L) bind to different sialylated ligands, often containing sialyl-LeX in a relatively selective manner and mediate interactions between platelets expressing P-selectin, leukocytes expressing L-selectin and endothelial cells expressing P-selectin and E-selectin.145 146 Expression of L-selectin on T cells can enhance cancer immunotherapy in mouse models.147 The P-selectin ligand carrying protein P-selectin glycoprotein ligand (PSGL)-1 was identified to be involved in T cell exhaustion,148 and was also shown to inhibit in anticancer immunity through its protein-protein interaction with the immune checkpoint VISTA, an inhibitory receptor expressed on T cells.149 However, these studies have not directly implicated interactions of the carbohydrate-modification of PSGL-1 to their effect on anticancer immunity and these effects are probably P-selectin independent. As sialoglycan-selectin interactions play an important role in leukocyte trafficking and mediation of immune responses, it is likely that sialogylcan-selectin interactions could influence anticancer immunity. Indeed, recent experiments demonstrate that modification of CAR T cells to bind to selectins can improve anticancer efficacy.150 151 However, further studies are needed to determine the role of selectins in anticancer immunity and how these interactions can be exploited for cancer immunotherapy.
Galectin-mediated interactions
Galectins are a class of carbohydrates-binding proteins capable of recognizing β-galactose via their carbohydrate-binding domain.152–154 Aberrant expression of galectins is frequent in cancer cells as well as in stromal cells and is associated with tumor progression.153 Importantly, galectins are involved in mediating interactions between tumor cells and innate and adaptive immune cells; upregulation of galectins by tumor cells is regarded as a mechanism of tumor immune escape,152–154 with some galectins being ligands for immune checkpoint receptors. For example, galectin-3 binds to cytotoxic T lymphocyte antigen 4 (CTLA-4) and lymphocyte activaiton gene 3 (LAG-3),155 156 whereas galectin-9 binds to T cell immunoglobulin and mucin-domain containing-3 (TIM-3).157
Galectin-1 is found upregulated in many different tumors and has been shown to antagonistically bind to the TCR, thus disrupting TCR signaling, and to determine T cell apoptosis, via redistribution of CD3 and CD45 clusters as well as CD7 and CD43 clusters.158–160 Early studies suggested that silencing expression of galectin-1 in tumor cells may be a strategy to enhance T-cell-mediated antitumor responses.161 Recently, a novel Gal-1-targeting DNA aptamer (AP-74 M-545) was developed and shown to suppress lung carcinoma growth in immunocompetent models. This was accompanied by an increase in CD4+ and CD8+ T cells, possibly by blocking the binding of galectin-1 to CD45.162 Nambial and colleagues showed that galectin-1 also prevents T cell migration into the tumor by upregulating PD-L1 and galectin-9 expression on endothelial cells. Blockade of galectin-1 resulted in increased T cell infiltration in multiple head and neck cancer mouse models as well as enhanced response to PD-1 blockade and radiotherapy combinations.58 Similarly, knockdown of galectin-1 in a mouse model of pancreatic ductal adenocarcinoma increased survival and enhanced T cell infiltration.163 In NSCLC, high expression of galectin-3 in the tumor microenvironment is associated with poor outcome164 and targeting of this galectin with an antagonist has been shown to inhibit lung adenocarcinoma growth and enhance response to PD-L1 blockade in vivo.165 Moreover, LAG-3 and interactions with galaectin-3 are also under investigation as a target for cancer immunotherapy in preclinical and clinical studies.156 166–168
Similarly, galectin-9/TIM-3 interactions are a recent target of immunotherapeutic intervention169 due to its suppressive properties of the antitumor immune response.168–170
Interactions with other lectins
C-type lectins are known to be involved in immunity, cell proliferation, tumor invasion and metastasis making them potential targets for cancer research such as selectins, DC-SIGN, Mincle, Dectin 1 and NKG2D.171 172 The C-type lectins are a superfamily of proteins that recognize a broad repertoire of ligands which the main feature is the C-type lectin-like domain. Cells of the adaptive and innate immune system commonly express c-type lectins including all myeloid cells, lymphocytes and dendritic cells.172–174 The lectin DC-SIGN can be recognized by glycosylated intercellular adhesion molecule 2 (ICAM-2) forming a DC-SIGN-ICAM-2 complex. This complex allows the maturation of dendritic cells that induces a specific cytotoxic T lymphocyte-modulated immune response promoting antitumor activity.171 Consisted with this, the DC-SIGN-Mac2 complex also inhibited the maturation of dendritic cells in colorectal carcinoma.175 DC-SIGN also has other ligands such as carcinoembryonic antigen and Le.176 A recent work has found that sialylation of antibodies could dampen autoimmune disease with potential consequences.177 Dectin-1 expressed on dendritic cells and macrophages is critical to the NK-mediated killing of tumor cells that expressed N-glycans in high levels.178 Furthermore, NK62DG is expressed on the surface of dendritic cells and its soluble ligands are very high in cancer leading to immunosuppression and poor prognosis in patients with cancer.179 180 In addition, the engagement of macrophage-inducible C-type lectin (Mincle) has been associated with immunosuppression and tumor progression.181
Impact of glycosylation on immune checkpoints
Glycosylation can mediate stability of different receptors and can influence cancer progression.12 Immune checkpoints are also glycosylated and targeting glycosylation of PD-1 can improve anticancer immunity.182 PD-1 in T cells is N-glycosylated and its N-glycosylation is critical for the stability on the cell surface.182 In addition, a glycosylation site on PD-1 was critical for interaction with PD-L1.182 On the other side, glycosylation of PD-L1 is also critical for its stability and targeting PD-L1 glycosylation could be used to improve anticancer immunotherapy.183 184 Moreover, the detection of PD-L1 in human cancers is dependent on glycosylation and could influence the predictive power of PD-L1 staining and the use of PD-(L)1 blocking antibodies in patients.185 Also, CTLA-4-mediated interactions are glycan-dependent and binding as well as stability could be influenced.186 187
Glycosylation and signaling pathways involved in resistance to immune checkpoint inhibitors
Several intracellular signaling pathways have been associated with resistance to ICI therapy including the activation of WNT/β-catenin signaling, MYC signaling and loss of phosphatase and tensin homolog (PTEN).188 Activation of the WNT/β-catenin signaling pathway has been shown to increase the production of immunosuppressive cytokines,189 prevention of the recruitment of BATF3+ dendritic cells190 191 and a shift towards an increase in regulatory T cells in the tumor.192 WNT/β-catenin signaling is also regulating glycosylation by promoting the expression of the enzyme DPAGT1 and thereby enhancing increased N-glycosylation.193 Increased N-glycosylation could further enhance immune evasion by providing ligands for immunomodulatory receptors such as Siglecs. MYC can influence the expression of PD-L1 and thereby influence the immune microenvironment in tumors.194 In addition, MYC signaling can directly mediate immune cell exhaustion in NSCLC.195 MYC is directly regulated by O-GlcNAcylation.196 O-GlcNAcylation was needed for proliferation and MYC stability.196
Conclusion and outlook
Recent discoveries and progress have revealed that tumor-associated carbohydrates are a interesting new target for cancer immunotherapy interventions. Many therapeutic opportunities are ready to be explored and targeting of TACA has just started as new field within cancer immunotherapy. Further studies are needed to determine patient populations that could benefit from such interventions. Whereas for proteins, the epidemiology and expression patterns are often well studied, the epidemiology of glycan expression will be important in order to determine the frequency of specific expression in cancer types and also during therapeutic interventions. In addition, glycan-mediated interactions with immunomodulatory lectins on leukocytes including Siglecs can directly affect anticancer immunity. Siglec-sialoglycan interactions have been described as potential new immune checkpoint. A first successful application in an early clinical trials with the Siglec-15 blocking agent (NC318) is promising. Besides the role as antigen and immunomodulatory factor, glycans can directly influence the stability and turnover of immune receptors including immune checkpoints.
We are confident that the field of glyco-immunology will enable us to improve cancer immunotherapy and help many of our patients by further studying mechanisms involved in glycan-mediated immune suppression and developing new approaches to target cancer-associated glycans.
References
Footnotes
NRM and MN contributed equally.
Contributors NRM, MN, AZ and HL have written the manuscript. All authors have approved the final version for publication.
Funding This work was supported by funding from the Goldschmidt-Jacobson Foundation, the Swiss National Science Foundation (SNSF grant #3 10 030–1 84 720), the Schoenemakers-Müller Foundation and the Cancer League of Basel (KlbB).
Competing interests HL received travel grants and consultant fees from Bristol Myers Squibb (BMS) and Merck, Sharp and Dohme (MSD). HL received research support from BMS and Palleon Pharmaceuticals.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.