Article Text

Abstract
Background Chemokine (C-X-C motif) ligand 13 (CXCL13) was known as a selective chemotaxis for B cells, a product of follicular helper CD4+T cells (TFH) and a contributor to tertiary lymphoid structures (TLS). Although secretion and function of CXCL13 produced by TFH have been deeply explored, the immune function and prognostic significance of CXCL13 secreted by CD8+T cells still remain unrevealed. This study aims to investigate the clinical merit of CXCL13+CD8+T cells in clear cell renal cell carcinoma (ccRCC).
Methods We analyzed prognostic value and immune contexture that associated with CXCL13+CD8+T cells infiltration level in a total of 755 patients from Zhongshan Hospital cohort (n=223) and The Cancer Genome Atlas cohort (n=532). In vitro analyses were conducted on 42 samples of resected tumor tissue from Zhongshan Hospital in order to detect the immune status of CXCL13+CD8+T cells and total CD8+T cells. Immunohistochemistry (IHC) and flow cytometry were applied to characterize immune cells and portray the tumor microenvironment (TME) in ccRCC.
Results Intratumoral CXCL13+CD8+T cells abundance was associated with inferior overall survival and disease-free survival. CXCL13+CD8+T cells possessed higher level of immune checkpoints like programmed cell-death protein 1 (PD-1), T-cell immunoglobulin mucin 3 (Tim-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), higher Ki-67 expression and lower tumor necrosis factor α (TNF-α), interferon γ (IFN-γ) expression. Total CD8+T cells in high-level CXCL13+CD8+T cells infiltration subgroup exhibited elevated exhausted markers (PD-1, Tim-3, TIGIT) and descended activated markers (TNF-α, IFN-γ) without quantity variance. Furthermore, the abundance of intratumoral CXCL13+CD8+T cell was correlated with immunoevasive TME accompanied by increased T helper 2 cells, tumor-associated macrophages, Foxp3+ regulatory T cells, TLS and decreased natural killer cells, GZMB+ cells.
Conclusions Intratumoral CXCL13+CD8+T cells infiltration indicated inferior clinical outcome in patients with ccRCC. CXCL13+CD8+T cells possessed increased exhausted markers, decreased effector molecules and better proliferation ability. CXCL13+CD8+T cells abundance impaired total CD8+T cells’ immune function. Intratumoral CXCL13+CD8+T cells abundance was associated with immunoevasive contexture. The abundance of CXCL13+CD8+T cells was an independent prognosticator and a potential immunotherapeutic target marker for ccRCC treatment.
- kidney neoplasms
- tumor microenvironment
- immunotherapy
- immune evasion
- CD8-positive T-lymphocytes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Renal cell carcinoma (RCC) was one of the most common types of malignant tumor in the adult urinary system and represented roughly 2–3% in all tumors around the world.1 One of the histological types of RCC, the clear cell renal cell carcinoma (ccRCC) made up approximately 75% of RCC.2 Surgical procedures worked well in treating localized RCC.3 However, most postoperative patients with metastasis or recurrences had grave prognosis and required further treatment.4 Tyrosine kinase inhibitors (TKI) was the first-line recommendation for metastatic renal cell carcinoma decades ago, but TKI therapy is now criticized for low response rate and long-term therapeutic resistance.5 Recent reports have shown that the novel immune checkpoint blockade (ICB) could lower the progression of tumor when TKI therapy fails.6 ICBs targeting programmed cell-death protein 1 (PD-1)/PD-L1 axis and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) were recommended as the first-line treatment in the intermediate/poor risk-advanced RCC according to National Comprehensive Cancer Network (NCCN) guideline 2020.2 version,7 but the response rate was still relatively low.8 In addition, patients were often classified by validated prognostic risk models such as the International Metastatic RCC Database Consortium (IMDC).9 However, with the approval of combined ICB therapy strategy for treating RCC, IMDC stratification seemed insufficient for the treatment selection due to its obsolete utility in ICB therapy classification.10 Hence, biomarkers and cell subsets that can predict outcome and immunotherapy efficacy are required for clinical use nowadays.
Analysis of tumor microenvironment (TME) has been a novel approach for the selection of ICB therapy in ccRCC recently.11 T cell exhaustion in TME might be responsible for the low response rate of ICB.12 T cell exhaustion generally meant that the state of CD8+T cells was converted from antineoplastic status to immune-functionally impaired status on account of long-term persistence of tumor antigens and/or the suppressive TME.13 Therefore, immunotherapy targeting conversion of T cell exhaustion into activated state has grown vigorous in ccRCC recently. CD8+T cells, one of the most essential cells in TME, were identified as the pioneer and the main force in antitumor effect for a long time.14 Unlike the vast majority of cancers, CD8+T cells infiltration level was reported as an adverse prognostic factor with a large amount of infiltration in ccRCC.15 We also found function of CD8+T cells paradoxical in ccRCC with regard to its antitumor effect in most solid tumors from prior studies.16 Well-validated markers of CD8+T cells that could be prognostic factors or revealers of connections with TME while treated with conventional ICB were entitled to an unmet need.
Especially in ccRCC TME, a great number of tumor-infiltrated lymphocytes, myeloid cells and specific structures presented highly heterogeneous phenotypes.17 As we previously reported, tumor-infiltrating cells, including macrophages,18 mast cells,19 B cells20 and neutrophils,21 could affect the balance of antitumor immunity and immune evasion in ccRCC. An accurate identification of CD8+T cell subsets was required for predicting clinical outcomes and exploring intrinsic mechanisms of antitumor immunity in patients with ccRCC.
Recent studies have demonstrated that chemokine (C-X-C motif) ligand 13 (CXCL13), a specific B-cell attracting chemokine22 as well as a contributor of tertiary lymphoid structure (TLS) formulation,23 serves as a significant factor in the process of tumor proliferation and migration.24 CXCL13 was identified as a potential biomarker for prognosis prediction in RCC,24 colorectal cancer,25 prostate cancer,26 lymphoma27 and non-small cell lung carcinoma.28 ROC curves indicated that CXCL13 could be used to distinguish ccRCC samples from normal kidney samples as well as assessing prognosis and recurrence.24 Recent discoveries have confirmed that CXCL13 could be secreted by T cells like T follicular helper cells (TFH) and tumor-infiltrating CD8+T cells.29 CXCL13 expression was recognized as a poor prognostic factor; however, CXCL13+TFH density was a good prognostic factor,30 which suggested a complicated role of CXCL13 in TME. Prior research put excessive attention on CXCL13 secreted by TFH, while the impact of tumor-infiltrating CXCL13+CD8+T cell subtype still required further exploration.
In this study, we found CXCL13 was highly expressed in exhausted CD8+T cells. We revealed CXCL13+CD8+T cells as a prognostic predictor for inferior survival, covering 755 patients with ccRCC from two cohorts. We also demonstrated the dysfunctional state of intratumoral CXCL13+CD8+T cells in ccRCC. Global CD8+T cells characterization was function-impaired in high CXCL13+CD8+T cells subgroup. CXCL13+CD8+T cells abundance yielded an immunoevasive TME. This study first elaborated the clinical significance and immune correlation of CXCL13+CD8+T cells in ccRCC.
Materials and methods
Study cohort and experimental specimens
The clinicopathological data of 223 consecutive patients with ccRCC from the Department of Urology, Zhongshan Hospital, Fudan University (Shanghai, China) between February 2005 and June 2007 were retrospectively analyzed in this study. All involved patients signed informed consent. Here were the inclusion criteria we followed: (1) undergone radical or partial nephrectomy at our institute; (2) pathologically diagnosed ccRCC; and (3) adults with age ≥18 years. The exclusion standards were: (1) accompanied with other malignant tumors; (2) preoperative systemic treatment; and (3) without complete/available follow-up/clinical/pathological data. Clinicopathological variables included age, gender, tumor size, TNM stage, Eastern Cooperative Oncology Group Performance Status and presence of tumor necrosis. Metastasis or recurrences were defined by images and histopathology information. Several urological pathologists reassessed the tumor histological type, differentiation and stage according to the 2004 WHO criteria31 and the American Joint Committee on Cancer 2010 TNM classification.32 Disease progression was identified via RECIST1.1 criteria.33 The time between curative surgery and recurrence or metastasis was defined as recurrence-free survival (RFS). Overall survival (OS) was defined as the time from the date of surgery to the date of death or last visit. The last collection of follow-up data was performed in June 2019. Archived tissue samples and surgical specimens of all patients were collected for the study. We randomly divided Zhongshan Cohort into two independent sets, that is, Discovery Cohort (n=112) and Internal Validation Cohort (n=111). We also enrolled patients from The Cancer Genome Atlas (TCGA) cohort as External Validation Cohort (n=532). Patient characteristics of TCGA data set were obtained from UCSC Xena (https://xenabrowser.net/datapages/) in January 2020. A total of 532 patients with ccRCC were feasible for the analysis with gene expression statistics, clinical data and follow-up information. Detailed patient characteristics of immunohistochemistry (IHC) analyses were presented in online supplemental table 1.
Supplemental material
In addition, we collected resected tumor specimen from 42 fresh RCC samples undergone radical or partial nephrectomy in stage I and II from March 2019 to August 2019. Of these samples, eight of them were paired with peripheral blood and paratumor tissue. These samples were performed with flow cytometry (FCM) examinations. Detailed patient characteristics of FCM analyses were presented in online supplemental table 2. Flowchart of the patient enrollment for IHC and FCM analysis was listed in online supplemental figure 1.
Immunohistochemistry
Tissue microarray (TMA) construction was described previously.16 34 Single and double color immunohistochemical staining were carried out according to the protocols detailed previously.16 34 TMA slides were scanned on NanoZoomer-XR (Hamamatsu). Two urological pathologists independently counted immune cell number while blinded to the clinical data. Immune cells were evaluated as the average of positive cell number in three representative and independent fields. Representative pictures for IHC were shown in online supplemental figure 2. Details of IHC antibodies were shown in online supplemental table 3.
Flow cytometry
Fresh tissues were collected and then examined as soon as the tumors were resected during surgery. Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized venous blood by lysing solution (BD Biosciences). Surgical tumor tissues and non-tumor tissues were minced and digested with collagenase IV (Sigma), and incubated by RBC lysis buffer (BD Biosciences). Then PBMCs and single cell suspensions were separately stained with membrane markers (monoclonal antibodies) for 40 min at 4℃. After disposed by Fixation/Permeabilization Solution Kit (BD Biosciences) according to manufacture protocol, intracellular protein was stained by corresponding antibodies. Cell suspensions and PBMC were stained with fluorochrome-labeled antibodies and preserved with cell staining buffer. FACS data were collected via BD FACSCelesta and analyzed via Flowjo V.10.0 (Tree Star). Details of FCM antibodies were provided in online supplemental table 4.
Bioinformatics analysis
In the TCGA cohort, the CXCL13+CD8+T cell signature comprised CD2, CD3E, CD5, CD7, CD8A, CD8B, CD27, CD28, HIF-1α, TGF-β1, TGF-β2, TGF-β3, CTGF, SMAD2, SMAD3, CXCL13 and CXCR5.35–37 Signature score was defined as the mean of normalized expression of related genes [log2 (FPKM +1)]. Gene signature was listed in online supplemental table 5. Absolute proportion of CD8+T cells in TCGA Cohort was obtained by CIBERSORT calculation.
The cut-off values were the median values in three cohorts. For CXCL13+CD8+T cells density in Discovery Cohort,≥40/ mm2 was defined as high and <40/ mm2 was defined as low. For holistic CD8+T cells density in Discovery Cohort, ≥86/ mm2 was defined as high and <86/ mm2 was defined as low. We directly used this threshold to test in Internal Validation Cohort. The cut-off value of CD8+T cell proportion and CXCL13+CD8+T cell signature level was 9.20% and 3.1493, respectively, in External Validation Cohort (TCGA Cohort). The cut-off value of TLS was defined as absence or presence. For FCM analyses, we also used the median proportion of CXCL13+CD8+T cells/ CD8+T cells (0.389) as the cut-off value.
Statistical analysis
Continuous variables were analyzed with Mann-Whitney test as appropriate. The relationship between CXCL13+CD8+T cell density and patient characteristics was evaluated by χ2 test. The correlation analyses were evaluated with non-parametric (Mann-Whitney U test and Spearman’s test) tests. Wilcoxon matched-pairs signed rank test was used for non-parametric pairing t-tests. Kruskal-Wallis test was applied to detect the association between CXCL13+CD8+T cells infiltration level and tumor stage. Results were shown in mean±SD. Kaplan-Meier method and Log rank test were applied to demonstrate survival curves between different groups in the cohort. Multivariate analyses of cox regression model were applied to estimate HRs and 95% CIs. All statistical analyses were performed with SPSS V.19.0 (IBM), R software V.4.0.2 (R Foundation for Statistical Computing, http://www.r-project.org/), Graphpad V.8.0 and Medcalc V.15. The heatmap was performed with use of https://software.broadinstitute.org/morpheus/. All of these analyses were performed at the statistically significant level of 5% (p<0.05) and two tailed.
Results
Tumor-infiltrated CXCL13+CD8+T cells are enriched in ccRCC tissue
The large amount of tumor-infiltrated CD8+T cells in ccRCC could exhibit various immune functional phenotypes. First, we found that exhausted T cells could be defined via expression levels of inhibitory receptors PD-1 and Tim-3. More importantly, CXCL13 expression was upregulated in these exhausted T cells (online supplemental figure 3), so we put the emphasis on CXCL13+CD8+T cells.
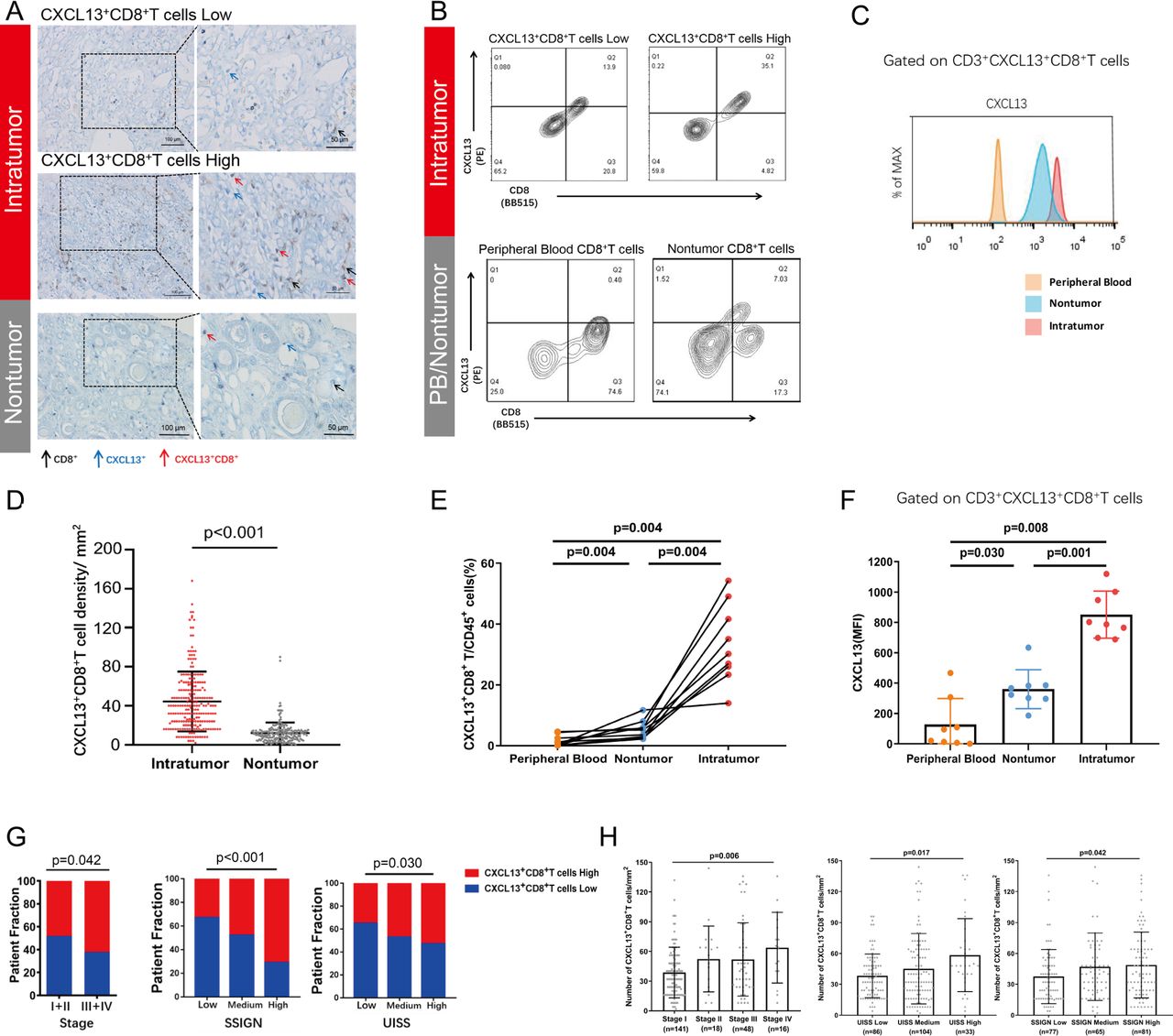
The existence of CXCL13+CD8+T cells was confirmed by double-stained IHC. IHC staining images that represented different infiltration levels of CXCL13+CD8+T cells in intratumor tissue and nontumor tissue (black arrow: CD8+T cells, blue arrow: CXCL13+ cells, red arrow: CXCL13+CD8+T cells) through two enlargement factors (200×, 400×) were presented (figure 1A). We found that CXCL13+CD8+T cells were more abundant in intratumoral tissues compared with nontumor tissues (p<0.001, figure 1D). We also conducted the flow cytometry analyses of fresh ccRCC specimens (figure 1B,C). Similar results were obtained through comparison of cell proportion and median fluorescence intensity (MFI) level (figure 1E,F). IHC data also revealed the positive correlation between the infiltration level of CXCL13+CD8+T cells and tumor stage (p=0.046), UCLA Integrated Staging System (UISS) stage (p<0.001), and the stage size grade and necorsis score (SSIGN) grade (p=0.030) (figure 1G). More CXCL13+CD8+T cells were infiltrated in tumor tissue in later tumor stage (p=0.006), later UISS stage (p=0.017) and higher SSIGN grade (p=0.042) in ccRCC (figure 1H). Conclusively, these results suggested that tumor-infiltrating CXCL13+CD8+T cells were potentially involved in ccRCC tumor progression.

Intratumoral CXCL13+CD8+T cells accumulate in clear cell renal cell carcinoma (ccRCC) and correlate with tumor progression. (A) Images at 200×, 400× magnification showing the residence of single-stained CD8+T (black arrow), CXCL13+ (blue arrow) and doubled-stained CXCL13+ CD8+ T cells (red arrow) for different infiltration levels of CXCL13+CD8+T cells in ccRCC tissue and non-tumor tissue. Scale bars: 100 µm, 50 µm. (B, C) Representative images of proportion/median fluorescence intensity (MFI) level of CXCL13+CD8+ T cells in different infiltration level in tumor tissue and in peripheral blood, non-tumor tissue. Cells were pregated on CD3, CD45 and CD8. (D) Quantification of CXCL13+CD8+T cells infiltrated in intratumoral tissue (n=223) versus corresponding non-tumoral tissue (n=215) in ccRCC. (E) CXCL13+CD8+T cells proportion of CD45+ cells in peripheral blood, non-tumor tissue and tumor tissue via flow cytometry (FCM) analyses. (F) MFI level of CXCL13 in peripheral blood, non-tumor tissue and tumor tissue via FCM analyses. (G) The distribution of high and low CXCL13+CD8+T cells in different stages of patients with ccRCC (classified by tumor stage, SSIGN grade and UISS stage). (H) The numbers of double stained CXCL13+CD8+ T cells/ mm2 in different stages of patients with ccRCC (classified by tumor stage, SSIGN grade and UISS stage). Mann-Whitney U test, Kruskal-Wallis test, Wilcoxon matched-pairs signed rank test and χ2 test were applied. Data were shown as mean±SD. All reported p values were two sided.
Accumulation of CXCL13+CD8+T cells determines poor prognosis in patients with ccRCC
Since the infiltration level of CXCL13+CD8+T cells might relate to tumor progression, the clinical merit of this specific CD8+T subtype required further exploration. Next, we employed Kaplan-Meier analyses to investigate the prognostic value of CXCL13+CD8+T cells in patients with ccRCC from three independent cohorts (Discovery cohort, Internal Validation Cohort and External Validation Cohort). CD8+T cells alone failed to define the prognosis of patients with ccRCC in these three cohorts (discovery OS p=0.164, RFS p=0.200, figure 2A; internal validation OS p=0.340, RFS p=0.190, figure 2C; external validation OS p=0.784, RFS p=0.770, figure 2E), which revealed high heterogeneity for CD8+T cells in ccRCC. However, we observed that high level of tumor-infiltrating CXCL13+CD8+T cells represented significantly shorter OS and RFS than low level of tumor-infiltrating CXCL13+CD8+T cells in patients with ccRCC (OS p=0.008; RFS p=0.029; figure 2B) in discovery cohort. Shorter OS in CXCL13+CD8+T cell high subgroup was observed in two validation cohorts as well (internal validation OS p=0.018, external validation OS p=0.010, figure 2D). Although the CXCL13+CD8+T cell signature failed to predict RFS in external validation cohort (RFS p=0.085, figure 2F), high infiltration of CXCL13+CD8+T cells indicated shorter RFS in internal validation cohort (RFS p=0.011, figure 2D). Cox regression multivariate analysis demonstrated the infiltration level of CXCL13+CD8+T cells as an independent prognostic factor in three cohorts (online supplemental table 6). Conclusively, these results above proved that infiltration level of CXCL13+CD8+T cells could serve as an independent prognosis predictor for poor outcomes in ccRCC.

The prognostic merit of intratumoral CXCL13+CD8+T cells in patients with clear cell renal cell carcinoma (ccRCC). (A, C, E) overall survival (OS) curves and recurrence-free survival (RFS) curves according to CD8+T cells infiltration level of patients in three independent sets. (B, D, F) OS curves and RFS curves according to CXCL13+CD8+T cells infiltration level of patients in three independent sets. Log-rank test was performed for Kaplan-Meier curves. Log-rank p values were shown. All reported p values were two sided.
Intratumoral CXCL13+CD8+T cells exhibit highly exhausted signature in ccRCC
In order to explain the prognostic merit of CXCL13+CD8+T cells, we felt the necessity to explore the status of intratumoral CXCL13+CD8+T cells. We conducted FCM analyses on 42 fresh ccRCC tumor tissues in order to detect the immune function of intratumoral CXCL13+CD8+T cells. CXCL13+CD8+T cells within ccRCC tissues exposed higher level of immune checkpoints (PD-1, Tim-3, T cell immunoreceptor with Ig and ITIM domains (TIGIT) and CTLA-4) compared with CXCL13−CD8+T cells (PD-1 p=0.002, Tim-3 p<0.001, TIGIT p=0.002; CTLA-4 p=0.007; figure 3A). Additionally, CXCL13+CD8+T cells had relatively better proliferation ability (Ki67 p=0.008, figure 3B). Besides, we found that intratumoral CXCL13+CD8+T cells produced fewer effective molecules (tumor necrosis factor (TNF)-α p=0.026, interferon (IFN)-γ p=0.028, figure 3C), which might explain the exhausted state of CXCL13+CD8+T cells. The expression of cytotoxic molecules (PRF-1, GZMB) and degranulation marker (CD107a), however, showed no significant difference between CXCL13+CD8+T cells and CXCL13−CD8+T cells (PRF-1 p=0.447, GZMB p=0.994, CD107a p=0.504, figure 3C). In addition, the proportion of CXCL13+CD8+T cells was correlated positively with the proportion of Tim-3+CD8+T cells and negatively with the proportion of IFN-γ+CD8+T cells (Tim-3 p=0.005, Spearman r=0.558; IFN-γ p=0.047, Spearman r=−0.489, figure 3D). The relationship of CXCL13+CD8+T cells proportion and other markers of CD8+T cells (PD-1, TIGIT, CTLA-4, Ki67 and TNF-α) were shown in online supplemental figure 4). Conclusively, these findings revealed that the specific subset, CXCL13+CD8+T cells, exhibited highly exhausted status, diminished antitumor function and better proliferation ability.
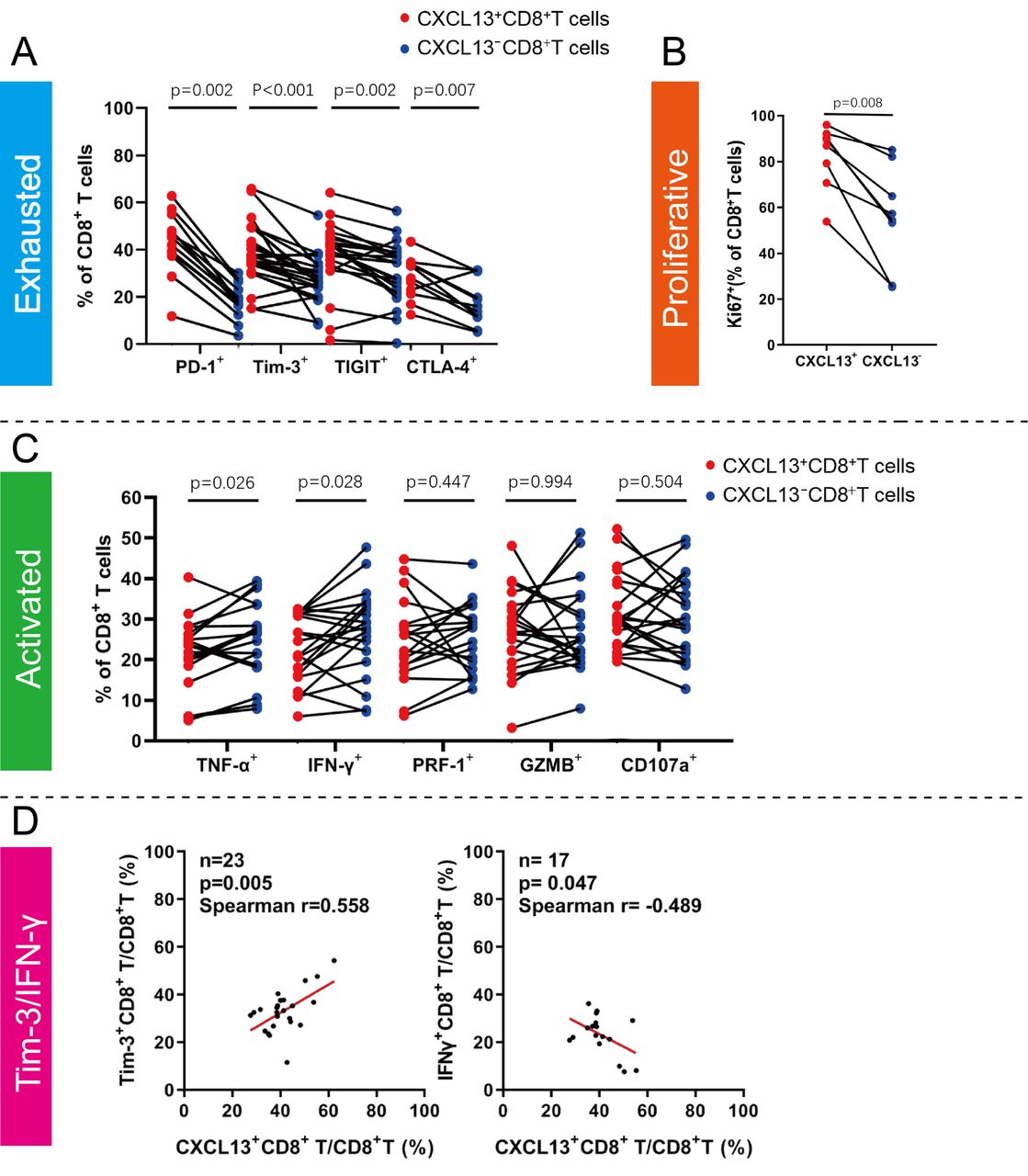
Intratumoral CXCL13+CD8+T cells expose a distinct subset with exhausted features in clear cell renal cell carcinoma (ccRCC). (A) Flow cytometry (FCM) analysis of inhibitory receptors (programmed cell-deathprotein 1 (PD-1), Tim-3, T cell immunoreceptor with Ig and ITIM domains (TIGIT) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)) expression in CXCL13+CD8+T cells within ccRCC fresh tumor tissues. (B) FCM analysis of proliferation molecules (Ki-67) expression in CXCL13+CD8+T cells within ccRCC fresh tumor tissues. (C) FCM analysis of effector markers and degranulation activity (tumor necrosis factor (TNF)-α, interferon (IFN)-γ, PRF-1, GZMB and CD107a) expression in CXCL13+CD8+T cells within ccRCC fresh tumor tissues. (D) Correlation assessed by Spearman’ s correlation analysis and linear regression analysis between proportion of CXCL13+CD8+T cells and Tim-3+CD8+T cells, IFNγ+CD8+T cells. Wilcoxon matched-pairs signed rank test was applied. All reported p values were all two sided.
Intratumoral CXCL13+CD8+T cells abundance are associated with dysfunctional CD8+T cells infiltration in ccRCC
Based on the results on the immune function of intratumoral CXCL13+CD8+T cells and the ‘paradoxical’ role of CD8+T cells in antitumor immunity, we assumed that the immune function of overall CD8+T cells might get restrained in ccRCC. The global characterization of CD8+ T cells was subsequently investigated based on CXCL13+ CD8+ T cell infiltration level. CD8+T cells in CXCL13+CD8+T cells high infiltration group possessed elevated immune checkpoints including PD-1, Tim-3 and TIGIT (p<0.001, p=0.023, p=0.024, respectively, figure 4B). Additionally, effective molecules like IFN-γ and TNF-α expressed by CD8+T cells were fewer in CXCL13+CD8+T cells high infiltration group (p=0.029, p=0.010, respectively, figure 4C). Though these CD8+T cells possessed increased exhausted markers and decreased activated molecules, we found the proportion, cytotoxic molecules (GZMB and PRF-1), degranulation capability (CD107a) and proliferation ability (Ki-67) of CD8+T cells showed no significant difference in CXCL13+CD8+T cells high/low infiltration group (figure 4A,C,D). As we mentioned above in figure 2, infiltration level of CD8+T cells alone could not define the prognosis of ccRCC patients. Merged survival curves, divided via CXCL13+CD8+T cells infiltration level and CD8+T cells infiltration level, indicated that combined division by CXCL13+CD8+T cells level and CD8+T cells level could serve as a prognosticator for patients with ccRCC as exhibited in figure 4E (p<0.001). We found that CD8+T cells abundance possessed the ability to independently define poor prognosis in CXCL13+CD8+T cells high infiltration group (p=0.009, online supplemental figure 5A). Nevertheless, CD8+T cells alone still could not define the prognosis in CXCL13+CD8+T cells low infiltration group (p=0.141, (online supplemental figure 5B). Conclusively, these results revealed that intratumoral CXCL13+CD8+T cells abundance might correlate with dysfunctional CD8+T cells infiltration, which led to poor outcomes in patients with ccRCC.
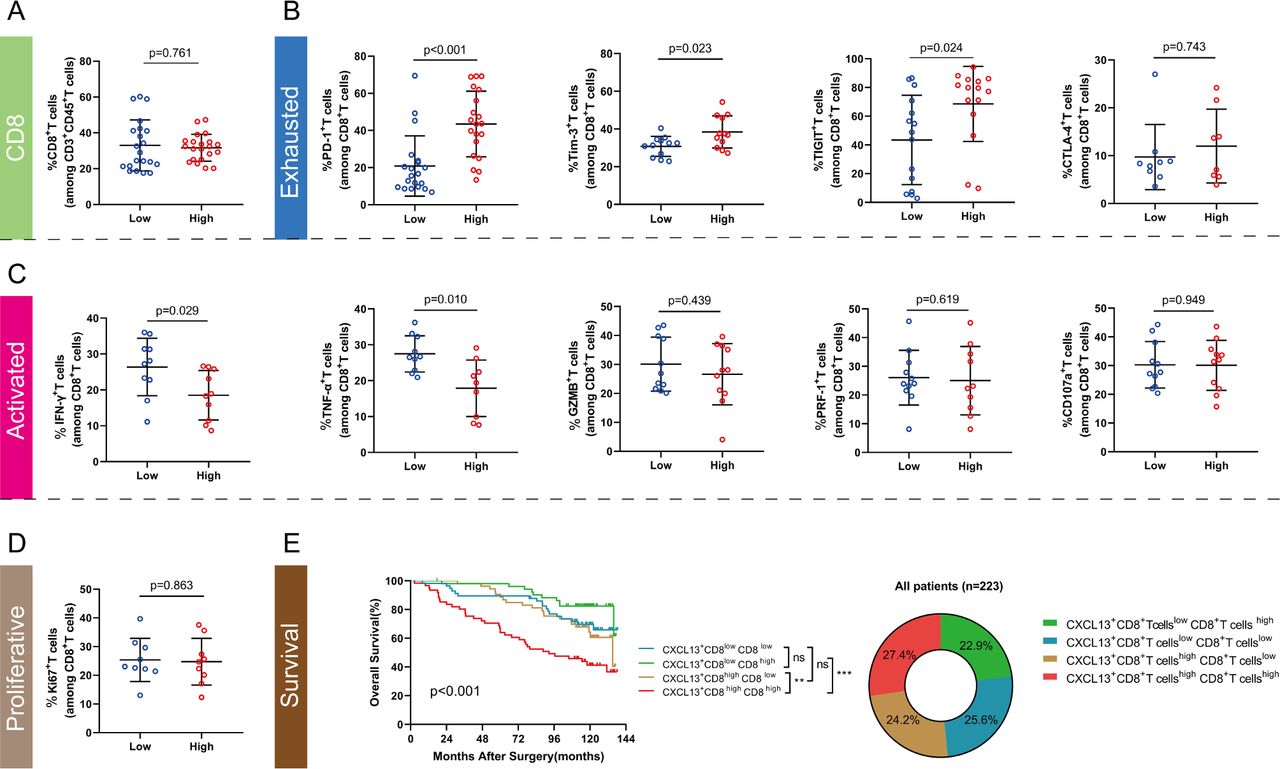
Intratumoral CXCL13+CD8+T cell high infiltration impairs total CD8+T cell immune function in patients with clear cell renal cell carcinoma (ccRCC). (A) CD8+T cell infiltration level in CXCL13+CD8+T cell high/low subgroup. (B, C, D) Expression of immune exhausted markers, activated markers, cytotoxic molecules, degranulation molecule and proliferative marker for total CD8+T cells in CXCL13+CD8+T cell high/low subgroup. (E) Merged Kaplan-Meier curves of overall survival (OS) for the combination of CXCL13+CD8+T cells with CD8+T cells infiltration. Mann-Whitney U test was applied. Log-rank test was applied for Kaplan-Meier curves. All reported p values were all two sided. IFN, interferon; PD-1, programmed cell-death protein 1.
Intratumoral CXCL13+CD8+T cells infiltration is associated with immunoevasive contexture in ccRCC microenvironment
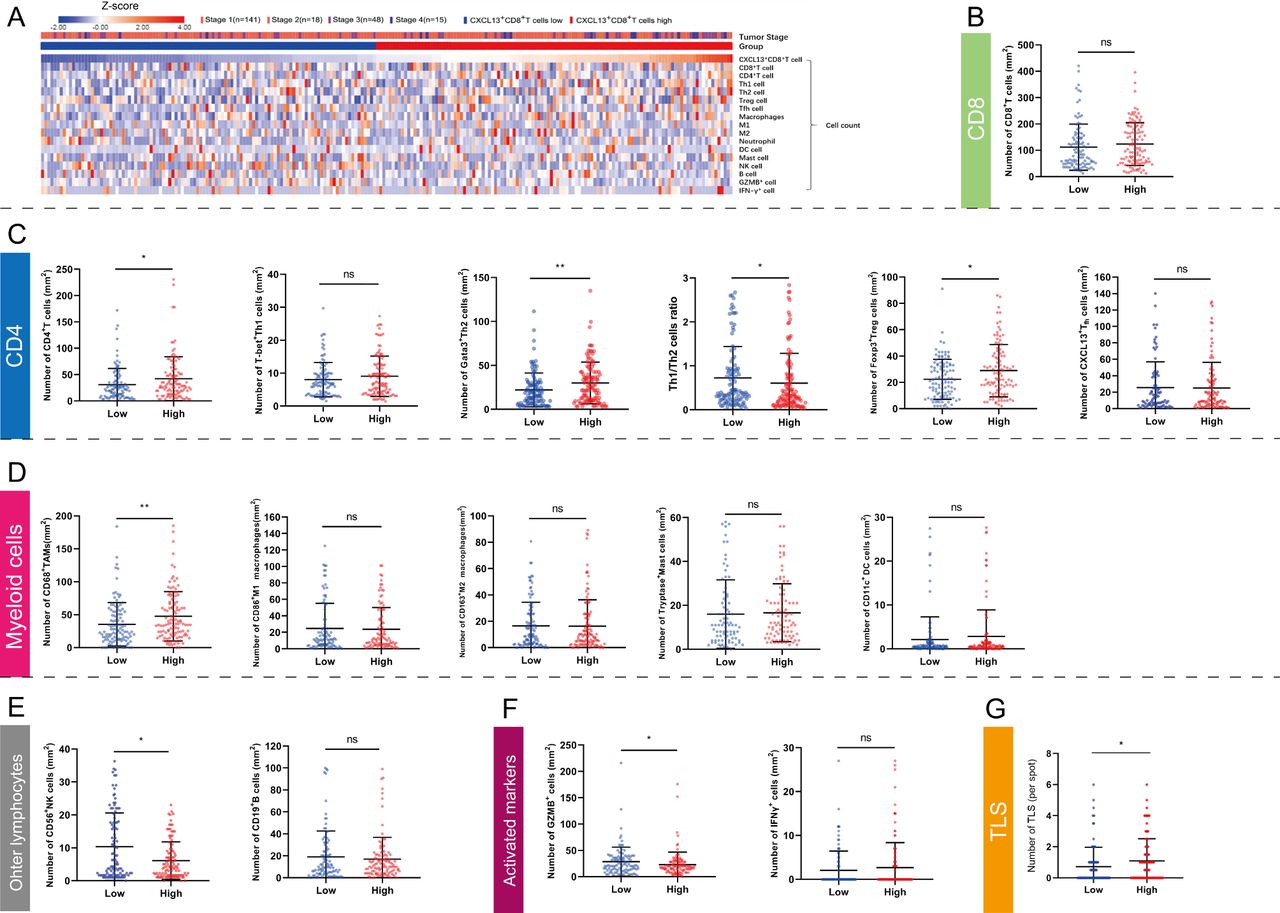
We found that tumor-infiltrating CXCL13+CD8+T cells represented a highly exhausted subtype. Therefore, the specific TME that induced the switch of exhausted T cells provoked our interest. Heatmap was used for intuitionistically exhibiting the relationship between the level of CXCL13+CD8+T cells and tumor stage, immune-related cells (CD8+T cells, CD4+T cells, Th1 cells, Th2 cells, Treg cells, TFH cells, TAMs, M1 macrophages, M2 marcophages, neutrophils, dendritic cells [DC], mast cells, natural killer [NK] cells and B cells) and immune-activated markers (GZMB, IFN-γ) (figure 5A). The number of total CD8+T cells was not related with CXCL13+CD8+T cells infiltration level (figure 5B), which was corresponded with results above in figure 4. Intriguingly, we found that microenvironment with high level of intratumoral CXCL13+CD8+T cells contained more CD4+T cells, Th2 cells (Gata-3+CD4+T cells), Treg cells (Foxp3+CD4+T cells) and TAMs (CD68+ cells) (figure 5C,D). The level of intratumoral CXCL13+CD8+T cells showed negative connection with Th1/Th2 ratio (figure 5C). Additionally, microenvironment with high level of intratumoral CXCL13+CD8+T cells possessed decreased NK cells and GZMB+ cells (figure 5E,F). The density of CXCL13+CD8+T cells was correlated positively with the amount of TLS (p=0.026, figure 5G). Though TLS was recognized as a trigger for antitumor response,38 we found that TLS amount could not indicate clinical outcomes for patients with ccRCC (online supplemental figure 6). Conclusively, tumor-infiltrating CXCL13+CD8+T cells were assumed to interrelate with immunoevasive contexture in ccRCC TME.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Intratumoral CXCL13+CD8+T cells demonstrate an immunoevasive landscape of tumor microenvironment in clear cell renal cell carcinoma (ccRCC). (A) Heatmap showing infiltrating levels of immune cells and molecules according to CXCL13+CD8+T cells. Expression was normalized via Z-score. (B) Quantification of CD8+T cells in CXCL13+CD8+T cell high/low subgroup. (C, D, E) Quantification of CD4+T cell subsets, myeloid cell subsets and other lymphocytes in CXCL13+CD8+T cell high/low subgroup. (F) Quantification of activated markers in CXCL13+CD8+T cell high/low subgroup. (G) Quantification of tertiary lymphoidstructures (TLS) in CXCL13+CD8+T cell high/low subgroup. Mann-Whitney U test was applied. All reported p values were two sided. *P<0.05, **p<0.01 and ***p<0.001, ns refered to not statistically significant.
Discussion
CD8+T cells, one of the most essential cells in TME, were identified as antitumor cells for a long time.14 Though a large number of CD8+T cells were infiltrated in ccRCC, studies reported that these CD8+T cell did not show functional status. Unlike the vast majority of cancers, CD8+T cell infiltration level was reported as an adverse prognostic factor in ccRCC.15 High degree of CD8+T cell infiltration was correlated with an increased expression of immune evasive markers like PD-1, PD-L1, PD-L2 and CTLA-4,39 and an elevated number of immune cells like M2-polarized macrophages, resting mast cells and resting memory CD4+T cells.17 CD8+T cells presented heterogeneous subtypes and showed inextricable connections with TME in ccRCC. Further studies on subpopulations of CD8+T cells and their connections with immune contexture were still necessary for thorough understanding. In this study, we focused on the subset CXCL13+CD8+T cells and elaborated the clinical significance and immune correlation of CXCL13+CD8+T cells in ccRCC.
In CD8+ inflamed ccRCC TME, high CXCL13+CD8+T cell level presented poor prognosis. In addition, in TME with high CXCL13+CD8+T cell infiltration, copious CD8+T cells presented poor prognosis. This could be the result of immune dysfunctional CD8+T cells when an elevated amount of CXCL13+CD8+T cells infiltrated in tumor tissue. However, when there were few CXCL13+CD8+T cells with high CD8+T cell infiltration, patients seemed to have the longest survival, which meant that when there were few CD8+T cells in aggregate, CXCL13+CD8+T cells infiltration level seemed to lose its capability of indicating survival for patients. As to recent studies, subtypes of ccRCC could be quartered via both immune level and stromal level.38 Patients with a relatively small amount of CD8+T cells infiltrated in ccRCC tumor tissue might indicate CD8- inflamed (more fibroblasts or endothelial cells infiltrated) or immune desert (metabolic or vascular endothlial growth factor [VEGF] immune desert) microenvironment.40 These statuses of ccRCC TME show fewer connections with tumor-infiltrated CD8+T cells but might be more sensitive to other cells or signal pathways. This result proved that combining CXCL13+CD8+T cells infiltration level and CD8+T cells level could be a good stratification strategy in further judgment for patients.
Although CXCL13+CD8+T cells expressed high level of immune checkpoint receptors, these cells possessed relatively fewer effective molecules. CD8+T cells formulated its exhaustion states continuously on account of the exposure to suppressive gradients in TME.41 As a marker of exhausted CD8+T cells, CXCL13 might serve as a feedback of immunosuppressive microenvironment and T cell dysfunction. Thommen et al reported that CXCL13 expression of PD-1high CD8+ T cells could be a sign for potential response to PD-1 blockade in NSCLC.42 In addition, for pretreatment, but not post treatment, CXCL13 was differentially expressed by specific CD8+T cells and TME in responders versus non-responders in patients with melanoma.43 Furthermore, though density of CXCL13+CD8+T cells correlated positively with TLS presence, this could not reverse the immunosuppressive tendency in ccRCC TME. Taking the positive association of CXCL13+CD8+T cell infiltration and TLS amount into account, we assumed that a specific subset of exhausted CD8+T cells secreted CXCL13 to recruit other activated lymphocytes and thus to postpone the tumor progression in TME.
CXCL13 expression was elevated in tumor tissues and recognized as potential prognostic predictor in various tumors.25 28 However, the mechanism of CXCL13 upregulation in tumor tissues remained obscure. HIF-1-TGF-β pathway was reported to stimulate activated CD8+T cells to secret CXCL13.44 CD4+T cells (not TH1) and TAMs were the main force to produce TGF-β in TME.45 46 Studies also reported that CXCL13 promoted progression through PI3K/AKT pathway in ccRCC.47 CXCL13 interacted with its receptor chemokine (C-X-C motif) receptor 5 (CXCR5) to regulate CD8+T cell immunity.47 The immunoevasive contexture and T cell function were reversed by the inhibition of CXCL13 through CXCR5/ERK signaling in breast cancer.48 On our study on TME in ccRCC, we found that the infiltration level of CXCL13+CD8+T cells was positively correlated with the infiltration level of CD4+T cells, Th2 cells, Treg cells and TAMs, which was consistent with prior findings. We assumed that TGF-β might serve as a crucial molecule in connecting CXCL13+CD8+T cells and TME as well as stimulating CXCL13 secretion by CD8+T cells in ccRCC.
Honestly, our study still had flaws and required follow-up research. Our study was retrospective and needed a larger validation cohort to confirm our conclusions. The switch of TME in ccRCC was multilayered, multicell participated and was a sophisticated system,11 which deserved further exploration. Myeloid cells and CD4+T cell subsets’ role on CXCL13+CD8+T cells remained obscure. Studies can also focus on what effect might take place with the inhibition of CXCL13 in ccRCC to testify the immune target function of CXCL13+CD8+T cells. Considering the close relationship between CXCL13 secretion and PD-1 expression in CD8+T cells, we assumed that ICB+ anti-CXCL13 combination therapy might serve better in treating patients with ccRCC.
Conclusion
In summary, our study first identified the infiltration level of CXCL13+CD8+T cells as an independent prognosticator for poor OS and RFS in ccRCC. We also confirmed that CXCL13+CD8+T cells were a highly exhausted subtype of intratumoral CD8+T cells with impaired immune function and exhausted markers. High infiltration of CXCL13+CD8+T cells could also demonstrate the immunoevasive contexture with an impaired CD8+T cell immunity, more pro-tumor cells and fewer antitumor factors. These results indicated the clinical significance of CXCL13+CD8+T cells in ccRCC.
Acknowledgments
The authors thank Dr Lingli Chen (Department of Pathology, Zhongshan Hospital, Fudan University, Shanghai, China) and Dr Yunyi Kong (Department of Pathology, Fudan University Shanghai Cancer Center, Shanghai, China) for their excellent pathological technology help.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SD, HZ, ZL and KJ contributed equally.
Contributors SD, HZ, ZL and KJ for acquisition of data, analysis and interpretation of data, statistical analysis and drafting of the manuscript. WJ, ZW, ZL, YX, JW, YC, QB, YX, LL, YZ and LX for technical and material support. YQ, JG and JX for study concept and design, analysis and interpretation of data, drafting of the manuscript, obtained funding and study supervision. All authors read and approved the final manuscript.
Funding This study was funded by grants from National Natural Science Foundation of China (31770851, 81772696, 81872082, 81902563, 81902898, 81974393, 82002670), Shanghai Municipal Natural Science Foundation (19ZR1431800), Shanghai Sailing Program (18YF1404500, 19YF1407900, 20YF1406100, 20YF1406200), Shanghai Municipal Commission of Health and Family Planning Program (20174Y0042, 201840168) and Fudan University Shanghai Cancer Center for Outstanding Youth Scholars Foundation (YJYQ201802). All these study sponsors have no roles in the study design, in the collection, analysis and interpretation of data.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study followed the Declaration of Helsinki and was approved by the Clinical Research Ethics Committee of Zhongshan Hospital of Fudan University. The Clinical Research Ethics Committee of Zhongshan Hospital approved the use of human specimens in this study (Registration No. B2015-030). Signed informed consent was obtained from each patient.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data generated that are relevant to the results presented in this article are included in this article. Other data that were not relevant for the results presented here are available from the corresponding author Dr Xu on reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.