Article Text

Abstract
Background As heterogeneous tumors develop in the face of intact immunity, tumor cells harboring genomic or expression defects that favor evasion from T-cell detection or elimination are selected. For patients with such tumors, T cell-based immunotherapy alone infrequently results in durable tumor control.
Methods Here, we developed experimental models to study mechanisms of T-cell escape and demonstrated that resistance to T-cell killing can be overcome by the addition of natural killer (NK) cells engineered to express a chimeric antigen receptor (CAR) targeting programmed death ligand-1 (PD-L1).
Results In engineered models of tumor heterogeneity, PD-L1 CAR-engineered NK cells (PD-L1 t-haNKs) prevented the clonal selection of T cell-resistant tumor cells observed with T-cell treatment alone in multiple models. Treatment of heterogenous cancer cell populations with T cells resulted in interferon gamma (IFN-γ) release and subsequent upregulation of PD-L1 on tumor cells that escaped T-cell killing through defects in antigen processing and presentation, priming escape cell populations for PD-L1 dependent killing by PD-L1 t-haNKs in vitro and in vivo.
Conclusions These results describe the underlying mechanisms governing synergistic antitumor activity between T cell-based immunotherapy that results in IFN-γ production, upregulation of PD-L1 on T-cell escape cells, and the use of PD-L1 CAR-engineered NK cells to target and eliminate resistant tumor cell populations.
- combined modality therapy
- immunotherapy
- adoptive
- immunotherapy
- head and neck neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Immunotherapy is now a powerful standard-of-care tool used in the treatment of solid cancers. Pembrolizumab, a monoclonal antibody which inhibits the programmed death receptor-1 (PD-1)/programmed death ligand-1 (PD-L1) axis, has gained US Food and Drug Administration approval for the first-line treatment of numerous recurrent or metastatic solid epithelial tumors, including head and neck squamous cell carcinoma (HNSCC).1 However, overall response rates remain low at 15%–35%. Additional immunotherapy approaches including therapeutic cancer vaccines and adoptive T-cell transfer are being clinically studied in many solid cancers and hold promise for the treatment of HNSCC.2–5 Notably, existing immunotherapies in clinical use are based on and limited by the ability of T cells to detect and kill cancer cells.
For a tumor to be established and clinically detectable, it must first evade the immune system. Mutations and expression defects that favor escape from T-cell detection and elimination are selected for in the face of immune pressure in progressing tumors.6 This process, termed cancer immunoediting, is largely driven by the ability of T cells to recognize and kill tumor cells that present tumor antigens via human leukocyte antigen (HLA) restriction elements.7 Mutations and expression defects in antigen processing and presentation machinery (APPM) are associated with resistance to immune checkpoint blockade and adoptive transfer of T cell receptor (TCR)-engineered T cells.8–10 Considering the subclonal structure and extensive intratumor heterogeneity reported in different carcinomas,11 12 heterogeneous expression of APPM genes may give rise to the presence of one or more tumor cell subpopulations that can escape baseline and immunotherapy-enhanced T-cell detection or elimination. In such tumors, T cell-based immunotherapy alone may offer little to no chance of cure.
One approach to overcome this mechanism of resistance to T cell-based immunotherapy may be the addition of immunotherapy that does not rely on intracellular antigen processing and presentation. The combination of T cell-based and natural killer (NK) cell-based immunotherapy may represent a rational alternative treatment strategy as NK cells can detect and kill tumor cells independent of antigen and HLA.13 The recent clinical development of irradiated NK cell lines (high-affinity NK cells (haNKs)), which can be engineered to increase cytotoxic potential and target-cell specificity, may provide a source of ‘off-the-shelf’ cellular therapy available to whole populations.14 These cultured NK cells can also be engineered to express a chimeric antigen receptor (CAR) to aid in targeting and potency.15 16 Because PD-L1 is expressed on antigen-positive and antigen-negative tumor cells within a heterogeneous tumor and generally has low expression on normal tissues in the absence of interferon,17 PD-L1 is a compelling therapeutic target.
To study mechanisms of APPM defect-mediated T-cell escape, we developed an experimental model using carcinoma cells that were engineered to be either sensitive or resistant to T-cell killing through control of antigen and HLA expression. Using our models of T-cell escape, we demonstrated synergistic activity with combination T and CAR-engineered NK cell therapies in multiple models of HNSCC. Following demonstration of T-cell escape of APPM defective tumor cells, we demonstrated the ability of irradiated haNKs engineered to express a CAR targeting PD-L1 (PD-L1 t-haNK) to prevent the clonal selection of T-cell escape variants in vitro and in vivo. Mechanistically, the ability of PD-L1 t-haNKs to eliminate T-cell escape variants was dependent on PD-L1 expression induced by T-cell interferon gamma (IFN-γ). The ability of PD-L1 t-haNKs to prevent the clonal selection of T-cell escape variants was abrogated when tumor cells that escaped T-cell killing lacked PD-L1 or STAT1. These results suggest that T cell-based immunotherapy, through induction of IFN-γ and upregulation of PD-L1 on T-cell escape variant tumor cells, can prime heterogeneous tumors to treatment with PD-L1 t-haNKs. Our results demonstrate the efficacy of combination T and CAR-engineered NK cell therapy and provide the preclinical rationale for the clinical evaluation of the safety and tolerability of this strategy in patients with advanced or relapsed solid tumors.
Methods
Bioinformatic analysis
Published single-cell RNA-seq data were obtained from and processed as described by Puram et al12; processed expression data were downloaded from Gene Expression Omnibus (GSE103322) and subjected to log2 transformation after adding one to each value. Analysis was performed using R V.3.6.2; heatmaps were generated using pheatmap.18 19
Multiplex immunofluorescence
Tumor biopsies from advanced stage (III/IV) patients with human papillomavirus (HPV)-negative HNSCC were obtained at the time of standard-of-care surgical resection under institutional review board-approved protocol NCT03429036 with informed consent. Sections were stained using Opal multiplex kits, according to the manufacturer’s protocol (PerkinElmer), for a panel of 4′,6-diamidino-2-phenylindole (DAPI), pan-cytokeratin (clone AE1/AE3), HLA class I (EMR8-5) and CD3 (SP7). Positive and negative controls included normal human tonsil and unstained sections (no primary antibody), respectively. After optimization, staining of all analyzed sections was done in a single automated assay. Images were captured by a PerkinElmer Vectra Polaris. Multispectral fluorescent images were analyzed using QuPath software.20 Following annotation of entire tumor sections into tumor and stromal tissue based on cytokeratin staining, HLA positivity and CD3+ cells were quantified using automated detection based on universally applied fluorescence detection thresholds.
Cell lines
UM-SCC-1, UM-SCC-47 and UM-SCC-109 cells were obtained from Drs T E Carey, M E Prince, and C R Bradford at the University of Michigan (Ann Arbor, Michigan, USA). JHU-029 cells were obtained from Dr D Sidransky at Johns Hopkins University (Baltimore, Maryland, USA). All cell lines were passaged two times per week, used at a low (<30) passage number and serially verified to be free from mycoplasma infection.
Retroviral transduction of cell lines
Retroviral expression constructs (pMSGV2) encoding HPV 16 E7 with an NGFR tag (in the same open reading frame) or HLA-A*02 were generated. These plasmids and plasmids encoding VSV-G were cotransfected into 293GP cells; 48-hour viral supernatant was transduced into target cells. For all engineered cell lines, verification of transduction of HLA-A*02 was performed by flow cytometry, and verification of HPV 16 E7 expression was performed by flow cytometry (NGFR tag) and quantitative PCR (qPCR) for HPV 16 E7 transcripts. For each model, fluorescent-activated cell sorting (FACS) was used to enrich to purity of >99%.
CRISPR Cas9 gene editing
CRISPR Cas9 gene editing of genes corresponding to IFN-γ receptor 1 (IFNGR1), STAT1, B2M, and PD-L1 was performed commercially by Synthego (Redwood City, California, USA). Sanger sequencing was performed to validate clonal knockout efficiency. FACS was used to enrich to purity of >99% when needed.
Quantitative PCR
RNeasy Mini Kits (Qiagen, Catalog #74104) were used to isolate RNA according to the manufacturer’s protocol. cDNA libraries were created using the SuperScript IV First-Strand Synthesis System (Thermo Fisher, catalog number 18091050). qPCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad Catalog #1725121) on an Applied Biosystems Quant Studio 6 Flex. Primers for HPV 16 E7 were synthesized (forward 5′-CAGCTCAGAGGAGGAGGATG-3′, reverse 5′-TGGAAGACCTGTTAATGGGC-3′) and expression levels were compared with a housekeeping gene (GAPDH).
HPV 16 E7 TCR T cells
Healthy donor peripheral blood mononuclear cells (PBMCs) were retrovirally transduced to express an HPV 16 E711-19-specific, HLA-A*02-restricted TCR as described previously.21 All in vitro and in vivo experiments were performed with the same batch of expanded T cells. Cryopreserved stocks of T cells were thawed and rested overnight in 50/50 AIM-V/RPMI-1640 with 5% human serum and 3000 IU/mL interleukin (IL)-2 for 24 hours prior to use in all experiments.
NK cell therapy products
haNKs and PD-L1 CAR-engineered high-affinity NK cells (PD-L1 t-haNKs)15 16 were obtained from NantKwest through a Cooperative Research and Development Agreement. Cells were irradiated (15 Gy) prior to cryopreservation and shipment from NantKwest to the NIH for experimental use.
Impedance analysis
Real-time impedance analysis was performed using the xCELLigence RealTime Cell Analysis platform per manufacturer recommendations. Tumor cells were plated (1×104 to 10.5×104/well) and allowed to adhere and expand for 24 hours prior to the addition of effector cells. Triton X-100 (0.1%) was used as a positive lysis control. Mean values without error bars are shown on impedance plots for clarity. All experiments were performed in technical triplicate and replicated in at least two independent experiments. Percentage loss of target cell index was quantified as 1−(experimental cell normalized cell index/control cell normalized cell index) for a given timepoint. Unless otherwise noted, impedance assays in this study were normalized to use an effector to target ratio (E:T) of 2:1 for HPV 16 E7 T cells and PD-L1 t-haNKs.
Flow cytometry
Cells were suspended in a 1% bovine serum albumin solution, and Fc receptor (CD16) blockade (Human TruStain FcX, BioLegend) was performed for 10 min. Cells were then stained on ice with anti-NGFR (clone ME20.4), HLA-A*02 (BB7.2), HLA-ABC (W6/32), CD3 (SK7), and PD-L1 (29E.2A3) primary antibodies directly conjugated to fluorophores for 30 min. Non-viable cells were excluded with SYTOX Blue (Thermo Fisher) or Zombie NIR (Biolegend) staining. All analyses were performed on a BD Fortessa analyzer running FACSDiva software and interpreted using FlowJo V.X10.0.7r2.
IFN-γ assays
IFN-γ ELISA was performed according to manufacturer recommendations.
Animal studies
Tumors were established in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory) via subcutaneous transplantation of 3×106 tumor cells into the left flank (n=6–7 mice per group per experiment). E7 TCR-engineered T cells were adoptively transferred via intraperitoneal injection (1×107 cells in phosphate buffered saline) 10 days after tumor implantation. PD-L1 t-haNKs were adoptively transferred via intraperitoneal injection (1×107 cells in phosphate buffered saline) beginning 10 days after T-cell adoptive transfer. IFN-γ depletion was initiated day 7 following T-cell adoptive transfer (200 µg clone XMG 1.2, Bio X Cell, intraperitoneal) two times per week for a total of five doses. Tumor volumes were collected twice weekly and calculated as (length×width2)/2.
Statistical analysis
Correlation between tumor cell HLA class I expression and CD3+ cell localization was determined with two-tailed Pearson correlation coefficient analysis. Models which compared means of two independent groups were tested using Student’s t-test with a two-tailed distribution at a confidence level of 95%. Comparison of more than two independent groups is done using analysis of variance with Tukey’s multiple comparison adjustment. All error bars indicate SD unless otherwise indicated. Tests are reported as p values with our significance cut-off set at p<0.05. All analysis was performed using GraphPad Prism V.7.
Results
APPM are heterogeneously expressed in HNSCC
Previous work has demonstrated heterogeneity in the expression of HLA and APPM genes in HNSCC.22 23 To further explore these findings in independent cohorts of HNSCC clinical samples, tumor cell-specific expression of a panel of APPM genes8 10 24 25 was determined in individual tumor cells subjected to single-cell RNA sequencing.12 Single tumor cell transcriptomes from six separate HNSCCs displayed substantial heterogeneity in the expression of APPM genes mechanistically linked to the detection of tumor cells by T cells (figure 1A). To evaluate heterogeneous APPM gene expression at the protein level, a separate cohort of 20 HNSCC clinical samples collected from patients undergoing surgical resection were assessed for HLA class I expression by immunofluorescence (online supplemental table S1). In figure 1B, the top panels demonstrate a representative HNSCC with absent tumor cell HLA class I expression. The bottom panels demonstrate a representative HNSCC with HLA class I expression in a subset of tumor cells. Across over 720 000 individual tumor cells analyzed in single tumor sections from 20 patients (range 4519–139 471 tumor cells/section, median 19 610), an average of 13.6% (range 0.1%–76%, median 7.3%) of all cytokeratin positive tumor cells within each tumor section were positive for HLA class I expression as determined by immunofluorescence (figure 1C).
Supplemental material
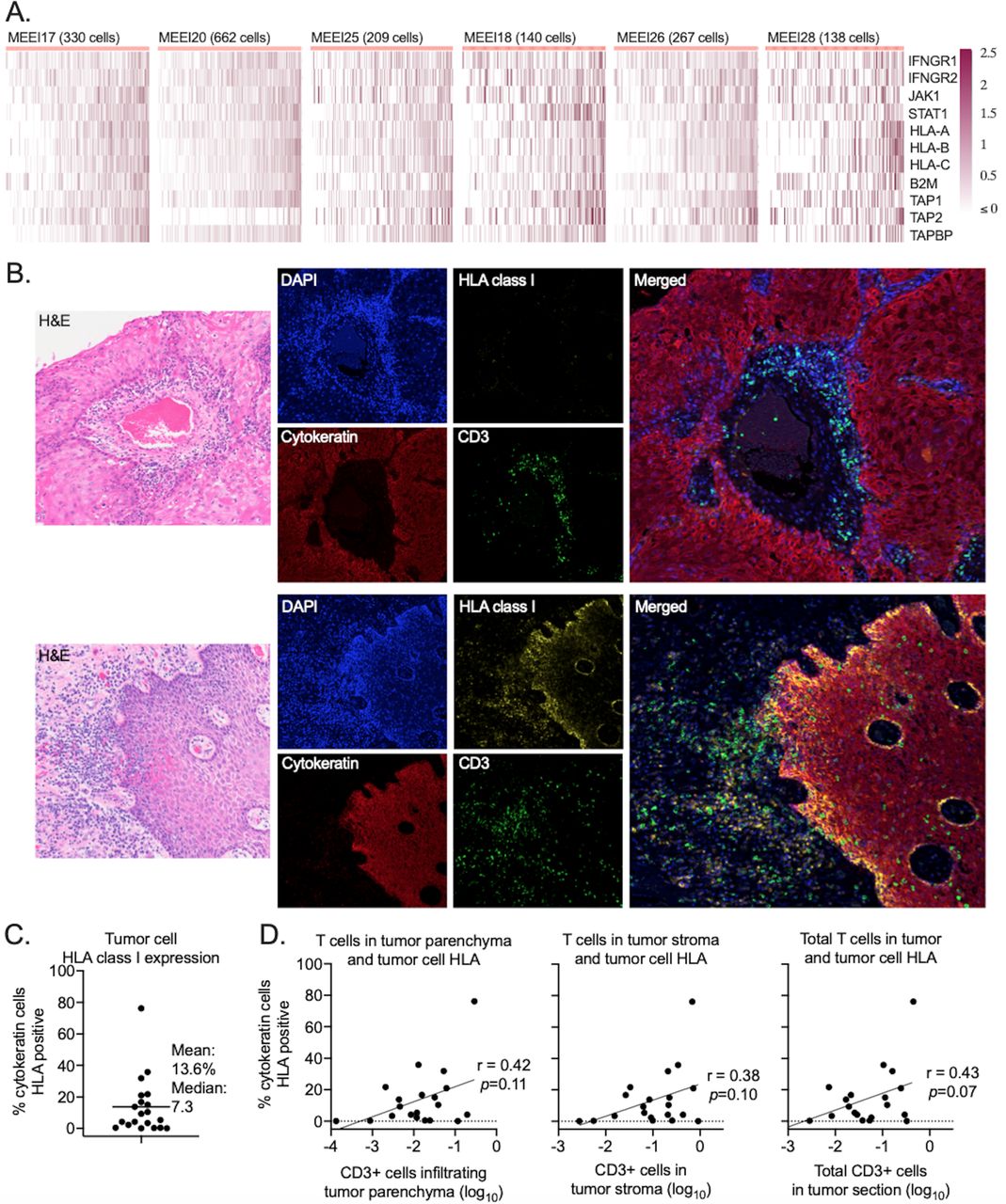
Head and neck squamous cell carcinoma tumor cells displayed heterogeneous expression of APPM. (A) Heatmaps show row standardized expression values of APPM genes (rows) within individual HNSCC cells (columns) obtained from six separate HNSCC tumors. Cells were ordered from right to left based on the average gene expression. All values above 2.5 were mapped to 2.5. (B) Representative photomicrographs from two tumors of H&E, single color, and merged immunofluorescence images assessing the expression of HLA class I (yellow) or cytokeratin (red)-positive tumor cells and localization of CD3 (green) T cells. (C) Quantification of the percentage of cytokeratin-positive tumor cells that are HLA class I positive. All tumor cells from each section (n=20 individual tumors) were scored. (D) Assessment of the relationship (Pearson correlation coefficient) between tumor cell HLA positivity and CD3+ T-cell quantity and location. The x-axis of each panel is the log10 transformed ratio of the number of CD3+ cells to the number of cytokeratin-positive tumor cells (tumor parenchyma, left panel), to the total number of stromal cells (middle panel) or to the total number of cells in the tumor section (right panel). APPM, antigen processing and presentation machinery; HLA, human leukocyte antigen; HNSCC, head and neck squamous cell carcinoma; 4′,6-diamidino-2-phenylindole, DAPI.
Genomic alterations in APPM genes are relatively infrequent events in HNSCCs.26 Since HLA class I expression on tumor cells can be induced by IFN-γ, costaining of HLA class I with CD3 was performed to determine if the presence of HLA class I tumor cell expression spatially correlated with the location of CD3+ T cells. Analysis of annotated multiplex immunofluorescence images revealed that although there was a trend toward positive correlation between greater tumor cell HLA class I expression and CD3+ tumor cells localized within the tumor parenchyma, stroma or entire tumor section, these correlations failed to reach statistical significance (figure 1D). Significant heterogeneity in the relationship between CD3 location and expression of tumor cell major histocompatibility class (MHC) class I was apparent between and within individual HNSCC clinical samples. There were areas of high CD3+ cell infiltration into tumor parenchyma with little to no HLA class I expression, and other areas of very high tumor cell HLA class I expression with no CD3+ cell infiltration (online supplemental figure S1). Together, these data demonstrated the heterogeneous tumor cell expression of APPM, including HLA class I in HNSCCs. Further, given infrequent genomic alterations in APPM genes and the lack of a clear relationship between the localization of tumor infiltrating CD3+ T cells and tumor cell HLA class I expression, these data suggested complex in vivo regulation of APPM expression on HNSCC tumor cells.
Supplemental material
TCR-engineered T cells efficiently kill HLA and antigen positive tumor cells
Although HNSCCs harbor infrequent genomic alterations in APPM genes, gene editing provides a powerful tool for proof-of-concept mechanistic studies exploring the effects of loss of gene expression. To further explore effects of APPM defects and to model the process of T-cell escape of tumor cells lacking T-cell antigen or APPM expression, we generated tumor cells susceptible to killing by TCR-engineered T cells. Introduction of genes encoding HLA or antigen required for TCR engagement was introduced into cell lines to maintain absolute control over gene expression for mechanistic studies. JHU-029 and UM-SCC-1 head and neck carcinoma cells were engineered via retroviral transduction to express HPV 16 E7 (E7) and HLA-A*02 (A*02) (figure 2A,B). Naturally HPV-positive UM-SCC-47 head and neck carcinoma cells were engineered to express A*02 (figure 2C), and naturally A*02-positive UM-SCC-109 head and neck carcinoma cells were engineered to express E7 (figure 2D). A*02 and E7 expression was validated by flow cytometry and qPCR (online supplemental figure S2A–D). T cells engineered to express a TCR specific for HPV 16 E711-19 restricted to HLA-A*02 were assessed for their ability to kill parental or engineered A*02 and E7-positive tumor cells via real-time impedance analysis.21 Dose-dependent killing of A*02 and E7-positive tumor cells was observed (figure 2A–D, right panels), with E:T of 2:1 resulting in complete elimination of all A*02 and E7 double-positive cancer cells within 12 hours of coculture. Parental cell lines lacking A*02 or E7 or both were not eliminated by E7 TCR-engineered T cells.
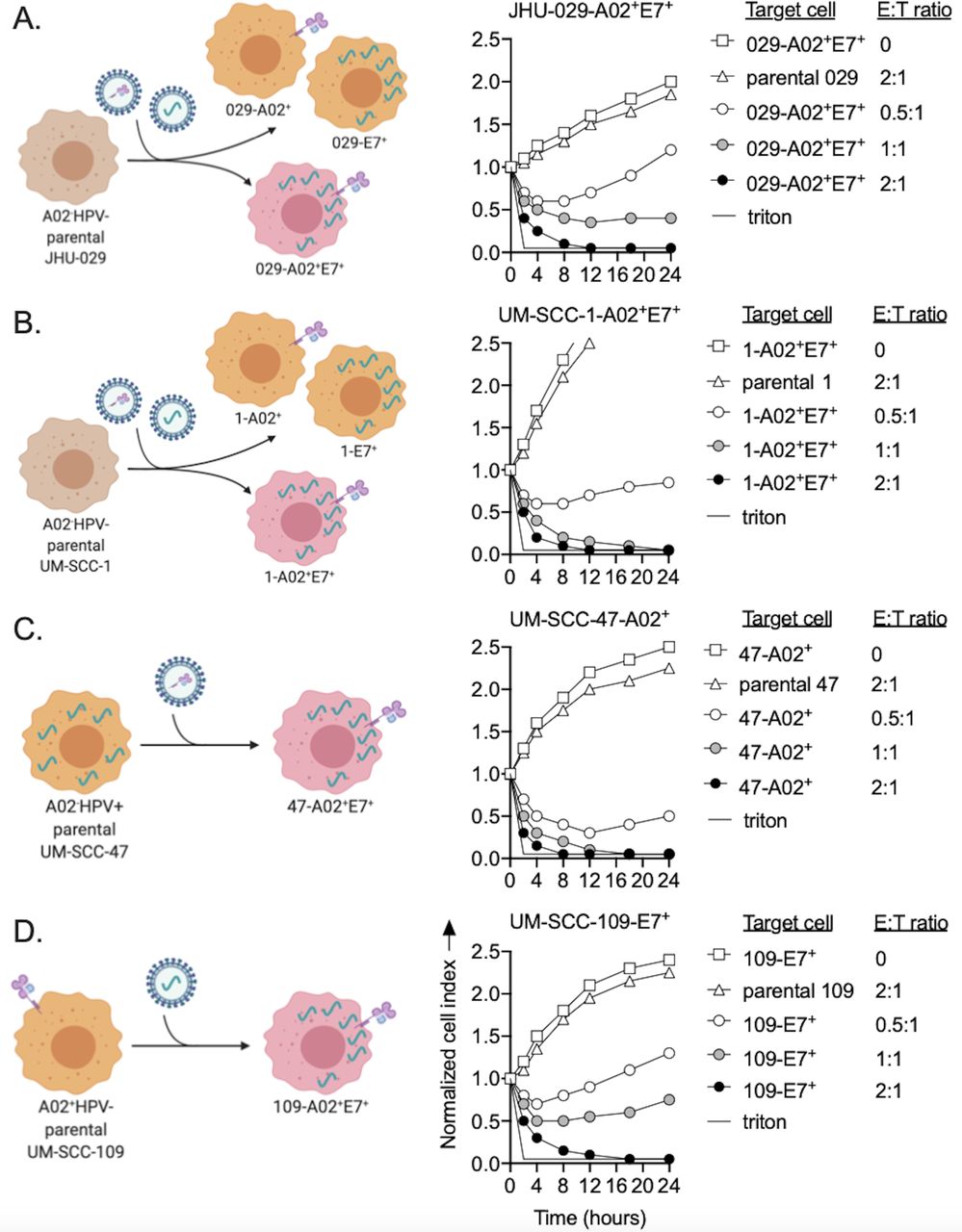
TCR-engineered T cells killed HLA-A*02 and HPV 16 E7 positive tumor cells. Left panels: illustrations depicting A*02−HPV− (A) JHU-029 and (B) UM-SCC-1 cells, (C) A*02−HPV+ UM-SCC-47 cells, and (D) A*02+HPV− UM-SCC-109 cells engineered to be double positive for A*02 and HPV 16 E7 via retroviral transduction. Right panels: real-time impedance analysis was used to determine the ability of E7 TCR-engineered T cells to kill each parental or engineered double-positive cell line. Target cells and E:T ratios are shown in the impedance plot legend. Time 0 on the x-axis is the time at which E7 TCR-engineered T cells were added to the coculture. Representative impedance plots from one of at least three independent experiments with similar results are shown. E:T, effector to target; HLA, human leukocyte antigen; HPV, human papillomavirus; TCR, T-cell receptor.
Tumor cells lacking HLA or antigen escape T-cell detection and killing
Cancer cells expressing or lacking A*02 or E7 were mixed in different proportions and used to determine if tumor cells lacking the A*02 and E7 could escape detection and killing and be selected on coculture with TCR-engineered T cells (figure 3A). After initial growth control on coculture with E7 TCR-engineered T cells, populations composed of 75% A*02 or E7-negative tumor cells escaped T cell (2:1 E:T ratio) control and gained impedance, indicating the presence of viable, expanding tumor cells (figure 3B and online supplemental figure 3A). Similarly, cells deficient in β2M via CRISPR/Cas9 gene editing were resistant to E7 TCR-engineered T cells. Cells lacking β2M failed to express baseline or IFN-γ-inducible cell surface A*02 but retained IFN-γ-inducible PD-L1 expression, demonstrating HLA-specific effects of β2M knockout (online supplemental figure 4A,B). These results suggested that coculture of TCR-engineered T cells with mixtures of tumors cells expressing and lacking A*02 or E7 could lead to the selection of T cell-resistant tumor cells.

Tumor cells lacking APPM that escape T-cell killing expressed increased PD-L1. (A) Illustrations depicting experiments combining different proportions of A*02+E7+ tumor cells with tumor cells lacking A*02 or E7, or genetically lacking β2M. (B) These heterogeneous tumor cell mixtures were assessed for their susceptibility to killing by E7 TCR-engineered T cells via real-time impedance analysis. Proportion of the tumor cell population lacking A*02, E7 or β2M is shown in each legend. Time 0 on the x-axis is the time at which E7 TCR-engineered T cells were added to the coculture. T cells were added at a fixed 2:1 E:T ratio. Representative impedance plots from one of at least three independent experiments with similar results are shown. (C) Following 24 hours of coculture with T cells or media alone, mixtures of tumor cells composed of 50% of cells expressing and 50% of cells lacking A*02, E7 or β2M were harvested and assessed by flow cytometry for expression of A*02, E7 (NGFR) and PD-L1. Non-adherent cells were washed away before tumor cell harvest. Representative dot plots of viable (sytox blue negative) tumor cells are shown. Pie graphs depicting the frequency of HLA or antigen-negative tumor cells (orange) relative to positive tumor cells (blue) are inset in each dot plot. (D) Representative histograms and quantification of MFI of PD-L1 expression on tumor cells with and without exposure to T cells are shown in the right panels. Experiments were performed in technical triplicate and reproduced in at least two independent experiments. ***P<0.001, t-test. APPM, antigen processing and presentation machinery; E:T, effector to target; HLA, human leukocyte antigen; MFI, median fluorescent intensity; NGFR, nerve growth factor receptor; PD-L1, programmed death ligand-1; TCR, T-cell receptor.
Tumor cell variants that escape T-cell killing lack HLA or antigen and express greater PD-L1
To further characterize tumor cells that escaped T-cell elimination, flow cytometry was used to determine the expression of A*02 or E7 in the remaining viable tumor cells following 24 hours of coculture with E7 TCR-engineered T cells. Most selected cells (95%–98%) displayed the corresponding cell line-specific A*02 or E7 deficiency (figure 3C and online supplemental figure 3B). Similar patterns of T-cell selection of tumor cells lacking A*02, E7 or β2M were observed across all models.
Tumor cells that escaped T-cell elimination were further assessed for expression of PD-L1. Consistent with patterns observed in prior reports,17 PD-L1 expression on T cell-escape variant tumor cells lacking A*02, E7 or β2M was significantly greater after coculture selection by T cells compared with before selection by T cells (figure 3C and online supplemental figure 3C). T cells cocultured with a pure or mixed population of tumor cells expressing both A*02 and E7 expressed greater levels of IFN-γ compared with T cells cocultured with tumor cells lacking A*02 and E7 (online supplemental figure 4C,D). Levels of IFN-γ produced in vitro during coculture approximated intratumoral IFN-γ levels following adoptive transfer of antigen-specific T cells into mice.27 These results demonstrated that selection of tumor cells lacking A*02 or E7 by antigen-specific T cells can be effectively modeled in vitro, and that increased PD-L1 expression on T-cell escape variant tumor cells could represent a therapeutic target for rationally designed combination immunotherapy.
PD-L1 t-haNKs prevent clonal selection of T-cell escape variants
Given increased PD-L1 expression on tumor cells that escaped T cell killing, haNK cells expressing a CAR targeting PD-L1 (PD-L1 t-haNKs) were assessed for their ability to kill these escape variant cells in the presence or absence of IFN-γ pretreatment. At baseline, PD-L1 t-haNKs killed tumor cells to a greater degree than haNKs lacking the PD-L1 CAR (online supplemental figure 5A–D). Further, IFN-γ pretreatment of escape variant tumor cells increased cell surface PD-L1 expression and enhanced killing by PD-L1 t-haNKs but did not enhance killing by haNKs lacking the PD-L1 CAR. These data demonstrated the ability of PD-L1 t-haNKs to kill escape variant tumor cells lacking A*02 or E7 expression as a monotherapy.
PD-L1 t-haNKs were next assessed for their ability to eliminate T-cell escape variant tumor cells following selection by TCR-engineered T cells. The addition of PD-L1 t-haNKs 12 hours after coculture of a tumor cell mixture consisting of 75% A*02, E7 or β2M-deficient tumor cells with TCR-engineered T cells led to a rapid reduction in cell number and prevented the escape of viable tumor cells observed with T-cell treatment alone (figure 4A–C and online supplemental figure 6A–C). Decreased cell viability was also observed following the addition of PD-L1 t-haNKs to heterogeneous populations of tumor cells that were not previously exposed to T cells. However, significantly greater reduction in cell viability following the addition of PD-L1 t-haNKs to tumor cell populations pretreated with T cells compared with untreated tumor cells demonstrated greater sensitivity of tumor cells that escaped T-cell killing to PD-L1 t-haNK killing (online supplemental figure 6D–G).

PD-L1 t-haNKs prevented clonal outgrowth of escape variant tumor cells selected by TCR-engineered T cells. Mixtures of (A) JHU-029 tumor cells expressing (25%) or lacking (75%) A*02, (B) UM-SCC-1 cells expressing or lacking E7, or (C) UM-SCC-109 cells expressing or lacking β2M were assessed for their susceptibility to killing by TCR-engineered T cells with or without PD-L1 t-haNKs via real-time impedance analysis. Heterogeneous mixtures of tumor cells expressing (25%) or lacking (75%) (D) A*02 or (E,F) E7 were assessed for their susceptibility to killing by TCR-engineered T cells with or without PD-L1 t-haNKs via real-time impedance analysis. In some conditions (solid triangles), PD-L1 was genetically deleted (PD-L1ko) from the A*02 or E7-negative tumor cells. Time 0 on the x-axis is the time at which TCR-engineered T cells were added to the coculture. PD-L1 t-haNKs were added to the coculture 12 hours after the addition of TCR-engineered T cells (red lines). All effector T and NK cells were added at a 2:1 E:T ratio. Representative impedance plots from one of at least three independent experiments with similar results are shown. Quantification of % loss of target cell index at 20 hours (8 hours after the addition of PD-L1 t-haNKs) in all assessed models is shown. *P<0.05, **P<0.01, ***P<0.001; analysis of variance. E:T, effector to target; NK, natural killer; PD-L1, programmed death ligand-1; TCR, T-cell receptor.
PD-L1 t-haNK elimination of T-cell escape variants is PD-L1 dependent
PD-L1 t-haNKs can kill target tumor cells through intrinsic antitumor activity following activation of receptors such as NKp46 and NKG2D or through PD-L1 CAR-mediated activation.15 16 To determine if PD-L1 elimination of T-cell escape variants was due to intrinsic NK activity or mediated through the PD-L1 CAR, CRISPR/Cas9 gene editing was used to generate PD-L1 knockout tumor cells that escaped T-cell killing via lack of expression of A*02 or E7. Knockout of baseline and IFN-γ-inducible PD-L1 expression was verified by flow cytometry. Although PD-L1 t-haNKs efficiently eliminated T-cell escape variants when PD-L1 expression was intact, this effect was subtotally abrogated when T-cell escape tumor cells lacked PD-L1 (figure 4D–F). PD-L1 t-haNKs demonstrated negligible control of PD-L1 knockout T-cell escape variants in two of three models, demonstrating intrinsic antitumor NK activity independent of the PD-L1 CAR. These data indicated that the ability of PD-L1 t-haNKs to eliminate T-cell escape variants was primarily mediated through the PD-L1 CAR and not through intrinsic NK antitumor activity.
Combination immunotherapy demonstrated synergistic antitumor efficacy in vivo through IFN-γ-dependent and PD-L1-dependent mechanisms
Two engineered variants of the UM-SCC-1 model were selected for in vivo study (figure 5A). NSG mice bearing 10-day-old subcutaneous tumors established from transplanting a pure population of UM-SCC-1 cells expressing both A*02 and E7 were cured following a single adoptive transfer of E7 TCR-engineered T cells. The same treatment of mice bearing tumors derived from a mixture of either 75% A*02-negative (figure 5B) or 75% E7-negative cells (figure 5C) resulted in progressive tumor growth after an initial delay, demonstrating escape of these tumors from T cell-mediated antitumor immunity. Initiation of 2 weeks of PD-L1 t-haNK treatment two times per week 10 days after the T-cell transfer in similar tumor-bearing mice resulted in significant tumor growth delay. A similar growth delay was not observed in mice bearing similar tumors treated with PD-L1 t-haNKs that did not receive T cell adoptive transfer. These data demonstrated the ability of PD-L1 t-haNKs to induce significant tumor growth control in mice bearing tumors that escaped rejection by antigen-specific T cells.
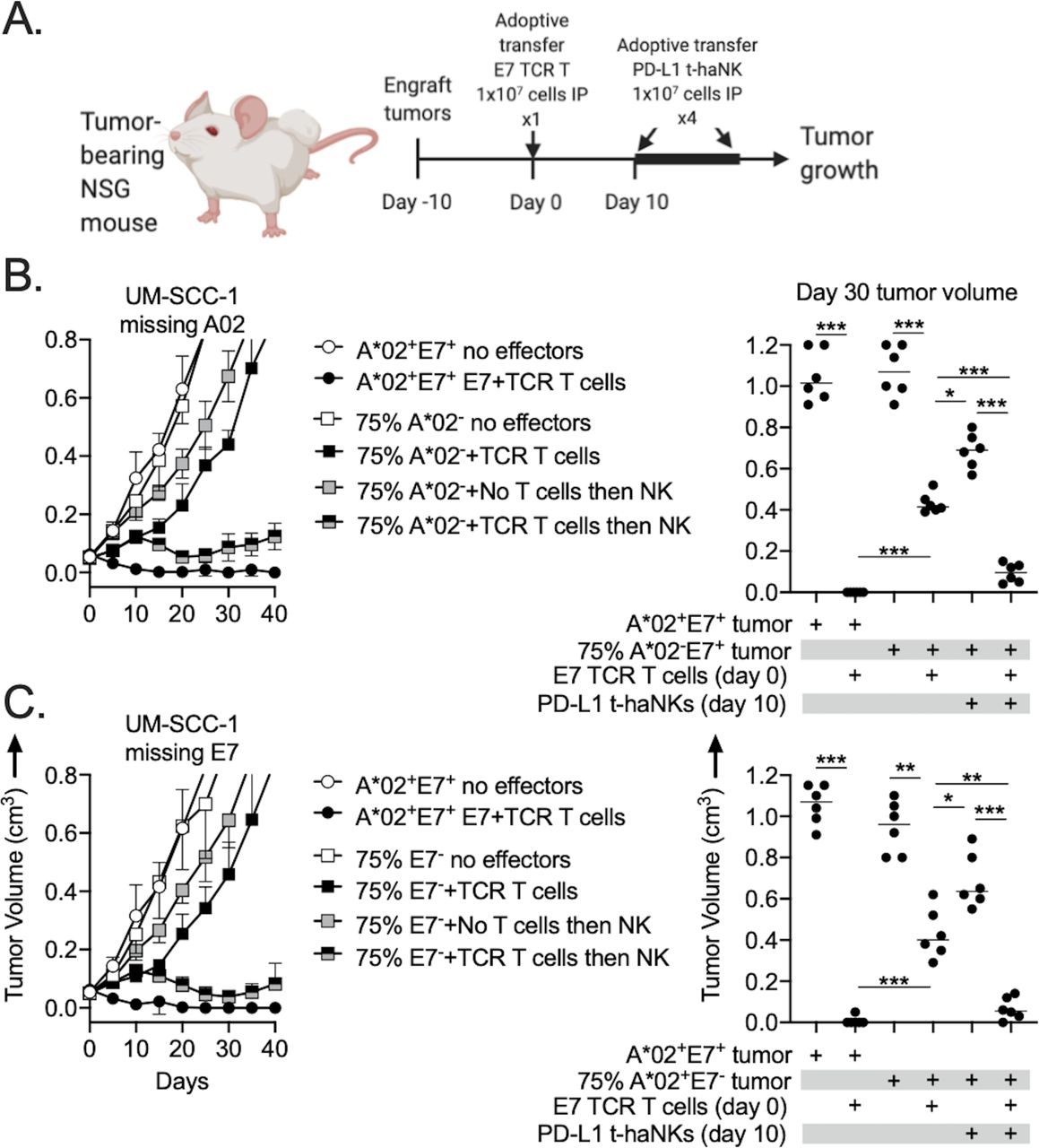
Combination antigen-specific T cell and PD-L1 t-haNK treatment demonstrated antitumor efficacy in vivo. (A) Illustration depicting the E7 TCR-engineered T cell and PD-L1 t-haNK treatment schema. NSG mice bearing 10-day-old tumors (n=6–7 mice/group) composed of 100% A*02+ and HLA-E7+ tumor cells or 75% A*02− (B) or E7− tumor cells (C) were treated with a single intraperitoneal adoptive transfer of 1×107 E7 TCR-engineered T cells alone or in combination with PD-L1 t-haNKs (1×107 cells two times per week intraperitoneally for four total treatments) starting 10 days after the T-cell adoptive transfer. Representative summary growth curves (mean±SE of the mean) are shown on the left from one of two independent experiments. Tumor volumes 30 days after the T-cell adoptive transfer are shown on the right. *P<0.05, **P<0.01, ***P<0.001; analysis of variance. NSG, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; PD-L1, programmed death ligand-1; TCR, T-cell receptor.
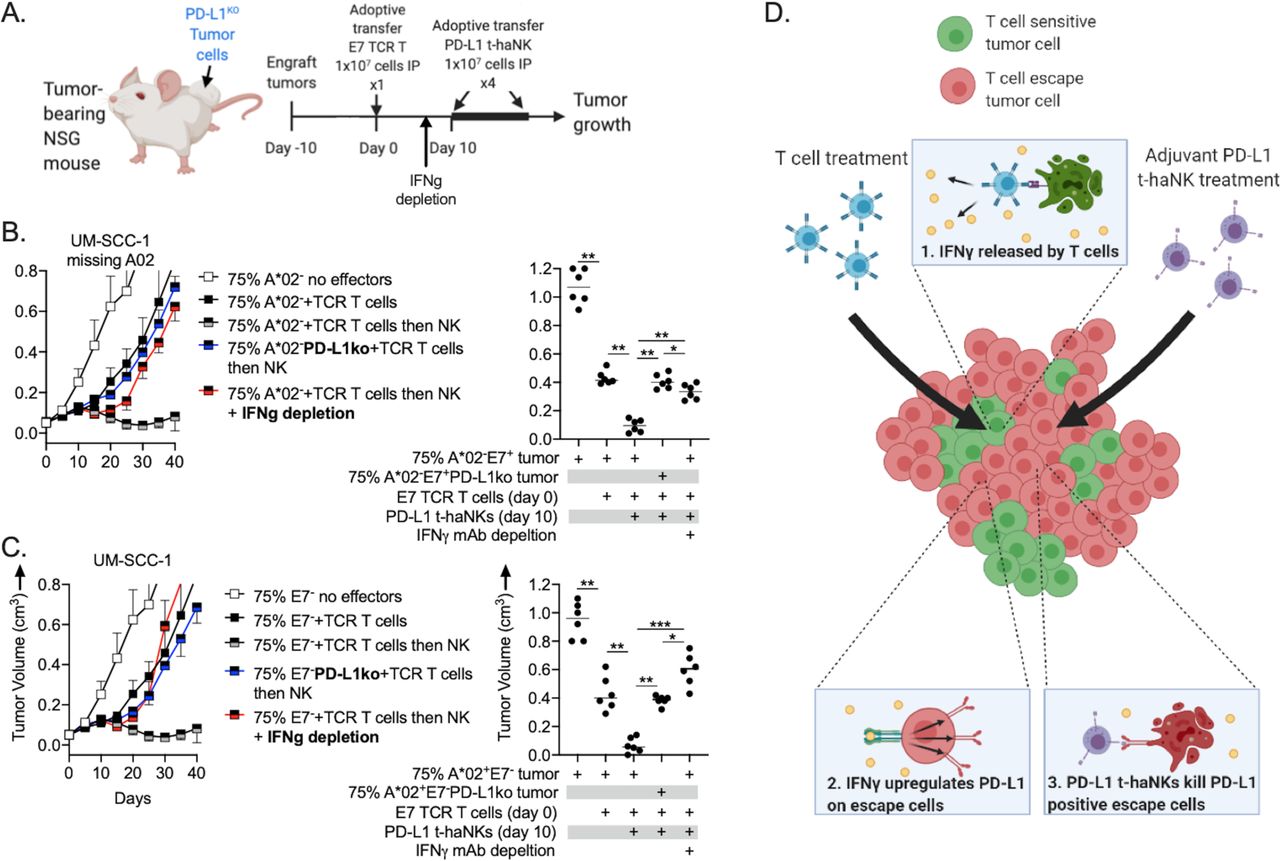
To explore in vivo mechanisms of enhanced tumor growth control with TaNK therapy, similar experiments were performed using PD-L1 knockout A*02 or E7-negative tumor cells or in vivo IFN-γ depletion (figure 6A). In mice bearing tumors derived from 75% A*02 (figure 6B) or E7-negative tumor cells (figure 6C) lacking PD-L1, the ability of PD-L1 t-haNKs to induce tumor growth control when administered 10 days after the T-cell adoptive transfer was abrogated. Further, in mice bearing tumors derived from 75% A*02 or E7-negative tumor cells with intact PD-L1, the ability of PD-L1 t-haNKs to induce tumor growth control when administered 10 days after the T-cell adoptive transfer was substantially reduced in the presence of IFN-γ depletion initiated 3 days before the start of PD-L1 t-haNK treatment. Together, these results indicated that the ability of PD-L1 t-haNKs to exert tumor growth inhibition in tumors that have escaped T-cell control is dependent on both IFN-γ and the ability of antigen or HLA-negative tumor cells to express PD-L1. Treatment of a heterogeneous tumor composed of a mixture of T cell-sensitive and T cell-resistant tumor cells with a T cell-based immunotherapy appears to sensitize the tumor to control by PD-L1 t-haNKs. A graphical representation of the proposed mechanism is provided in figure 6D.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Combination treatment efficacy in vivo was through IFN-γ and PD-L1-dependent mechanisms. (A) Illustration depicting the E7 TCR-engineered T cell and PD-L1 t-haNK treatment schema. NSG mice bearing 10-day-old tumors (n=6–7 mice/group) composed of 75% A*02− (B) or E7- tumor cells (C) were treated with a single IP adoptive transfer of 1×107 E7 TCR-engineered T cells alone or in combination with PD-L1 t-haNKs (1×107 cells two times per weel intraperitoneally for four total treatments) starting 10 days after the T-cell adoptive transfer. In some conditions, A*02− or E7− tumor cells were CRISPR/Cas9 gene edited to lack PD-L1 (blue lines). In other experiments, IFN-γ monoclonal depleting antibody (clone XMG 1.2) was injected intraperitoneally beginning 7 days after T-cell adoptive transfer and two times per week for five total doses (red lines). Representative summary growth curves (mean±SE of the mean) are shown on the left from one of two independent experiments. tumor volumes 30 days after the T-cell adoptive transfer are shown on the right. (D) Graphical mechanism for combination antigen-specific T cell and PD-L1 t-haNK antitumor activity. *P<0.05, **P<0.01, ***P<0.001; analysis of variance. IFN-γ, interferon gamma; NSG, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; PD-L1, programmed death ligand-1; TCR, T-cell receptor.
Loss of tumor cell interferon (IFN) signaling conveys resistance to PD-L1 t-haNKs but not T cells
Previous studies have reported that loss of IFN-γ response pathway components including IFNGR1, JAK1/2, and STAT1 convey resistance to T cell-based immunotherapy.8 24 25 IFNGR1 or STAT1 knockout via CRISPR/Cas9 gene editing abrogated the ability of tumor cells to upregulate HLA class I molecules in response to IFN-γ but did not affect baseline HLA class I expression in either engineered A*02+ JHU-029 cells or naturally A*02-positive UM-SCC-109 cells (online supplemental figure 7A–D). Loss of IFGR1 or STAT1 expression did not confer resistance to killing by TCR-engineered T cells, perhaps due to constitutive baseline A*02 expression.
IFN-γ is also a key regulator of PD-L1 expression.28 IFNGR1 or STAT1 knockout abrogated both baseline and IFN-γ-inducible PD-L1 expression in JHU-029 and UM-SCC-109 cells (online supplemental figure 7A–D). Coculture of a population of 75% JHU-029 cells lacking both E7 and STAT1 revealed escape from T-cell killing similar to that observed with 75% JHU-029 cells lacking E7 alone. However, PD-L1 t-haNKs were unable to effectively prevent escape of the JHU-029 cells lacking both E7 and STAT1 (online supplemental figure 7E). These data suggested that in tumor cell populations that escape T-cell detection and elimination through APPM-deficiency, PD-L1 t-haNKs depend on functional tumor cell IFN-γ signaling and robust upregulation of PD-L1 expression for efficient tumor cell targeting and elimination.
Discussion
PD-L1 t-haNKs are derived from a human, allogeneic NK cell line (NK92) that has been engineered to express endoplasmic reticulum-retained IL-2 and a second-generation CAR targeting PD-L1.15 16 PD-L1 t-haNKs kill a wide variety of tumor cell targets at low E:T ratios through both CAR-dependent and independent mechanisms.15 16 PD-L1 t-haNKs are irradiated prior to use, similar to NK92 cells, which prevents engraftment. Irradiated CAR-engineered NK cells can be administered repeatedly and stopped with early signs of off-tumor toxicity. However, the short half-life of PD-L1 t-haNKs may also be a limitation as cells require repeat administration to replenish and persist in the tumor microenvironment.15
Here, we demonstrated that adjuvant administration of irradiated PD-L1 t-haNKs following T cell-based therapy can overcome the clonal outgrowth of cancer subpopulations that escape T-cell detection and elimination. Our data suggest that PD-L1 t-haNKs are particularly effective at targeting T-cell escape variants because of an IFN-γ-dependent increase in PD-L1 expression on escape tumor cells following selection by T cells. More generally, these experiments provide experimental evidence that the activation of T cell-based antitumor immunity with resulting production of IFN-γ may sensitize tumors to the antitumor activity of PD-L1 t-haNKs. Whether tumors from patients that have failed T cell-based immunotherapy display increased PD-L1 expression, or increased expression of other IFN-responsive genes, requires further study and likely depends on treatment timing and the persistence of functional T cells within a tumor. It is possible that more complete proteomic or transcriptomic profiling of T-cell escape variant tumor clones could reveal additional cell markers to target with CAR-engineered effector immune cells.
Resistance to immune checkpoint blockade has been associated with defects in APPM gene expression.8 10 Tumor subpopulations harboring these defects undergo clonal selection due to selective pressure by the immune system.29 Immunofluorescence of 20 HNSCC specimens revealed significant heterogeneity in expression with only a minority of tumor cells expressing HLA class I, consistent with data reported from other tumor types.30 Yet bulk genetic analyses of large cohorts of tumors have detected mutations in β2M or HLA alleles in only 7%–11% of HNSCC.26 Our single-cell transcriptome analysis of six HNSCC revealed significant intratumoral heterogeneity in the expression of APPM genes. This suggests that non-genetic mechanisms may account for the high degree of heterogeneity in APPM genes observed at the single tumor cell level. One explanation could be heterogeneous T-cell infiltration resulting in variable IFN-γ levels across distinct topographic tumor regions.27 This hypothesis was not supported by our data exploring colocalization between CD3+ cells and tumor cell HLA class I expression. Another possible explanation includes epigenetic regulation and/or autophagy of APPM genes, as has been reported by others.31 32
Combination T cell-based immunotherapy and CAR-engineered NK cell therapy is a promising strategy to overcome the selection of T cell-resistant tumor subclones observed with T cell-based immunotherapy alone. As a proof of concept, we used genetic models of antigen, HLA, and β2M loss.8 24 25 33 34 HLA-A*02 loss mimics loss of heterozygosity at the HLA locus leading to loss of expression of a specific HLA allele.35 Similarly, loss of B2M abrogates expression of class I HLA molecules.8 10 36 Lastly, cancer cells expressing the most immunogenic antigens may be eliminated in the face of immune pressure during tumor development or T cell-based immunotherapy, leading to tumor cells lacking antigen or harboring weak antigens.6 37 This was modeled by the absence of HPV 16 E7 in our work. Loss of other APPM machinery, such as the transporter associated with antigen processing 1/2 or endoplasmic reticulum aminopeptidase 1/2, may also result in antigen loss and be clinically relevant, but were not studied here.38–40
Tumor-infiltrating T cells typically accumulate in tumor areas with high antigen expression.41 However, CD8 T cell-derived IFN-γ spreads from the site of antigen presentation in vivo to distances of over 800 µm.17 41 In heterogenous tumors, an IFN-γ response can be elicited in the entire tumor cell population if there are both tumor-specific CD8 T cells and a subpopulation of antigen-presenting tumor cells.17 Therefore, CD8 T cell-derived IFN-γ activity can modulate the phenotype of a large tumor mass when only a portion of tumor cells are capable of mediating T-cell activation. Our work demonstrated the ability of antigen-specific T cells to induce IFN-γ-dependent upregulation of PD-L1 on cells that evade T cell detection and elimination. PD-L1 genetic deletion and IFN-γ depletion abrogated the antitumor efficacy of PD-L1 t-haNKs following escape from T cell killing, indicating that IFN-γ dependent PD-L1 upregulation was responsible for the combinatorial effect of this treatment.
Clinically, patients with tumors harboring intact IFN-γ signaling are more likely to respond to immunotherapy compared with patients harboring tumors with one or more IFN-γ signaling defects.10 24 25 42 IFNGR1 and STAT1 knockout did not convey direct resistance to T-cell killing in our experimental models. We noted that IFNGR1 and STAT1 knockout abrogated IFN-γ-inducible but not baseline HLA-A*02 expression. One explanation for why defects in IFN-γ signaling predict an inability to respond to T cell-based immunotherapy is that bystander killing of antigen negative cells by IFN-γ may play a role in select tumors. Different tumor cells have different responses to IFN-γ; some cell lines are killed by IFN-γ concentrations as low as 0.5 ng/mL, whereas others are unaffected at concentrations exceeding 50 ng/mL in vitro.41 Another possibility is that the baseline expression of MHC class I varies among cancers. The MHC class I expression on JHU-029 and UM-SCC-109 was relatively high at baseline; cancers that express low levels of MHC class I at baseline may be dependent on IFN-γ upregulation of APPM for T-cell detection and elimination. Our work also demonstrated that IFN-γ pathway defects reduced PD-L1 expression and abolished the ability of PD-L1 t-haNKs to eliminate T-cell escape tumor cells. These potentially important results indicate that careful genomic analyses should be performed in patients who receive PD-L1 t-haNK immunotherapy to identify if IFN signaling defects are enriched in non-responders.
Our data suggest that the important antitumor activity conferred by PD-L1 t-haNKs is mediated through the PD-L1 CAR. Several properties of CAR-engineered NK cells may offer a clinical advantage over CAR-engineered T cells. NK92-derived cells can be expanded in culture to large numbers, bypassing the costly and complex process of isolating, expanding, and genetically engineering autologous effector immune cells from individual patients.43 haNK cells are also resistant to acute hypoxia and can maintain cytolytic function in the hypoxic tumor microenvironment.44 Further, haNKs are generally well tolerated clinically and have not been reported to induce cytokine storm, which can occur following adoptive transfer of CAR-T cells.45
Several limitations of this study exist. First, our work did not delineate the minimum fraction of HLA or antigen-positive tumor cells needed to induce levels of IFN-γ sufficient to induce PD-L1 expression on T-cell escape tumor cells. Work from others suggests that a tumor consisting of 10% T cell-sensitive tumor cells was sufficient to induce a widespread IFN-γ response after adoptive T-cell transfer.17 We also did not delineate the kinetics of in vivo tumor cell PD-L1 expression following adaptive T-cell transfer. NK-cell transfer 10 days after T-cell transfer induced greater tumor growth inhibition compared with T-cell transfer alone, but it is possible that earlier NK-cell transfer could induce greater tumor growth inhibition. Multiplex immunofluorescent analysis of PD-L1 and E7 TCR-engineered T cells could also characterize the spatial relationship between T-cell location and tumor cell PD-L1 in addition to the kinetics of PD-L1 expression. These assays are being developed and validated for future use. Next, our work did not explore the combination of PD-L1 t-haNKs with other T cell-based immunotherapies. E7 TCR-engineered T cells are in clinical development (NCT02858310) but represent a very specific therapy for HPV-positive malignancies. Previous work has demonstrated the efficacy of combining PD-L1 t-haNKs with anti-PD-1 antibody and IL-15 cytokine treatment in syngeneic murine models.15 Our work did not assess the ability of haNK cells combined with a PD-L1 mAb capable of inducing antibody-dependent cell-mediated cytotoxicity to eliminate E7 TCR-engineered T-cell escape variants. Additionally, some in vitro and in vivo experiments did not use haNKs as a control for the activity of PD-L1 t-haNKs. Lastly, our use of human cell lines and immunocompromised mice precluded our ability to study the effect of PD-L1 t-haNKs on the local tumor immune microenvironment. Previous studies have shown that PD-L1 t-haNKs may preferentially target myeloid cell populations with constitutively high PD-L1 expression, which may alter the effects of PD-L1 t-haNK in vivo.15 16
In conclusion, through our ability to model T-cell escape variants, our work demonstrated the efficacy and mechanisms underlying enhanced antitumor control of heterogeneous tumors with combination T and CAR-engineered NK-based immunotherapy. Our study provides the preclinical rationale for future clinical trials combining T cell-based with CAR-engineered NK cell-based immunotherapy. PD-L1 t-haNKs may be an effective tool for such combination therapies as they efficiently target T-cell escape variant tumor cells due to increased PD-L1 expression following selection by antigen-specific T cells. Careful clinical study of PD-L1 t-haNKs in combination with T cell-based immunotherapy is warranted.
Acknowledgments
The authors thank Dr Scott Norberg and Dr Jason Redman for their critical review of this article.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @MaxwellYLee, @HsaterMD
Contributors MYL: methodology, investigation, data curation, and writing (original draft); YR, CSie, JF, PEC, and CSil: methodology, investigation, and data curation; HAS: methodology and data curation; NJ and ET: conceptualization, resources, and writing (review and editing); PS-S: methodology, resources, and writing; MRP: methodology and resources; JS, JH, and CH: conceptualization, methodology, resources, and writing (review and editing); CA: conceptualization, methodology, investigation, data curation, and writing (review and editing).
Funding This work was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Institute on Deafness and Other Communication Disorders (project number ZIA-DC00008). This research was also made possible through the NIH Medical Research Scholars Program, a public–private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation (DDCF Grant #2014194), the American Association for Dental Research, the Colgate-Palmolive Company, Genentech, Elsevier, and other private donors.
Competing interests PS-S is an employee of NantKwest.
Patient consent for publication Not required.
Ethics approval Human tissues were acquired under institutional review board-approved protocol NCT03429036 with informed consent; all animal studies were conducted under approved Animal Care and Use Committee protocol #1364-18.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. All data are presented in the main and supplemental results.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.