Article Text

Abstract
Background Current clinical trials are using radiation therapy (RT) to enhance an antitumor response elicited by high-dose interleukin (IL)-2 therapy or immune checkpoint blockade (ICB). Bempegaldesleukin (BEMPEG) is an investigational CD122-preferential IL-2 pathway agonist with prolonged in vivo half-life and preferential intratumoral expansion of T effector cells over T regulatory cells. BEMPEG has shown encouraging safety and efficacy in clinical trials when used in combination with PD-1 checkpoint blockade. In this study, we investigated the antitumor effect of local RT combined with BEMPEG in multiple immunologically ‘cold’ tumor models. Additionally, we asked if ICB could further enhance the local and distant antitumor effect of RT+BEMPEG in the setting of advanced solid tumors or metastatic disease.
Methods Mice bearing flank tumors (B78 melanoma, 4T1 breast cancer, or MOC2 head and neck squamous cell carcinoma) were treated with combinations of RT and immunotherapy (including BEMPEG, high-dose IL-2, anti(α)-CTLA-4, and α-PD-L1). Mice bearing B78 flank tumors were injected intravenously with B16 melanoma cells to mimic metastatic disease and were subsequently treated with RT and/or immunotherapy. Tumor growth and survival were monitored. Peripheral T cells and tumor-infiltrating lymphocytes were assessed via flow cytometry.
Results A cooperative antitumor effect was observed in all models when RT was combined with BEMPEG, and RT increased IL-2 receptor expression on peripheral T cells. This cooperative interaction was associated with increased IL-2 receptor expression on peripheral T cells following RT. In the B78 melanoma model, RT+BEMPEG resulted in complete tumor regression in the majority of mice with a single ~400 mm3 tumor. This antitumor response was T-cell dependent and supported by long-lasting immune memory. Adding ICB to RT+BEMPEG strengthened the antitumor response and cured the majority of mice with a single ~1000 mm3 B78 tumor. In models with disseminated metastasis (B78 primary with B16 metastasis, 4T1, and MOC2), the triple combination of RT, BEMPEG, and ICB significantly improved primary tumor response and survival.
Conclusion The combination of local RT, BEMPEG, and ICB cured mice with advanced, immunologically cold tumors and distant metastasis in a T cell-dependent manner, suggesting this triple combination warrants clinical testing.
- radiotherapy
- cytokines
- combined modality therapy
- translational medical research
- costimulatory and inhibitory T-cell receptors
Data availability statement
Data are available upon reasonable request. Please connect with the first author with any data requests.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- radiotherapy
- cytokines
- combined modality therapy
- translational medical research
- costimulatory and inhibitory T-cell receptors
Introduction
Cancer immunotherapy is an integral component of cancer care. However, more work is needed to find effective immunotherapy regimens for immunologically ‘cold’ cancers.1 The tumor characteristics that are used to delineate cold tumors (low mutation burdens, low levels of recognizable neoantigens, and/or low baseline levels of T-cell infiltrates) are associated with worse clinical responses to immune checkpoint blockade (ICB).2–4 Preclinical evidence suggests that local radiation therapy (RT) can help turn some immunologically cold tumors ‘hot’.2 5 In preclinical models, dosing RT prior to ICB can result in a systemic antitumor response when either treatment alone is ineffective.6 This has led to several clinical trials testing the combination of RT with ICB.7 8
We have used immunomodulatory RT to enhance the antitumor response to intratumoral (IT) injections of immunocytokine (IC), a monoclonal antibody targeting disialoganglioside, GD2, fused with interleukin (IL)-2 molecules.9 In some preclinical models, the combination of RT+IT IC is capable of curing mice of their tumors as a T cell-dependent in situ vaccine.10–14 However, drawbacks of this approach include the requirement of a tumor-selective antigen to be targeted with the IC and an easily accessible tumor nodule for administering multiple IT injections.
High-dose IL-2 has been used in the clinical setting because of its ability to stimulate T-cell activation and proliferation through its interaction with the interleukin-2 receptor (IL-2R).15 Naïve T cells, memory T cells, and natural killer (NK) cells express the intermediate-affinity heterodimeric IL-2R, which is composed of the beta (CD122) and gamma (CD132) subunits. The high-affinity trimeric IL-2R, composed of the alpha (CD25), beta, and gamma subunits, is transiently expressed on T effector cells (Teffs) following TCR activation and is constitutively expressed on T regulatory cells (Tregs).16 Bempegaldesleukin (BEMPEG, NKTR-214) is an investigational CD122-preferential IL-2 receptor agonist that leverages the clinically validated IL-2 pathway to stimulate an antitumor immune response.17 BEMPEG consists of recombinant human IL-2 conjugated with an average of six releasable molecules of polyethylene glycol (PEG) and circumvents the limitations of traditional high-dose systemic IL-2 immunotherapy (ie, dose-limiting toxicity, activation of immune suppressive Tregs, and a short in vivo half-life).18 Relative to native IL-2, PEGylation reduces BEMPEG’s affinity for CD25, leading to preferential expansion of the intermediate-affinity IL-2Rs on Teffs and NK cells in the tumor microenvironment (TME).19 As a result, BEMPEG preferentially activates Teffs over Tregs. In clinical and preclinical studies, BEMPEG monotherapy has been shown to increase IT Teffs without increasing IT Tregs.18 20 21
Here we report a cooperative interaction between RT and BEMPEG in multiple murine models, resulting in a strong, adaptive antitumor immune response consistent with an in situ vaccine. Additionally, we show that ICB can further enhance this response in the setting of large, locally advanced tumors and systemic disease.
Materials and methods
Mice
Female C57BL/6 and BALB/c mice 7–8 weeks old were purchased from Taconic (Germantown, New York, USA). T cell receptor alpha knockout (TCR alpha KO) mice were a gift from Dr Marcel Wuethrich. Mice were housed in accordance with the Guide for Care and Use of Laboratory Mice. Experiments were performed under an animal protocol approved by the Institutional Animal Care and Use Committee.
Tumor cells
B78-D14 (B78) melanoma is derived from B16 melanoma and was obtained from Ralph Reisfeld.22–24 B16-F10 melanoma was obtained from American Type Culture Collection (ATCC). B16-F10 cells were transduced to express luciferase (B16-Luc) via lentiviral transduction for in vivo imaging. The 4T1 triple-negative breast cancer cell line was obtained from ATCC. The MOC2 head and neck squamous cell carcinoma cell line was a kind gift from Dr Ravindra Uppaluri. B78, B16, 4T1, and MOC2 cells were grown in vitro as previously described.9 25–27 Mycoplasma testing via PCR was routinely done.
In vivo models
B78 cells (2×106) and MOC2 (2×106) cells were injected intradermally into the right flank of C57BL/6 mice, and 4T1 cells (2×105) were injected into BALB/c mice. Tumors were measured two times per week with calipers. Tumor volumes were calculated as previously described.9 Once average tumor volumes reached the target size, mice were randomized into their treatment groups so that each group had a similar average starting tumor volume. Mice were removed from the study and euthanized when the longest tumor dimension reached 20 mm or the mouse became moribund due to its metastatic disease. Some mice that were cured of their tumor burden were rechallenged with a second inoculation (on the abdomen) of the same dose of tumor cells 90–120 days after treatment began.
For depletion experiments, anti-NK1.1 (100 μg), anti-CD4 (400 μg), anti-CD8 (400 μg), or rat IgG (400 μg) was given via intraperitoneal injection on days 1, 6, 13, 20, and 27 of the experiment. Depletion of immune subsets was confirmed as described further.
Radiation therapy
In vivo RT was dosed to flank tumors using an X-Rad 320 irradiator as previously described (Precision X-Ray).9 RT was delivered in one fraction totaling either 8 Gy (4T1, MOC2 models) or 12 Gy (B78 models). The day of RT was defined as day 0 of treatment.
Immunotherapy and antibodies
BEMPEG was provided by Nektar Therapeutics. Anti-CTLA-4 (clone 9D9, IgG2c) was provided by Bristol Myers Squibb. Anti-PD-L1 (clone 10 F.G92) was purchased from Bio X Cell. IL-2 (teceleukin) was provided by the National Institutes of Health. NK 1.1 depleting antibody (clone PK136), CD4 depleting antibody (clone GK1.5), and CD8 depleting antibody (clone 2.43) were purchased from Bio X Cell. Rat IgG was purchased from Sigma Aldrich.
Intravenous injections of immunotherapy were given by retro-orbital injection. Intravenous injections of 12 μg (4T1 model) or 16 μg (B78, MOC2 models) BEMPEG dissolved in 100 μL BEMPEG buffer (buffer) were given on days 5, 14, and 23. Intravenous and IT injections of 9.375 μg IL-2 in 100 μL phosphate-buffered saline (PBS) were given on days 5–9. In experiments where BEMPEG was compared with IL-2, each cohort of mice received the IL-2 equivalent of ~750 000 U of IL-2 total per mouse. Anti-CTLA-4 (200 μg) was administered by intraperitoneal injections on days 2, 5, and 8. Anti-PD-L1 (200 μg) was administered by intraperitoneal injections on days 1, 3, and 7.
Peripheral blood analysis to confirm immune depletion
Peripheral blood (10 μL) was collected in EDTA-treated tubes from mice via facial vein on days 13 (prior to scheduled dose of depletion antibody) and 34. Red blood cell (RBC) lysis was performed with ammonium-chloride-potassium (ACK) lysis buffer (Thermo Fisher). Cells were incubated with anti-CD16/32 (Biolegend, Clone 93) for 10 min at room temperature and then stained for 30 min at 4°C with the following antibodies: CD45 BV510 (30-F11), CD4 BV785 (GK1.5), CD8 APC-R700 (53–6.7), and NK1.1 PE-CF594 (PK136) all from BD Biosciences. 4’,6’-diamindino-2-phenylindole (DAPI) was used for live/dead staining. All data were collected on an Attune flow cytometer (Thermo Fisher) and analyzed with FlowJo V.10 software (BD).
Splenocyte analyses
B78 tumor-bearing mice were randomized into no treatment or RT groups when tumors were ~150 mm3 (n=5 per group). On day 5 after RT, mice were euthanized, and spleens were harvested and disaggregated between two microscope slides. Cells were prepped, Fc blocked, and stained for live/dead as described previously. Each sample was stained with the following antibodies in addition to those listed previously: CD3PE-Cy5 (145–2 C11) and CD122 PE (TM-β1) all from BioLegend and CD25 BB515 (PC61) from BD Biosciences. Samples were fixed and permeated overnight at 4°C with the FoxP3/Transcription Factor Staining Buffer Set from eBioscience following kit instructions. Samples were then stained with FoxP3 PE-Cy7 (FJK-16s) from eBioscience for 30 min at 4°C. Data were collected and analyzed as described previously for blood analysis.
CD4+ T cells with Tregs (CD45+ CD4+ CD25+ FoxP3+) gated out are referred to as ‘CD4+ T-helper cells’ (acknowledging that this population can contain other non-Treg CD4+ cells).
Tumor-infiltrating immune cell analyses
B78 tumor-bearing mice were randomized into four treatment groups (n=5 per group). Groups were treated with buffer, BEMPEG, RT, or RT+BEMPEG as described previously. On day 14 after treatment, mice were euthanized, and tumors were excised and digested into a single-cell suspension using a Miltenyi gentleMACS Octo Dissociator (2.5 mL of complete RPMI media, 2.5 mg of collagenase, and 250 μg of DNAase per tumor). After tumor disassociation, tumor contents were passed through a 70 μm filter. In addition to those described for blood and spleen cells, the following antibodies were used: CD19 PE-Cy5 (6D5), Ly6G Alexa647 (1A8), and OX40 PE (OX-86), all from BioLegend; and Ly6C BV605 (AL-21) and CD11b V450 (M1/70), all from BD Biosciences.
Tregs and T-helper cells were gated separately within the CD4+ population.
Statistical analyses
Average group tumor volumes are plotted showing mean±SEM. Tumor volume plots were summarized by time-weighted average (area under the volume–time curve, calculated using trapezoidal method). Time-weighted averages were compared between treatment groups overall by Kruskal-Wallis tests. If significance was found using Kruskal-Wallis test, then pairwise comparisons were conducted using Mann-Whitney tests. Survival data were plotted using Kaplan-Meier methods and analyzed using log-rank comparisons. Despite the large number of tests, no p value correction methods were used to account for inflated type I error; no corrections for multiple hypothesis testing were made. Flow cytometry results are plotted as mean±SEM. Flow results were analyzed using an unpaired t-test or one-way analysis of variance analysis with Tukey’s multiple comparisons test (as clarified in the figure legends). χ2 analysis was used to compare rechallenge rejection results. P values of <0.05 were considered significant and were indicated in all figures as follows: *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; ns indicates non-significance.
Results
RT+BEMPEG demonstrates a cooperative effect in multiple immunologically cold tumor models
Based on our previously published data showing a synergistic interaction between RT and intratumoral injectioned of immunocytokine (IT-IC),9 we hypothesized a similar interaction would occur between RT and BEMPEG, without the need for IT delivery. Mice bearing B78 tumors were randomized when average tumor volumes were 100–150 mm3 and were treated with intravenous injections of buffer, BEMPEG, local external beam RT, or RT+BEMPEG.
Compared with the vehicle control, RT alone and BEMPEG alone significantly slowed tumor progression (figure 1A) and improved survival, and achieved 0% or 20% complete regression, respectively (figure 1B). The combined RT+BEMPEG therapy enhanced treatment efficacy, with significant reductions in the tumor growth rate (figure 1A) and significantly improved survival (figure 1B). The majority of mice (67%) treated with RT+BEMPEG were cured of their tumor burden (figure 1B and online supplemental file 1), and all cured mice rejected the B78 tumor rechallenge, thus demonstrating immune memory (online supplemental figure S1B).
Supplemental material
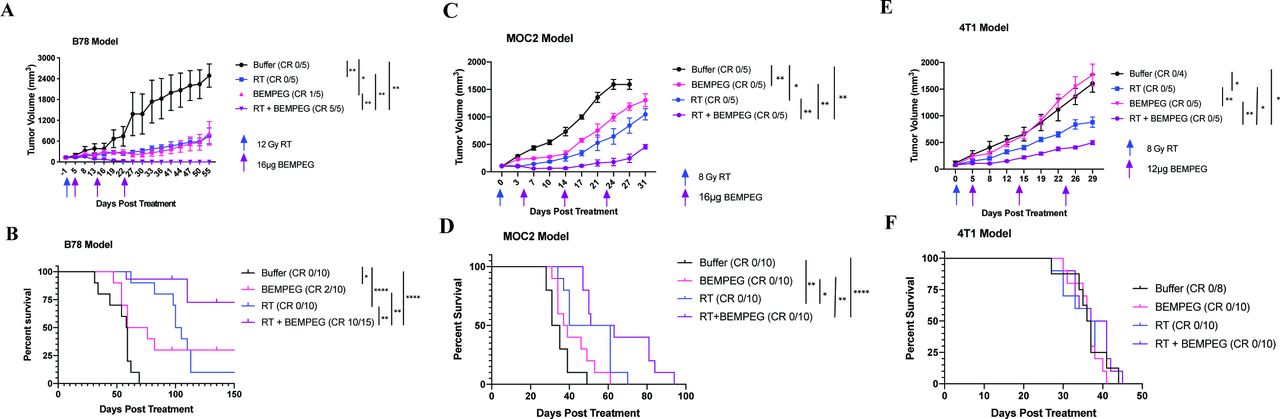
External beam radiation and BEMPEG are cooperative in multiple tumor models. Mice bearing B78 melanoma flank tumors (A,B), 4T1 flank tumors (C,D), or MOC2 flank tumors (E,F) were treated with buffer (black), RT alone (blue), BEMPEG alone (pink), or RT+BEMPEG (purple). Average tumor volume plots (A,C,E) from a representative experiment in each model and combined overall survival (B,D,F) from two independent experiments in each model are shown. The number of mice that demonstrated a CR out of the total number in each group is shown in parenthesis (ie, CR 0/5). Tumor-free mice were censored from the Kaplan-Meier plot when they were rechallenged with a second tumor cell injection, which is indicated by the tick marks. As described in the statistical subsection of the Materials and methods section, p values for average tumor volume plots were calculated using time-weighted analysis. P values for overall survival were calculated via log rank test. *P≤0.05, **P≤0.01, ≤, ****P≤0.0001. For comparisons where there is no p value shown, the p value was not significant. BEMPEG, bempegaldesleukin; CR, complete response; RT, radiation therapy.
In the MOC2 model (average starting tumor volume ~100 mm3), BEMPEG alone and RT alone significantly slowed tumor progression compared with the vehicle control (figures 1C and online supplemental figure S1D). RT alone significantly improved overall survival (figure 1D). RT+BEMPEG significantly improved tumor growth inhibition over the three other groups and displayed a significant survival benefit over BEMPEG alone and vehicle control.
In the 4T1 model (average tumor starting tumor volume 25–75 mm3), BEMPEG alone provided minimal benefit in tumor control, while RT alone significantly slowed tumor progression. RT+BEMPEG proved to be significantly more effective in controlling tumor progression than the separate treatments (figure 1E and online supplemental figure S1C). However, RT+BEMPEG did not improve overall survival in the 4T1 model (figure 1F). The mice in all groups required euthanasia at similar times due to respiratory failure resulting from metastatic disease spread.
RT increases CD122 expression on T cells in the spleen
Since the administration of BEMPEG after RT induced antitumor activity in multiple tumor models, we hypothesized that local RT was priming the immune system for BEMPEG. To investigate this, we randomized mice bearing 100–150 mm3 B78 flank tumors and treated half with local flank RT. On day 5 after RT, spleens were excised from control and RT-treated mice.
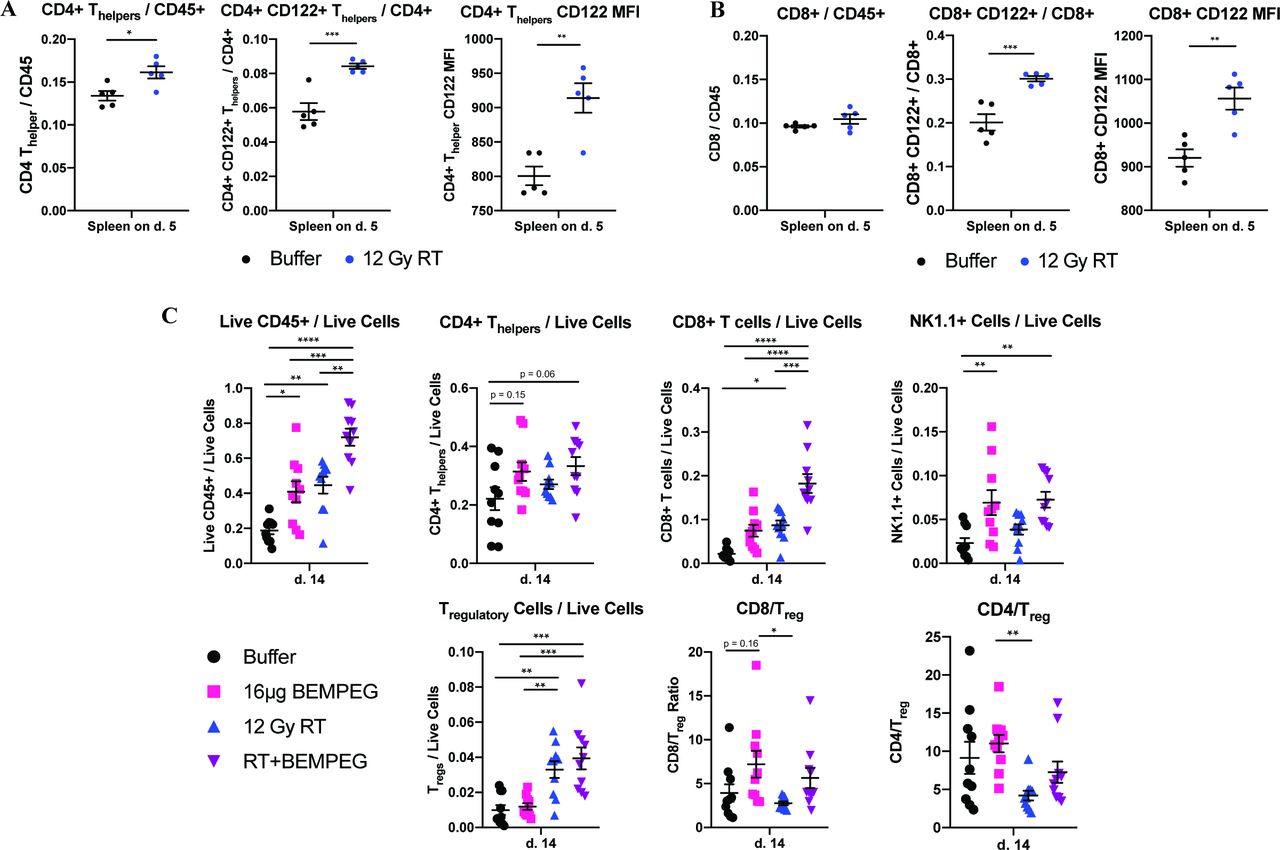
Mice treated with RT had an increased ratio of CD4+ T-helper/CD45+ cells in the spleen and similar CD8+:CD45+ ratios as control mice (figure 2A,B and online supplemental figure S2). The ratio of CD4+ and CD8+ T cells that were copositive for CD122 significantly increased following RT (figure 2A,B). These increased ratios were accompanied by significant increases in CD122 median fluorescent intensity (MFI) on both the CD8+ T cells and CD4+ T helpers in the spleen following RT (figure 2A,B). Given BEMPEG’s preferential activity towards the CD122 subunit (the intermediate affinity, heterodimeric IL-2R), these data indicate RT is priming peripheral T cells for activation by BEMPEG.
Supplemental material

External beam radiation primes B78 tumor-bearing mice for BEMPEG treatment by increasing CD122 expression on CD8+ and CD4+ T-helper cells in the spleen; RT+BEMPEG increases the number of TILs in the B78 melanoma model. (A) Immune cell profile in the spleen 5 days after RT showing CD4+ T helpers/CD45+, CD4+ CD122+ T helpers/CD4+ T helpers, and CD122 MFI on CD4+ T helpers. (B) Immune cell profile in the spleen 5 days after RT showing CD8+/CD45+, CD8+ CD122+/CD8+, and CD122 MFI on CD8+T cells. A separate independent experiment (with a smaller sample size) demonstrated similar trends as the data presented in (A,B) (analyses here done by unpaired t-test). (C) The combined results from two independent flow experiments (n=5 per group, per experiment) showing the number of TILs as a ratio of live cells (or as a ratio to T regulatory cells for the last two panels) in the tumor following treatment with buffer (black), RT alone (blue), BEMPEG alone (pink), or RT+BEMPEG (purple) on day 14 after treatment began. Each symbol represents the TIL or splenic immune cell profile from one mouse (analyses done by one-way ANOVA). P values calculated using one-way ANOVA with Tukey’s multiple comparison tests. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001. For comparisons where there is no p value shown, the p value was not significant. ANOVA, analysis of variance; BEMPEG, bempegaldesleukin; RT, radiation therapy; TIL, tumor-infiltrating lymphocyte.
RT+BEMPEG increases tumor-infiltrating lymphocytes (TILs) in the B78 model
Next, we investigated the type of immune cells that RT+BEMPEG recruits to the TME. Mice bearing 100–150 mm3 B78 flank tumors were treated with buffer, BEMPEG (day 5), RT (day 0), or RT+BEMPEG.
RT +BEMPEG significantly increased CD45+ immune cell infiltration compared with all other treatment groups, while BEMPEG and RT alone significantly increased the CD45+ immune infiltrate over the buffer control (figure 2C). There was a trend toward increased CD4+ T-helper infiltrate with RT+BEMPEG (p=0.06 vs buffer) and BEMPEG monotherapy (p=0.15 vs buffer). CD8+ T-cell infiltration was significantly greater in mice treated with RT+BEMPEG when compared with all other groups, while BEMPEG monotherapy and RT alone both demonstrated a modest increase in CD8+TIL frequency over control. Both groups treated with BEMPEG demonstrated a significant increase in NK cell tumor infiltration compared with buffer.
BEMPEG monotherapy did not significantly increase tumor Treg cell frequency over baseline. RT alone significantly increased the frequency of tumor Tregs, but RT+BEMPEG did not result in a further increase in Tregs over RT alone (figure 2C).
BEMPEG monotherapy and RT+BEMPEG both demonstrated CD4+ T-helper:Treg and CD8+:Treg ratios that were not significantly different from those for buffer. BEMPEG monotherapy significantly increased the CD8+:Treg and CD4+ T-helper:Treg ratios compared with RT alone. While RT+BEMPEG had mean CD8+:Treg and CD4+ T-helper:Treg ratios that appeared higher than RT alone, these were not significant (figure 2C).
Taken together, RT+BEMPEG increases the tumor-infiltrating CD8, T helper, and NK cell frequencies, and the increase in these populations is not accompanied by an equivalent increase in Tregs.
T cells are required for curing mice with RT+BEMPEG
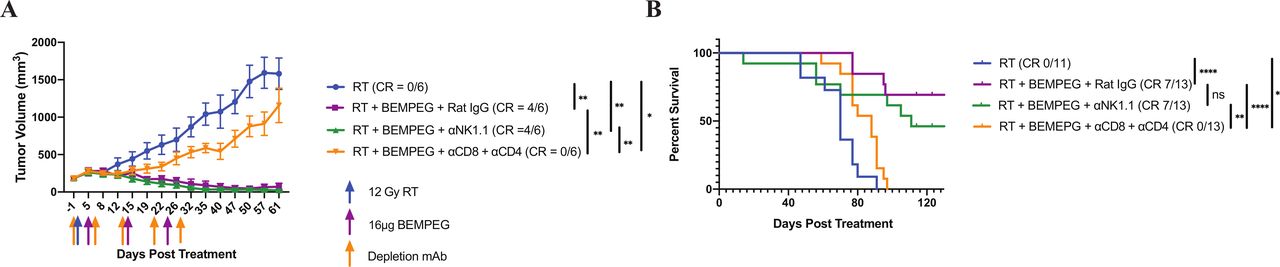
We conducted NK-cell and T-cell depletion experiments in the B78 model to better understand the role these cells have in the antitumor effect of RT+BEMPEG. Mice bearing ~150 mm3 B78 tumors were treated with RT, RT+BEMPEG+rat IgG, RT+BEMPEG+αNK1.1, or RT+BEMPEG+αCD4+αCD8. Depletion efficacy was confirmed via peripheral blood collection on days 13 and 34 (online supplemental figure S4).
Supplemental material
The cohort of mice receiving RT+BEMPEG+αNK1.1 antibodies displayed similar tumor volume changes, complete response (CR) rate, and overall survival compared with the group treated with RT+BEMPEG+rat IgG (figure 3A,B). Additionally, depletion of NK cells during the treatment phase of the experiments did not negatively impact the long-term immune memory in the animals that were cured of their disease burden (online supplemental figure S5).
Supplemental material

NK cells are not critical for the antitumor effect of RT+BEMPEG in the B78 model, while CD4 and CD8 T cells play an important role in curing mice of their disease burden. (A) Average tumor volume plot from a representative experiment and (B) combined overall survival from two independent experiments, showing group responses to RT alone (blue), RT+BEMPEG+Rat IgG (purple), RT+BEMPEG+NK cell depletion (green), and RT+BEMPEG+CD4 and CD8 T-cell depletion (orange), along with the number of mice in the experiment that demonstrated a CR. Tumor-free mice were censored from the Kaplan-Meier plot when they were rechallenged with a second B78 injection, which is indicated by the tick marks. P values for average tumor volume plots calculated using time-weighted analysis. P values for overall survival calculated via log-rank test. *P≤0.05, **P≤0.01, ≤, ****P≤0.0001. For comparisons where there is no p value shown, the p value was not significant. BEMPEG, bempegaldesleukin; CR, complete response; ns, not significant; RT, radiation therapy.
Mice treated with RT+BEMPEG+αCD4+αCD8 demonstrated a modest, but significant, slowing of tumor progression compared with the RT control, resulting in a significant survival benefit over RT alone (figure 3A,B) and suggesting that RT+BEMPEG is stimulating an antitumor immune effect outside of the T-cell compartment. Mice treated with RT+BEMPEG+rat IgG demonstrated significantly better tumor control and overall survival than the T-cell depletion cohort. The necessary role of T cells in the antitumor effect of RT+BEMPEG in the B78 model is supported by the lack of tumor regression following RT+BEMPEG in T cell-deficient TCR alpha KO mice (online supplemental figure S6).
Supplemental material
RT+BEMPEG generates stronger antitumor memory response than RT+IL-2
Since the combination of RT and high-dose IL-2 is being tested in the clinic, we compared the antitumor strength of RT+BEMPEG to RT+IL-2 in our B78 melanoma model. Mice were randomized (average B78 starting tumor volume 100–150 mm3) and treated with RT, RT+intravenous IL-2, RT+IT IL-2, or RT+BEMPEG. Equivalent amounts of IL-2 were dosed between groups receiving IL-2 or BEMPEG.
Dosing intravenous IL-2 after RT had little effect on average tumor volume and survival (figure 4A,B). One out of 10 mice treated with RT+intravenous IL-2 completely cleared its initial tumor burden (online supplemental figure S7B). This cured mouse failed to demonstrate immune memory on rechallenge (figure 4C).
Supplemental material

RT+BEMPEG results in a stronger antitumor memory response than does RT+intravenous IL-2 or RT+IT IL-2 in the B78 melanoma model. (A) Average tumor volume plot from a representative experiment and (B) combined overall survival from three independent experiments (the one shown in A and the two additional experiments shown in online supplemental figure S7A), showing group responses to RT alone (blue), RT+intravenous IL-2 (orange), RT+IT IL-2 (red) and RT+BEMPEG (purple), along with the number of mice in the experiment that demonstrated a CR, shown in parentheses. Mice were censored from the Kaplan-Meier plot when they were rechallenged with a second B78 injection on the abdomen, indicated by the tick marks. (C) The percent of cured mice following RT+intravenous IL-2 (0/1), RT+IT IL-2 (5/9), and RT+intravenous BEMPEG (16/16) that rejected engraftment of the tumor rechallenge is shown and compared with untreated naïve mice receiving the same tumor inoculum on the same day where 0/10 rejected the tumor. P values for average tumor volume plots were calculated using time-weighted analysis. P values for overall survival calculated via log-rank test. P values comparing tumor rechallenge rejection rates were calculated using χ2 analysis. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001. For comparisons where there is no p value shown, the p value was not significant. BEMPEG, bempegaldesleukin; CR, complete response; IL, interleukin; IT, intratumoral; RT, radiation therapy.
RT+IT IL-2 caused similar tumor regression and survival as RT+BEMPEG. When the results of three independent experiments were included, there was a suggestion that RT+BEMPEG could be more effective at curing animals of their disease: 16 of 21 (76%) for RT+BEMPEG vs 9 of 15 (60%) for RT+IT IL-2 (figure 4A and online supplemental figure S7A,B). However, this difference was not significant (p=0.298). Nevertheless, RT+BEMPEG did activate a stronger, long-lasting immune memory response than RT+IT IL-2. Namely, when cured mice were rechallenged with B78 melanoma, 100% of the RT+BEMPEG-cured mice rejected the rechallenge, compared with only 56% of the RT+IT IL-2-cured mice (p<0.01) (figure 4C).
CTLA-4 ICB further potentiates therapy of advanced B78 tumors with RT+BEMPEG
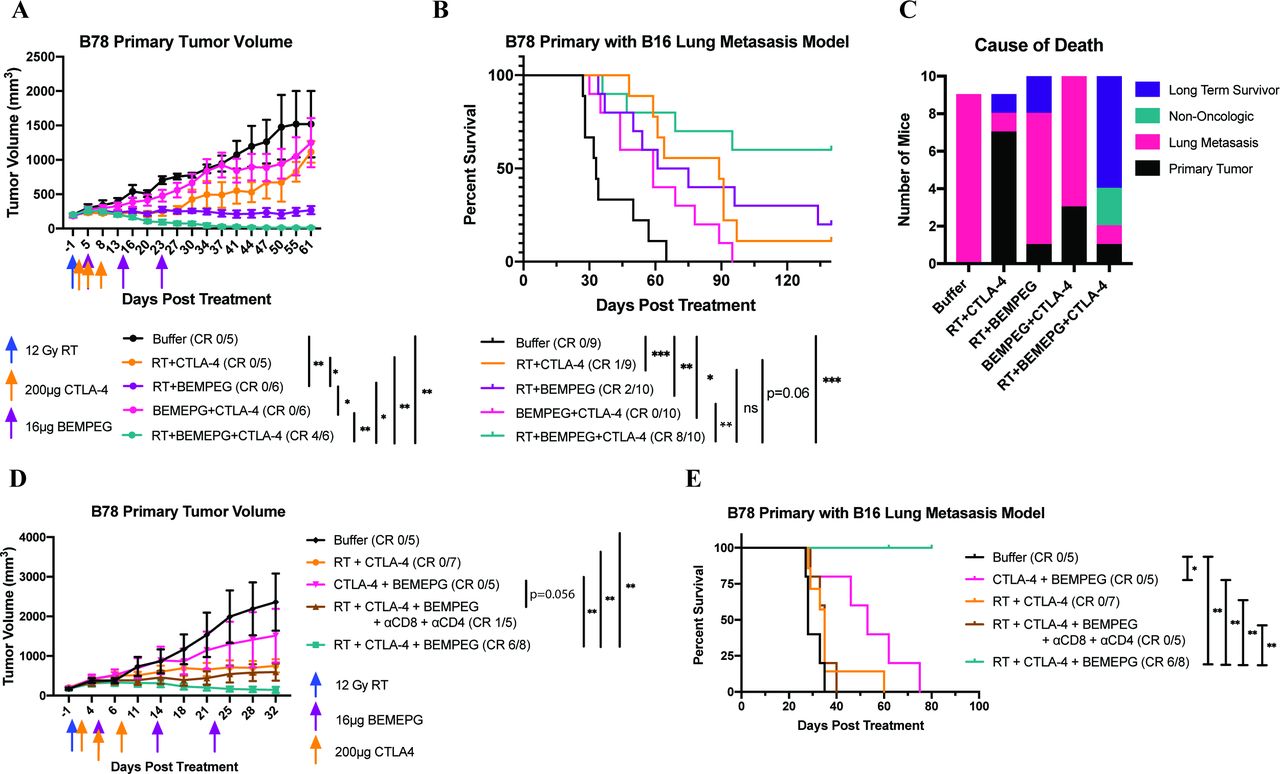
Recent reports have shown that smaller baseline tumor size is a predictor of improved overall survival in patients treated with ICB.28–30 We tested RT+BEMPEG under conditions where tumors had a larger starting volume and, therefore, were more difficult to treat. Mice bearing larger B78 tumors were randomized when average tumor volumes were 400 or 1000 mm3, and each cohort received RT, BEMPEG, and/or α-CTLA-4.
In the model with 400 mm3 tumors, RT+α-CTLA-4 provided no improvement in tumor volume control or survival over RT alone. RT+BEMPEG controlled tumor progression better than RT alone or RT+α-CTLA-4, resulting in a significantly improved overall survival (figure 5A,B and online supplemental figure S8). RT+BEMPEG+α-CTLA-4 demonstrated similar tumor volume control and overall survival as RT+BEMPEG.
Supplemental material
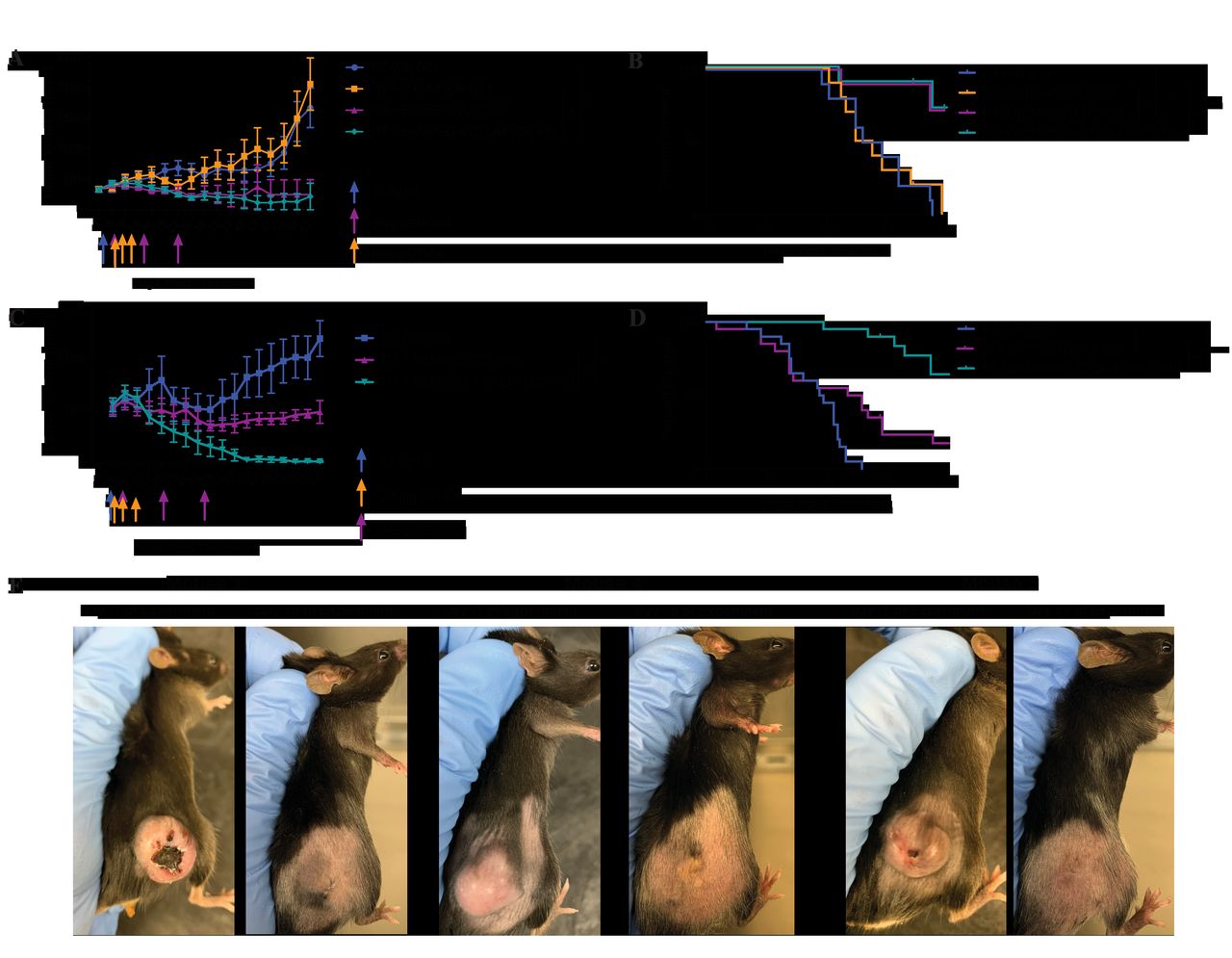
Addition of α-CTLA-4 to RT+BEMPEG is necessary when treating 1000 mm3, but not 400 mm3, advanced solid tumors. Mice bearing 400 mm3 B78 flank tumors (A–B) or 1000 mm3 B78 flank tumors (C–D) were treated with RT alone (blue), RT+α -CTLA-4 (orange), RT +BEMPEG (purple), or RT +BEMPEG+α -CTLA-4 (teal). average tumor volume plots (A, C) from a representative experiment and combined overall survival from two (B) and four (D) independent experiments are shown. the number of mice that demonstrated a CR is shown in parenthesis, and tumor-free mice were censored from the Kaplan-Meier plot when they were rechallenged with a second B78 injection on the abdomen, indicated by tick marks. (E) Photographs of three representative mice (of the 11 cured in figure 5D) bearing 1000 mm3 B78 tumors before treatment with RT+BEMPEG+α-CTLA-4 (day -1) and showing tumor resolution >1 month after treatment (day 66). P values for average tumor volume plots calculated using time-weighted analysis. P values for overall survival calculated via log-rank test. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001. For comparisons where there is no p value shown, the p value was not significant. BEMPEG, bempegaldesleukin; CR, complete response; RT, radiation therapy.
At a still higher B78 tumor burden of 1000 mm3 tumors (figures 5C–E and online supplemental figure S9), RT+BEMPEG slowed tumor progression compared with RT alone. Mice treated with RT+BEMPEG showed a 20% CR rate (figure 5D and online supplemental figure S9). Remarkably, RT+BEMPEG+α-CTLA-4 more than doubled the CR rate in this large tumor burden model, providing 55% (11/20) CR with improved tumor control and overall survival compared with RT+BEMPEG. The images in figure 5E highlight the striking effect of triple therapy on tumor burden, comparing day 1 vs day 66 post-treatment.
Supplemental material
RT+BEMPEG+α-CTLA-4 cures local and distant disease in the B78 primary with B16 metastasis model
The goal of an in situ vaccine is to create a systemic tumor-specific memory response that mediates an antitumor effect at all sites of disease. To investigate the strength of RT+BEMPEG+α-CTLA-4 as an in situ vaccine, we tested the regimen in an induced metastatic model. There are limitations to preclinical models studying abscopal responses; in particular, patients have increased levels of tumor heterogeneity that are difficult to replicate in orthotopic preclinical models.31 We attempted to recapitulate tumor heterogeneity by injecting B16 cells, the parental line to B78, intravenously into mice bearing a single ~150 mm3 B78 flank tumor (primary) 1 day before mice were randomized and treated. Mice were treated with buffer or a combination of local flank RT, α-CTLA-4, and BEMPEG.
Untreated mice in the B78 primary with B16 metastasis model succumb to pulmonary compromise from their metastatic disease burden prior to reaching the flank tumor size endpoint. In this model, BEMPEG+α-CTLA-4 significantly prolonged survival compared with the buffer control (figure 6B). This effect was due to BEMPEG+α-CTLA-4 slowing the progression of lung metastasis, as BEMPEG+α-CTLA-4 failed to control flank tumor progression (figure 6A,C). RT+α-CTLA-4 improved survival over buffer, largely due to its effect on slowing the progression of metastatic disease. The majority (77%) of the mice treated with RT+α-CTLA-4 were euthanized due to primary tumor progression. RT+BEMPEG significantly improved survival over buffer by controlling both flank tumor and lung metastasis progression, although the majority of mice still required euthanasia because of metastatic lung progression (figure 6A–C). Under similar starting tumor volumes, fewer mice in the B78 primary with B16 metastasis model demonstrated complete clearance of their flank tumor following RT+BEMPEG than in the B78 primary alone model (20% vs 67% in figure 6A,B vs figure 1A,B).
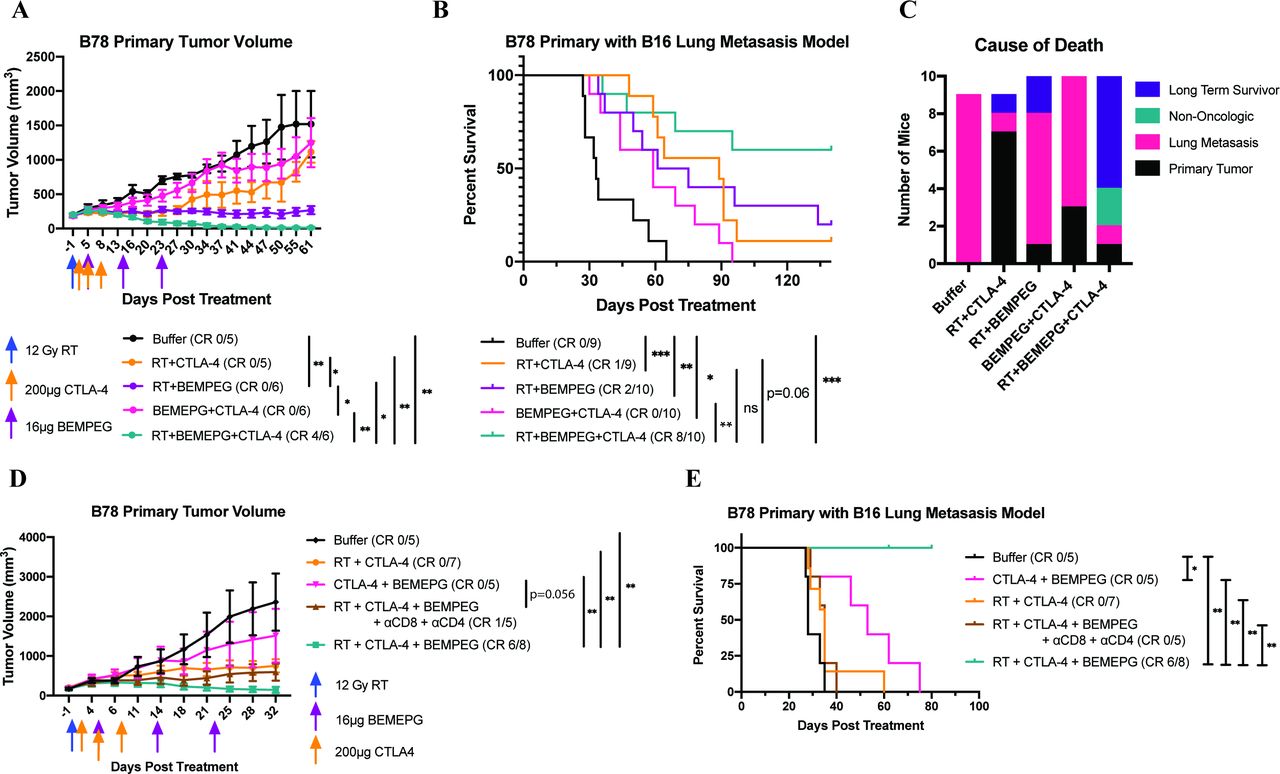
RT+BEMPEG+α-CTLA-4 prolongs survival through T cell-mediated control of metastasis. Mice bearing B78 flank tumors were injected intravenously in the tail vein with B16 cells 1 day prior to starting treatment with buffer (black), RT+α-CTLA-4 (orange), RT+BEMPEG (purple), α-CTLA-4+BEMPEG (pink), or RT+BEMPEG+α-CTLA-4 (teal). Average primary tumor volume plot from a representative experiment (A) and combined Kaplan-Meier survival curve from two independent experiments (B) in the B78 primary tumor with B16 metastases model. Mice that demonstrated a CR (no residual primary tumor and no death due to metastasis as of day 135) are shown in parentheses. A bar graph is provided for each treatment group (C) showing the number of long-term survivors and the cause of death of each mouse that died in the B78 primary with the B16 metastases model, based on the data presented in (B). In a separate experiment in the B78 primary with the B16 metastasis model, mice were treated with buffer (black), RT+α-CTLA-4 (orange), α-CTLA-4+BEMPEG (pink), RT+BEMPEG+α-CTLA-4 (teal), or RT+BEMPEG+αCTLA-4+CD4 and CD8 depletion antibodies (brown). The average primary tumor volume plot (D) and Kaplan-Meier survival plot (E) are presented. on day 62 of this experiment, a cage housing three mice in the RT+BEMPEG+α-CTLA-4 group was flooded due to an equipment malfunction. These three mice were censored from the Kaplan-Meier plot on this day (indicated by the tick mark). Of the three mice, two had cleared their flank tumor burden by day 62 and all three mice did not have visible lung metastases on autopsy. P values for average tumor volume plots were calculated using time-weighted analysis. P values for overall survival calculated via log-rank test. *P≤0.05, **P≤0.01, ***P≤0.001, ≤. For comparisons where there is no p value shown, the p value was not significant. BEMPEG, bempegaldesleukin; CR, complete response; RT, radiation therapy.
Importantly, RT+BEMPEG+α-CTLA-4 clearly provided superior primary tumor control and an overall survival benefit compared with all other treatment combinations (figure 6A,B and online supplemental figure S10). Even in the context of lung metastasis, 80% of mice showed complete clearance of the flank B78 tumors, and 60% of the mice were long-term survivors (figure 6B,C). Two mice were euthanized due to complications resulting from ulcerative dermatitis (UD). We believe that this infrequent side effect of the treatment regimen may reflect a predisposition of this particular group of C57BL/6 mice; other mice in this study also developed UD but required euthanasia because of tumor burden prior to the lethal progression of UD.32
Supplemental material
Control of lung metastasis progression by RT+BEMPEG+α-CTLA-4 was T cell mediated (figure 6D,E and online supplemental figure S13). Depletion of CD4+ and CD8+ T cells reduced the effectiveness of RT+BEMPEG+α-CTLA-4, particularly against metastatic disease; all mice that received T-cell depletion died of lung metastases.
Supplemental material
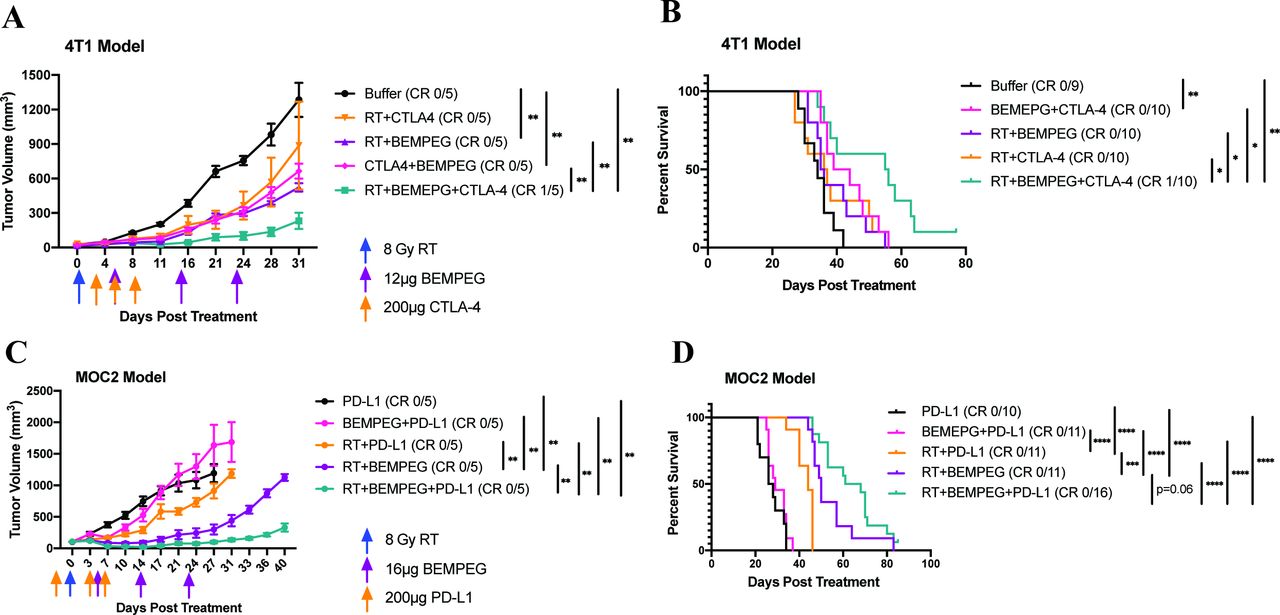
RT+BEMPEG+α-CTLA-4 prolongs survival in the 4T1 breast cancer model
In the 4T1 model, RT+BEMPEG and BEMPEG+α-CTLA-4 significantly slowed flank tumor progression compared with buffer, but RT+α-CTLA-4 did not (figure 7A and online supplemental figure S11). However, among these groups, only BEMPEG+α-CTLA-4 demonstrated a significant survival benefit over buffer, indicating BEMPEG+α-CTLA-4 showed some enhanced effect on metastatic progression (figure 7B). The triple combination of RT+BEMPEG+α-CTLA-4 controlled primary tumor progression and improved survival better than all other groups in the study. In a preliminary flow study on day 7 after RT, we did not observe any significant cell frequency differences in the CD8+, CD4+, Treg, myeloid-derived suppressor cell (MDSC) or macrophage populations between PBS control mice and RT+BEMPEG+CTLA-4 treated mice (data not shown). One mouse in the RT+BEMPEG+α-CTLA-4 demonstrated a CR at the primary tumor site and was a long-term survivor past 180 days. Since all the other mice in the 4T1 model were euthanized because of pulmonary complications resulting from lung metastases, the prolonged survival shown in figure 7B indicates that this triple therapy controlled the progression of spontaneous lung metastases better than all other immunotherapy combinations tested.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
RT+BEMPEG+checkpoint blockade improves local tumor control and prolongs survival in the 4T1 and MOC2 models. Mice bearing 4T1 flank tumors were treated with buffer (black), RT+α-CTLA-4 (orange), RT+BEMPEG (purple), α-CTLA-4+BEMPEG (pink), or RT+BEMPEG+α-CTLA-4 (teal). Average tumor volume plot from a representative experiment (A) and combined Kaplan-Meier survival curve from two independent experiments (B) in the 4T1 model are shown. Mice that demonstrated a CR are shown in parentheses. In this 4T1 model, all of the mice requiring euthanasia do so because of spontaneous symptomatic pulmonary metastases rather than flank tumors meeting endpoint criteria. Mice bearing MOC2 flank tumors were treated with α-PD-L1 (black), BEMPEG+α-PD-L1 (pink), RT+α-PD-L1 (orange), RT+BEMPEG (purple), and RT+BEMPEG+α-PD-L1 (teal). Average tumor volume plot from a representative experiment (C) and combined Kaplan-Meier survival curve from two independent experiments (D) in the MOC2 model are shown. Mice that demonstrated a CR are shown in parentheses. P values for average tumor volume plots were calculated using time-weighted analysis. P values for overall survival calculated via log-rank test. *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001. For comparisons where there is no p value shown, the p value was not significant. BEMPEG, bempegaldesleukin; CR, complete response; RT, radiation therapy.
RT+BEMPEG+α-PD-L1 improves local tumor control and overall survival in the MOC2 head and neck squamous cell carcinoma model
Finally, we evaluated the addition of α-PD-L1 to RT+BEMPEG to test whether RT+BEMPEG could be combined with different checkpoint blockade mechanisms in multiple preclinical models. Local antitumor immune effects of RT have been shown to be dependent on induction of interferon (IFN) in the TME, and PD-L1 expression in the MOC2 model has been shown to be IFN dependent.33 34 In the MOC2 head and neck squamous cell carcinoma model (HNSCC), flank tumors grew progressively when treated with α-PD-L1 alone (figure 7C,D and online supplemental figure S12). Adding BEMPEG to α-PD-L1 failed to provide additional flank tumor control or survival benefit. Adding RT to α-PD-L1 improved local tumor control and significantly increased overall survival compared with α-PD-L1 alone and BEMPEG+α-PD-L1. Importantly, RT+BEMPEG+α-PD-L1 significantly slowed flank tumor progression better than all other groups (figure 7C) and resulted in a survival benefit that was significant over all other combinations tested, except for RT+BEMPEG, where there was a trend (p=0.0604) (figure 7D).
Supplemental material
Discussion
Our group has previously shown that the antitumor efficacy of IT injections of IC is greatly enhanced when combined with RT.9 Adding ICB, in the form of α-CTLA-4 further improves the antitumor immune response and cures animals of irradiated and distant non-irradiated B78 tumors.9 25 Given the efficacy of combining RT with an IL-2-based therapy in some of our preclinical models, we wanted to explore the antitumor potential of combing RT with BEMPEG, a novel CD122-preferential IL-2 pathway agonist. Rather than local delivery of IC, BEMPEG is dosed systemically, and its long in vivo half-life combined with a minimized toxicity allows for a patient-favorable dosing schedule and outpatient administration.
Herein, we demonstrate a cooperative interaction between local RT and BEMPEG in multiple immunologically cold tumor models. The findings show that the antitumor effect of RT+BEMPEG in the B78 model is T cell dependent and results in immune memory. RT acts to prime the immune system, in part, by significantly increasing CD122 expression on peripheral Teffs; RT+BEMPEG increases tumor-infiltrating CD8+ T cells, CD4+ T-helper cells, and NK cells. Importantly, the increase in TILs following RT+BEMPEG is not accompanied by an increase in IT Tregs over the level present following RT alone. These results are consistent with what others have published regarding BEMPEG’s effect on tumor Tregs, and its preferential expansion of CD8, CD4 T helpers, and NK cells in mice and in patients with cancer.16 17 20 21 35 Additionally, we demonstrate that adding ICB, in the form of α-CTLA-4 or α-PD-L1, to RT+BEMPEG further enhances the antitumor immune response in preclinical models that more closely resemble conditions of high tumor burden and metastases in human patients.
In the B78 melanoma model, BEMPEG monotherapy occasionally cures animals of smaller (~100 to 150 mm3) primary tumors, but the responses are heterogeneous. When examined in the context of other published work, our data suggest that these strong antitumor responses with BEMPEG monotherapy are due to an increase in TILs and/or T-cell receptor clonality.18 21 Local RT prior to starting BEMPEG therapy significantly improves the antitumor response in the B78, 4T1, and MOC2 tumor models. While some studies have shown local RT can mobilize immune-suppressive populations like MDSCs and Tregs to the TME,25 36 other studies have reported on the immunological benefits of RT to include increased antigen presentation in the tumor draining lymph node, increased MHC-I and FAS/CD95 expression on tumor cells, activation of a type I IFN response, and trafficking of immune cells to the TME.5 37 38 RT may therefore help turn an immunologically cold tumor into one that is immunologically hot, thus making these tumors more likely to respond to ICB or cytokine therapy.2 Our data agree with this RT effect, as best demonstrated in the 4T1 model. BEMPEG monotherapy in the immunologically cold 4T1 model had a negligible effect on slowing tumor progression, presumably due to a lack of pre-existing tumor-specific TILs. There was, however, a measurable benefit of BEMPEG therapy after 4T1 tumors were treated with single fraction RT, suggesting BEMPEG therapy is more effective when treating tumors that have increased TIL levels.
In some instances, RT alone can act as an in situ vaccine, activating tumor-specific T cells and resulting in T cell-mediated tumor shrinkage of the treated tumor and distant metastasis.39 40 This systemic effect of RT was demonstrated in our B78 primary with the B16 metastasis model. The addition of RT to BEMPEG+α-CTLA-4 not only improved the local antitumor response but also enhanced the response to distant non-irradiated B16 lung metastasis (figure 6A–C); 70% of mice treated with BEMPEG+α-CTLA-4 were euthanized due to lung metastasis vs 10% of mice treated with RT+BEMPEG+α-CTLA-4.
Additional enthusiasm around RT was generated when preclinical and clinical data emerged, demonstrating a benefit of combining RT and ICB.6 41 This resulted in several clinical trials testing various combinations of RT and ICB, which have had mixed success.41–43 RT has also been combined with high-dose IL-2 therapy in clinical trials. In one phase I clinical trial, 8 of 12 patients with metastatic melanoma or renal cell carcinoma (RCC) achieved a CR or partial response following treatment with RT and intravenous IL-2.44 These results encouraged researchers to move forward with this treatment combination into a phase II trial for patients with metastatic melanoma and RCC (NCT02306954). However, high dose IL-2 therapy is still limited by its short in vivo half-life and its adverse side effect profile. Our preclinical results in the melanoma model comparing RT+BEMPEG with RT+intravenous IL-2 would suggest that substituting high-dose IL-2 with BEMPEG might result in more favorable patient outcomes. Additionally, the long half-life and decreased IL-2 associated toxicity of BEMPEG should allow these patients to receive their BEMPEG therapy in an out-patient setting.17 20
In preclinical experiments that more closely model the conditions in human patients (ie, large, locally advanced tumors or systemic/metastatic disease), the combination of RT+BEMPEG activated a systemic anti-tumor response sufficient to cure some mice of their disease (20% CR in 1000 mm3 B78 model, figure 5D; 20% CR in B78 primary with B16 metastasis model, figure 6B). These results align with a recent publication that demonstrated a synergistic effect of RT+BEMPEG in the immunogenic, CT26 and MCA-205 two-flank tumor mouse models.45 However, in our difficult-to-treat preclinical models (figures 1C–E, 5C,D and 7A–D), RT+BEMPEG was insufficient to eradicate tumor burden in the majority of treated mice and required combination with ICB therapy.
Others have published a synergistic interaction between BEMPEG and ICB showing an increase in T-cell clones, T-cell activation, and tumor control.21 The preliminary results of the PIVOT-02 clinical trial suggest BEMPEG+α-PD-1 ICB could be beneficial for some patients with metastatic melanoma, RCC, and small cell lung cancer.20 In our metastatic disease models, we demonstrate a similar benefit when BEMPEG is combined with α-CTLA-4 or α-PD-L1 ICB. The adverse side effects observed in the PIVOT-02 trial do not appear to be significantly different from those observed with α-PD-1 therapy alone.20 46 Therefore, we cautiously predict combing BEMPEG with α-CTLA-4 in the clinic would not significantly increase the adverse side effect profile over treatment with α-CTLA-4 alone.
Importantly, we show for the first time that the combination of RT+BEMPEG+ICB was more effective than any double combination of these approaches. The triple combination cured large, locally advanced primary tumors and distant metastatic disease in our melanoma models. Additionally, the triple combination demonstrated greater activity, particularly against metastatic disease, in the 4T1 and MOC2 models. Our data indicate that all three components of this regimen play a necessary, non-redundant role in these systemic responses. We suggest the combination may be working as follows: RT augments immunogenicity, acting as ‘ignition’ to prime the immune system through previously discussed mechanisms; BEMPEG provides the necessary ‘gas’ to the immune response to expand and maintain the antigen specific T-cell response,21 while ICB removes the ‘brakes’, allowing the tumor-specific T cells to mediate and maintain their cytotoxic killing at both local and distant sites of disease.47
Conclusion
The preclinical data presented here show that the triple combination of RT+BEMPEG+ICB has benefit over the combination of any two of these modalities. Ongoing clinical trials are testing the effectiveness of RT+high-dose IL-2, RT+ICB, and BEMPEG+ICB. Our preclinical data suggest that a regimen that combines these approaches, to include RT, BEMPEG, and ICB, merits clinical exploration.
Supplemental material
Supplemental material
Data availability statement
Data are available upon reasonable request. Please connect with the first author with any data requests.
Ethics statements
Ethics approval
The research contained in this article does not use patient data in any form. Animal studies were conducted according to a protocol approved by the University of Wisconsin Institutional Animal Care and Use Committee.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @PiepsTweeets
Contributors AAP was responsible for experimental design, execution, and analysis of data. AAP, and created final versions of all figures and drafted the manuscript. DS and WJJ collected and analyzed experimental data. RBP collected tissue for flow cytometry analysis. PMC and RBP designed and tested the antibody panel used for the flow cytometry experiments. JB conducted and confirmed statistical analysis of experimental data. PMS, ALR, RBP, DC, WWO, and ZM contributed to experimental design, thorough edits, and review of the manuscript. ALR, JH, and AE contributed to experimental design, data collection, mouse colony maintenance, and literature research. PMS provided experimental design and review of data. All authors provided thorough review and editing of the manuscript draft.
Funding This work was supported by Midwest Athletes Against Childhood Cancer, Stand Up 2 Cancer, the St. Baldrick’s Foundation, the Crawdaddy Foundation, and the University of Wisconsin Carbone Cancer Center. This research was also supported in part by public health service grants TR002373, U54-CA232568, R35-CA197078, 5K08CA241319, 1DP5OD024576, U01-CA2331 02, P50 DE026787, and P01 CA250972 from the National Cancer Institute, the National Institutes of Health (NIH) and the Department of Health and Human Services. AAP was supported by NIH award TL1 TR002375; PMC was supported by the NIH-NCI award F30CA228315 and NIH award TL1 TR002375. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.