Article Text

Abstract
Background GWN323 is an IgG1 monoclonal antibody (mAb) against the glucocorticoid-induced tumor necrosis factor receptor-related protein. This first-in-human, open-label phase I/Ib study aimed to investigate the safety and tolerability and to identify the recommended doses of GWN323 with/without spartalizumab, an anti-programmed cell death receptor-1 agent, for future studies. Pharmacokinetics, preliminary efficacy and efficacy biomarkers were also assessed.
Methods Patients (aged ≥18 years) with advanced/metastatic solid tumors with Eastern Cooperative Oncology Group performance status of ≤2 were included. GWN323 (10–1500 mg) or GWN323+spartalizumab (GWN323 10–750 mg+spartalizumab 100–300 mg) were administered intravenously at various dose levels and schedules during the dose-escalation phase. Dose-limiting toxicities (DLTs) were assessed during the first 21 days in a single-agent arm and 42 days in a combination arm. Adverse events (AEs) were graded per National Cancer Institute-Common Toxicity Criteria for Adverse Events V.4.03 and efficacy was assessed using Response Evaluation Criteria in Solid Tumors V.1.1.
Results Overall, 92 patients (single-agent, n=39; combination, n=53) were included. The maximum administered doses (MADs) in the single-agent and combination arms were GWN323 1500 mg every 3 weeks (q3w) and GWN323 750 mg+spartalizumab 300 mg q3w, respectively. No DLTs were observed with single-agent treatment. Three DLTs (6%, all grade ≥3) were noted with combination treatment: blood creatine phosphokinase increase, respiratory failure and small intestinal obstruction. Serious AEs were reported in 30.8% and 34.0%, and drug-related AEs were reported in 82.1% and 77.4% of patients with single-agent and combination treatments, respectively. Disease was stable in 7 patients and progressed in 26 patients with single-agent treatment. In combination arm patients, 1 had complete response (endometrial cancer); 3, partial response (rectal cancer, adenocarcinoma of colon and melanoma); 14, stable disease; and 27, disease progression. GWN323 exhibited a pharmacokinetic profile typical of mAbs with a dose-dependent increase in the pharmacokinetic exposure. Inconsistent decreases in regulatory T cells and increases in CD8+ T cells were observed in the combination arm. Gene expression analyses showed no significant effect of GWN323 on interferon-γ or natural killer-cell signatures.
Conclusions GWN323, as a single agent and in combination, was well tolerated in patients with relapsed/refractory solid tumors. The MAD was 1500 mg q3w for single-agent and GWN323 750 mg+spartalizumab 300 mg q3w for combination treatments. Minimal single-agent activity and modest clinical benefit were observed with the spartalizumab combination.
Trial registration number NCT02740270.
- antibodies
- neoplasm
- clinical trials as topic
- drug therapy
- combination
- therapies
- investigational
Data availability statement
All data relevant to the study are included in the article or uploaded as supplemental information. Novartis will not provide access to patient-level data, if there is a reasonable likelihood that individual patients could be reidentified. Phase I studies, by their nature, present a high risk of patient reidentification; therefore, patient individual results for phase I studies cannot be shared. In addition, clinical data, in some cases, have been collected Patient to contractual or consent provisions that prohibit transfer to third parties. Such restrictions may preclude granting access under these provisions. Where codevelopment agreements or other legal restrictions prevent companies from sharing particular data, companies will work with qualified requestors to provide summary information where possible.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
With the success of targeted antibody therapies against cytotoxic T-lymphocyte antigen-4 or programmed cell death receptor-1 (PD-1),1–3 anticancer immunotherapy has evolved and led the way for new and promising approaches that involve activating costimulatory pathways to improve antitumor immune responses. One such strategy targets the costimulatory molecules on T cells, such as the glucocorticoid-induced tumor necrosis factor receptor (GITR), CD40, CD27, 4-1BB and OX40.4–6
GITR is a type 1 transmembrane protein belonging to the tumor necrosis factor receptor superfamily that modulates both the adaptive and innate immune responses. It is constitutively expressed at high levels on activated CD4+CD25− effector T (Teff) cells, Foxp3+ regulatory T (Treg) cells, CD8+ Teff cells, B cells, monocytes and macrophages, natural killer (NK) cells, plasmacytoid dendritic cells, mature dendritic cells, mast cells, eosinophils, basophils and leukocytes.7–9 On activation, GITR has the capacity to promote the function of Teff cells and to inhibit Treg cells.10 11 This shift in the balance between the Teff and Treg cells increases the activity of the immune system, making it more effective at tumor cell destruction.10 11 Studies in GITR transgenic mice suggest that the GITR engagement may increase the levels of memory CD4+ cells (CD44+/CD62L−).12
GWN323 is an agonistic Humaneered anti-GITR IgG1 monoclonal antibody (mAb) that binds specifically and with high affinity to the human GITR.10 GWN323, a human and cynomolgus monkey cross-reactive mAb, showed functional activity in vitro in human T-cell assays and in vivo in syngeneic tumor models in hGITR.hGITRL dKI mice. Preclinical mouse models have demonstrated that GITR agonists have a synergistic antitumor effect when combined with other anticancer therapies, especially PD-1 inhibitors.9 10 13 14 Spartalizumab is a humanized IgG4 anti-PD-1 mAb that binds to PD-1 and blocks its interaction with programmed death-ligand (PD-L) 1 and PD-L2. Spartalizumab has previously shown favorable pharmacokinetics (PK) and safety and preliminary antitumor activity in patients with anaplastic thyroid cancer and other advanced solid tumors.15 16
Here, we report the results of a first-in-human, phase I/Ib, multicenter, open-label study of GWN323 as a single agent and in combination with spartalizumab in adult patients with relapsed or refractory solid tumors and lymphomas. The primary objective was to characterize the safety and tolerability of GWN323 as a single agent and in combination with spartalizumab and to identify recommended doses and schedules for future studies. The secondary objectives were to characterize the PK, assess the pharmacodynamic (PD) effects and evaluate the preliminary antitumor activity of GWN323 as a single agent and in combination with spartalizumab.
Methods
Patient population
Inclusion criteria
The study enrolled patients aged ≥18 years with histologically confirmed advanced/metastatic solid tumors or lymphomas; with an Eastern Cooperative Oncology Group (ECOG) performance status of ≤2 with measurable or non-measurable disease, as determined by the Response Evaluation Criteria in Solid Tumors (RECIST) V.1.1; and who had progressed on or were intolerant to standard treatment or for whom no standard treatment existed. Eligible patients had a site of disease amenable to biopsy.
Exclusion criteria
Key exclusion criteria included symptomatic central nervous system (CNS) metastases or CNS metastases requiring local CNS-directed therapy, diagnosis of T-cell lymphomas, prior allogeneic transplants, prior anti-GITR therapy or history of severe hypersensitivity reactions to other mAbs. Patients intolerant to prior immunotherapy (unable to continue/receive owing to immune-related adverse events (AEs)); patients with active HIV, hepatitis B virus or hepatitis C virus infections; and patients with impaired cardiac function and inadequate bone marrow or end-organ function during screening were also excluded.
Study design and treatment
The study consisted of two phases: a dose-escalation phase to establish the maximum tolerated dose (MTD) and/or recommended phase 2 dose (RP2D) and a dose-expansion phase at the RP2D. The dose-escalation phase included two parallel dose escalations (GWN323 single agent and combination of GWN323 and spartalizumab (GWN323+spartalizumab) with staggered starts. Patients received GWN323 or GWN323+spartalizumab via intravenous infusion over 30 min once in every 21-day cycle (every 3 weeks (q3w)) until disease progression per the immune-related response criteria (irRC), unacceptable toxicity or treatment discontinuation at the investigator’s or patient’s discretion. Premedication per institutional standard of care was allowed (except on cycle 1 day 1) at the discretion of the treating physician if the patient experienced infusion reactions.
In the single-agent arm, groups of three to six patients were enrolled in seven dosing cohorts from 10 mg to 1500 mg of GWN323 on day 1 and q3w. The starting dose was 10 mg; selected based on the predicted human minimum anticipated biological effect level using the preclinical PK/PD data and in vitro toxicology studies of GWN323. Similarly, in the combination arm, eight dosing cohorts were evaluated with GWN323 doses ranging from 10 mg to 750 mg and spartalizumab doses ranging from 100 mg to 300 mg. Dose escalations were guided using the Bayesian logistic regression model following escalation with overdose control principle. Dose escalation in the combination arm started only after the completion of the first two dose-level cohorts of the single agent in the q3w schedule. Patients were followed up for 90 days after the last dose in the single-agent arm and for 150 days in the combination arm for safety evaluations.
MTD determination
The MTD in single-agent arm was defined as the highest drug dosage not expected to cause a dose-limiting toxicity (DLT) in ≥33% of the treated patients in the 21 days following the first dose of GWN323 treatment. In the combination arm, MTD was the highest combination drug dose not expected to cause a DLT in ≥33% of the treated patients in the 42 days following the first treatment with the combination. A DLT was defined as any grade ≥3 AE that occurred within the first 21 days of GWN323 treatment or within 42 days of GWN323+spartalizumab treatment, as assessed using the National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI-CTCAE), unless it could be clearly attributed to another cause.
Safety and efficacy assessments
Safety was assessed according to the NCI-CTCAE V.4.03 and included incidence and severity of treatment-emergent adverse events (TEAEs) and serious adverse events (SAEs), including changes in laboratory parameters, vital signs and electrocardiograms. Dose interruptions, reductions and intensity were also assessed.
The efficacy assessments included best overall response (BOR) per RECIST V.1.1 and irRC, measured from treatment initiation until disease progression and summarized by treatment arm.
Pharmacokinetic assessments
Blood samples were collected on day 1 of cycle 1 (predose and end of infusion) and during the study visits on days 2, 4, 8 and 15 of cycle 1; on day 1 (predose and end of infusion) of cycles 2 and 3; on days 1 (predose and end of infusion), 2, 4, 8 and 15 of cycle 4; on day 1 (predose) of every subsequent cycle; and at the final visit. The PK parameters were determined using non-compartmental methods for GWN323 and spartalizumab.
Biomarker and PD assessments
Tumor biopsy samples (new or recent (≤3 months from registration) plus two additional biopsies during the course of the study) and peripheral blood mononuclear cells (PBMCs) were used for biomarker and PD assessments. The timings of the tumor sample collections were flexible (at screening, between cycle 2 day 1 and cycle 2 day 15, and between cycle 4 day 1 and cycle 6 day 20). Expression and localization of the biomarkers, including Foxp3, CD8 and PD-L1, were measured using immunohistochemistry or RNA sequencing. In addition, the effector:Treg cell ratio at screening and during treatment was measured using flow cytometry to assess the PD effect of GWN323 alone and in combination with spartalizumab in the PBMC samples.
Statistical analyses
The safety and efficacy analysis population included all patients who received ≥1 full or partial dose of GWN323 or spartalizumab (full analysis set (FAS)). The dose-determining set included all patients from the FAS who completed the minimum exposure requirement (one full dose in the q3w and two-thirds of the planned doses of GWN323 in the weekly schedule plus a full dose of spartalizumab in the combination arm) or had a DLT during the first cycle (21 days) in the single-agent arm or during cycles 1 and 2 (42 days) in the combination arm. Patients who received ≥1 of the planned treatments and provided ≥1 primary PK parameter were included in the PK analysis set.
Descriptive statistics was used to summarize demographic and other baseline data (including disease characteristics and duration of exposure) by treatment arm. All safety assessments were summarized descriptively. DLTs and their incidence were summarized by primary system organ class, preferred term, type and grade of AE and treatment arm. The overall response rate (ORR, defined as the proportion of patients with a BOR of complete response (CR) or partial response (PR)) and disease control rate (DCR, defined as the proportion of patients with a BOR of CR or PR or stable disease (SD)) were presented by treatment arm. Descriptive statistics (mean, SD, coefficient of variation (CV) %) were presented for all serum PK parameters, study arms and study cycles/days.
Results
A total of 92 patients with relapsed/refractory solid tumors and lymphomas were enrolled between July 2016 and November 2018 in Canada, Israel, Japan, Singapore, Spain, the USA and the UK. Owing to minimal antitumor activity (and not owing to safety concerns), the enrollment in the study was terminated at the end of the dose-escalation phase, and the dose-expansion phase was not initiated. Based on the early signal of antitumor activity during initial dose escalation, the GWN323 150 mg+spartalizumab 300 mg dose level in the combination arm was enriched for patients with high microsatellite instability (MSI-H) cancers and for patients with melanoma.
Patient characteristics
Overall, 92 patients were treated with GWN323 as a single agent (n=39) or in combination with spartalizumab (n=53) and were included in the FAS. The baseline characteristics and demographics of the patients are listed in table 1. The median age of the patients in the single-agent arm was 61 (range 31–79) years; 56.4% of the patients were female and 61.5% were Caucasian. Most had an ECOG performance status of 0 (53.8%) or 1 (43.6%). The most common cancer types included were colorectal cancer (12.8%) and ovarian cancer (10.3%). All the patients (100%) in the single-agent arm received ≥1 regimen of antineoplastic therapy before entering the study; 28.2% of the patients had two prior antineoplastic regimens, and 82.1% had ≥2 prior antineoplastic regimens. The median age in the combination arm was 60.0 (range 34–84) years; 56.6% of the patients were male and 67.9% were Caucasian (table 1).
Demographic and baseline characteristics by treatment arm
Of the 39 patients in the single-agent arm, 33 (84.6%) discontinued treatment owing to progressive disease (online supplemental table 1). The major reason for discontinuation in the post-treatment follow-up phase was completion of study treatment (12 (30.8%)). In the combination arm, 41 patients (77.4%) discontinued owing to progressive disease (online supplemental table 2). Of the 40 patients who entered the post-treatment follow-up phase, 28 (52.8%) discontinued owing to new therapy for study indication.
Supplemental material
Treatment exposure
The median (range) duration of exposure to GWN323 was 9 (2–72) weeks in the single-agent arm and 12 (3–139) weeks in the combination arm. Most patients (41.0%) in the single-agent arm were exposed to GWN323 for 6–12 weeks. In the combination arm, 35.8% of the patients had an exposure of ≥18 weeks for GWN323. The median (range) exposure to spartalizumab in the combination arm was 12 (3–139) weeks.
MTD and maximum administered dose (MAD)
No DLTs were observed with the single-agent treatment, and MTD was not reached (table 2). MAD was GWN323 1500 mg q3w for the single-agent treatment as further dose escalation was not deemed to significantly increase GITR receptor occupancy and elevate biological response.
Summary of DLTs
DLTs were reported in three patients (6%) in the combination arm; blood creatine phosphokinase increase (grades 3–4) in the GWN323 10 mg+spartalizumab 200 mg dose-escalation cohort, respiratory failure (grade 3) in the GWN323 30 mg+spartalizumab 100 mg dose-escalation cohort and small intestinal obstruction (grade 3) in the GWN323 150 mg+spartalizumab 300 mg dose-escalation cohort (table 2).The number of DLTs in these cohorts did not exceed the threshold set for MTD (>33%), and MTD was not reached. GWN323 750 mg+spartalizumab 300 mg q3w was determined as MAD for the combination treatment. Owing to minimal antitumor activity, no dose escalation beyond GWN323 750 mg was carried out in the combination arm.
Safety
All 39 patients (100%) in the single-agent arm had ≥1 TEAE, including 17 (43.6%) with grade ≥3 TEAEs (table 3). The most frequently reported TEAEs were constipation (30.8%), fever (28.2%) and fatigue (25.6%). In the combination arm, 51 patients (96.2%) reported ≥1 TEAE, including 25 patients (47.2%) with grade ≥3 TEAEs; the most frequently reported TEAEs were fatigue (28.3%), decreased appetite (26.4%) and nausea (24.5%; table 3).
Summary of adverse events reported in ≥10% of the patients
SAEs occurred in 30.8% and 34.0% of the patients in the single-agent and combination arms, respectively (online supplemental tables 3 and 4). Most common SAEs were anemia, pulmonary embolism, nausea, vomiting and small intestinal obstruction in the single-agent arm (5.1% each), and abdominal pain, sepsis and vomiting in the combination arm (5.7% each, online supplemental table 4). No SAEs in the single-agent arm were related to the study drug. However, 7.5% of the patients in the combination arm reported SAEs related to the study drug (infusion-related reaction (1.9%), small intestinal obstruction (1.9%), type II respiratory failure (1.9%) and abdominal pain (1.9%)). AEs suspected to be related to the study drug were reported in 82.1% and 77.4% of the patients (online supplemental tables 3 and 5) and AEs leading to treatment discontinuation were reported in 7.7% and 5.7% of the patients in the single-agent and combination arms, respectively. Most common AEs suspected to be related to the study drug were fever (25.6%), chills (17.9%) and myalgia (15.4%) in the single-agent arm and fatigue (18.9%), fever (18.9%) and chills (15.1%) in the combination arm (online supplemental table 5); 10.3% and 20.8% of the patients in the single-agent and combination arms, respectively, required no dose reduction. One patient died on treatment owing to renal failure and one died owing to disease progression in the single-agent and combination arms, respectively. However, neither death was suspected to be related to the study drug (online supplemental table 3).
Efficacy
None of the patients in the single-agent arm had CR or PR. Of the 39 evaluable patients, 7 (17.9%) achieved SD (patients remained free of disease progression for a period of 1.4–5.8 months after showing SD) and 26 (66.7%) had disease progression per RECIST V.1.1. DCR was 17.9% (95% CI 7.5% to 33.5%, figure 1).

Best percentage change and best overall response by investigator assessment (RECIST V.1.1) in patients with target lesions in the single-agent arm. n is the number of patients with ≥1 baseline and postbaseline assessment of target lesions. CR, complete response; GWN, GWN323; PD, progressive disease; PR, partial response; q3w, every 3 weeks; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease; UNK, unknown.
In the combination arm, one patient (1.9%) with Lynch syndrome (PMS2 mutation) with MSI-H endometrial cancer (PD-1/PD-L1 naïve) had CR, and three patients (5.7%) had PR. Tumor type and pretreatment were (1) MSI-H rectal cancer, PD-1/PD-L1 naïve; (2) Lynch syndrome with poorly differentiated adenocarcinoma of the colon (MSI-H adenocarcinoma of the colon), PD-1/PD-L1 naïve; and (3) melanoma pretreated with two different lines of PD-1/PD-L1 immune checkpoint inhibitors (pembrolizumab and nivolumab). The disease was stable in 14 patients (26.4%) and progressed in 27 patients (50.9%). The ORR was 7.5% (95% CI 2.1% to 18.2%), and DCR was 34% (95% CI 21.5% to 48.3%, figure 2). Efficacy results based on the irRC were identical to those observed with RESIST V.1.1.
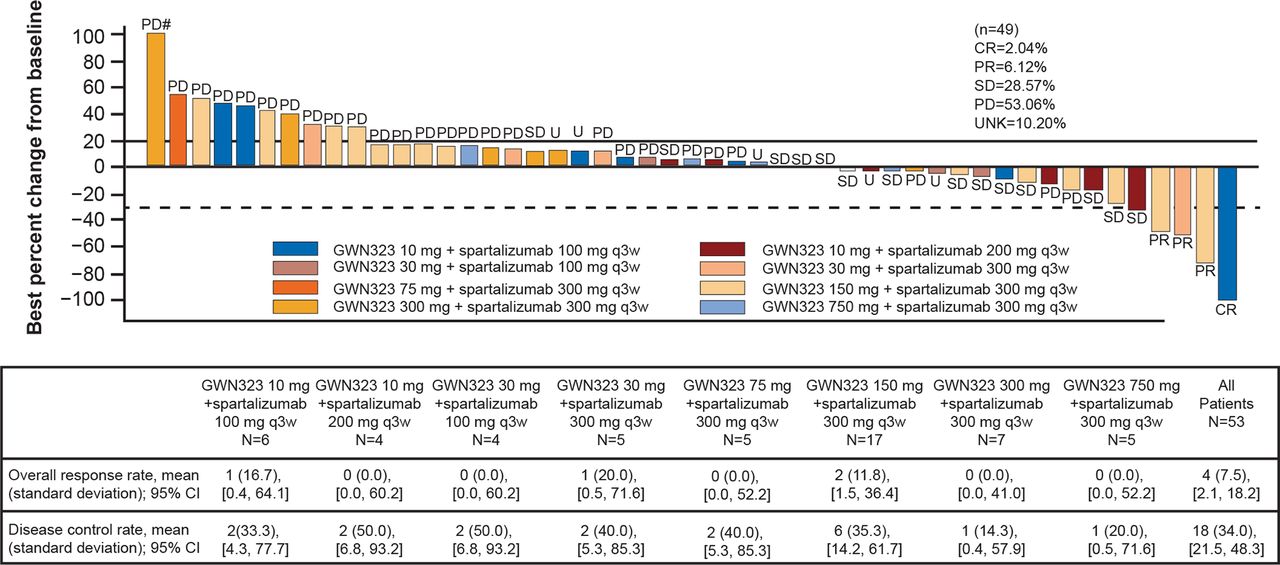
Best percentage change and best overall response by investigator assessment (RECIST V.1.1) in patients with target lesions in the combination arm. # indicates percentage changes from baseline of >100% are set to 100%. n indicates the number of patients with ≥1 baseline and postbaseline assessment of target lesions. CR, complete response; GWN, GWN323; PD, progressive disease; PR, partial response; q3w, every 3 weeks; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease; UNK, unknown.
After the inclusion of the enrichment cohort, of the nine patients treated for microsatellite instability tumors, one had CR (MSI-H endometrial cancer treated with GWN323 10 mg+spartalizumab 100 mg) and two had PR (both MSI-H colorectal cancer treated with GWN323 150 mg+spartalizumab 300 mg). The three patients were anti-PD-1/PD-L1 treatment naïve. Of the eight patients with melanoma, one patient had PR (previously treated with immune checkpoint inhibitors and was treated at a dose of GWN323 150 mg+spartalizumab 300 mg) and one had unconfirmed PR (treated with GWN323 10 mg+spartalizumab 200 mg).
Pharmacokinetics
The PK variability for single-agent GWN323 was moderate at cycle 1 day 1, as illustrated by the between-patient variability (CV%) for maximum concentration (Cmax, 8.6%–54.3%) and area under the curve from time=0 to last measurable concentration (AUClast, 22.6%–45.9%). The median terminal half-life (t½) of GWN323 was 7.3 days (range 5.6–9.5 days) in the single-agent arm (online supplemental table 6). A higher PK exposure was observed at cycle 4 day 1 compared with cycle 1 day 1. The t½ of GWN323 in the combination arm was 8.7 days (range 4.6–12.6 days, online supplemental table 7). A dose-dependent increase in PK exposure (Cmax and AUClast) was observed with the increasing dose of GWN323. Similarly, the PK exposure in the combination arm increased with increasing GWN323+spartalizumab doses. The PK exposure of GWN323 in the combination setting was comparable to that of the single agent, indicating no significant drug–drug interaction between GWN323 and spartalizumab.
Biomarkers
Flow cytometry of the PBMC samples was conducted to examine several immune cell subsets. Transient increases in proliferating CD8, NK and effector memory CD8 cells were observed during treatment in patients in both the single-agent and combination arms (figure 3A,B). Some patients in the single-agent arm exhibited on-treatment decreases in peripheral effector Treg cells (online supplemental figure 1), although these changes were not dose dependent. No effects of GWN323 treatment on the total Treg cells were observed by flow cytometry analysis (data not shown).

Effect of treatment on (A) proliferating CD8 and NK cells and (B) effector memory CD8 cells (flow cytometry data). Blood was collected and PBMCs were isolated at screening, C1D8 and C2D1. PBMCs were stained to identify the percentages of proliferating CD8 and NK cells (A) as well as effector memory T cells (B). BOR categories are indicated by shape, and doses of GWN±spartalizumab are color coded. BOR, best overall response; C, cycle; CR, complete response; D, day; GITR, glucocorticoid-induced tumor necrosis factor receptor; GWN, GWN323; NK, natural killer; PBMC, peripheral blood mononuclear cell; PD, progressive disease; PR, partial response; SD, stable disease; UNK, unknown.
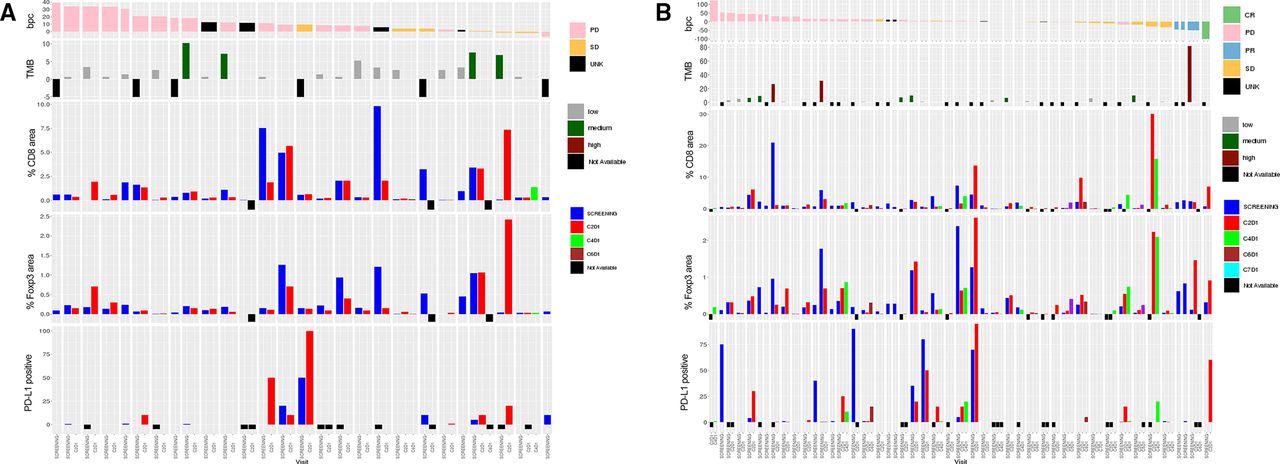
Immunohistochemistry was performed to assess the levels of the PD-L1, Foxp3 and CD8 markers in paired tumor biopsies. The CD8 levels were low in most patients; only five patients in the single-agent arm exhibited CD8 levels of >2% at baseline (figure 4A). Similarly, low levels of Foxp3 staining were observed in most patients, with four patients exhibiting >1% Foxp3 levels at baseline. Of these four patients, three exhibited an on-treatment decrease in the Foxp3 level. Levels of PD-L1 were low in most patients. One patient with SD had a modest on-treatment increase in the levels of all three markers (figure 4A). Most patients in the combination arm had generally low baseline CD8 (<2%) and Foxp3 (<1%) levels (figure 4B). Three patients with >1% Foxp3 level at baseline exhibited an on-treatment decrease in this marker (figure 4B). One patient in the combination arm with a confirmed CR exhibited on-treatment increases in the levels of all three markers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immunohistochemistry data for the (A) single-agent and (B) combination arms. Immunohistochemistry was performed on paraffin-embedded sections of the tumor samples collected at screening, C2D1 and C4/5/6D1. Visits are color coded. BPC, best percent change in tumor size; C, cycle; CR, complete response; D, day; PD, progressive disease; PR, partial response; SD, stable disease; TMB, tumor mutational burden; UNK, unknown.
Combined RNA sequencing data from all the patients in the single-agent arm (n=15 paired samples) indicated no significant correlations between GWN323 dosing and changes in T-cell function (as measured by the interferon (IFN)-γ expression levels) or NK cell signatures (online supplemental figure 2). Analysis of paired samples from the combination arm (n=13 paired samples) indicated that IFN-γ and Treg signatures were upregulated in patients who had a 30% on-treatment decrease in tumor volume (online supplemental figure 3).
Discussion
Agonistic antibodies targeting GITRs are expected to have a dual mechanism of action, involving both elimination of Treg cells and enhanced activation of Teff cells.10 11 Several human GITR agonists are currently in early clinical development for the treatment of solid tumors, for example, TRX518, INCAGN01876, AMG 228, MEDI1873 and MK-4166 among others.9 10 17–19 However, most GITR agonists failed to show sustainable antitumor activity, especially as monotherapy, in phase I trials.9 10 Here, we report the results of a first-in-human phase I/Ib study of GWN323, a highly selective anti-GITR antibody. In this study, GWN323 was well tolerated at all dose levels, including the highest dose level (1500 mg q3w). Although no formal MTD was reached in the study for either single-agent or combination treatments, MADs of GWN323 1500 mg q3w for single-agent treatment and GWN323 750 mg+spartalizumab 300 mg q3w for combination treatment were considered to be tolerated based on the nature and severity of the DLTs observed at that dose level.
The AE profile of GWN323 observed in this study as monotherapy or in combination was as expected and consistent with that observed in the early clinical studies of other GITR agonists.17–19 The safety profile of GWN323 in combination with spartalizumab at all dose levels was tolerable and identical to that of spartalizumab alone.15 No on-treatment deaths related to the study drug were reported.
Single-agent GWN323 exhibited a PK profile that is typical of a mAb with a dose-dependent increase in the PK exposure and a higher PK exposure at cycle 4 day 1 compared with cycle 1 day 1. The variability of the PK exposure was moderate. The PK exposure of GWN323 in the combination setting was comparable to that of a single agent, while the PK exposure of spartalizumab in the combination setting was comparable to the published data of single-agent spartalizumab study (data not shown).15 Taken together, these observations suggest that no significant drug–drug interaction exists between GWN323 and spartalizumab.
During the dose escalation, no patients had objective responses with GWN323 monotherapy. The limited antitumor activity of GWN323 monotherapy with overall good tolerability observed in this study is consistent with early clinical data available for the other GITR agonists, MEDI1873, TRX518 and AMG 228, in patients with advanced solid tumors.17 19 20 Dose-escalation results from the AMG 228 studies showed no antitumor activity but good overall tolerability in patients with advanced solid tumors.21 One patient in the combination arm achieved CR; 3 patients achieved PR; and 14 had SD. However, the clinical activity observed in the combination arm was mostly in patients with PD-1/PD-L1-sensitive tumors who had not received prior checkpoint inhibitor therapy, and we were unable to ascertain the additional benefit with GWN323. The antitumor activity observed in the enrichment cohort with immune checkpoint inhibitor-responsive tumors did not clearly demonstrate additional benefit from GWN323 in combination with spartalizumab.
It was hypothesized that treatment with GWN323 would suppress the Treg cells and enhance the activation of Teff cells in the tumor microenvironment. However, based on the current biomarker data, conclusive evidence of T-cell activation with GWN323 monotherapy was not observed. This result is consistent with the observation that GITR agonists are not effective as monotherapy to produce robust antitumor effects compared with that of combination therapy with a PD-1 antibody.9 Recent studies on GITR agonists MK-4166, MK-1248 and BMS-986156 showed good tolerability in patients with advanced solid tumors alone and in combination with checkpoint inhibitors, but objective responses were seen only for combination treatment.22–24
The flow cytometry analysis showed modest reductions in peripheral Teff cells in patients with single-agent treatment; however, these changes were not dose dependent and were not observed in all patients. The decreases in Teff cells and increases in proliferating NK and CD8+ cells observed in the combination arm could potentially be attributed to spartalizumab treatment. The gene expression analysis by RNA sequencing in paired tumor samples from the single-agent arm showed lack of correlation between GWN323 treatment and upregulation of IFN-γ or NK signatures. An upregulation of IFN-γ and Treg signatures was observed in patients in the combination arm who showed a decrease in tumor volume; however, this could be due to the effects of spartalizumab. Importantly, immunohistochemistry staining of paired tumor biopsies showed very low levels (<1%) of the Foxp3 marker at baseline in most patients in both arms. On-treatment decreases in Foxp3 were observed in only three patients treated with single-agent GWN323; all of these patients had >1% Foxp3 staining at baseline. This observation highlights the potential explanation for the lack of effect of GWN323 on Treg cell levels in the tumor; if most patients have no Foxp3 Treg cells in the tumor microenvironment at baseline, it may be impossible to test the hypothesis that GWN323 decreases Treg cells. In addition, several patients demonstrated, by RNA sequencing analysis, higher levels of inhibitory markers (ie, indoleamine 2,3-dioxygenase 1). It is possible that the complex pattern of immune stimulatory/inhibitory factors in combination with tumor heterogeneity may have contributed to limited clinical efficacy. Future efforts to select patients with higher baseline levels of Treg cells may allow us to test the effects of GWN323 more effectively.
Conclusions
In patients with relapsed or refractory solid tumors and lymphomas, GWN323 q3w monotherapy was well tolerated at all dose levels tested, including the highest dose level (1500 mg q3w). GWN323 in combination with spartalizumab was tolerable up to the highest tested dose (GWN323 750 mg+spartalizumab 300 mg q3w). MAD was GWN323 1500 mg q3w for single-agent treatment and GWN323 750 mg+spartalizumab 300 mg q3w for combination treatment with spartalizumab. GWN323 exhibited a PK typical of mAbs with no drug–drug interaction between GWN323 and spartalizumab. No evidence of T-cell activation or antitumor activity with GWN323 monotherapy was reported.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplemental information. Novartis will not provide access to patient-level data, if there is a reasonable likelihood that individual patients could be reidentified. Phase I studies, by their nature, present a high risk of patient reidentification; therefore, patient individual results for phase I studies cannot be shared. In addition, clinical data, in some cases, have been collected Patient to contractual or consent provisions that prohibit transfer to third parties. Such restrictions may preclude granting access under these provisions. Where codevelopment agreements or other legal restrictions prevent companies from sharing particular data, companies will work with qualified requestors to provide summary information where possible.
Ethics statements
Ethics approval
The study protocol and amendments were reviewed by an independent ethics committee or institutional review board for each site. The study was conducted in compliance with the Declaration of Helsinki and applicable regulatory requirements. Written informed consent was obtained from all patients.
Acknowledgments
The authors thank the patients participating in this clinical trial and their families, as well as the staff at each participating institution. The authors thanks Himabindu Gutha, PhD, of Novartis Healthcare Pvt Ltd, Hyderabad, India, who provided medical editorial support for the manuscript; Abdelkader Seroutou, who assisted with statistical inputs; Deborah Knee for preclinical research; Erica Vieira, Liza Morgan and Gregor Balaburski for clinical operations; Neelesh Sharma (former clinical program lead), Sushil Sharma (global program manager), Becker Hewes, Cinara Dias (patient safety physician) and Sabina Hernandez Penna (regulatory affairs) for their contributions in the development of GWN323 and the design and execution of the trial.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @OsamaRahma2, @jasonlukemd
Contributors AX and LN were responsible for the study conception and design and development of methodology. SAP-P, RG, JJL, PLB and TJT were responsible for the clinical data collection. OR, XC, SAP-P, RG, AX, LN, JJL, PLB and TJT performed the analysis and interpretation of data. All authors were responsible for the writing, review and/or revision of the manuscript. All authors read and approved the final manuscript.
Funding This study was supported by Novartis Oncology.
Competing interests SAP-P reports other from AbbVie, Inc., other from ABM Therapeutics, Inc., other from Acepodia, Inc., other from Alkermes, other from Aminex Therapeutics, other from Amphivena Therapeutics, Inc., other from BioMarin Pharmaceutical, Inc., other from Boehringer Ingelheim, other from Bristol Myers Squib, other from Cerulean Pharma, Inc., other from Chugai Pharmaceutical Co., Ltd., other from Curis, Inc., other from Daiichi Sankyo, Inc., other from Eli Lilly, other from ENB Therapeutics, other from Five Prime Therapeutics, other from Gene Quantum, other from Genmab A/S, other from GlaxoSmithKline, other from Helix BioPharma Corp., other from Incyte Corp., other from Jacobio Pharmaceuticals Co., Ltd., other from Medimmune, LLC., other from Medivation, Inc., other from Merck Sharp and Dohme Corp., other from Novartis Pharmaceuticals, other from Pieris Pharmaceuticals, Inc., other from Pfizer, other from Principia Biopharma, Inc., other from Puma Biotechnology, Inc., other from Rapt Therapeutics, Inc., other from Seattle Genetics, other from Silverback Therapeutics, other from Taiho Oncology, other from Tesaro, Inc., other from TransThera Bio, grants from NCI/NIH P30CA016672 - Core Grant (CCSG Shared Resources) outside the submitted work. RG reports honoraria (self) from BMS, Lilly, Medison, Roche, Novartis, Janssen, Takeda, MSD, Pfizer, Merck; advisory/consultancy to EISAI, AstraZeneca, Bayer, MSD, Novartis, BI. BOL Pharma, Roche; research grant/funding (institution): educational grant to the research unit—Novartis; travel/accommodations/expenses: Merck, Bayer, BMS, Medison. TT reports grants, personal fees and non-financial support from AstraZeneca; personal fees from Roche, Novartis, Pfizer, DKSH Singapore; other from Immunomedics, outside the submitted work. DWTL reports grants from Bristol-Myers Squibb; personal fees from MSD, Boehringer-Ingelheim, Pfizer; non-financial support from Astra-Zeneca, outside the submitted work. CH reports grants from Merck; personal fees from MSD, Lilly, and Merck, outside the submitted work. TD reports grants from Lilly, Merck Serono, Pfizer, Quintiles (IQVIA), and Eisai; grants and personal fees from MSD, Daiichi Sankyo, Sumitomo Dainippon, Taiho, Novartis, Janssen, Boehringer Ingelheim, Bristol-Myers Squibb, Abbvie; personal fees from Amgen, Takeda, Chugai Pharma, Bayer, Rakuten Medical, Ono Pharmaceutical, Astellas Pharma, Oncolys BioPharma, Otsuka Pharma, outside the submitted work. OR reports personal fees from Imvax, GSK, Bayer, Gennentech, Sobi, Puretech, Maverick Therapeutics, Merck, outside the submitted work. In addition, OR has patent methods of using pembrolizumab and trebananib pending. AL reports grants from Novartis, during the conduct of the study; grants from Bristol Myers Squib, personal fees from Trillium Therapeutics, grants, personal fees and non-financial support from Pfizer, grants and personal fees from Janssen, outside the submitted work. In addition, AL has a patent US20150037346A1 with royalties paid. JL reports Scientific Advisory Board: (no stock) 7 Hills, Spring bank (stock) Actym, Alphamab Oncology, Arch Oncology, Kanaph, Mavu, Onc.AI, Pyxis, Tempest; Consultancy with compensation: Abbvie, Alnylam, Array, Bayer, Bristol-Myers Squibb, Checkmate, Cstone, Eisai, EMD Serono, KSQ, Janssen, Inzen, Macrogenics, Merck, Mersana, Nektar, Novartis, Pfizer, Regeneron, Ribon, Rubius, Silicon, Synlogic, TRex, Werewolf, Xilio, Xencor; Research Support: (all to institution for clinical trials unless noted) AbbVie, Agios (IIT), Array (IIT), Astellas, Bristol-Myers Squibb (IIT & industry), Corvus, EMD Serono, Immatics, Incyte, Kadmon, Macrogenics, Merck, Moderna, Nektar, Numab, Replimmune, Rubius, Spring bank, Synlogic, Takeda, Trishula, Tizona, Xencor; Patents: (both provisional) Serial #15/612,657 (Cancer Immunotherapy), PCT/US18/36052 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof). JO is a full-time employee and stockholder of Novartis Pharmaceuticals Corp. LN, AS, AX, XC and JM are full-time employees and stockholders of Novartis Pharmaceuticals Corp. PLB reports grants from Novartis, during the conduct of the study; grants from BristolMyersSquibb, grants from Sanofi, grants from Genentech/Roche, grants from Novartis, grants from GlaxoSmithKline, grants from Nektar Therapeutics, grants from Merck, grants from Lilly, grants from Servier, grants from PTC Therapeutics, grants from SeaGen, grants from Sanofi, grants from Mersana, grants from Amgen, grants from Zymeworks, grants from VelosBio, outside the submitted work; and uncompensated advisory boards for BristolMyersSquibb, Sanofi, Pfizer, Genentech/Roche, Amgen, Lilly, SeaGen, Merck; Past Chair, Investigational New Drug Committee, Canadian Clinical Trials Group; executive board member, Breast International Group; steering committee member, American Association for Cancer Research Project GENIE; member, NCI-BIO Breast Cancer Immunotherapy Task Force; Interface Committee, Alexandria Phase III Trial.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.