Article Text

Abstract
Background CD47 is a broadly expressed cell surface glycoprotein associated with immune evasion. Interaction with the inhibitory receptor signal regulatory protein alpha (SIRPα), primarily expressed on myeloid cells, normally serves to restrict effector function (eg, phagocytosis and immune cell homeostasis). CD47/SIRPα antagonists, commonly referred to as ‘macrophage checkpoint’ inhibitors, are being developed as cancer interventions. SRF231 is an investigational fully human IgG4 anti-CD47 antibody that is currently under evaluation in a phase 1 clinical trial. The development and preclinical characterization of SRF231 are reported here.
Methods SRF231 was characterized in assays designed to probe CD47/SIRPα blocking potential and effects on red blood cell (RBC) phagocytosis and agglutination. Additionally, SRF231-mediated phagocytosis and cell death were assessed in macrophage:tumor cell in vitro coculture systems. Further mechanistic studies were conducted within these coculture systems to ascertain the dependency of SRF231-mediated antitumor activity on Fc receptor engagement vs CD47/SIRPα blockade. In vivo, SRF231 was evaluated in a variety of hematologic xenograft models, and the mechanism of antitumor activity was assessed using cytokine and macrophage infiltration analyses following SRF231 treatment.
Results SRF231 binds CD47 and disrupts the CD47/SIRPα interaction without causing hemagglutination or RBC phagocytosis. SRF231 exerts antitumor activity in vitro through both phagocytosis and cell death in a manner dependent on the activating Fc-gamma receptor (FcγR), CD32a. Through its Fc domain, SRF231 engagement with macrophage-derived CD32a serves dual purposes by eliciting FcγR-mediated phagocytosis of cancer cells and acting as a scaffold to drive CD47-mediated death signaling into tumor cells. Robust antitumor activity occurs across multiple hematologic xenograft models either as a single agent or in combination with rituximab. In tumor-bearing mice, SRF231 increases tumor macrophage infiltration and induction of the macrophage cytokines, mouse chemoattractant protein 1 and macrophage inflammatory protein 1 alpha. Macrophage depletion results in diminished SRF231 antitumor activity, underscoring a mechanistic role for macrophage engagement by SRF231.
Conclusion SRF231 elicits antitumor activity via apoptosis and phagocytosis involving macrophage engagement in a manner dependent on the FcγR, CD32a.
- FC receptor
- oncology
- tumours
- lymphoma
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
CD47 is a ubiquitously expressed transmembrane protein with pleiotropic roles in immune homeostasis, innate and adaptive immune cell activation, and leucocyte recruitment.1–3 CD47 was originally identified as a tumor antigen, OA3, overexpressed in human ovarian cancer4 and as integrin-associated protein that copurified with certain integrins.5 Many tumor types overexpress CD47 protein, and clinical prognostic as well as nonclinical functional data suggest that this upregulation may allow tumors to evade innate immune cell destruction via phagocytosis.6–9 Signal regulatory protein alpha (SIRPα), an immunoreceptor tyrosine-based inhibitory motif-containing inhibitory signaling protein expressed on myeloid cells,10 11 is a well-known binding partner of CD47 that restricts effector functions on CD47/SIRPα engagement.12 Because of these properties, disrupting the CD47/SIRPα axis is a target for therapeutic intervention. In addition to CD47/SIRPα blockade, some CD47 targeting agents also engage Fc effector function to varying degrees, which is believed to play an important role in eliciting antitumor effects.7 13 14 While initiation of tumor cell phagocytosis has long been a focus of CD47 targeting agents, engagement of cell death pathways downstream of CD47 on the tumor cell is another possible mechanism of action of some of these agents that could be exploited clinically.6 15–18
Targeting CD47 as an approach to treat cancer is under investigation clinically (NCT03512340). Investigational methods to antagonize the CD47/SIRPα axis as a therapeutic intervention include CD47 and SIRPα monoclonal antibodies (mAbs),19–21 SIRPα-Fc fusion protein,13 high-affinity SIRPα variants22 and CD47/tumor-antigen bispecific antibodies.23 While CD47 is often highly expressed on tumor cells,7 9 24–26 it is also expressed on several other non-malignant cell types, including red blood cells (RBCs), where it plays a role in the regulation of RBC lifespan.27 Furthermore, many anti-CD47 mAbs induce RBC hemagglutination.28 Clinical hemagglutination could result in hemolysis and potential arterial thrombotic events. Therefore, agents that target CD47 without hemagglutination could be clinically significant.
The generally accepted ‘eat-me/don’t-eat-me’ model of CD47/SIRPα regulation of phagocytosis is a two-signal model, where macrophages require the absence of SIRPα signaling (signal 1) as well as the presence of an activating or ‘eat-me’ signal (signal 2). This two-signal model has been established with CD47/SIRPα antagonists in combination with IgG1-bearing tumor opsonizing antibodies such as rituximab, trastuzumab and cetuximab,7 20 22 29 and with a CD47/SIRPα antagonist containing the IgG1 Fc itself.28 30 However, how mAbs with IgG4 isotypes can provide this second signal has not been well described, and the IgG4 isotype has seemingly been selected to minimize the recruitment of antibody Fc-dependent effector functions.19
SRF231, an investigational fully human IgG4 (hIgG4) anti-CD47 antibody was selected for development in part for its lack of RBC hemagglutination activity. SRF231 binds specifically to human CD47, blocks the CD47/SIRPα interaction and leads to induction of tumor cell phagocytosis and tumor-intrinsic cell death. Both activities depend on myeloid cell-expressed activating Fc receptor CD32a, accentuating the importance of the Fc region of SRF231 for its activity. SRF231 is currently being evaluated in a phase 1 clinical trial across multiple tumor types (NCT03512340).
Materials and methods
Generation of anti-human CD47 antibody
Anti-CD47 antibodies were created by immunization of H2L2 transgenic Harbour MiceTM (Harbour BioMed) with a human CD47-hIgG1Fc fusion protein. Following immunization, a panel of hybridomas expressing human antibodies was obtained that bound specifically to CD47. Based on binding data, high-affinity antibodies were selected for hybridoma expansion, production and characterization. SRF2.3D11 and SRF4.2C11 were selected for further characterization, and SRF2.3D11 was subsequently engineered to have a wild-type (WT) hIgG4 Fc domain, resulting in SRF231.
Cell lines and tissues
Jurkat (E6.1), Raji, HL-60, SU-DHL4 and TK-1 cell lines were obtained from American Type Culture Collection. Ri-1 and OPM-2 were obtained from DSMZ. The Jurkat CD47 knockout (KO) cell lines were generated via CRISPR/CAS9 technology (Applied StemCell). Primary bone marrow-derived acute myeloid leukemia (AML) samples were purchased (Cureline or Conversant). All cell lines were maintained in R10 medium (Roswell Park Memorial Institute [RPMI] 1640 + 10% FBS+penicillin [100 U/mL] and streptomycin [100 µg/mL]) at 37°C, 5% CO2. RBCs were isolated from fresh human ethylenediaminetetraacetic acid (EDTA) anti-coagulated whole blood (Research Blood Components) via centrifugation and subsequent washing in PBS+2 mM EDTA. RBC suspensions were stored at 4°C at 10% v/v in PBS+2 mM EDTA and used within 3 days.
Antibodies and tool reagents
The following antibodies were generated in-house: antidinitrophenyl hIgG4 isotype control, SRF231 (hIgG4), SRF231 F(ab’)2 (full-length antibody incubated on Immobilized FABricator enzyme [Genovis A2-FR2-1000]), Fc byproduct removed by incubation with MabSelect SuRe [GE 17-5438-03] and F(ab’)2 purified by collection of elution fragments and verified by analytical size exclusion chromatography), SRF231mut (S228P and L235E mutations introduced into SRF231 to reduce Fc-FcR binding and stabilize half-antibody formation)31 and SRF4.2C11 (hIgG1). B6H12 (mIgG1) and respective isotype control were purchased from BD Biosciences; polyclonal hIgG was purchased from BioXCell. hIgG4 or SRF231-Alexa Fluor 647 (AF 647) conjugates were generated using AF 647 Antibody Labeling Kit (ThermoFisher) or custom ordered from BioLegend. For macrophage detection, anti-hCD14-Allophycocyanin (APC), clone M5E5 (BD Biosciences) or anti-mCD11b-AF 647, clone M1/70 (BioLegend) were used. For combination experiments, anti-CD20 antibody (rituximab; anti-hCD20-hIgG1; Invivogen) was used. For CD32a blocking experiments or flow-based detection, either unlabeled or fluorescein isothiocyanate labeled antibody (clone IV.3; StemCell Technologies), was used.
Hemagglutination assay
For whole blood smears, fresh human whole blood was incubated with antibody overnight at 37°C, 5% CO2. The following day, a drop of blood was smeared onto a glass slide using a glass ‘spreader’ slide. Smears were air-dried and imaged with an EVOS light microscope using the 40X objective. Images were analyzed qualitatively for evidence of RBC agglutination. For flow-based quantitative analyses, purified human RBCs were incubated with antibody for 0.5 hour at 37°C in a 96-well round-bottom plate (Corning), followed by incubation at 4°C overnight and flow cytometry-based assessment of RBC doublets.
Cell death assay
To mimic scaffolded antibody conditions, Protein G-coated 96-well plates (Thermo Fisher) were coated with antibody diluted in PBS overnight at 4°C. The following day, unbound antibody was washed away, and cells added at 1×105 cells/well in RPMI +1% FBS. Alternatively, isotype control or soluble antibodies were added to Jurkat cells in the soluble form. Plates were incubated overnight at 37°C. Cells were pipetted off the plate; cell death induction was evaluated using AnnexinV/Propidium Iodide (Life Technologies) via flow cytometry according to the manufacturer’s instructions.
Tumor xenograft studies
All animal studies were conducted according to guidelines established by the internal and external Institutional Animal Care and Use Committee. Female CB17 SCID or non-obese diabetic (NOD)/SCID mice aged 6–8 weeks (Charles River Laboratories) were implanted with either Raji (10×106), OPM-2 (10×106), Ri-1 (10×106), HL-60 (5×106), or human diffuse large B-cell lymphoma cell line (SU-DHL-4 [10×106]) cells in 0.2 mL 50% Matrigel (BD Biosciences) subcutaneously into the right flank. Antibody treatments were initiated when tumors averaged 100–200 mm3. For clodronate studies, mice were treated with 100 µg clodronate liposomes (Thermo Fisher Scientific) intravenously 3 times/week for 2 weeks. One day following the second clodronate treatment, mice were treated intraperitoneally (IP) with either 100 µg isotype control antibody or SRF231 3 times/week for 3 weeks. Mice were monitored, and tumor volume was measured twice weekly by electronic calipers (length × width2)×0.52. Mice received SRF231 IP in 0.2 mL at indicated doses, either once/week for 1 week, once/week for 2 weeks or once/week for 3 weeks depending on the tumor model. Rituximab was administered in 0.2 mL IP once/week for 3 weeks (days 1, 8 and 15).
Plasma and tumor cytokine and immunoglobulin measurement
Mice were terminally bled through cardiac puncture, and blood was collected into EDTA tubes. Samples were immediately centrifuged and aliquoted. Tumor tissues were collected in Lysing Matrix A, orange tube (MP Biomedicals). Lysis buffer was added, and tissues were homogenized using FastPrep (MP Biomedicals). Mouse macrophage inflammatory protein 1α (MIP-1α) and Mouse chemoattractant protein 1 (MCP-1) cytokines were assayed using multiplex V-Plex cytokine kit from (Meso Scale Discovery). Plates were read on a Meso Scale Discovery Imager.
Immunohistochemistry
Xenograft tumor tissues were collected, formalin-fixed and paraffin-embedded. Blocks were sectioned (Leica RM2145 microtome) at approximately 4 μm. Immunohistochemistry was performed with rat anti-mouse F4/80 (Bio-RAD). Slides were scanned (Aperio AT2 whole slide scanner). Image analysis was performed on digital slide images (Visiopharm software or QuPath imaging software).32 Tumor tissue regions of interest were identified in whole slide images and image analysis algorithms or tools applied to quantitate either F4/80 expression as a percent positive value within the tumor area or evaluate necrotic tumor area as a percentage of total tumor area (QuPath).
Macrophage generation
For human macrophages, CD14+ monocytes were isolated via negative selection from pooled buffy coats (n=3 donors, Research Blood Components). Primary monocytes were cultured for 5–7 days in the presence of 100 ng/mL human macrophage colony stimulating factor (hM-CSF) (Invitrogen). For mouse macrophages, bone marrow was collected by flushing the femur and tibia from Balb/c mice with cold R10. Bone marrow was passed through a 70 µm filter, spun down and resuspended in R10 with 100 ng/mL mouse macrophage colony stimulating factor (mM-CSF) (Invitrogen) and cultured in a T75 tissue culture treated flask for 6 days.
Phagocytosis assay
Macrophages were seeded in 96-well plates (Corning). The following day, target cells were carboxyfluorescein succinimidyl ester (CFSE)-labeled according to manufacturer’s protocol (Life Technologies) and cocultured with macrophages at a 2:1 ratio for 2 hours at 37°C in the presence of antibody. In some cases, macrophages were pretreated with a CD32a (IV.3) blocking antibody prior to addition of target cells. For RBC phagocytosis the target:effector ratio was 10:1, and for primary AML samples it was 1:1. Cells were trypsinized and stained with a macrophage-specific marker (CD14-APC for human and CD11b-APC for mouse macrophages). Flow cytometry was performed, followed by doublet exclusion analysis and phagocytosis reported as a percentage of CD14+ macrophages that were CFSE+. For analysis of cell death in this assay, a live/dead dye (Life Technologies) was added to the CD14-APC staining cocktail. Cell death was identified within the non-phagocytosed target cell population.
Phospho-array screening
Jurkat cells were cocultured with primary human macrophages in the presence of 10 µg/mL hIgG4 isotype control, SRF231 or SRF231 F(ab’)2 for 10 min at 37°C. Following antibody treatment, Jurkat cells were separated from macrophages by gentle pipetting, and each cellular fraction was assayed independently according to manufacturer’s instructions from the phospho-immunoreceptor array kit (R&D Systems, Bio-Techne Corporation). Phospho-signals were detected by chemiluminescence and HLImage++ software used to quantitate and normalize phospho-signals.
On-cell SIRPα/CD47 blocking assay
Jurkat cells were pretreated with either isotype or anti-CD47 antibodies for 20 min at 4°C, washed and incubated with biotinylated human recombinant SIRPα-Fc protein (R&D Systems) for another 20 min at 4°C. After washing, remaining biotinylated SIRPα-Fc was detected with streptavidin-APC (Life Technologies), and geometric mean fluorescence intensity values were captured by flow cytometry excluding dead cells using a Live/Dead dye (Invitrogen). Inhibition of SIRPα-Fc binding to Jurkat cells correlates with decreased fluorescence intensity.
Statistics
Data were presented as mean±SD, and statistical analysis was performed using Prism V.8 software. A Student’s t-test was used to determine the p value at the indicated day, and p values were considered significant below 0.05 (*p≤0.05; **p≤0.01; ***p≤0.001; ****p≤0.0001).
Results
SRF231 binds specifically to human CD47 and induces tumor cell phagocytosis
SRF2.3D11 was identified from a panel of IgG transgenic hybridomas expressing antibodies with human variable regions and was subsequently engineered as a hIgG4 and named SRF231. SRF231 specifically binds to CD47 as indicated by surface staining of CD47-expressing WT Jurkat cells but not CD47 KO Jurkat cells (figure 1A). Multiple human tumor cell lines of hematopoietic lineage are sensitive to SRF231-mediated phagocytosis by primary human macrophages (figure 1B). Additionally, significant enhancement of phagocytosis of primary AML bone marrow cells is observed in the presence of SRF231 (figure 1C). As expected, SRF231 cooperates with the anti-CD20 antibody (rituximab) resulting in enhanced antibody-dependent cellular phagocytosis (ADCP) of malignant B-cell lymphoma cell lines, Raji and SU-DHL-4 (figure 1D).

SRF231 binds to human CD47 and induces phagocytosis of tumor cells in vitro. (A) Jurkat wild-type (WT) and CD47 knockout (KO) lines were stained with 10 µg/mL AF647-labeled SRF231 or isotype control (hIgG4) and analyzed by flow cytometry. Data from 1 of 2 independent KO lines is shown. (B) CFSE-labeled tumor cell lines or (C) primary bone marrow samples from AML patients were evaluated in a phagocytosis assay in the presence of SRF231. For cell lines (B), duplicate data points are shown, with non-linear regressions as connecting lines. For AML samples (C), each line represents an individual AML donor, p=0.002, paired Student’s t-test, n=11. (D) SRF231+anti-CD20 (anti-hCD20-IgG1) combination activity was evaluated in a phagocytosis assay. Dotted lines denote single-agent activity with anti-CD20 antibody alone. SRF231 was added at the indicated concentrations; anti-CD20 was added at a fixed concentration of 0.625 µg/mL for Raji cell targets (left panel) and 0.039 µg/mL for SU-DHL-4 cell targets (right panel). AML, acute myeloid leukemia; CFSE, carboxyfluorescein succinimidyl ester; hIgG4, human IgG4.
SRF231-mediated phagocytosis is dependent on CD32a FcγR expression
To evaluate the distinct mechanism of action of SRF231-mediated phagocytosis, SRF231 (whole IgG) and SRF231 F(ab’)2 fragments were evaluated in phagocytosis assays using Jurkat target cells with either human or mouse macrophage effectors. Despite the ability of SRF231 to block the human CD47/human SIRPα interaction (figure 2A), pure blockade of the CD47/SIRPα interaction using a SRF231 F(ab’)2, capable of saturating CD47 binding sites (online supplementary figure S1), was insufficient for phagocytosis induction in a fully human system (figure 2B). In contrast, pure blockade of the human CD47/mouse SIRPα interaction using the SRF231 F(ab’)2 was sufficient to induce mouse macrophage-mediated phagocytosis (figure 2C). Additionally, a ~10-fold shift in the potency of SRF231 was observed when mouse versus human macrophage effector cells were used (with mouse effectors being more potent), highlighting species considerations in probing CD47/SIRPα biology. The affinity of the interaction between WT mouse SIRPα and human CD47 is substantially weaker than the affinity between the NOD SIRPα variant and human CD47, while the human CD47/human SIRPα affinity is intermediate to the two.33 When macrophages from different mouse backgrounds were used as effectors in the phagocytosis assay, differences were observed in the potency of the ability of SRF231 to induce phagocytosis of human targets (Jurkat cells). Bone marrow-derived macrophages from Balb/c mice were more sensitive to the induction of phagocytosis by SRF231, in full length or F(ab’)2 form, than macrophages from NOD mice (online supplementary figure S2A). However, this difference was not observed when mouse targets (TK-1 cells) and an anti-mouse CD47 antibody (clone miap410) were used (online supplementary figure S2B), implicating the affinity difference between WT and NOD SIRPα for human CD47 in the differences observed.
Supplemental material
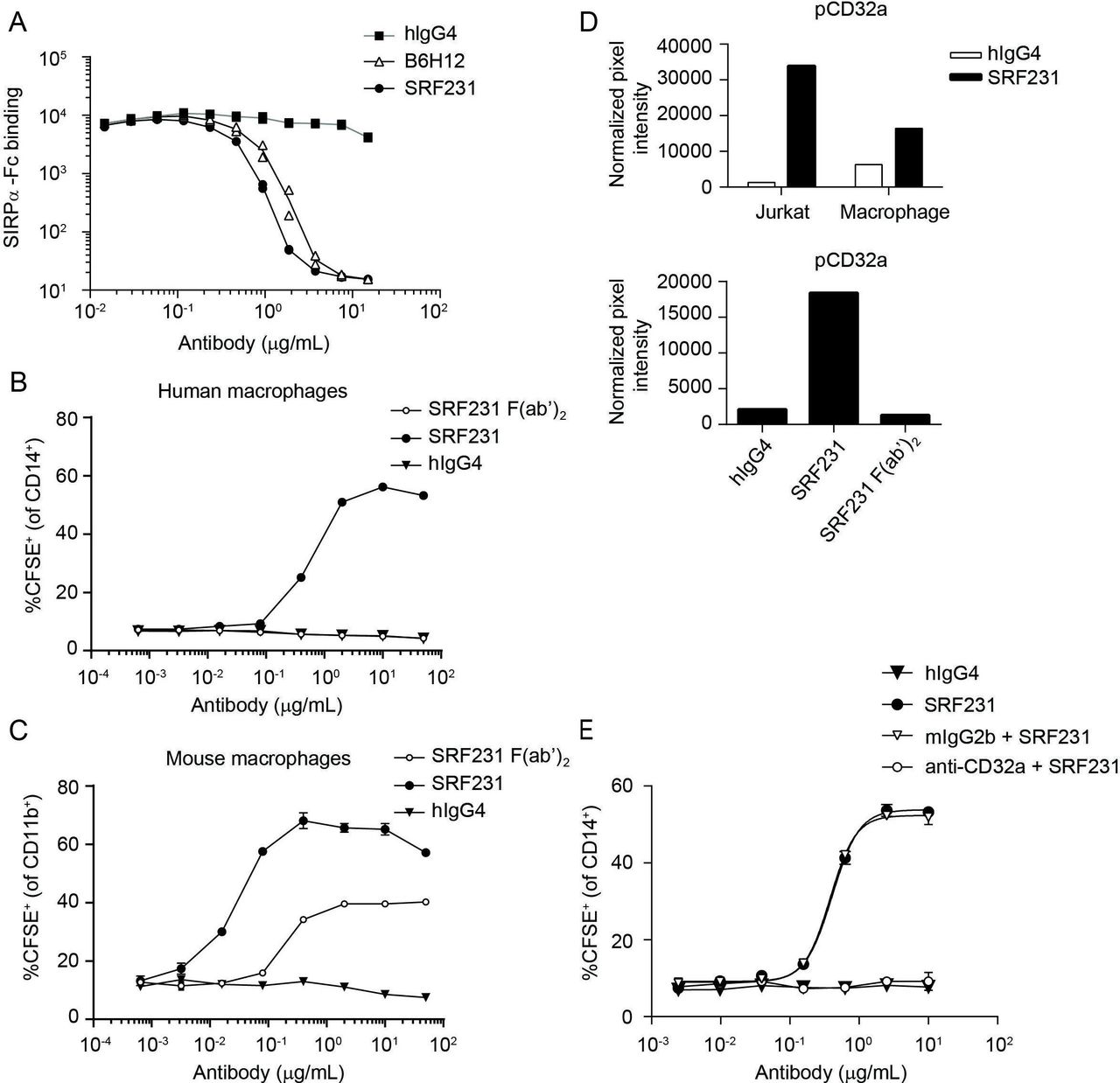
Induction of human macrophage-mediated phagocytosis by SRF231 is dependent on CD32a engagement. (A) Jurkat cells expressing endogenous CD47 were pretreated with the indicated antibodies, whose ability to block the CD47/SIRPα interaction was then tested by incubation with biotinylated SIRPα-Fc, followed by streptavidin-APC detection. Antibody-dependent blockade of the CD47/SIRPα interaction corresponds with reduced mean fluorescence intensity. Duplicate data points are plotted; connecting lines represent the means of these data points. (B, C) SRF231 (whole IgG) and F(ab’)2 fragments were evaluated in parallel in phagocytosis assays using Jurkat target cells cocultured with either human (B) or mouse (C) macrophages at a 2:1 ratio. Mean and SD of triplicate values are plotted. (D) Phospho-CD32a mean pixel intensity was obtained from chemiluminescence phospho-array imaging of lysates obtained from Jurkat/macrophage coculture +/-SRF231 (each fraction assayed independently, top panel) or the Jurkat fraction from cocultures treated with either hIgG4, SRF231 or SRF231 F(ab’)2 (bottom panel). (E) Primary human macrophages were pretreated with anti-CD32a blocking antibody (IV.3) or mouse IgG2b isotype control (mIgG2b) prior to conducting the phagocytosis assay with Jurkat targets with or without hIgG4 or SRF231. Mean and SD of triplicate values are plotted, with non-linear regressions as connecting lines. APC, allophycocyanin; CFSE, carboxyfluorescein succinimidyl ester; hIgG4, human IgG4; SIRPα, signal regulatory protein alpha.
Since CD47/SIRPα blockade is sufficient to induce phagocytosis in a mouse/human system yet insufficient in a fully human system, other prophagocytic signal(s) are likely necessary for the phagocytosis-inducing effects of SRF231 in the human setting. To search for additional signals that may mediate the effects of SRF231, phospho-immunoreceptor array screening was conducted within the context of the phagocytosis assay. Jurkat cells were separated from human macrophages after SRF231 treatment, and each fraction assayed independently. This screening approach led to the identification of FcγRIIa (CD32a) phosphorylation on treatment with SRF231 in both Jurkat and macrophage fractions (figure 2D). This signaling event was not observed with the SRF231 F(ab’)2 fragment (figure 2D, bottom panel), which emphasizes the requirement for the antibody Fc region for CD32a phosphorylation and suggests that CD32a phosphorylation may be linked to Fc engagement of the FcR and subsequent phagocytosis induction. The 2 cell types used within this assay, Jurkat cells and human macrophages, were profiled for surface expression of CD32a. As expected, macrophages showed robust CD32a expression while Jurkat cells were negative (online supplementary figure S3A). Since the Jurkat fraction contained <10% macrophage contamination (data not shown) in the phospho-array screening assays, it is possible that the Jurkat subset is enriched for a fraction of activated macrophages that have already undergone conjugate formation with Jurkat cells in the presence of SRF231. The requirement for CD47 on the target cell for both CD32a phosphorylation and phagocytosis induction by SRF231 was evaluated using Jurkat WT and Jurkat CD47 KO cells. These data reveal that CD47 is required for both CD32a phosphorylation and phagocytosis induction, as CD47 Jurkat KO cells are insensitive to SRF231 in both assays (online supplementary figure S3B,C). The functional significance of CD32a in the phagocytosis assay was evaluated by blocking this receptor on macrophages prior to their coculture with Jurkat target cells. Pre-incubation of macrophages with a CD32a-specific function-blocking antibody led to abrogation of SRF231 activity within the phagocytosis assay (figure 2E) reinforcing the functional significance of CD32a in SRF231-mediated phagocytosis. In summary, while pure CD47/SIRPα blockade is sufficient for phagocytosis using mouse macrophage and human targets, pure blockade of the CD47/SIRPα interaction is insufficient in a fully human system where Fc/FcR engagement is required for functional activity.
SRF231 induces tumor cell death in a CD32a-dependent manner
In addition to promoting macrophage-mediated phagocytosis, SRF231 is capable of inducing tumor cell death. This cell death phenotype was initially observed within the context of the phagocytosis assay, in which both SRF231 and another anti-human CD47 mAb, B6H12, induced phagocytosis of Jurkat target cells, commensurate with the loss of tumor cell viability in the non-phagocytosed target cell population in a dose-dependent manner (figure 3A). This phenomenon was also observed in phagocytosis assays using mouse macrophages (online supplementary figure S4A). As macrophage-derived CD32a had been shown to be essential for SRF231-mediated phagocytosis by human macrophages, the significance of CD32a was also evaluated with respect to SRF231-induced cell death observed within macrophage/Jurkat coculture. CD32a blockade abrogated SRF231-mediated tumor cell death in the non-phagocytosed Jurkat population (figure 3B) as did the lack of CD47 expression on the target population (online supplementary figure S4B). Transwell analyses also revealed that this cell death phenomenon is both contact dependent and independent of the phagocytosis event (data not shown). These data implicate a dual role for CD32a in both SRF231-mediated phagocytosis and direct tumor cell death induction. To further assess the importance of CD32a-expressing macrophages in SRF231-mediated cell death, Jurkat cells were treated with soluble SRF231 and cell death was evaluated. Unlike anti-CD47 mAb clone CC2C6, which induced Jurkat cell death in the soluble form,18 soluble SRF231 did not induce Jurkat cell death (figure 3C, top panel). While CD32a engagement leads to downstream signaling within macrophages that may be involved in SRF231-mediated phagocytosis, it may also serve as a scaffold for SRF231, permitting a particular antibody orientation that allows for CD47 cross-linking on the tumor cells leading to tumor-intrinsic apoptosis. To mimic the scaffolding function of macrophages, SRF231 or hIgG4 isotype control antibody were immobilized on protein G-coated plates prior to addition of Jurkat cells. Under these conditions, scaffolded SRF231 was capable of inducing tumor cell death (figure 3C, bottom panel). Scaffolded SRF231-induced cell death was not limited to Jurkat cells and was also observed in AML and ovarian cell lines (online supplementary figure S4C). A panel of primary, non-malignant cells were evaluated for sensitivity to scaffolded SRF231-mediated cell death. CD47-high Jurkat cells were more susceptible to cell death compared with non-malignant cells, which uniformly expressed lower levels of surface CD47 and showed little to no susceptibility to cell death induction by SRF231 (online supplementary figure S4D).

SRF231 induces caspase-independent tumor cell death that is dependent on antibody scaffolding. (A) Phagocytosis induction (left panel) evaluated in the presence of SRF231 or B6H12 using Jurkat cell targets and human macrophages. Anti-CD47 mAb-induced cell death (right panel) measured as % of dead cells (as identified by uptake of LIVE/DEAD dye) within the non-phagocytosed (CFSE+CD14-) target cell population. (B) Macrophages were pretreated with 10 µg/mL CD32a blocking antibody prior to addition of the indicated concentrations of hIgG4 or SRF231. SRF231-induced cell death was evaluated as described in figure 3A. (C) Jurkat cell death induction (measured by Annexin V/PI) on 24 hours treatment with 10 µg/mL soluble (top panel) or protein G-bound (bottom panel) anti-CD47 mAb or isotype controls. (D) Jurkat cells were pretreated for 30 min at 37°C with 100 µM pan-caspase inhibitor (Z-VAD-FMK) prior to culture with protein G-bound SRF231. CFSE, carboxyfluorescein succinimidyl ester; DMSO, dimethyl sulfoxide; hIgG4, human IgG4; mAb, monoclonal antibody; mIgG1, mouse IgG1; PI, propidium iodide.
CD47 perturbation has been implicated in a unique caspase-independent cell death pathway implicating phospholipase C gamma 1.6 15–17 To probe the molecular mechanism of SRF231-induced cell death, Jurkat cells were pretreated with a pan-caspase inhibitor (Z-VAD-FMK) prior to incubation with scaffolded SRF231. The addition of 100 µM Z-VAD-FMK (a pan-caspase inhibitor) did not appreciably abrogate SRF231-induced Annexin V staining (figure 3D), in contrast to doxorubicin-induced cell death, which is partially dependent on caspase induction (data not shown).
SRF231 lacks RBC hemagglutination and phagocytosis capabilities
SRF231 binds to CD47-expressing RBCs in a dose-dependent manner (figure 4A), but does not cause hemagglutination (figure 4B,C) in contrast to B6H12 and SRF4.2C11, a tool antibody generated in a similar fashion to SRF231. Similarly, SRF231 does not induce RBC phagocytosis as is observed with B6H12 (figure 4D). Despite binding to RBCs, SRF231 displays a distinct, RBC-sparing functional profile in vitro.

SRF231 does not induce RBC aggregation or RBC phagocytosis. (A) Human whole blood was stained with the indicated concentrations of SRF231 or isotype control (hIgG4) and analyzed by flow cytometry gating specifically on the RBC population. (B) Human whole blood was incubated with 100 µg/mL of the indicated antibody, overnight at 37°C, after which blood smears were performed on glass slides and analyzed by light microscopy. scale bar=100 µm. (C) Human RBCs were incubated with the indicated concentrations of antibody and evaluated by flow cytometry. Data are presented as the percent doublets gated on a side scatter-height versus side scatter-width plot. Data points for 2 individual donors are plotted. (D) Human RBCs were CFSE labeled and incubated with human macrophages in the phagocytosis assay, as described in the materials and methods. CFSE, carboxyfluorescein succinimidyl ester; hIgG4, human IgG4; RBC, red blood cell.
SRF231 has antitumor activity across a range of hematologic tumor xenograft models
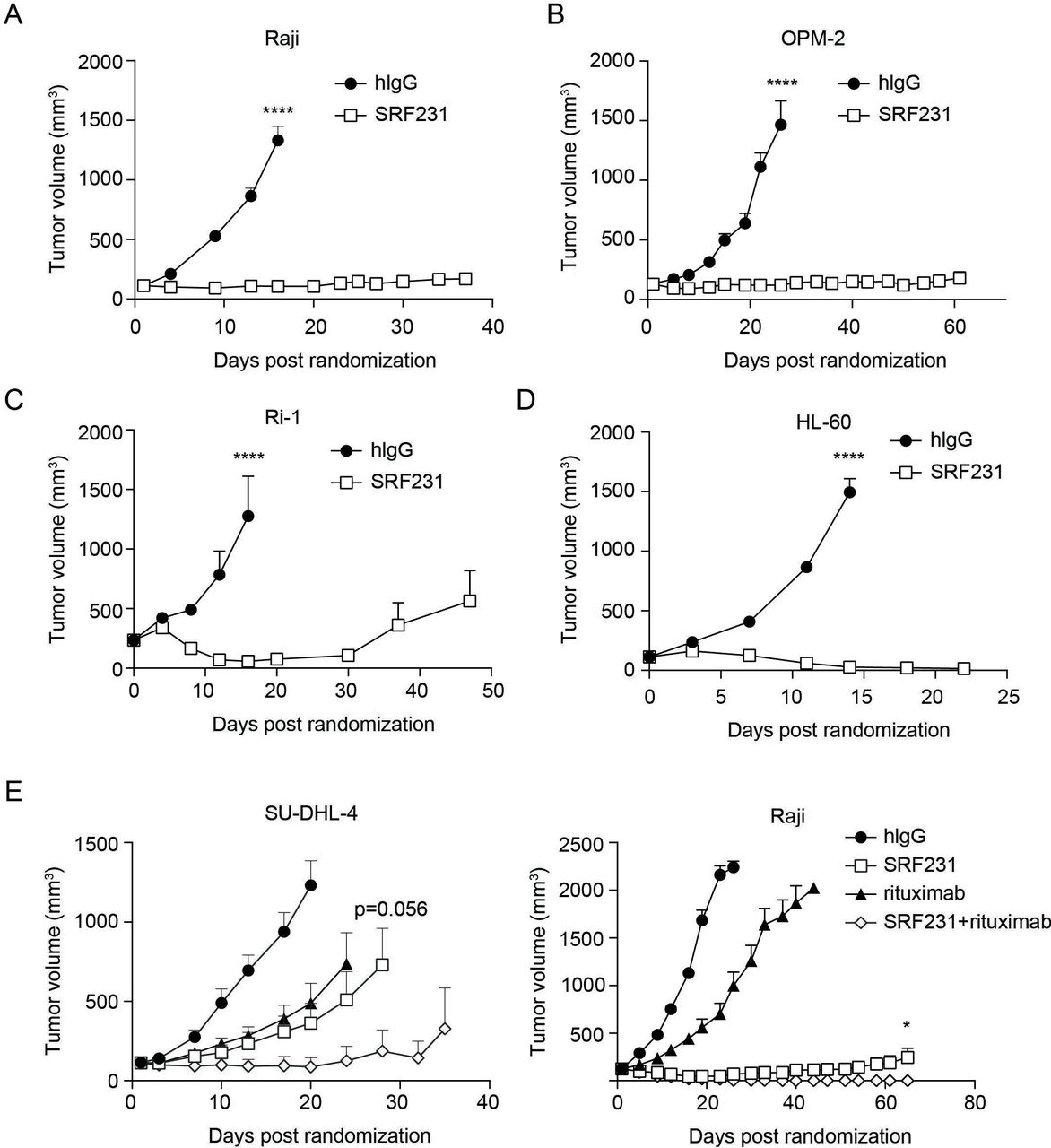
SRF231 does not cross-react with mouse CD47 (data not shown). Therefore, the therapeutic activity of SRF231 was evaluated in multiple hematological xenograft models, including Burkitt’s lymphoma (Raji), multiple myeloma (OPM-2), B-cell lymphoma (Ri-1) and AML (HL-60) (figure 5A–D). In all models evaluated, SRF231 showed significant monotherapy antitumor activity (p<0.0001), and sustained tumor regressions were observed. To address whether Fc receptor interactions were important for SRF231 antitumor activity in the mouse, a variant of SRF231 with a mutation that results in reduced Fc-receptor binding (termed SRF231mut) was engineered. When tested in the Raji xenograft model, SRF231mut showed reduced antitumor activity compared with SRF231, suggesting that Fc-receptor engagement, at least in part, contributes to the antitumor activity of SRF231 (online supplementary figure S2C). To confirm that the reduced activity with SRF231mut was not due to reduced antibody exposure, a pharmacokinetic analysis was performed. Serum exposures were comparable for both SRF231 and SRF231mut across the time points evaluated (data not shown). Consistent with in vitro phagocytosis assays (online supplementary figure S2), efficacy was more readily achieved in CB17 SCID (Balb/c background) recipients of Raji tumors as opposed to NOD SCID animals, possibly due to the increased affinity of NOD SIRPα for human CD47 (online supplementary figure S2D). Collectively, these data suggest that in a mouse-human interaction, despite the absence of CD32a Fc-receptor engagement is important for optimal macrophage activation and phagocytic activity.
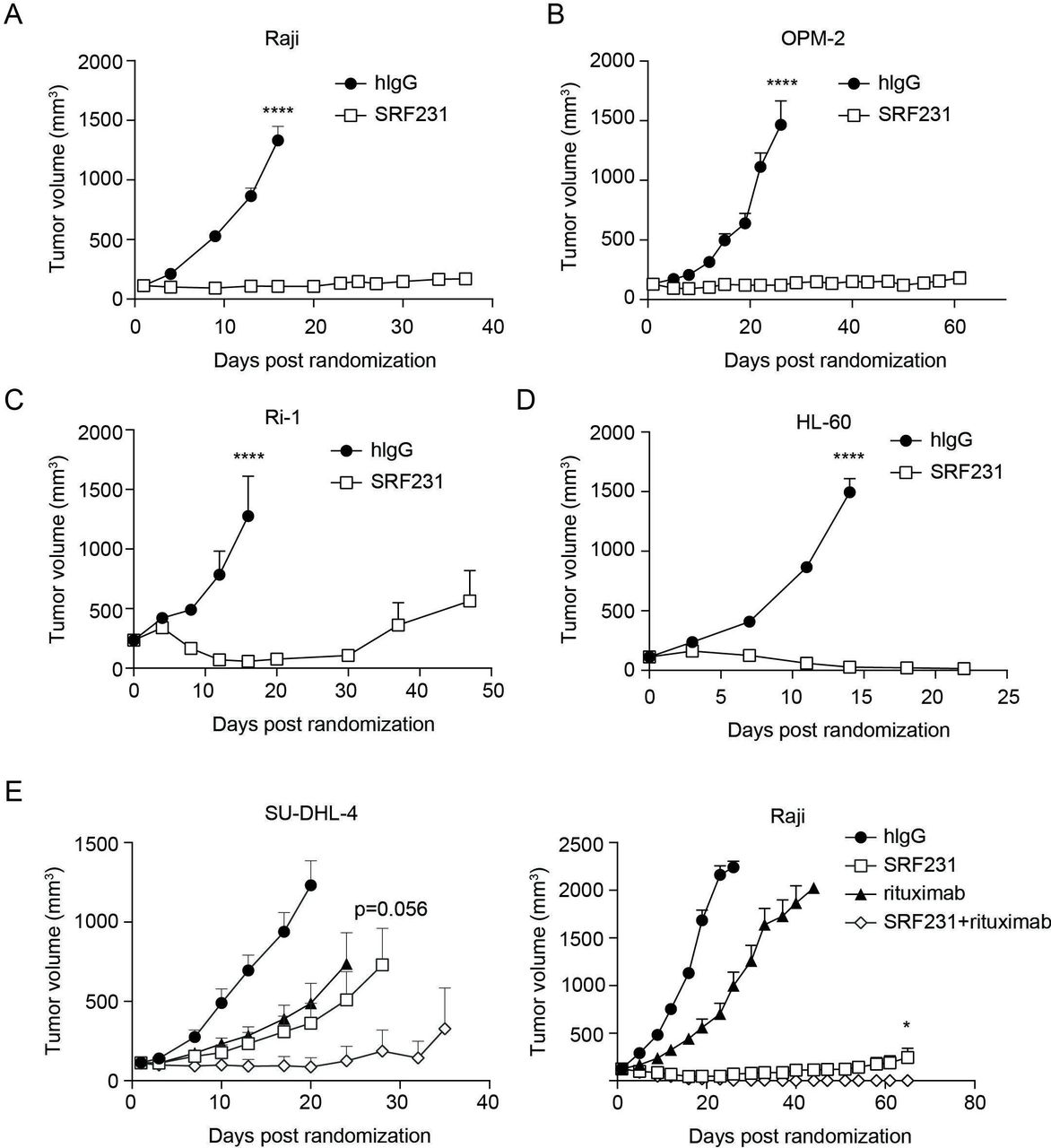
Therapeutic activity of SRF231 in hematologic xenograft models. CB.17 SCID mice were subcutaneously engrafted with: (A) Raji cells or (B) OPM-2 cells. Mice (n=8/group for Raji, n=6/group for OPM-2) were treated IP with 100 µg isotype control (hIgG) or SRF231 3 times/week for 3 weeks; (C) Ri-1 cells, mice (n=7/group) were treated IP with 100 µg hIgG or SRF231 on days 0, 3 and 7 postrandomization; (D) HL-60 cells, mice were treated IP with 100 µg hIgG or SRF231 on days 0 and 7 postrandomization (****p<0.0001 for SRF231 vs hIgG in all 4 models); (E, left panel) SU-DHL-4 cells, mice (n=10/group) were treated IP with 100 µg hIgG, 100 µg SRF231 (3 times/week for 3 weeks), 200 µg rituximab (once/week for 3 weeks) or the combination; p=0.0004 and 0.0016 for SRF231 vs hIgG and rituximab vs hIgG, respectively, on day 20; p=0.056 for SRF231 vs SRF231 +rituximab on day 28; (E, right panel) Raji cells, mice (n=10/group) were treated with 100 µg hIgG, 100 µg SRF231 (3 times/week for 3 weeks), 100 µg rituximab (once/week for 3 weeks) or the combination; p=0.0272 for SRF231 vs SRF231 +rituximab on day 65. Data are shown as mean tumor volumes±SEM, hIgG, human IgG; IP, intraperitoneally.
The efficacy of SRF231 in combination with the anti-CD20 antibody rituximab (Invivogen) was also evaluated in 2 lymphoma xenograft models. SU-DHL-4 is a diffuse large B-cell lymphoma model of the germinal center subtype containing a 14:18 chromosomal translocation resulting in Bcl-2 over-expression. As figure 5E (left panel) indicates, both SRF231 and rituximab showed single-agent antitumor activity in this model (p=0.0004 and 0.0016, respectively, versus isotype control on day 20). The combination of SRF231 with rituximab had a trend toward greater antitumor activity than SRF231 alone (p=0.056 on day 28 with 7/10 tumor-free animals in the SRF231 +rituximab combination group vs 1/10 or 0/10 tumor-free animals in the rituximab or SRF231 single agent arms, respectively, on day 24 onward). Increased antitumor activity was also observed in the Raji Burkitt’s lymphoma model when SRF231 and rituximab were co-administered (figure 5E; p=0.0272 for SRF231 vs SRF231+rituximab on day 65). In this case, complete regression of tumors in mice treated with the combination of SRF231 and rituximab was observed.
Macrophages contribute to SRF231 antitumor activity in vivo
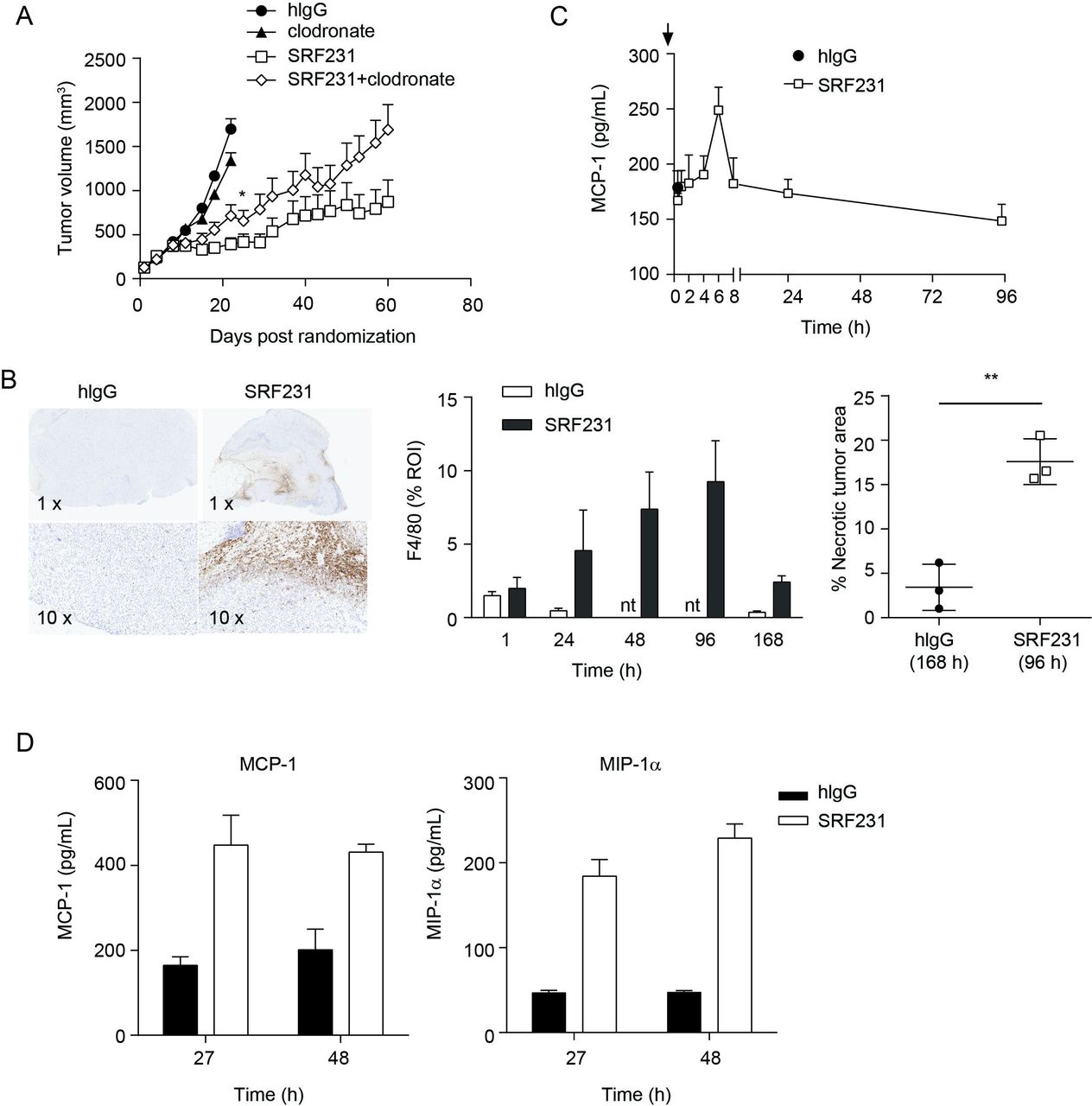
Since macrophages were involved in both in vitro antitumor mechanisms induced by SRF231, namely tumor cell phagocytosis and induction of cell death, the in vivo role of macrophages as effectors of SRF231 activity was investigated. Macrophages were depleted using clodronate liposomes in a Raji xenograft efficacy study. Treatment of Raji tumor-bearing animals with clodronate alone resulted in minimal tumor growth inhibition relative to the control group (p=0.0299; day 22). However, clodronate treatment prior to SRF231 administration reduced the antitumor effects of SRF231 (p=0.0451; day 22) (figure 6A). To determine whether macrophages were acting directly in the tumor, immunohistochemistry (IHC) was performed on Raji tumors from mice treated with a single dose of isotype control antibody or SRF231 and collected at different time points after treatment was performed. Tumors were stained with the murine macrophage marker F4/80. Treatment of Raji xenografts with SRF231 resulted in recruitment of F4/80+ macrophages into the tumor (figure 6B, left and middle panels). This macrophage infiltration was most prominent at the tumor margin and in the tumor core. In addition to infiltration of macrophages, SRF231 antitumor activity was also associated with significantly increased (p=0.0035) areas of tumor necrosis compared with isotype control treated animals (figure 6B, right panel). MCP-1 is a well-described monocyte-attracting chemokine that contributes to the recruitment of blood monocytes into tumors and sites of inflammation.34 To determine if SRF231 could induce MCP-1 in vivo, Raji tumor-bearing mice were treated with a single dose of isotype control antibody or SRF231 and plasma was collected and analyzed at various timepoints following treatment. As shown in figure 6C, mouse MCP-1 levels spiked in plasma from SRF231 treated mice approximately 6 hours after dosing and decreased to baseline thereafter. Macrophage cytokines were also evaluated in an HL-60 xenograft model in which tumor lysates were generated at 27 and 48 hours after a single administration of isotype control antibody or SRF231. In this study, both MCP-1 and a related macrophage chemoattractant, MIP-1α, were increased in tumor lysates from SRF231 treated mice by 27 hours and still elevated at 48 hours (figure 6D). Collectively, these data suggest that SRF231 may promote macrophage recruitment and activation that contributes to antitumor activity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Macrophages contribute to SRF231 antitumor activity. (A) CB17.SCID mice were inoculated with Raji cells subcutaneously. When tumors reached 100–150 mm3, day 0, mice (n=10/group) were treated with 100 µg clodronate liposomes IV 3 times/week for 2 weeks. On day 4, mice were treated IP with either 100 µg isotype control antibody (hIgG) or SRF231 IP 3 times/week for 3 weeks. Data are shown as mean tumor volumes±SEM; p=0.0451 for SRF231 vs SRF231+clodronate on day 22. (B, C) CB17.SCID mice bearing subcutaneous Raji tumors were treated IP with a single 100 µg dose of hIgG or SRF231. Mice were euthanized and tumors and plasma were collected at 1, 24, 48, 96 and 168 hours after treatments (n=3/group). (B) Representative Raji tumor sections from isotype-treated (168 hours) and SRF231-treated (96 hours) animals were stained with F4/80 (left panel, 1X and 10X magnification). F4/80 percent positive expression (middle panel) and necrotic tumor area percentage (right panel; **p=0.0035) was quantified with digital image analysis software applied to tumor images from both isotype and SRF231-treated animals at the defined timepoints. (C) Plasma MCP-1 expression from Raji tumor-bearing mice at the indicated times following a single 100 µg dose of hIgG or SRF231. (D) HL-60 tumor-bearing mice were treated with a single 100 µg dose of hIgG or SRF231. After 27 and 48 hours, mice (n=5/group) were euthanized, tumors were collected, and tumor lysates were analyzed for expression of mouse MCP-1 (left panel) and MIP1α (right panel). hIgG, human IgG; IV, Intravenous; IP, intraperitoneally; MCP1, Mouse chemoattractant protein 1; MIP1α, macrophage inflammatory protein 1α; nt, not tested; ROI, region of interest.
Discussion
In this study, the mechanism of action and antitumor potential of SRF231, an investigational fully human anti-human CD47 IgG4 antibody, is described. SRF231 blocks the CD47/SIRPα interaction, does not cause hemagglutination and promotes macrophage-mediated tumor cell phagocytosis over a broad range of tumor cell lines and primary AML cells. SRF231 also induces tumor cell death in a caspase-independent manner. Both induction of macrophage-mediated phagocytosis and cell death by SRF231 are dependent on the low-affinity, activating FcγR CD32a, which is expressed predominantly by myeloid cells.35 In vivo, SRF231 elicits profound antitumor activity across multiple hematologic xenograft models in a manner involving macrophages.
A leading therapeutic hypothesis around the mechanism of action of CD47 antibodies is that blockade of CD47/SIRPα is central to lowering the threshold for tumor cell phagocytic uptake by macrophages and other phagocytic cells that express SIRPα.36 However, a number of reports have emerged to suggest that CD47/SIRPα blockade alone is not sufficient to trigger phagocytosis induction.3 Moreover, the growing number of CD47 antibodies and CD47 targeting strategies described also highlight the unique and overlapping properties of these agents. A better mechanistic understanding of distinct CD47 blocking agents is warranted and may have clinical implications.
Studies described here demonstrate that factors beyond CD47/SIRPα blockade contribute to the mechanism of action of SRF231. SRF231 acts through at least 2 distinct, macrophage-driven mechanisms: (1) induction of tumor cell phagocytosis and (2) induction of tumor-intrinsic cell death. Both of these events are dependent on the ability of SRF231 to bind CD47 on the tumor cell and CD32a on the myeloid effector cell.
In the case of phagocytosis induction by macrophages, phosphorylation of CD32a is observed and is likely important for activation of macrophage effector function. CD47/SIRPα blockade alone by SRF231 F(ab’)2 was not sufficient to drive phagocytosis when effector cells were human monocyte-derived macrophages. In this system, CD32a engagement was required, suggesting that SRF231 acts in part as an opsonizing antibody to drive classic ADCP. However, CD47/SIRPα blockade by SRF231 may play a more dominant role in driving antitumor activity in other cellular contexts. For example, CD47/SIRPα blockade can augment ADCP when co-administered with opsonizing antibodies.7 Additionally, interruption of CD47/SIRPα signaling has been linked to enhanced immunosurveillance via myeloid polarization37 38 and antigen presentation.39 Therefore, by blocking the CD47/SIRPα interaction in addition to engaging CD32a, the dual mechanistic properties of SRF231 may be advantageous in driving antitumor activity via multiple mechanisms of action that are context dependent.
The observation that phagocytosis induction with SRF231 F(ab’)2 was observed only when mouse, but not human macrophages were used as effector cells (figure 2B), could be a result of several factors. Mouse macrophages may be more dependent on SIRPα than human macrophages to inhibit phagocytosis at baseline and the two-signal model requiring both an ‘eat-me’ and a ‘don’t-eat-me’ signal may be less dominant in the mouse setting.40 In addition, the relative affinity differences between mouse and human SIRPα to human CD47 may influence the degree of phagocytosis in these different settings.41 This was observed when comparing phagocytosis using macrophages from Balb/c versus NOD mice (online supplementary figure S2A). It is worth noting that SRF231 F(ab’)2 potency was still greatly reduced relative to the full-length molecule in mouse macrophage-driven phagocytosis, suggesting that FcR engagement is still functionally relevant in the mouse, despite the lack of CD32a. It is likely that one or more mouse FcR can functionally substitute for CD32a and further studies will be required to confirm this.
While the ability of anti-CD47 molecules to induce apoptosis when immobilized and cross-linked has been previously described,17 42 the physiological relevance of this phenomenon deserves emphasis. In the case of SRF231-mediated tumor cell death induction, the presence of CD32a could be replaced by immobilizing antibody in the correct orientation, suggesting that CD32a is likely playing a scaffolding function for this cellular outcome. This highlights the independence of cell death induction from blockade of the CD47/SIRPα interaction and is consistent with the notion that antibody scaffolding via Fc/FcR engagement is supported in FcR- and phagocyte-rich tumor microenvironments.
The phagocytosis and cell death assays comparing SRF231 to other CD47 blocking antibodies underscore the complexity associated with differential properties of these antibodies. Unlike SRF231, only CC2C6 induced cell death in a soluble form (figure 3C). Cell death induction for CC2C6 was reported to be linked to shorter incubation times, but whether this phenomenon was associated with CD47 internalization, downstream signaling or binding mode remains poorly understood.18 It is likely that many unique antibody attributes contribute to the distinct behaviors of different CD47 antibodies. In addition to Fc-isotype, factors such as binding kinetics (on-rates and off-rates), relative affinity/avidity parameters and epitope are likely to also influence overall activity. Moreover, the relative expression of prophagocytic receptors on various tumor cells that counterbalance CD47 could influence the degree of phagocytosis or the requirement for additional macrophage activation through Fc-receptor engagement. This confounds the comparison of CD47 antibody properties across different assay systems. While multiple mechanisms of antitumor activity have been proposed for CD47/SIRPα antagonists, including SRF231, the context dependency and relative contributions of these mechanisms to overall antitumor activity warrants further investigation.
A defining feature of SRF231 is its ability to bind to CD47 with high affinity without inducing hemagglutination or RBC phagocytosis, in contrast to 4.2C11 or B6H12. The binding mode(s) that allows for SRF231 RBC binding without hemagglutination is not well understood and requires further study. One possibility may be that orientation of the 2 Fab arms of the bivalent SRF231 molecule are not permissive for simultaneous binding of 2 RBCs, which is required to seed the lattice formation effect leading to hemagglutination.
The contribution of murine macrophages to overall antitumor activity of SRF231 was addressed in vivo. Macrophage depletion using clodronate liposomes led to a partial reduction of SRF231 antitumor activity in a Raji lymphoma xenograft model. The observation that tumor growth inhibition was only partially blocked could be due to several factors. On the one hand, it suggests that other effector cells in the tumor microenvironment are also important for SRF231 activity. Alternatively, clodronate treatment may have resulted in only partial macrophage depletion, still allowing remaining macrophages to exert antitumor effects in response to SRF231.
Multiple hematological tumor cell lines were responsive to SRF231 treatment in a subcutaneous xenograft setting (figure 5). While several of these cell lines were sensitive to SRF231-mediated phagocytosis in vitro (figure 1B), tumor cell death is also evident in vivo by the increased presence of necrotic areas in tumors treated with SRF231 (figure 6B). Additionally, the induction of myeloid-derived chemokines MCP-1 and MIP-1α in response to SRF231 treatment in vivo is consistent with the idea of further myeloid cell recruitment and has been previously reported for other CD47-targeting agents.43 44 Thus, we propose that macrophages (and possibly other myeloid effector cells) are central to SRF231-driven antitumor efficacy via induction of tumor cell phagocytosis, cell death induction and chemokine-driven amplification of myeloid cell-driven responses. In addition, the observation that SRF231mut (a variant of SRF231 with reduced Fc-receptor engagement; data not shown) showed decreased antitumor activity relative to SRF231 (online supplementary figure S2C), supports a requirement for FcR effector function for mediating optimal antitumor responses.
The dependence of an IgG4 isotype antibody on FcR-mediated functions is notable. Therapeutic mAbs that drive antibody‐dependent cell‐mediated cytotoxicity (ADCC) and ADCP responses are typically an IgG1 isotype to maximize engagement of CD16 (FcγRIIIa), an activating receptor expressed on natural killer cells that can mediate induction of ADCC (eg, alemtuzumab, rituximab).7 20 22 29 Furthermore, human macrophages express activating FcγRs CD32a, CD16a and CD64,45 all of which have submicromolar affinity for hIgG1,46 and have been linked to phagocytosis effector function in the context of a SIRPα-Fc IgG1 fusion protein.13 On human macrophages, the relative abundance of CD32a is greatest compared with other activating FcγRs, suggestive of its dominant role in mediating FcR-driven effector function.45 Antibodies that are hypothesized to exert therapeutic activity primarily through ADCC generally have Fc regions with high FcR engagement such as IgG1.46 47 In contrast, antibodies that are thought to work mainly through receptor/ligand blockade are often selected to have Fc regions with limited FcR engagement, such as IgG4 (eg, natalizumab, nivolumab).48 49 As expected, the IgG4 Fc of SRF231 has undetectable affinity for CD16 and does not lead to CD16-driven ADCC in vitro, while affinity for CD32a is low, but measurable (data not shown). It is likely that with targets such as CD47, where receptor density is sufficiently high and/or clustered,50 dense accumulation of IgG4 molecules is able to overcome a certain affinity threshold to allow for CD32a engagement.51
Conclusions
In summary, the investigational anti-CD47 antibody SRF231 lacks hemagglutination properties and elicits antitumor activity via both phagocytosis and cell death. The activating FcγR, CD32a, is central to the mechanism of action of SRF231 despite the hIgG4 Fc. Recent literature has implicated an important role for Fc/FcγR engagement for therapeutic antibodies targeting T-cell antigens such as CTLA-4.52–55 Therefore, activating FcγR engagement is becoming increasingly appreciated even for targets in which the mechanism is thought to be primarily driven through ligand/receptor blockade. A further understanding of how CD47/SIRPα blockade and concurrent Fc/FcR engagement contribute toward overall biological activity of CD47/SIRPα antagonists is warranted.
Acknowledgments
We are grateful to Rob Ross for intellectual input, Karen D’Amour for editing, Matt Rausch for interpretation of in vivo data and input on statistical analyses, and Krisha Panchalingam for sample and laboratory management. We would also like to thank Alexander Rudensky for critical review of the manuscript.
References
Footnotes
MOP and AA contributed equally.
Contributors AMP, MOP, AA, PMH, VJP and EN conceived and designed the study experiments. MOP, AA, CMA, LZ, RWO and ACL developed assays, performed experiments and/or analyzed data. AMP and MOP wrote the manuscript, and PMH, AA, BHL, VJP, ACL and JAH revised and edited the manuscript. SCC was involved in the synthesis of SRF231 and control antibodies. All authors read and approved the final manuscript.
Funding This work was funded by Surface Oncology.
Competing interests All authors are current or former employees and shareholders of Surface Oncology.
Patient consent for publication Not required.
Ethics approval All animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health under protocols approved by the Charles River Accelerator and Development Lab (CRADL) Institutional Animal Care and Use Committee (Protocol Number: CR-008) and all efforts were made to minimize suffering.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.