Article Text

Abstract
Background In patients with hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC), virus-specific cytotoxic T lymphocytes (CTLs) fail to eliminate HCC cells expressing HBV antigens. As the expression of viral antigen in HBV-associated HCC may decrease to allow tumor to escape immune attacks, we hypothesized that an HBV surface antigen (HBsAg)-specific affinity-improved-T-cell receptor (TCR) will enable T cells to target HCC more effectively than corresponding wild-type-TCR. We also postulated that TCR promiscuity can be exploited to efficiently capture HBV variants that can hinder CTL-based therapeutics.
Methods We applied flexi-panning to isolate affinity-improved TCRs binding to a variant antigen, the human leukocyte antigen (HLA)-A*02:01-restricted nonapeptide HBs371-379-ILSPFLPLL, from libraries constructed with a TCR cloned using the decapeptide HBs370-379-SIVSPFIPLL. The potency and safety of the affinity-improved-TCR engineered T-cells (Ai-TCR-T) were verified with potentially cross-reactive human and HBV-variant peptides, tumor and normal cells, and xenograft mouse models.
Results Ai-TCR-T cells retained cognate HBV antigen specificity and recognized a wide range of HBV genotypic variants with improved sensitivity and cytotoxicity. Cell infusions produced complete elimination of HCC without recurrence in the xenograft mouse models. Elevated accumulation of CD8+ Ai-TCR-T cells in tumors correlated with tumor shrinkage.
Conclusion The in vitro and in vivo studies demonstrated that HBsAg-specific Ai-TCR-T cells had safety profiles similar to those of their wild-type counterparts and significantly enhanced potency. This study presents an approach to develop new therapeutic strategies for HBV-related HCC.
- CD8-Positive T-lymphocytes
- cell engineering
- immunohistochemistry
- immunotherapy
- adoptive
- liver neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Hepatocellular carcinoma (HCC) ranks as the sixth most common cancer and fourth leading cause of cancer-related death worldwide, accounting for nearly 800,000 deaths per year.1 2 Among HCC cases, almost 80% are associated with chronic hepatitis B or C virus (HBV or HCV, respectively) infection.3 4 Current treatments for HCC have a 10-year actual survival rate of only 7.2%,5 and the relapse rate for HBV-positive HCC is high following liver transplantation.6 There is a great demand for potent and tolerable therapies in clinics worldwide.
Although immunotherapies have been considered for HCC, virus-specific T cells are deleted or dysfunctional, and immune attack to eliminate cancer cells is often retarded in patients with HBV-related HCC.7 This problem may be overcome with T-cell receptor (TCR)-engineered T cell (TCR-T cell) therapy,8 because the viral DNA frequently integrates into the liver cell genome.9 10 Whether HCC cells can present any HBV peptide antigens remains controversial, as there is often no HBV protein detected in HCC cells with immunohistochemistry (IHC).11 However, Tan et al 12 recently demonstrated that HBV-related HCC cells might not express whole HBV antigens but could transcribe short HBV messenger RNAs encoding epitopes recognized by HBV-specific T cells. Additionally, HBV surface antigen (HBsAg) has been used as a target for TCR-T cells to treat HBV-related HCC.13 An HBs183-191-specific wild-type (WT) TCR, which was isolated from a human leukocyte antigen (HLA)-A2+ patient who had resolved an HBV infection, was shown to recognize HCC cells integrated with natural HBV-DNA and mediate HBV-specific T cell immunity in vitro and in vivo.14–16 As a result, clinical studies of HBsAg-specific T cell therapy have demonstrated good tolerability and safety with wild-type T-cell receptor (TCR)-engineered T cells (WT-TCR-T). The efficacy of the treatment revealed a volume reduction of several pulmonary HCC metastatic lesions in one of the two patients.12 17 18
Although HBV antigen-negative HCC cells can present HBV peptide antigens, the density may be relatively low.19 20 TCR-T cells engineered with a wild-type TCR, whose natural affinities are limited to ~1 to 100 µM, may not effectively target cells with downregulated tumor antigen presentation.21–26 However, affinity-improved TCRs can recognize tumor cells presenting low-density antigens.24 On the other hand, liver transplantation-treated HCC patients still have a large number of liver tumor cells outside the liver, which may present sparse antigens and contribute to HCC relapse during a later stage. Therefore, we hypothesized that in the treatment of HBV-related HCC, especially in liver-transplanted patients, affinity-improved TCR-engineered T-cells (Ai-TCR-T) will produce better outcomes than WT-TCR-T.
Furthermore, nine HBV genotypes and many variants that have different peptide antigens have been identified to date,27 and it would be valuable to create one TCR that can specifically recognize several HBV-variant peptide antigens. It is well documented that a TCR can show promiscuity for recognizing a set of variant antigens,28–31 but the T cell maintains physiological specificity in the peripheral immune system. We then questioned whether this feature can be harnessed for the generation of affinity-improved TCRs that are capable of recognizing several HBV variants.
To address both questions, we constructed TCR libraries with a wild-type TCR that was isolated initially with an HLA-A*02:01-restricted HBV genotype-A HBs370-379 (SIVSPFIPLL, named vrt0) decameric peptide antigen complex from a resolved HBV patient.14 16 Because HBV genotype C is closely related to an elevated HCC risk,32–35 we isolated high-affinity TCRs binding to a genotype B/C/D HBs371-379 (ILSPFLPLL, named vrt1) nonameric peptide antigen from the TCR libraries. An affinity-improved mutant TCR could retain HBsAg specificity similar to the specificity of the wild-type TCR. The mutant TCR constructed Ai-TCR-T cells showed enhanced sensitivity and cytotoxicity in vitro. In addition, Ai-TCR-T cells recognized not only vrt1 and vrt0 but also nine other peptide variants found at high frequencies in HBV species. However, both WT-TCR-T cells and Ai-TCR-T cells were hardly activated by irrelevant peptide-pulsed or potentially cross-reactive human peptide-pulsed cells, human normal primary cells and healthy peripheral blood mononuclear cells (PBMCs), indicating that the Ai-TCR-T cells have a good safety profile. The Ai-TCR-T cells were also demonstrated to eliminate HCC tumors without recurrence in vivo.
Methods
HBs peptides and HBV genotypes
HBs371-379 and HBs370-379 epitope sequences were retrieved from 5855 unique full-length HBV surface antigen (HBs) polyproteins (389, 399 and 400AA HBs polyprotein sequences) collected from UniProt around May 2019. First, there were 134 unique nonameric or 190 unique decameric peptides extracted respectively from HBs371-379 and HBs370-379 epitope sequences with Strawberry Perl 5.26.1.1 (http://strawberryperl.com) after removing duplication. Second, the most common 20 peptides of HBs371-379 and HBs370-379 were statistically analyzed from unique 5657 (97% of 5855) and 5528 (94% of 5855) full-length HBs polyprotein sequences separately. Finally, the most common 20 peptides that already categorized the HBV genotypes in UniProt were statistically analyzed with HBV genotypes. The corresponding HBV genotypes of the most common eight peptides of HBs371-379 (vrt1 to vrt8) and HBs370-379 (vrt0, vrt9 to vrt16) were analyzed with Package ‘pheatmap’
(https://cran.r-project.org/web/packages/pheatmap/index.html) based on statistical software R V.3.6.1 (https://www.r-project.org/).
Identification of potential cross-reactive human peptides of vrt1 and vrt0
Potential cross-reactive human peptides were identified by Expitope webserver 2.0 (http://webclu.bio.wzw.tum.de/expitope2/) designed to predict the similar peptides and its potential presentation by the HLA allele of interest.36 37
For identifying vrt1 and vrt0 related human cross-reactive peptides, we combined the results of the alanine scanning to determine the critical TCR binding position of vrt1 and vrt0. The maximum allowed number of mismatches were set as three for vrt1 and four for vrt0 for the primary screening. The chosen allele was HLA-A*02:01. The default parameters were used for the other options. We also carried out a second screening without fixing the critical positions, and the maximum allowed number of mismatches were set two for vrt1 and three for vrt0. Those potential peptides derived from proteins with expression data were proceeded the third step of T cell activation assays.
Antigen peptides
The 16 most frequent HBs371-379 (HBs371) and HBs370-379 (HBs370) antigenic peptides were retrieved from 5855 full-length HBsAg polyproteins. There were eight HBs371 nonameric peptides, that is, vrt1-ILSPFLPLL, vrt2-ILSPFIPLL, vrt3-ILSPFMPLL, vrt4-ILNPFLPLL, vrt5-IVSPFIPLL, vrt6-TLSPFLPLL, vrt7-ILNPFIPLL and vrt8-IVRPFIPLL, and eight HBs370 decameric peptides, that is, vrt9-NILSPFLPLL, vrt10-NILSPFIPLL, vrt11-SILSPFLPLL, vrt12-NILSPFMPLL, vrt13-NILNPFLPLL, vrt14-SILSPFIPLL, vrt0-SIVSPFIPLL and vrt16-NILNPFIPLL, that were analyzed (online supplemental figure S1, tables S1.1 and S1.2).
Supplemental material
The irrelevant peptide-HLA complexes (pHLAs) (online supplemental table S2) were used to verify the specificity of the TCR mutants. Alanine-scanning peptides and potentially cross-reactive human peptides related to vrt1 and vrt0 (online supplemental tables S3.1 and S3.2) were used to test the binding site and TCR specificity, respectively. All peptides were purchased from Genscript and validated with a mass spectrometer, and the purities were confirmed to be greater than 95%.
Cell lines, PBMC and human normal primary cells
T2, J.RT3-T3.5 cell, 293T and HepG2 were purchased from ATCC. HCC cell lines, PLC/PRF/5 were provided by Dr Xiaoping Chen from Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences. PBMC, provided by informed consent healthy donors, are listed in online supplemental table S4 and 12 sets of human primary cells from ScienCell are listed in online supplemental table S5, including human bronchial smooth muscle cells, human aortic smooth muscle cells, human meningeal cells, human renal mesangial cells, human gastric smooth muscle cells, and human renal epithelial cells. Cell culture dish was treated with Poly-L-Lysine before culturing. All cell lines were authenticated using short tandem repeat profiling and regularly tested negative for mycoplasma contamination throughout the whole duration of this study.
HLA-A2 negative HCC cell line PLC/PRF/5 has multiple, random integrations of HBV DNA and can express HBsAg. PLC/PRF/5 cells were transduced with HLA-A*02:01 subtype with pLenti-CMV-puro lentivirus vectors to constructed HLA-A*02:01+ cell lines. The transduced cells were maintained with the selection of 2 µg/mL puromycin (BD Biosciences). Cells were stained with APC-conjugated anti-human-HLA-A2 monoclonal antibody (BioLegend) and positive cells were isolated by sorting on a FACSAria III flow cytometer (BD Biosciences).
Generation and analysis of affinity-improved TCR mutants
We constructed Vα and Vβ chain single-chain TCR variable domain (scTv) evolution libraries using TCR0, which was isolated previously with vrt0-SIVSPFIPLL. The nonapeptide variant vrt1-ILSPFLPLL HBs371-HLA-A*02:01 complex was used as the antigen for bio-panning to isolate affinity-improved TCR variants from the libraries using methods described previously.22 38 Briefly, the scTv libraries were displayed on the surface of the filamentous bacteriophage M13, and affinity-improved scTv variants were selected with biotinylated vrt1-pHLA captured on streptavidin-coated Dynabeads (Life Technologies). Binding improved scTvs were analyzed in soluble heterodimeric TCR molecules that were purified after refolding Escherichia coli-produced inclusion bodies. The TCR affinities were measured by surface plasma resonance with ProteOn and Biacore.
TCR-T cell construction
TCR α/β chain transgene lentiviral vectors contained a picornavirus 2A sequence. In addition, the mouse TCR constant domains replaced the human constant domains to reduce mispairing.
PBMCs, provided by healthy human volunteer donors under Institutional Review Board approved informed consent, were prepared with human Lymphoprep reagent (Axis-Shield). Peripheral blood lymphocytes (PBLs) prepared with the PBMCs were activated with Human T-Activator CD3/CD28 Dynabeads (Life Technologies) at a bead-to-cell ratio of 1:1 in RPMI 1640 medium supplemented with 10% fetal bovine serum and 100 IU recombinant human IL-2. Lentiviral vectors for WT HBsAg-TCR, affinity-improved HBsAg-TCRs and control-TCR-A6 (a TCR specific for the HTLV-1 Tax11-19 (LLFGYPVYV)-HLA-A*02:01 complex) were transduced into PBLs, which were supplemented with protamine (10 µg/µl). After proliferation, T cells were stained with FITC-conjugated or APC-conjugated antibodies (BioLegend) for human CD3, CD8, TCR Vβ13.1 or mouse TCR β-chain, and a PE-conjugated HTLV-1 Tax11-19-pHLA or vrt1-pHLA tetramers to verify TCR expression.
T cell activation
Interferon gamma (IFN-γ) secretion was measured with ELISPOT kits (BD Biosciences). Briefly, 2×103 positive TCR-T cells were incubated with T2 cells (HLA-A*02:01+) pulsed with vrt0-vrt16 or irrelevant peptides from human antigens (ranging from 10−6 to 10−13 M), HCC cells (HLA-A*02:01− PLC/PRF/5 or HLA-*02:01+ PLC/PRF/5 cells), PBMCs or human normal primary cell lines at an effector-to-target (E:T) ratio of 1:10. To compare specificity between WT and affinity-improved TCRs, TCR-T cells were incubated with T2 cells pulsed with the irrelevant antigen SLLMWITQC-HLA-A*02:01 in a range of 10−6 to 10−13 M. The other two negative controls were unpulsed T2 cells and HLA-A*02:01+ HBsAg− HepG2 cells.
T-cell-mediated cytotoxicity
Cell lysis was determined by releasing lactate dehydrogenase (LDH) with a CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega). T2 cells (5×104) pulsed with vrt0-vrt16 (ranging from 10−6 to 10−13 M) were incubated with an equal amount of TCR-T cells overnight. Unpulsed T2 and 10−6 M irrelevant SLLMWITQC-peptide-pulsed T2 cells were set as negative controls. Cell-line assays were carried out with the HCC cell line PLC/PRF/5 transduced with HLA-A*02:01 at an E:T ratio of 1:1 and 3:1, and the controls were HLA-A*02:01− PLC/PRF/5 cells and HBsAg− HLA-A*02:01+ HepG2 cells. Experiments were performed in triplicate.
Mouse xenograft models
Immunodeficient male NOD/SCID IL2rg−/− (NSI) mice aged 6 to 9 weeks were provided by Dr Peng Li at the Guangzhou Institutes of Biomedicine and Health, CAS.39 Mice were maintained with daily monitoring under controlled sterile environment conditions on a 12 hours light/dark cycle, and received a rodent diet sterilized with 60Co-γ-ray irradiation and sterilized water. Experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC). Each mouse was injected subcutaneously with 3.0×106 HLA-A*02:01+ PLC/PRF/5 HCC cells on day 0. After the tumors reached nearly 60 mm3 (day 13), the mice were randomly assigned to groups and treated with phosphate-buffered solution (PBS), 3.0×107 TCR-A6-T cells, 3.0×107 TCR0-T cells or 3.3×106, 1.0×107 or 3.0×107 TCR15-T cells via tail vein injection. Tumor volumes were measured every 3 days. From the criteria of IACUC, for subcutaneous tumors the maximum allowable size is 20 mm in diameter for a mouse. So, on day 34, the tumor-bearing mice were euthanized by cervical dislocation, and tumor tissues were resected and weighed.
Detection of human T cells in tumors
Histological HCC tumor sections were stained with H&E or antibodies for IHC. Primary anti-human CD3 (Dako) and anti-human CD8 (ZSGB-BIO) monoclonal antibodies and Dako REAL EnVision Detection System were used for IHC, and DAPI (blue) stained nuclei. The sections were imaged with a LAIKA2500, and cell numbers were calculated with Image-Pro Plus 6.0.
Results
Selection and analysis of affinity-improved TCRs
Commonly, phage display bio-panning for affinity improvement applies the antigens used for the isolation of WT molecules to maintain specificity. However, TCR binding shows a certain degree of promiscuity. Therefore, the nonameric antigen vrt1-ILSPFLPLL pHLA, the variant of decameric antigen vrt0-SIVSPFIPLL used to isolate the WT TCR, was employed for affinity evolution, and we named this kind of selection flexi-panning. Nineteen TCR variants with CDR3α mutations were identified and showed affinity-improved binding to the vrt1-pHLA (table 1).
The affinities of mutated TCRs isolated from TCR0 (WT) mutation libraries*
Compared with TCR0-T cells (WT), TCR6-T, TCR11-T, TCR14-T, TCR15-T, TCR17-T and TCR19-T cells were Ai-TCR-T cells that could be specifically activated by T2 cells pulsed with relatively low-level antigens (figure 1A,B). Four TCR variants demonstrated stronger capabilities than WT TCR0 for redirecting T cells, with TCR14-T, TCR15-T, TCR17-T and TCR19-T cells showing significantly better half maximal effective concentration (EC50) values. TCR14-T cells and TCR15-T cells showed the best activation with the HLA-A*02:01+ PLC/PRF/5 cell line (figure 1C) despite showing relatively little improvement in the EC50 by peptide titration (online supplemental table S6).
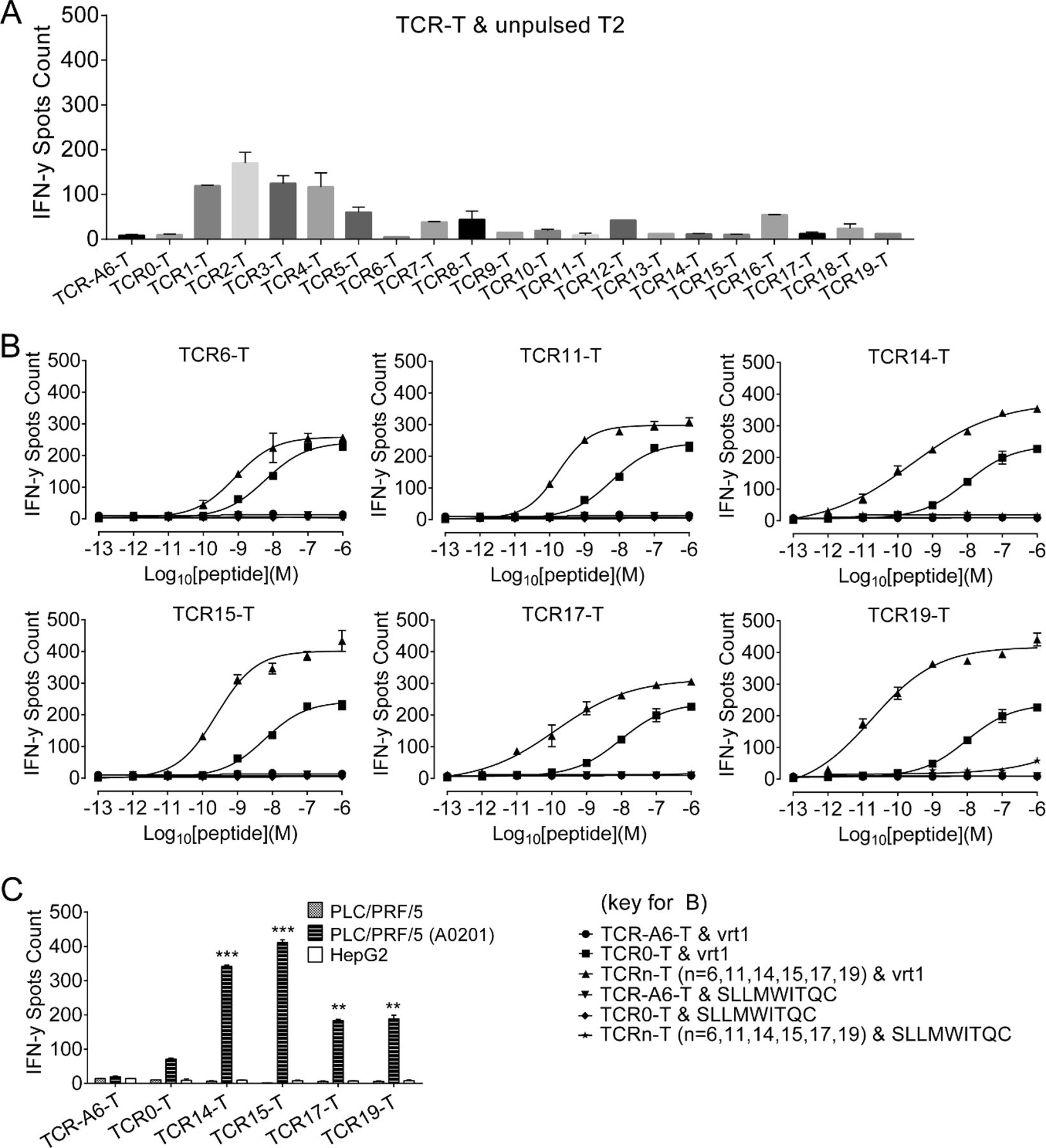
Analysis of TCR-T cell activation by measuring IFN-γ release. All samples were set up in triplicate and evaluated after coculturing cells overnight at an E:T ratio of 1:10. (A) To verify specificity, 19 affinity-improved TCR-engineered T cells (Ai-TCR-T cells) were incubated with unpulsed T2 cells overnight to detect IFN-γ release. (B) T2 cells pulsed with vrt1 (concentration range: 10−6 to 10−13 M) were incubated with Ai-TCR-T cells. T2 cells pulsed with an irrelevant peptide (10−6 to 10−12 M) were the negative control. (C) IFN-γ release was measured after TCR-T cells were incubated with HLA*A-02:01- PLC/PRF/5 or HLA-A*02:01+ PLC/PRF/5 HCC cells. The negative controls were HBsAg-negative HepG2 cells and TCR-A6-T cells. *p<0.05, **p<0.01, ***p<0.001. E:T, effector-to-target; HBsAg, HBV surface antigen; HBV, hepatitis B virus; HLA, human leukocyte antigen; IFN-γ, interferon gamma; TCR, T-cell receptor; TCR-T, TCR-engineered T cell.
Characteristics of TCR15 and enhanced cytotoxicity of TCR15-T cells
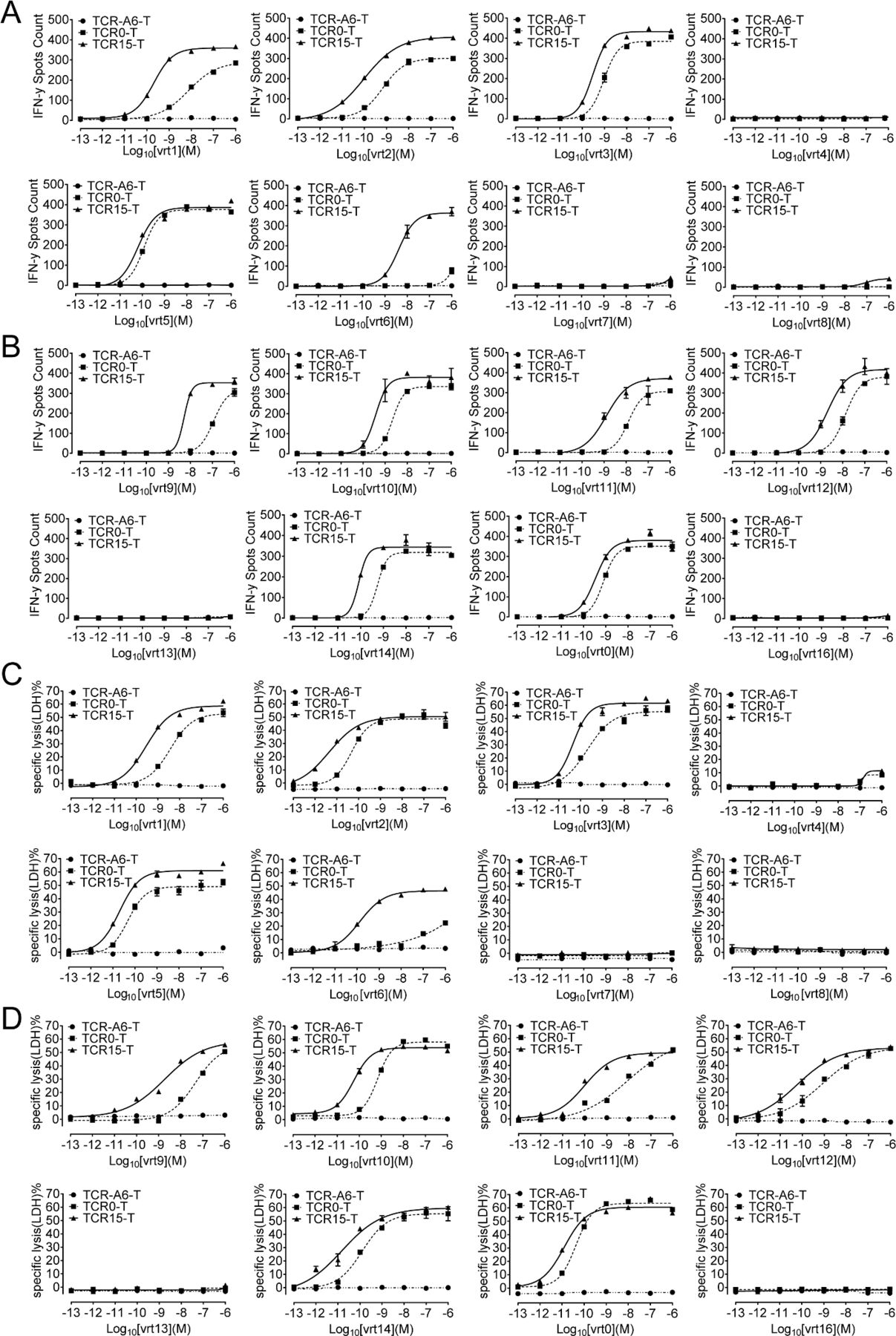
TCR14-T and TCR15-T cells exhibited strong cytotoxicity, as measured by assessing the lysis of vrt1-pulsed T2 cells (figure 2A,B). However, only TCR15-T cells showed specific cytotoxicity with minimal background cytotoxicity. The EC50 of vrt1-pulsed T2 cell lysis by TCR15-T cells was nearly 38-fold better than that by TCR0-T cells (online supplemental figure S2). When HBsAg-specific cytotoxicity was investigated by targeting HLA-A*02:01+ PLC/PRF/5 HCC cell lines, both WT-TCR-T cells and Ai-TCR-T cells exhibited dose-dependent target cell lysis, and TCR15-T cells caused significantly enhanced LDH release compared with TCR0-T cells (figure 2C).

Detection of TCR-T cell cytotoxicity. (A) Cytotoxicity assays were performed by incubating TCR-T cells with T2 cells pulsed with vrt1 (10−6 to 10−13 M) at an E:T ratio of 1:1. (B) To verify specificity, LDH release was also measured for unpulsed and irrelevant peptide-pulsed T2 cells. (C) Cytotoxicity of LDH release was determined after incubating TCR-T cells with HLA*A-02:01- or HLA*A-02:01+ PLC/PRF/5 HCC cell lines at an E:T ratio of 1:1 or 3:1. The negative controls were HBsAg-negative HepG2 cells and TCR-A6-T cells. All assays were performed in triplicate. *p<0.05, **p<0.01, ***p<0.001. E:T, effector-to-target; HBsAg, HBV surface antigen; HBV hepatitis B virus; LDH, lactate dehydrogenase; TCR, T-cell receptor; TCR-T, TCR-engineered T cell.
These data demonstrated TCR15 transduced T cells with acceptable activation, killing potency and specificity. Peptide vrt0-isolated TCR0 was compared with vrt1-pHLA-isolated TCR15 for binding to the two distinct antigens by Biacore. The mutant TCR15 had over sevenfold and fivefold higher binding affinities for the variant vrt1-pHLA and the original vrt0-pHLA antigens, respectively (table 2). TCR15 also demonstrated modest affinity enhancement for binding to vrt0-pHLA, despite vrt1-pHLA being used as the ligand for molecular evolution. This result revealed that flexi-panning could maintain the original affinity pattern of the TCR.
The affinities of TCR0 and TCR15 for the vrt1-pHLA and vrt0-pHLA*
Specificity and safety of TCR15-T cells
The specificity of TCR0 and TCR15 was verified, and these TCRs showed no binding with a panel of irrelevant pHLAs (online supplemental table S2). A three-step approach for an in-depth study was carried out for potential cross reactivity on relevant pHLAs; the first step involved alanine scanning of vrt0 and vrt1, the second step involved searching for cross-reactive antigens in the available human proteome expression data (online supplemental tables S3.1 and S3.2), and the third step involved verifying these antigens with pulsed T2 cells. Safety was measured with a panel of normal cells. Based on the cell activation showed in the alanine scanning, the residue positions F5 and L8 of vrt1 and F6 and L9 of vrt0 are crucial for maintaining the interactions between the TCR and HBsAg peptide-pulsed HLA-A*02:01+ T2 cells (online supplemental figure S3A,B). We identified a total of 33 vrt1-related potentially cross-reactive human peptides, 1 of which contained 2 mismatch residues and 32 of which contained 3 mismatches plus invariable F5 and L8 residues. For vrt0, we identified 2 sequences containing 2 or 3 mismatches and 10 sequences containing 4 mismatches with fixed F6 and L9 residues. All 45 potentially cross-reactive candidates were verified for agonist capabilities on peptide-pulsed T2 cells over a concentration range from 10−6 to 10−8 M. We found that most of these peptides, except high concentration (10−6 and 10−7 M) vrt1-L1, vrt1-L5, vrt1-L12, and vrt1-L19, which mainly derived from poorly expressed proteins, showed no agonist capability (figure 3A,B, online supplemental figure S4). TCR0-T cells but not TCR15-T cells appeared to be activated by T2 cells pulsed with vrt1-L13 derived from a poorly expressed protein.
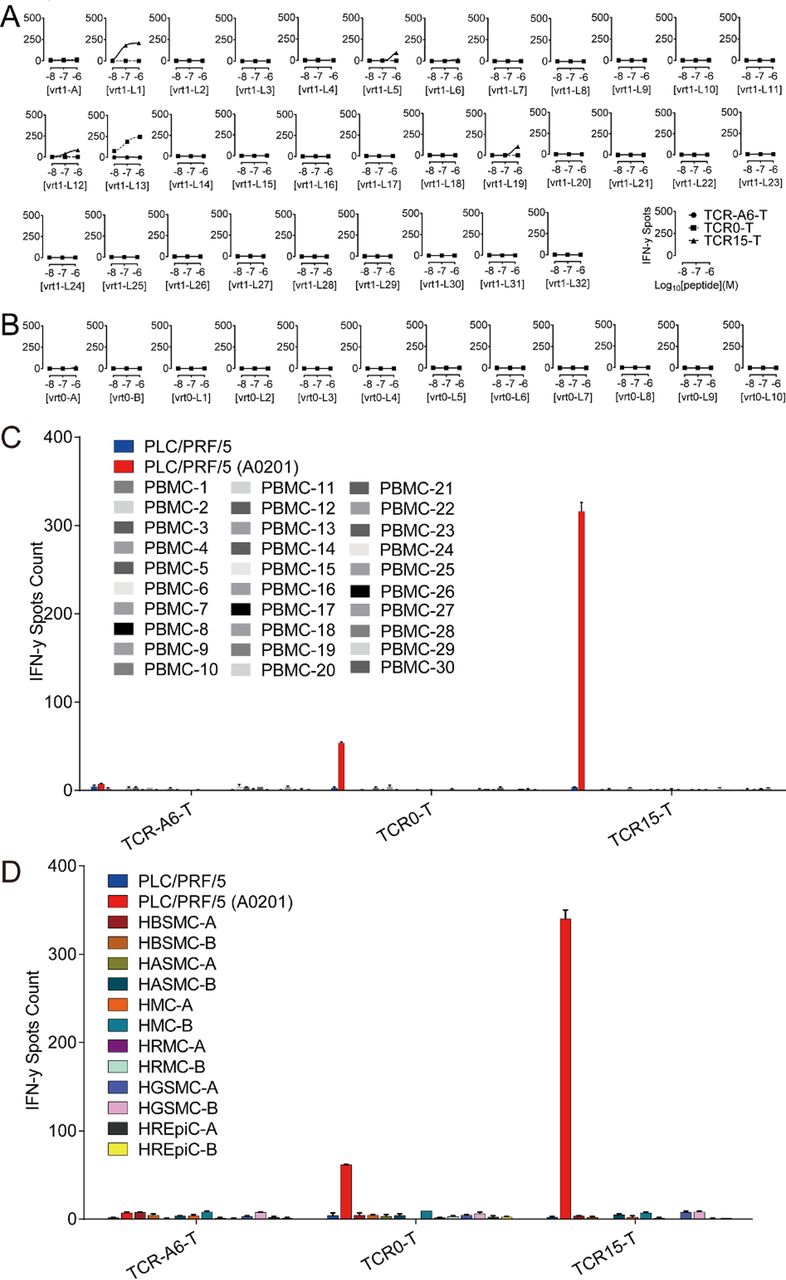
Verification of potentially cross-reactive human peptide antigens of vrt1 and vrt0, and interactions between TCR-T cells and PBMCs or human normal primary cells. After coculturing TCR-T cells with potentially cross-reactive human peptides (10−6 to 10−8 M)-pulsed T2 cells, PBMC and human normal primary cells in triplicate overnight at an E:T ratio of 1:10, IFN-γ release was detected. There were 33 peptides related to vrt1 (A),12 peptides related to vrt0 (B), 30 healthy PBMC sets (C) and 12 human normal primary cell sets (D). The negative and the positive controls were PLC/PRF/5 cells and HLA-A*02:01+ PLC/PRF/5 cells, respectively. E:T, effector-to-target; HASMC, human aortic smooth muscle cells; HBSMC, human bronchial smooth muscle cells; HGSMC, human gastric smooth muscle cells; HLA, human leukocyte antigen; HMC, human meningeal cells; HREpiC, human renal epithelial cells; HRMC, human renal mesangial cells; IFN-γ, interferon gamma; PBMCs, peripheral blood mononuclear cells; TCR, T-cell receptor; TCR-T, TCR-engineered T cell.
To verify TCR-T cell specificity with primary cells, TCR15-T and TCR0-T cells were tested and showed no activation by any PBMCs from 30 different healthy donors (figure 3C, online supplemental table S4). Further verification of the TCR15-T cell interactions with antigens presented by normal cells was carried out with 12 sets of human primary cells (online supplemental table S5). These normal cells could not activate TCR0-T or TCR15-T cells (figure 3D).
In summary, these results demonstrated that TCR15-T cells showed HBsAg specificity similar to that of WT-TCR-T cells and at least an equivalent safety profile under a range of measurements.
TCR15-T cells had improved recognition of HBs370 and HBs371 variants
The specificity of flexi-panning-generated TCRs was measured with HBs371 and HBs370 variants (online supplemental figure S1, tables S1.1 and S1.2). Approximately 5855 unique full-length HBsAg protein sequences were collected from UniProt, and 16 of the most frequent HBs371- and HBs370-variant peptides represented approximately 90% of the sequences, that is, vrt1 to vrt8 were nonamer HBs371 variants, and vrt0, vrt9 to vrt16 were decamer HBs370 variants. Results for IFN-γ (figure 4A,B) and LDH release (figure 4C,D) showed that both WT and affinity-improved TCRs were able to endow T cells with the ability to recognize 11 of the 16 variants, excluding vrt4, vrt7, vrt8, vrt13 and vrt16. Epitope sequence analysis of these five non-stimulating peptides indicated that the Ser at position three in the nonapeptide and position four in the decapeptide would be the key in the epitope. Ser is allowed at this position, while Asn or Arg will abolish the binding to the TCR. Additionally, comparing EC50 values made it very clear that the variant-mediated activation and cytotoxicity of TCR15-T cells were better than those of TCR0-T cells. This was consistent with the TCR affinity improvement (online supplemental table S7). Especially for vrt6, TCR15-T cells showed over 1000-fold enhancement than TCR0-T cells, which indicated no control of infection with HBV containing vrt6 variant. In conclusion, TCR15-T cells exhibited enhanced killing of T2 cells presenting the 11 most frequent variant peptides.

Determination of TCR-T cell activation by and cytotoxicity to T2 cells pulsed with viral peptide variants. T2 cells pulsed with an HBsAg epitope variant peptide (10−6 to 10−13 M) were incubated with TCR-T cells overnight at an E:T ratio of 1:10. IFN-γ release was detected for vrt1-8 (A) and vrt0, vrt9-16 (B) with an ELISPOT kit. Measuring LDH release showed the percentages of specific lysis of vrt1-8 (C) and vrt0, vrt9-16 (D) (10−6 to 10−13 M) pulsed T2 cell, which were incubated with TCR0-T or TCR15-T cells at an E:T ratio of 1:1 overnight. E:T, effector-to-target; HBsAg, HBV surface antigen; HBV, hepatitis B virus; IFN-γ, interferon gamma; LDH, lactate dehydrogenase; TCR,T-cell receptor; TCR-T, TCR-engineered T cell.
TCR15-T cells could eradicate HCC xenograft tumors
To verify whether improving TCR affinity affects in vivo tumor suppression, TCR0-T and TCR15-T cells were evaluated in xenograft mouse models established with HLA-A*02:01+ PLC/PRF/5 HCC cells, which expressed the HBs371 nonapeptide antigen vrt2 and HBs370 decapeptide antigen vrt10 (confirmed with reverse transcription-PCR, data not show) as the agonist antigens for both TCRs (figure 4). The mice bearing tumors were injected with TCR-T cells on day 13 (figure 5A). All tumors volume enlarged on the third day after the injection; however, on the ninth day, they showed different changes. The full-dose of TCR0-T cells (3.0×107) produced only slight tumor-suppressive effects that were similar to those found in the mice receiving the low-dose of TCR15-T cells (3.3×106). However, the medium-dose of TCR15-T cells (1.0×107) produced significant tumor suppression. The mice in this group had only residual tumors detected on day 25, despite the tumors increased to approximately 107 mm3 on day 34. On the other hand, more than 50% (7/14) and 85% (12/14) of the mice that received the full-dose of TCR15-T cells (3.0×107) showed no detectable tumors on day 23 and day 25, respectively. All tumors were eliminated by day 28 in the full-dose group, and no relapse occurred during an additional 17 days of monitoring. Thus, TCR15-T cells demonstrated superior efficacy and dose-dependent tumor suppression (figure 5B–E). These results confirmed that Ai-TCR-T cells (TCR15-T cells) had a significant advantage over WT-TCR-T cells (TCR0-T cells) in the suppression and elimination of HCC in vivo.

TCR15-T cells could eradicate HBV-related HCC in xenograft NSI mouse models. (A) Experimental setup. Tumor growth curves of individual mice grouped by treatment with (B) TCR0-T cells (3.0×107) or (C) TCR15-T cells (3.0×107), mice (n=14). (D) Mean tumor growth curves of six groups. Tumor volume was calculated every 3 days with the formula (length×width2 /2. PBS, 3.0×107 TCR-A6-T cells, 3.3×106 or 1.0×107 TCR15-T cells, mice (n=5). (E) Statistical comparison of tumor weights among the xenograft model groups. The pooled results of three independent experiments are shown. Error bars denote the SEM, and groups were compared with a two-sided, unpaired t-test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. HBV, hepatitis B virus; HCC, hepatocellular carcinoma; i.v., intravenous; PBL, peripheral blood lymphocyte; PBS, phosphate-buffered solution; s.c., subcutaneous; TCR, T-cell receptor; TCR-T, TCR-engineered T cell.
TCR15-T cells efficiently infiltrated tumors
Human CD3+ and CD8+ T cell infiltration was detected in tumor sites by IHC. Examining H&E-stained (figure 6A) and IHC (figure 6B,C) samples revealed that significant CD3+ and CD8+ T cell staining was observed in the tumor sections from mice treated with 3.3×106 (CD3+:~25.9% and CD8+:~16.8%) or 1.0×107 (CD3+:~57.1% and CD8+:~38.6%) TCR15-T cells. In contrast, the positive rates for CD3+ and CD8+ T cell populations detected in tumors were only ~12.1% and~4.1%, respectively, for mice receiving 3-fold or ~10-fold more TCR0-T cells (figure 6D). It was obvious that elevated TCR15-T cell accumulation occurred in a dose-dependent manner. Furthermore, we found it intriguing that the percent of CD8+ cells in the CD3+ cells population was significantly different between the groups. Only 35% of the human T cells detected in the tumor sections of the mice infused with 3.0×107 TCR0-T cells were CD8+ cells. On the other hand, 66% or 72% of the human T cells detected in the tumors of the mice infused with 3.3×106 or 1.0×107 TCR15-T cells, respectively, were CD8+ cells (figure 6E). However, the initial CD8+ percentages of all tested TCR-T cells, including TCR-A6-T cells, were very similar, with an approximate average of 65.4% (online supplemental figure S5). The increased CD8+ populations were consistent with the tumor reduction data for the models. We concluded that a large quantity of CD8+ TCR15-T cells accumulated in tumor sites.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Detection of human T cells in xenograft tumors by H&E and IHC. Tissue sections of xenograft tumors stained with H&E or antibodies; the tumors were harvested from animals treated with various reagents including PBS, 3.0×107 TCR-A6-T cells, 3.0×107 TCR0-T cells, and 3.3×106 or 1.0×107 TCR15-T cells. Representative images were acquired at 100× (scale bar, 300 µm) and 400×magnification (scale bar, 90 µm). (A) H&E staining showing classic HCC morphology. (B) Immunohistochemical staining with anti-CD3 and (C) anti-CD8 antibodies, with positive cells colored brown. (D) Percentages of CD3+ and CD8+ T cells in tumor sections. Average positive cells were calculated from six fields at 400×magnification (online supplemental table S8). (E) The percentage of CD8+ cells in the human T cell population detected in the sections. HCC, hepatocellular carcinoma; IHC, immunohistochemistry; PBS, phosphate-buffered solution; TCR, T-cell receptor; TCR-T, TCR-engineered T cell.
Discussion
Treating HBV-related HCC has been a major challenge in the clinic. There are many ways for HCC cells to evade available treatments. One of the fundamental problems is immune escape, which ultimately hinders HCC eradication. In particular, poorly differentiated cells can effectively escape by downregulating antigen presentation to avoid CTL attacks, so strategies are needed to address this issue. Immunotherapies have been considered for HCC, HBV antigen-specific TCR-T12 and AFP-specific TCR-T therapy40 41 are ongoing trials to address these clinic needs. We consider HBV-specific TCR-T can not only treat HCC, may also be used to treat HBV hepatitis with careful manipulated clinic practices.
Although the downregulation of pHLA presentation in tumor cells is well documented in many cancers,42–45 whether HCC cells present pHLAs at sufficient levels to activate a CTL response is still under debate.46–49 Recent studies by Dr Bertoletti’s team demonstrated that HBV antigens could be presented by HCC cells despite these antigens not being detectable by IHC techniques.12 In the current study, we demonstrated that an affinity-improved HBsAg-specific TCR could enable T cells to eliminate HCC completely in xenograft mouse models and prevent recurrence. Because high-avidity natural T cells are common for recognition of a viral antigen, with modest affinity enhancement over the HBsAg specific WT-TCR, TCR15 was sufficient to produce T cells with significantly improved potency that could eliminate HCC in mice. As the HBs-specific TCR-Ts are able to target HBV epitopes expressed on HBV-infected hepatocytes in addition to HCC cells, to avoid liver failure, HBs-specific TCR-Ts are suggested to treat or prevent HCC relapse in liver transplanted patients.
In an immune synapse, the threshold for triggering the T cell is determined by the available number of agonist ligands interacting with the TCR cluster formed at the T-cell-target-cell junction.50 Every TCR cluster unit requires two or four agonist pMHCs for reaching the activation threshold,50 51 which is a requirement that evolved to balance central tolerance towards self-antigens with wild-type TCRs possessing relatively low affinities for pMHCs (KD in the range of 1 to 100 µM).21 The EC50 of TCR0-T cell activation by vrt1-pulsed T2 cells was approximately 6.3 nM (online supplemental figure S2), and this result indicated that effective activation of this TCR might need 50 to 100 pHLAs presented per cell,52 which could be reduced further during the cancer development. It offers a chance for HCC to escape attacks by TCR0-T cells. However, it would be difficult for antigen downregulation to enable tumor cells escape from attacks by TCR15-T cells, as these cells had observed approximately 20-fold activation and 40-fold killing enhancements verified in the EC50 (online supplemental figure S2). It is intriguing that the infiltrated the per cent of CD8+ cells in the CD3+ cells population were ~1/3 for TCR0-T cell-treated animals and ~2/3 for TCR15-T cell-treated animals (figure 6D,E). This may imply that an appropriate TCR-pHLA interaction strength will assist T cell tumor homing and activation. These results also suggested that Ai-TCR-T cells, especially CD8+ cells, infiltrated better than low-affinity WT-TCR-T cells in vivo. The efficient expansion of CD8+ cells in mice resembled observations in clinical investigations with Ai-TCR-T cells targeting the NY-ESO-1 antigen (Yi Li, unpublished data). However, the relation between CD8+ T cell infiltration and tumor elimination is still unclear in the current study. Despite almost fourfold more CD8+ T cells detected with Ai-TCR-T cell treatment than with TCR0-T cell treatment, there was no significant improvement in tumor suppression in the mice infused with the low-dose of TCR15-T cells compared with those infused with TCR0-T cells (figures 5D,E and 6D).
It was interesting that the cured mice showed no recurrence during an additional 11-day monitoring after day 34 (figure 5). Another experiment showed no tumor detected on day 14 (more than required days to form detectable tumor mass) after re-inoculation of TCR15-T cell-cured mice with 3.0×106 HLA-A*02:01+ PLC/PRF/5 tumor cells (data not shown). This might indicate that the human T cells were well maintained during this period.
There are nine HBV genotypes identified, and genotypes A and C are the dominant variants in Europe and China, respectively. HBV genotype C has the highest risk of HCC conversion.32–35 53 To generate affinity-improved TCRs for the genotype C surface antigen, the dominant HBV genotype B/C/D HBs371-379 nonamer vrt1-pHLA was used as the ligand. TCR15 matched the specificity of TCR0, and could cross-react with 11 main HBV antigen variants but not with other potentially cross-reactive human peptides or irrelevant peptides. Based on the epitope frequencies of HBs371-379 and HBs370-379, TCR15 could recognize more than 85% of HBV variants. These results might help broaden the clinical use of TCR15-T cells.
TCR promiscuity is considered a mechanism for a relatively limited TCR repertoire to deal with the vast diversity of antigenic peptide repertoire. However, when the TCR is modified for affinity enhancement, T cells may show on-target and/or off-target toxicity.54 Extensive screening strategies are necessary to assess the specificity and safety of the TCR-T. In current study, we have used irrelevant peptides, alanine scanning, potentially cross-reactive human peptides, HBV-variant peptides, PBMC and human normal primary cells to verify the specificity and safety of TCR15-T. However, further verification should be in place for the safety of TCR15-T during preclinical study.55 56
Conclusion
In summary, the in vitro and in vivo studies demonstrate HBsAg-specific Ai-TCR-T with significantly enhanced potency of HCC treatment, and also showed good safety profiles. We found that Ai-TCR-T were safe with no activity against human normal primary cells, and had good potency showing effective tumor infiltration and tumor clearance in mouse xenograft models. In addition, flexi-panning can generate TCR-T cells that cover several HBV antigen variants, which should facilitate the development of TCR-T cell therapy for HCC in a larger population. This is the first example using affinity-improved-TCR to redirect HBV specific T cells for addressing the problem of HBV-related HCC. Our studies will provide supports for clinical investigations of the Ai-TCR-T for treating HBV associated HCC.
Acknowledgments
We acknowledge the advice of Dr Lin Chen and Dr Hongjun Zheng for protein sample assistance. We thank Dr Zhong Su for providing valuable proofreading of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Contributors QL conceived the study, designed experiment, performed experiments, analyzed data, and wrote the manuscript. YL directed the study and revised the manuscript. YT and YJL assisted with safety testing experiment and provided feedback. YYL, YJL, ZL, PZ and YZ assisted with cell and animal experiments and provided feedback. WZ, YT and YR assisted with hepatitis B virus variants analysis and provided feedback. WC assisted with molecular and biochemical experiment, and provided feedback. YB provided expertise and feedback.
Funding The National Key R&D Program, 2016YFC1303404; the Science and Technology Program of Guangzhou, 201704020220; the Frontier Research Program of the Guangzhou Regenerative Medicine and Health Guangdong Laboratory, 2018GZR110105010; Project SKLRD-QN-201915 supported by the State Key Laboratory of Respiratory Disease; Project SKLRD-MS-201902 supported by the State Key Laboratory of Respiratory Disease; and the Project of Guangdong Province, 2019A1515010235.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included in the article or uploaded as supplementary information. The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.