Article Text

Abstract
Background Children with recurrent and/or metastatic osteosarcoma (OS), neuroblastoma (NB) and glioblastoma multiforme (GBM) have a dismal event-free survival (<25%). The majority of these solid tumors highly express GD2. Dinutuximab, an anti-GD2 monoclonal antibody, significantly improved event-free survival in children with GD2+ NB post autologous stem cell transplantation and enhanced natural killer (NK) cell-mediated antibody-dependent cell cytotoxicity. Thus, approaches to increase NK cell number and activity, improve persistence and trafficking, and enhance tumor targeting may further improve the clinical benefit of dinutuximab. N-803 is a superagonist of an interleukin-15 (IL-15) variant bound to an IL-15 receptor alpha Su-Fc fusion with enhanced biological activity.
Methods The anti-tumor combinatorial effects of N-803, dinutuximab and ex vivo expanded peripheral blood NK cells (exPBNK) were performed in vitro using cytoxicity assays against GD2+ OS, NB and GBM cells. Perforin and interferon (IFN)-γ levels were measured by ELISA assays. Multiple cytokines/chemokines/growth factors released were measured by multiplex assays. Human OS, GBM or NB xenografted NOD/SCID/IL2rγnull (NSG) mice were used to investigate the anti-tumor combinatorial effects in vivo.
Results N-803 increased the viability and proliferation of exPBNK. The increased viability and proliferation are associated with increased phosphorylation of Stat3, Stat5, AKT, p38MAPK and the expression of NK activating receptors. The combination of dinutuximab and N-803 significantly enhanced in vitro cytotoxicity of exPBNK with enhanced perforin and IFN-γ release against OS, GBM and NB. The combination of exPBNK+N-803+dinutuximab significantly reduced the secretion of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), platelet-derived growth factor-BB (PDGF-BB), and stem cell growth factor beta (SCGF-β) from OS or GBM tumor cells. Furthermore, OS or GBM significantly inhibited the secretion of regulated on activation, normal T cell expressed and presumably secreted (RANTES) and stromal cell-derived factor-1 alpha (SDF-1α) from exPBNK cells (p<0.001) but significantly enhanced monokine induced by gamma interferon (MIG) secretion from exPBNK cells (p<0.001). N-803 combined with dinutuximab and exPBNK cells significantly extended the survival of OS, GBM or NB xenografted NSG mice.
Conclusions Our results provide the rationale for the development of a clinical trial of N-803 in combination with dinutuximab and ex vivo exPBNK cells in patients with recurrent or metastatic GD2+ solid tumors.
- immunotherapy
- killer cells
- natural
- sarcoma
- neuroblastoma
- brain neoplasms
- osteosarcoma
- glioblastoma
- dinutuximab
- expanded natural killer cells
- targeted immunotherapy
- ADCC
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunotherapy
- killer cells
- natural
- sarcoma
- neuroblastoma
- brain neoplasms
- osteosarcoma
- glioblastoma
- dinutuximab
- expanded natural killer cells
- targeted immunotherapy
- ADCC
Background
Osteosarcoma (OS) is the most common primary bone tumor in children, adolescents, and young adults.1 While the 5-year event free survival (EFS) in patients with localized OS remains around 70%–75%, for patients with metastatic disease at diagnosis and those with progressive or relapsed disease, the prognosis is dismal with <20% EFS.2 Among high-grade gliomas, childhood glioblastoma multiforme (GBM) is the most aggressive and patients’ survival is only 14.6 months, despite multimodal therapy with debulking surgery, concurrent chemotherapy and radiotherapy.3 Pediatric neuroblastoma (NB) is the most common extracranial solid tumor in children, with approximately 800 new cases diagnosed in the USA in 2015.4 Forty-five percent of children with NB have high-risk tumors at diagnosis, for which the 5-year EFS remains <50% despite combination therapy with myeloablative chemotherapy, radiotherapy, stem cell transplantation, isotretinoin, and anti-GD2 antibody immunotherapy.5 6 The prognosis for those with high-risk disease at diagnosis and those who relapse is dismal with <20% 5-year EFS.7 8 Therefore, novel therapies including combinatorial immunotherapy are desperately needed for patients with relapsed or metastatic/refractory OS, GBM and NB.
GD2 is highly expressed on neuroectoderm-derived pediatric tumors and sarcomas, including NB, OS, rhabdomyosarcoma, and Ewing sarcoma.9 10 Dinutuximab is a GD2-binding monoclonal antibody used in combination with granulocyte macrophage colony stimulating factor, interleukin-2 (IL-2) and isotretinoin, for the treatment of pediatric patients with high-risk NB5 and is undergoing investigation in patients with relapsed OS11 (NCT02484443).
Natural killer (NK) cells are an attractive candidate as a cellular therapy approach in patients with a variety of malignancies.12 Unlike T cells, NK cells kill tumor cells in a major histocompatibility complex independent manner without the need for prior sensitization.13 NK cells are easily isolated, expanded ex vivo and can be made available as an off-the-shelf allogeneic product for immediate clinical use in adoptive or autologous cell therapies.12 The barriers to NK cells therapy include small numbers of active circulating NK cells, poor persistence, lack of specific tumor targeting, exhaustion, inhibitory receptor induced inhibition, and poor trafficking and tumor infiltration.12 14 Our group and others have successfully expanded active NK cells in vitro by short-term culture with cytokines alone and coculture with engineered feeder cells.15 16 We have demonstrated that expanded peripheral blood NK cell (exPBNK) targeting specificity can be enhanced by engineering exPBNK cells to express chimeric antigen receptors (CAR) such as an anti-CD20 CAR against CD20+ B cell non-Hodgkin’s lymphoma.17 18
N-803 (formerly known as ALT-803) is a new interleukin-15 (IL-15) superagonist and was developed to increase NK persistence and activation in vivo.19 It consists of an IL-15 superagonist mutein (IL-15N72D) and a dimeric IL-15 receptor alpha (IL-15Rα)/Fc fusion protein (figure 1 (online supplemental file 1)). N-803 has 25 times greater in vivo activity and significantly longer serum half-life as compared with IL-15.20 N-803 is currently being investigated in clinical trials to treat patients with myeloma, melanoma and relapsed hematological malignancies, and is well tolerated, and has no dose-limiting toxicity.21 The efficacy of exPBNK in combination with N-803 and dinutuximab against GD2+ OS, GBM, and NB has not yet been investigated. We, therefore, investigated the combination of N-803, dinutuximab and exPBNK cells against GD2+ OS, GBM, and NB both in vitro and in vivo and the mechanism associated with this combinatorial immunotherapy.
Supplemental material
Materials and methods
Cell lines and reagents
U2OS (OS), M059K (GBM) and SKNFI (NB) cell lines were purchased from the American Type Culture Collection, Gaithersburg, Maryland. K526-mbIL21-41BBL cells were generously provided by Dean A. Lee, MD/PhD from Nationwide Children’s Hospital, Columbus, Ohio.22 Dinutuximab was generously provided by United Therapeutics, Silver Springs, Maryland. N-803 was generously provided by Hing Wong, PhD, Peter R. Rhode, PhD, John H. Lee, MD, and Jeffrey T. Safrit, PhD from ImmunityBio/Altor Bioscience, Culver City, California. Leukocytes were obtained after informed consent from healthy donors at the New York Blood Center, New York, New York. Peripheral blood mononuclear cells (PBMNCs) were obtained by Ficoll gradient (Amersham Biosciences, Piscataway, New Jersey, USA) separation as we previously described.17 U2OS, M059K and SKNFI cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) and antibiotics penicillin and streptomycin (100 µg/mL). K526-mbIL21-41BBL cells were cultured in complete medium (RPMI1640 medium supplemented with 10% FBS and penicillin and streptomycin (100 µg/mL)). NK cells were cultured in complete medium with 50 IU IL-2.
NK cell expansion
PBMNCs were stimulated with irradiated genetically modified K562-mbIL21 - 41BBL cells for 2 weeks as we previously described.22 Expanded PBNK cells were isolated by negative selection using Miltenyi NK cell isolation kit (Miltenyi Biotec, Cambridge, Massachusetts) as we have previously described.17
Bioluminescence based in vitro cytotoxicity
Bioluminescence (BLI) based in vitro cytotoxicity assays were performed as previously described with minor modification.23 Luciferase-expressing tumor cells were placed in 96–well flat bottom plates at a concentration of 3×105 cells/mL. Subsequently, effector cells were added at different effector-to-target (E:T) ratios with or without N-803 and dinutuximab and incubated at 37°C for 2–3 days. After incubation, the samples in the plates were spun and 75 µg/mL D-firefly luciferin potassium salt (PerkinElmer, Massachusetts, USA) with fresh media added to the cell pellets in each well. BLI was measured with a luminometer (Molecular Devices Multifilter F5 plate reader) as relative light units (RLU). Cells were treated with 1% Triton X-100 as a measure of maximal killing. Target cells incubated without effector cells were used to measure spontaneous death RLU. Percent lysis was calculated from the data with the following equation: % specific lysis = 100×(spontaneous death RLU – test RLU)/(spontaneous death RLU – maximal killing RLU). All tests were run in quadruplicate.
MTS assays
PBMNCs were stimulated with irradiated genetically modified K562-mbIL21 - 41BBL cells for 2–3 weeks. The same number of purified exPBNK cells were cultured in medium with 0.35 ng/mL (low) or 3.5 ng/mL (high) N-803, the same molar dose of immunoglobulin G (IgG), or medium only for 3 days. CellTiter 96 AQueous one solution cell proliferation assay (Promega, Madison, Wisconsin, USA)18 was used to determine the number of proliferating viable cells following the manufacturer’s instructions. Briefly, 104 NK cells/well were seeded in culture medium containing N-803, IgG or medium at 37°C and 5% CO2. At the end of 3 days incubation, CellTiter 96 AQueous one solution was added to each well and cells were incubated for 4 hours at 37°C and 5% CO2. Finally, spectrophotometrical absorbance was measured using a multifilter plate reader (Molecular Device, San Jose, California, USA) at OD490.
Flow cytometry analysis of intracellular proteins and phosphoproteins
Intracellular proteins and phosphoproteins were measured as we have previous described.18 Fixed and permeabilized cells were stained with fluorescent-dye conjugated anti-human antibodies: phospho-p38 MAPK-PE (ebioscience, #12-9078-41), phospho-Akt1-APC (ebioscience, #17-9715-41), phospho-Stat3-FITC (ebioscience, #11-9033-41), or phospho-Stat5-PE (ebioscience, #12-9033-41). Cells were analyzed using MACSQuant Analyzer (Miltenyi Biotec Cambridge, Massachusetts, USA). No stain, or isotype controls were used for gating.
Bio-Plex Pro human cytokines screening
Cell culture supernatants were collected after 3 days culture and were stored at −80 °C. The concentrations of cytokines/chemokines/growth factors were measured by the Bio-Plex Pro Human cytokines screening panel 48 cytokines assay (Bio-Rad Laboratories, Hercules, California, USA) according to the manufacturer’s instructions. In brief, 50 µL aliquot of sample was diluted 1:4 with sample diluent, incubated with antibody-coupled beads, biotinylated secondary antibodies, and followed by streptavidin-phycoerythrin. The beads were read on a Luminex System (Bioplex 200, Bio-Rad) and the data were analyzed using Bioplex Manager Software.
Enzyme-linked immunosorbent assay (ELISA)
Platelet-derived growth factor (PDGF)-AA (Raybiotech, # ELH-PDGFAA-1), PDGF-BB (Raybiotech, # ELH-PDGFBB-1), interferon (IFN)-γ (eBioscience, # KHC4021), and Perforin (Abcam, # ab46068) concentrations were analyzed by ELISA according to the manufacturer’s instructions. Briefly, recombinant standards were run with serial dilutions. Cell culture supernatants were diluted at 1:1 or 1:4 with assay diluent. 100 uL of diluted samples and standard were added to microwells simultaneously and incubated for 2–2.5 hours at room temperature. Biotin conjugated anti-human PDGF-AA, PDGF-BB, IFN-γ, or perforin antibody was used and incubated for 1 hour at room temperature. After washing, streptavidin-HRP solution was added for 30 min at room temperature. ELISA plates were developed with 100 uL TMB substrate reagents. TMB Stop Solution was added to halt the reaction. The absorbance at 450 nm was measured on a Molecular Devices Multifilter F5 plate reader.
Flow cytometry-based phenotyping of NK activating and inhibitory receptors
The exPBNK cells under different conditions were analyzed for phenotypic expression of inhibitory NK receptors (CD94, NKG2A), inhibitory NK killer cell immunoglobulin-like receptors (KIR) (CD158a, CD158b, CD158e), activating NK KIR (KIR2DSA), activating C-lectin NK receptors (NKG2C, NKG2D), and activating natural cytotoxicity receptors (Nkp46, NKp30, NKp44) by flow cytometry as we have previous described.17 Cells were analyzed using MACSQuant Analyzer (Miltenyi Biotec, Cambridge, Massachusetts, USA). No stain, or isotype controls were used for gating.
Xenograft models
Six to eight weeks old NOD/SCID/γ-chain-/- (NSG) mice were purchased from the Jackson Laboratory (Bar Harbor, Maine). The experimental protocol was conducted in accordance with the recommendations of the Guide for Care and Use of Laboratory Animals with respect to restraint, husbandry, surgical procedures, feed and fluid regulation, and veterinary care. The animal care and use program at New York Medical College is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Luciferase expressing U2OS-Luc (OS), M059K-Luc (GBM) and SKNFI-Luc (NB) cells were generated as we have previously described.17 4×106 of U2OS-Luc, M059K-Luc cells, or SKNFI-Luc cells were subcutaneously injected in NSG mice on day 0. After confirming the tumor engraftment at day 7, 1×107 exPBNK cells+15 µg IgG, 1×107 exPBNK cells+15 µg dinutuximab, 1×107 exPBNK cells+0.2 mg/kg N-803, 1×107 exPBNK cells+15 µg dinutuximab +0.2 mg/kg N-803, 15 µg dinutuximab +0.2 mg/kg N-803, or phosphate-buffered saline (PBS) was intraperitoneally injected to each mouse. NK cells were administered once a week for 3 weeks and IgG, dinutuximab and N-803 were given twice a week for 6 weeks. Tumor engraftment and progression were evaluated using the Xenogen IVIS-200 system (PerkinElmer, Shelton, Connecticut) as we have previously described.17 Tumor size was estimated according to the following formula: tumor size (cm3)=length (cm) × width2 (cm) × 0.5. Mice were followed until death or sacrificed if any tumor size reached 2 cm3 or larger.
Statistical analyses
Statistical analyses were performed using the InStat statistical program (GraphPad, San Diego, California, USA). Average values were reported as the mean±SEM. Results were compared using the one-tailed unpaired Student’s t-test with p<0.05 considered as significant. Probability of survival in animal studies was determined by the Kaplan-Meier method using the Prism program V.8.0 (GraphPad Software).
Results
N-803 increased the viability and proliferation of exPBNK with enhanced p-Stat3, p-Stat5, pAkt, p-p38MAPK and NK activating receptors
Expanded PBNK cells (exPBNK) were generated by coculturing PBMNCs with irradiated genetically modified K562-mbIL21 - 41BBL feeder cells for 2 weeks and isolated by negative selection using Miltenyi NK cell isolation kit as we previously described.22 To investigate if N-803 stimulates exPBNK cells viability and proliferation as compared with the same molar dose of IgG, the purified exPBNK cells without any feeder cells were cultured in medium with 0.35 ng/mL (low) or 3.5 ng/mL (high) N-803 or molar equivalent dose of IgG for 3 or 7 days. The exPBNK cells with N-803 at 0.35 ng/mL or 3.5 ng/mL had significantly higher viability as compared with IgG or medium controls (p<0.001). Furthermore, N-803 at 3.5 ng/mL significantly stimulated the proliferation of exPBNK cells as compared with N-803 at 0.35 ng/mL at day 3 (figure 1A) (p<0.001) and day 7 (figure 2 (online supplemental file 1)) by MTS assays. We also observed that sustained viability and proliferation of exPBNK cells stimulated by N-803 at 3.5 ng/mL with morphological changes in NK cell shape, size, and number which correlate to NK proliferation and activation as compared with NK cells cultured with IgG (figure 1B). Consistent with enhanced exPBNK proliferation and similar to IL-15, N-803 at 3.5 ng/mL significantly enhanced the phosphorylation of Stat 3, AKT1 and p38MAPK as compared with IgG (p<0.01) at day 3 (figure 1C).
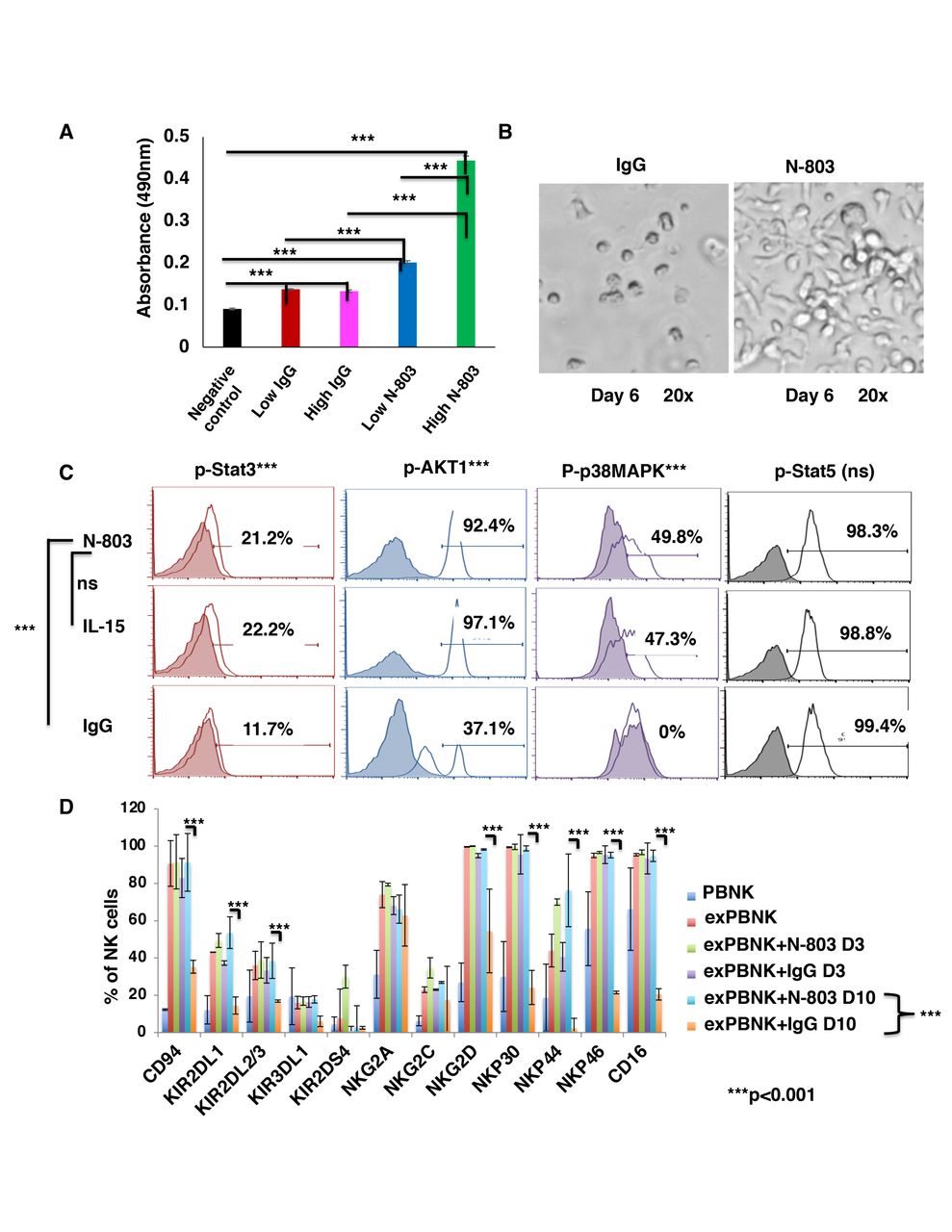
N-803 increased the viability and proliferation of exPBNK with enhanced p-Stat3, p-Stat5, pAKT, p-p38MAPK and NK activating receptors. PBMNCs were stimulated with irradiated genetically modified K562-mbIL21-41BBL cells for 2–3 weeks. (A) Purified exPBNK cells were cultured in complete medium with 0.35 ng/mL (low) or 3.5 ng/mL (high) N-803 or molar equivalent dose of IgG for 3 days. NK viability and proliferation were monitored by MTS assays. The amount of 490 nm absorbance is directly proportional to the number of living exPBNK cells in the culture. The exPBNK cells with N-803 at 0.35 ng/mL or 3.5 ng/mL have significantly higher viability as compared with IgG or medium controls (p<0.001) and N-803 at 3.5 ng/mL significantly stimulated the proliferation of exPBNK cells as compared with N-803 at 0.35 ng/mL (p<0.001). (B) ExPBNK cell phenotypic changes cultured in medium with IgG or N-803 under light microscopy (Axiovert 200M; Carl Zeiss) are shown at day 6 (original magnification 200 x). (C) Purified exPBNK cells were cultured in medium with 3.5 ng/mL N-803 or molar equivalent dose of IL-15 or IgG for 3 days. Intracellular phosphorylated STAT3 (p-Stat3), phosphorylated Akt1 (p-AKT1), phosphorylated p38MAPK (p-p38MAPK), and phosphorylated STAT5 (p-Stat5) were monitored by flow cytometry analysis. N-803 at 3.5 ng/mL significantly enhanced the phosphorylation of STAT 3, Akt1 and p38MAPK as compared with IgG (p<0.001) at day 3. (D) Purified exPBNK cells were cultured in medium with 3.5 ng/mL N-803 or molar equivalent dose of IL-15 or IgG for 3 days or 10 days. ExPBNK cells were stained with indicated monoclonal antibodies. The expression of receptors on viable exPBNK cells were compared by flow cytometry analysis. Purified non-expanded NK (PBNK) cells were used as controls. At day 10, the expression levels of NKG2D, NKp30, NKp44, NKp46 and CD16 were significantly enhanced in exPBNK with N-803 as compared with exPBNK with IgG. ***p<0.001, Data were presented as mean±SEM from three independent experiments. ExPBNK, expanded peripheral blood natural killer cell; IL-15, interleukin-15; ns, not significant; PBMNCs, peripheral blood mononuclear cells.
The expression of receptors on viable exPBNK cells was compared by flow cytometry analysis. Purified non-expanded NK (PBNK) cells were used as controls. N-803 at 3.5 ng/mL significantly enhanced the expression of NK activating receptors: NKG2D, NKp30, NKp44, NKp46 at day 10 as compared with IgG (p<0.001) (figure 1D). N-803 at 3.5 ng/mL also significantly enhanced the expression of CD16, known as FcγRIII, at day 10 as compared with IgG (p<0.001) (figure 1D), supporting in part the rationale for the combinatorial immunotherapy of monoclonal antibody with expanded NK cells and N-803.
The combination of dinutuximab and N-803 significantly enhanced in vitro cytotoxicity of exPBNK with enhanced perforin and IFN-γ release against OS, GBM and NB
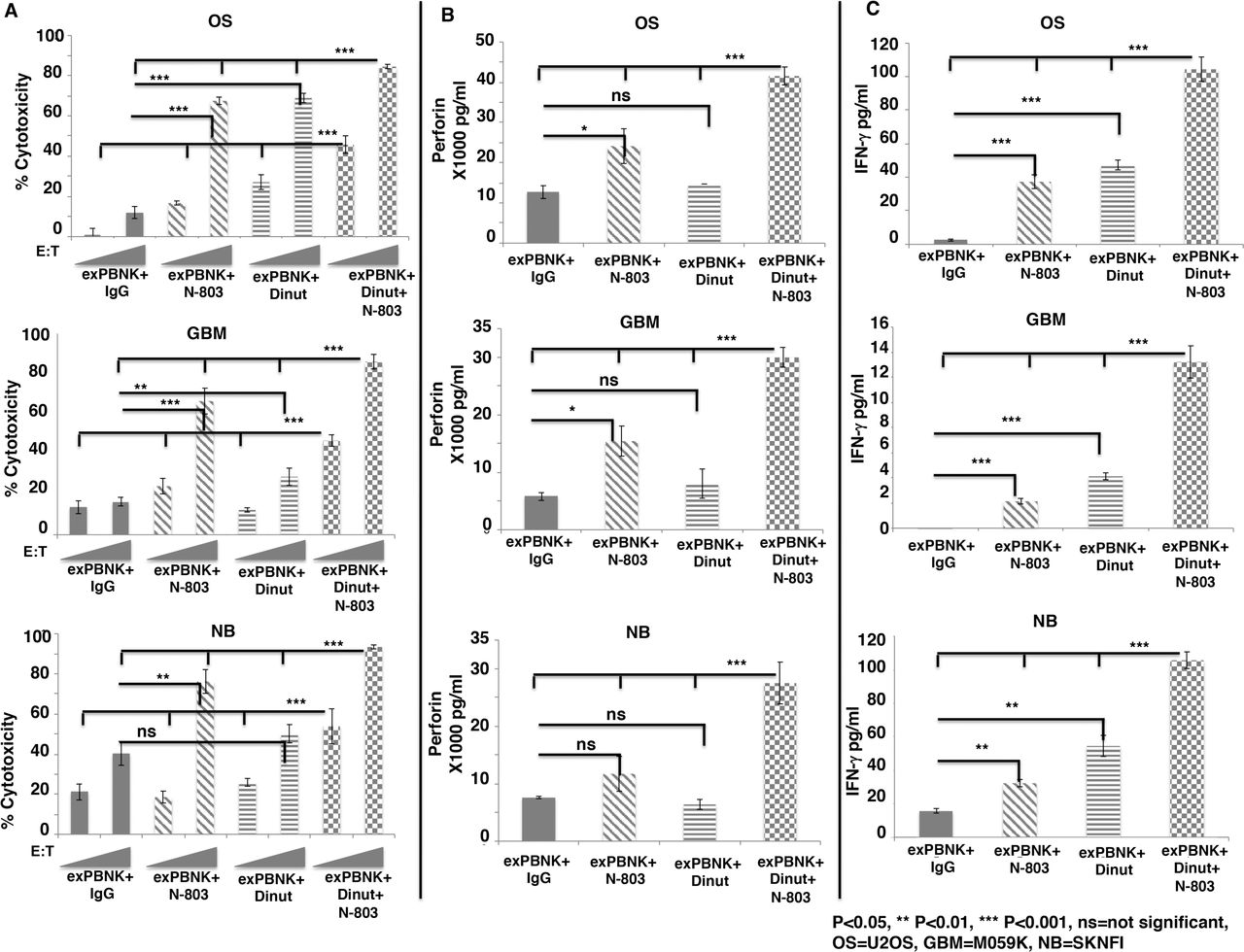
Since N-803 stimulated exPBNK cells to express high level of CD16 (figure 1D) and dinutuximab is an IgG1 type monoclonal antibody, we investigated whether the combination of N-803 and dinutuximab significantly stimulates the antibody dependent cellular cytotoxicity (ADCC) of exPBNK cells against OS, GBM, and NB. Tumor cell lines: U2OS, M059K, SKNFI cells express GD2 (figure 3 (online supplemental file 1)) and were treated with 1 µg/mL IgG +exPBNK, 3.5 ng/mL N-803 +exPBNK, 1 µg/mL dinutuximab +exPBNK or 3.5 ng/mL N-803 +1 µg/mL dinutuximab +exPBNK cells at E:T ratio=1:1 and 3:1. We found that the combination of N-803, dinutuximab and exPBNK significantly killed U2OS, M059K and SKNFI cells (p<0.001) as compared with other groups (figure 2A) in an E:T ratio dependent manner. The enhanced in vitro cytotoxicity of exPBNK cells was associated with significantly enhanced secretion of perforin (figure 2B) and IFN-γ (figure 2C) from exPBNK cells as compared with all other groups against U2OS, M059K and SKNFI cells (p<0.001) at E:T=3:1.

The combination of N-803 and dinutuximab significantly enhanced in vitro cytotoxicity of exPBNK with enhanced perforin and IFN-γ release against OS, NB and GBM cells. Expanded NK cells were isolated for invitro cytotoxicity assays against OS, NB and GBM cells. (A) The combination of exPBNK cells with N-803 +dinutiximab significantly killed U2OS (OS), M059K (GBM) and SKNFI (NB) cells as compared with exPBNK cells with single agent (IgG, N-803 or dinutuximab) at E:T ratios=1:1 or 3:1 at day 3. The ‘RAMPs’ stand for the E:T ratios at 1:1 and 3:1. (B) The combination of exPBNK cells with N-803+ dinutiximab significantly enhanced perforin release from exPBNK cells as compared with exPBNK cells with single agent (IgG, N-803 or dinutuximab) at E:T ratios=3:1. (C) The combination of exPBNK cells with N-803+ dinutiximab significantly enhanced IFN-γ release from exPBNK cells as compared with exPBNK cells with single agent (IgG, N-803 or dinutuximab) at E:T ratios=3:1. *P<0.05, **p<0.01, ***p<0.001, Dinut=dinutuximab. Data were presented as mean±SEM from four independent experiments. OS=U2OS cell line, GBM=M059K cell line, and NB=SKNFI cell line. E:T, effector-to-target; exPBNK, expanded peripheral blood natural killer cell; GBM, glioblastoma multiforme; IFN-γ, interferon-γ; NK, natural killer; ns, not significant; OS, osteosarcoma; NB, neuroblastoma.
Cytokines and growth factors screen of exPBNK cells against OS and GBM stimulated by N-803+ dinutuximab
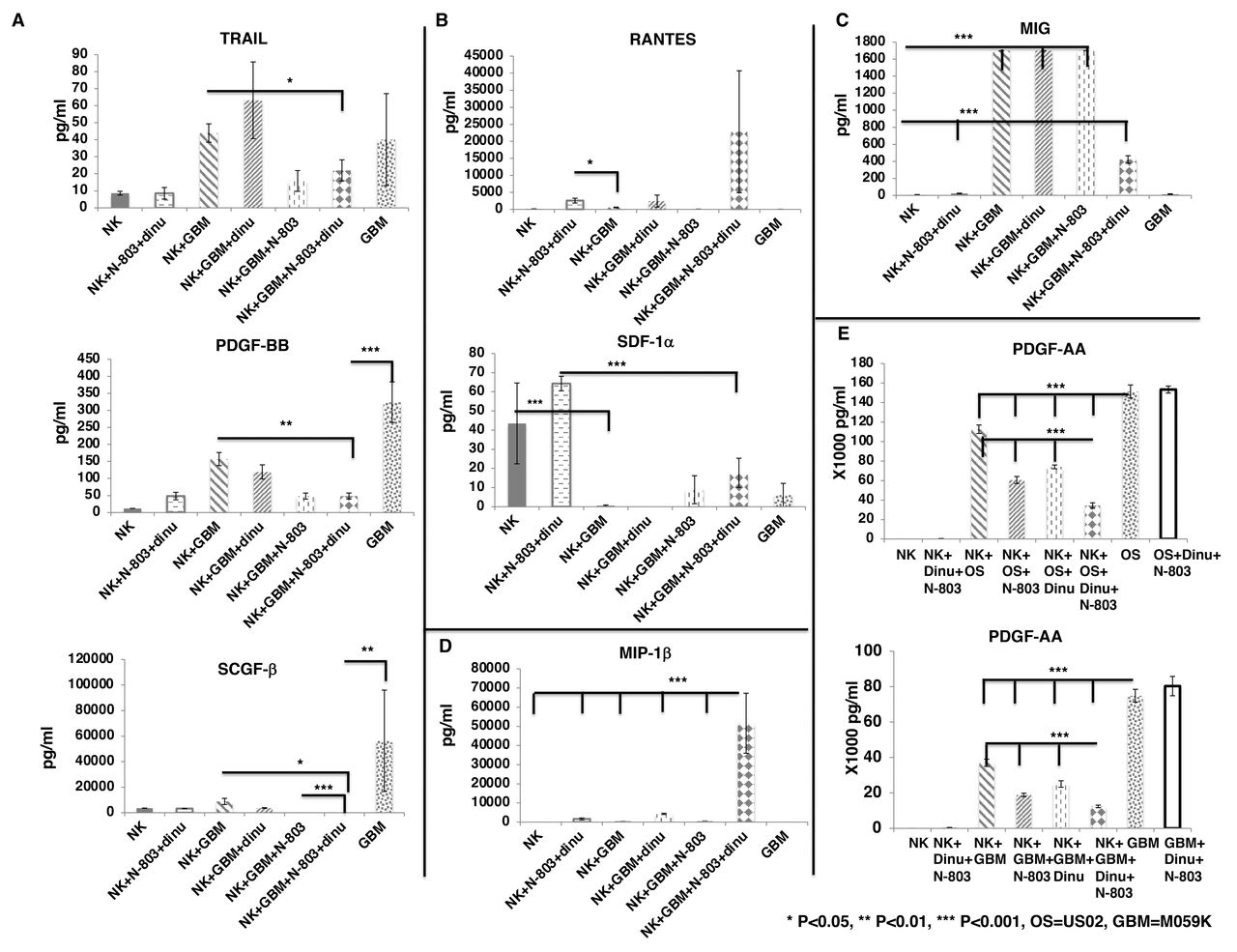
To investigate the cytokines and growth factors that are significantly secreted by exPBNK cells stimulated by N-803 and dinutuximab against tumor cells, exPBNK cells were cultured with N-803, dinutuximab or the combination of N-803 and dinutuximab with or without U2OS cells at E:T=3:1 for 3 days. The concentrations of cytokines/chemokines/growth factors in the supernatants were measured by the Bio-Plex Pro Human cytokines screening panel 48 cytokines assay. For the factors that are secreted by U2OS tumor cells such as tumor factor-related apoptosis-inducing ligand (TRAIL), platelet-derived growth factor-BB (PDGF-BB), and stem cell growth factor beta (SCGF-β), the combination of exPBNK +N-803+dinutuximab significantly reduced the secretion of these factors from U2OS tumor cells as compared with U2OS alone (p<0.001) and exPBNK +U2OS (p<0.01, p<0.05 and p<0.001, respectively) (figure 3A). For the factors that are secreted by exPBNK cells and can be further enhanced by N-803 and dinutuximab such as regulated on activation, normal T cell expressed and presumably secreted (RANTES) and stromal cell-derived factor-1 alpha (SDF-1α), U2OS significantly inhibited the secretion of these factors from exPBNK cells (p<0.001) (figure 3B). Monokine induced by gamma interferon (MIG) and interferon gamma-induced protein 10 (IP-10) are important ligands of CXCR3 that are pivotal for NK-cell migration towards tumor cells.24 We found that without U2OS tumor cells, exPBNK cells or exPBNK cells incubated with N-803 +dinutuximab do not secrete MIG. However, U2OS significantly increased the secretion of MIG and IP-10 from exPBNK cells (p<0.001) (figure 3C). Macrophage inflammatory proteins (MIP) 1 alpha and beta are members of the C-C motif subfamily of chemokines and both are ligands of C-C chemokine receptor type 5 receptor, which is essential for NK trafficking in host defense.25 We found that the combination of U2OS, N-803 and dinutuximab significantly enhanced the secretion of MIP-1beta from exPBNK cells (p<0.001) (figure 3D), while MIP-1alpha secretion by exPBNK cells was significantly enhanced by U2OS alone, N-803+ dinutuximab, or U2OS+N-803+dinutuximab (p<0.001) as compared with exPBNK cells (figure 3E).

Screening of cytokines and growth factors secreted from exPBNK regulated by OS cells with/without dinutuximab +N-803. Purified exPBNK cells were cultured in medium with U2OS (OS) tumor cells with/without IgG, dinutuximab, N-803, or dinutuximab+N-803 for 3 days. OS tumor cells cultured in medium were used as controls. The supernatants were collected after 3 days culture and used for Bio-Plex pro human cytokines screening panel 48 cytokines assay. (A) The combination of exPBNK+N-803+ dinutuximab significantly reduced the secretion of TRAIL, PDGF-BB, and SCGF-β from OS tumor cells (p<0.001). (B) OS significantly inhibited the secretion of RATNES and SDF-1α from exPBNK cells (p<0.001). (C) OS significantly enhanced MIG and IP-10 secreted from exPBNK cells (p<0.001). (D) The combination of OS, N-803 and dinutuximab significantly enhanced the secretion of MIP-1beta from exPBNK cells (p<0.001). (E) OS alone, N-803+ dinutuximab, or OS+N-803+dinutuximab significantly enhanced the secretion of MIP-1alpha from exPBNK cells (p<0.001). NK=exPBNK, Dinu=dinutuximab, OS=U2OS cell line. Data were presented as mean±SEM from three independent experiments. exPBNK, expanded peripheral blood natural killer cell; TRAIL, tumor factor-related apoptosis-inducing ligand; PDGF-BB, platelet-derived growth factor-BB; IP-10, interferon gamma-induced protein 10; MIG, Monokine induced by gamma interferon; MIP, macrophage inflammatory proteins; OS, osteosarcoma; RANTES, regulated uponon activation, normal T cell expressed and presumably secreted; SCGF, stem cell growth factor; SDF, stromal cell-derived factor.
We further confirmed our findings using M059K cells as tumor targets. We found that the combination of exPBNK +N-803+dinutuximab reduced the secretion of TRAIL (p<0.05), and significantly reduced the secretion of PDGF-BB (p<0.01), and SCGF-β (p<0.05) from M059K tumor cells as compared with exPBNK +M059K (figure 4A). M059K significantly inhibited the secretion of SDF-1α from exPBNK cells (p<0.001) (figure 4B) but significantly enhanced MIG secretion from exPBNK cells (p<0.001) (figure 4C). The combination of M059K, N-803 and dinutuximab significantly enhanced the secretion of MIP-1beta from exPBNK cells (p<0.001) as compared with all other groups (figure 4D).
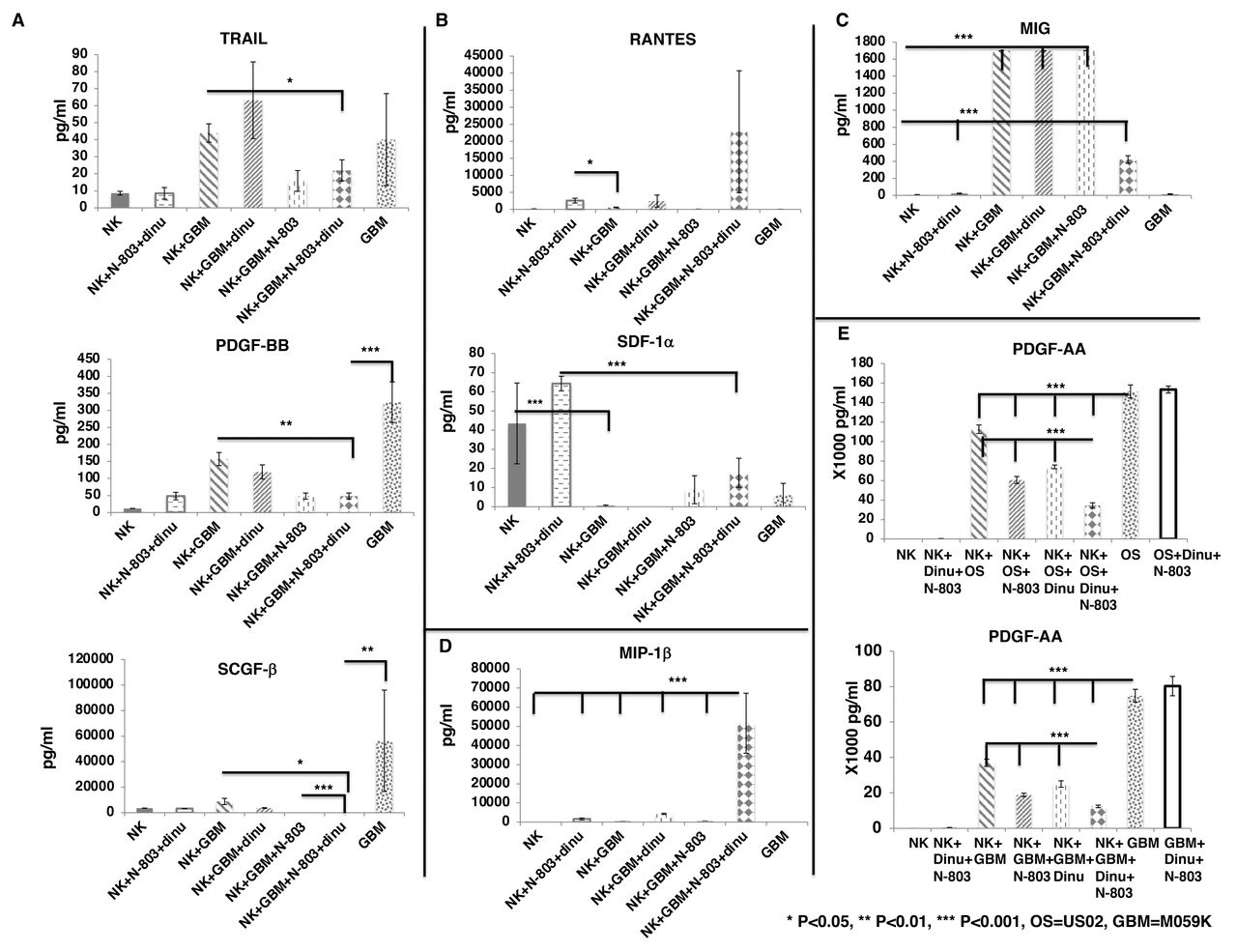
Screening of cytokines and growth factors secreted from exPBNK regulated by GBM cells with/without Dinutuximab +N-803. Purified exPBNK cells were cultured in medium with M059K (GBM) tumor cells with/without IgG, dinutuximab, N-803, or dinutuximab +N-803 for 3 days. GBM tumor cells cultured in medium were used as controls. The supernants were collected after 3 days culture and used for Bio-Plex pro human cytokines screening panel 48 cytokines assay. (A) The combination of exPBNK +N-803+ dinutuximab reduced the secretion of TRAIL, PDGF-BB, and SCGF-β from GBM tumor cells (p<0.001). (B) GBM significantly inhibited the secretion of RATNES and SDF-1α from exPBNK cells (p<0.001). (C) GBM significantly enhanced MIG secreted from exPBNK cells (p<0.001). (D) The combination of GBM, N-803 and dinutuximab significantly enhanced the secretion of MIP-1beta from exPBNK cells (p<0.001). (E) The combination of exPBNK+N-803+ dinutuximab significantly reduced the secretion of PDGF-AA from OS or GBM tumor cells (p<0.001). NK=exPBNK, Dinu=dinutuximab, OS=U2OS cell line, GBM=M059K cell line. Data were presented as mean±SEM from three independent experiments. GBM, glioblastoma multiforme; exPBNK, expanded peripheral blood natural killer cell; NK, natural killer; OS, osteosarcoma; TRAIL, tumor factor-related apoptosis-inducing ligand; PDGF, platelet-derived growth factor; RANTES, regulated uponon activation, normal T cell expressed and presumably secreted; SDF, stromal cell-derived factor; SCGF, stem cell growth factor; MIG, Monokine induced by gamma interferon; MIP, macrophage inflammatory proteins.
PDGF-BB is one of the five ligands of PDGF Receptor (PDGFR) α and PDGFRβ. PDGFRs and their ligands have been found to be overexpressed or mis-regulated in many cancers, such as gliomas and sarcomas26 and PDGFRs are also expressed on the non-cancerous cells of the tumor microenvironment to support the growth of cancer cells.27 These findings were similar when we assessed PDGF-AA that the combination of exPBNK +N-803+dinutuximab significantly reduced the secretion of PDGF-AA (p<0.001) from U2OS or M059K tumor cells as compared with the NK, NK +N-803, or NK +dinutuximab (figure 4E).
N-803 combined with dinutuximab and exPBNK cells significantly inhibited OS cells growth and extended the survival of OS xenografted NSG mice
To investigate if N-803 stimulates the proliferation of exPBNK in vivo and has anti-tumor effect with exPBNK cells, we generated luciferase expressing U2OS-Luc cells and xenografted U2OS-Luc cells to immunodeficient NSG mice.18 We found that the mice treated with exPBNK cells+N-803 have a significantly higher number of human NK cells as compared with the mice treated with exPBNK alone (figure 5A). Tumor burden was significantly reduced in mice treated with exPBNK cells+N-803 as compared with mice treated with exPBNK cells alone (figure 5B).
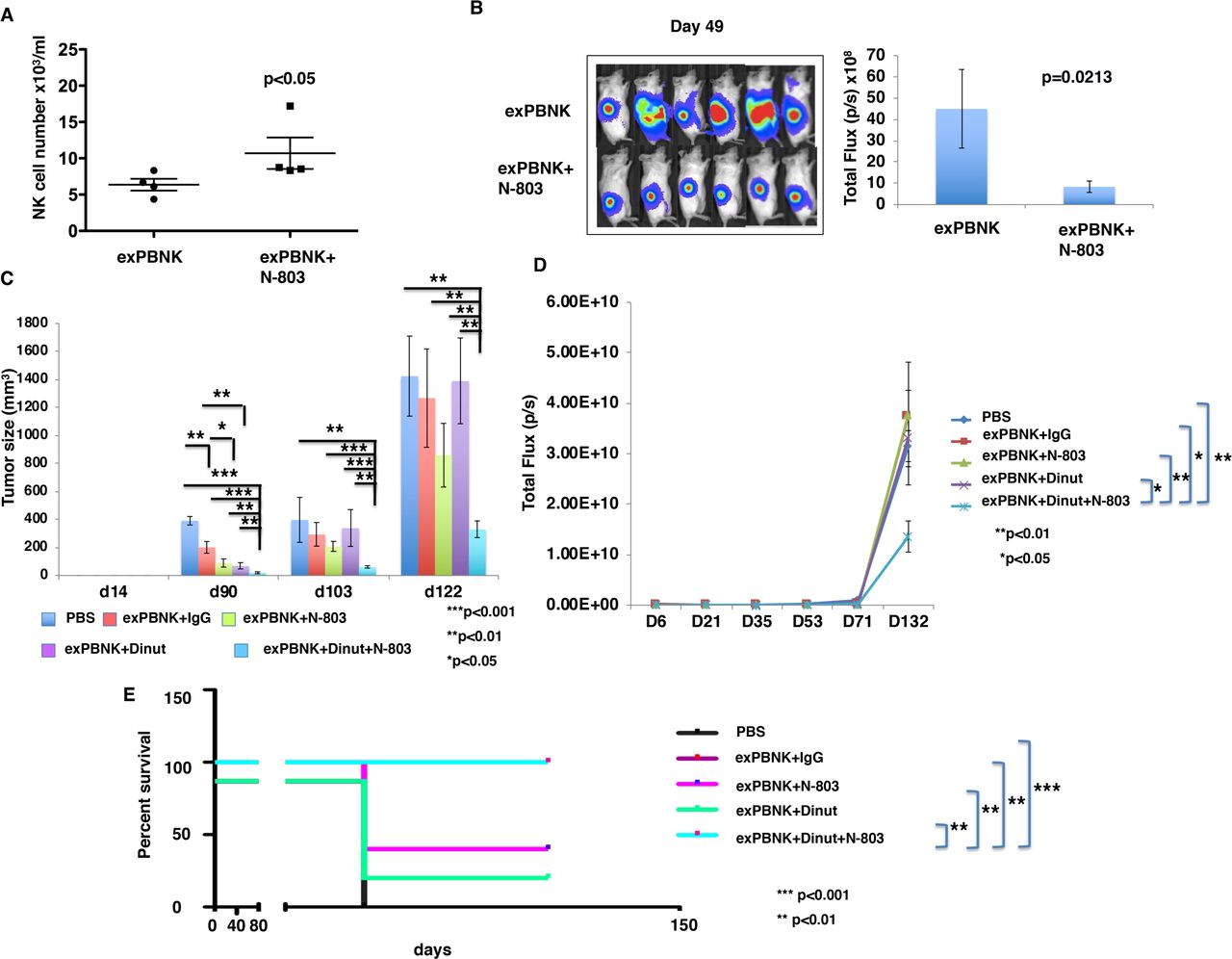
N-803 enhanced exPBNK cells numbers in vivo and the combination of exPBNK+N-803+ dinutuximab significantly inhibited OS cells growth and extended the survival of OS xenografted NSG mice. (A) After confirming tumor engraftment at day 7, 1×107 exPBNK cells or 1×107 exPBNK cells mixed with 0.2 mg/kg N-803 were intraperitoneally injected to each mouse once a week for 6 weeks. Two weeks after the last NK administration, blood was collected from the orbital sinus from each mouse and human NK cells were counted using flow cytometry. N-803 significantly enhanced human NK counts as compared with the mice injected with human NK cells without N-803 (each group n=4). (B) Whole mouse luciferase activity was measured once weekly at various time points. Photos at day 49 are shown in the left panel. photons emitted from luciferase-expression cells were measured in regions of interest that encompassed the entire body and quantified using the living image software. Signal intensities (total flux) are shown at the time points plotted as mean±SEM in the right panel (each group n=6). (C) 4×106 of luciferase expression U2OS-Luc (OS) cells were subcutaneously injected in NSG mice on day 0. After confirming the tumor engraftment at day 7, 1×107 exPBNK cells+15 ug IgG (n=5), 1×107 exPBNK cells+15 µg dinutuzimab (n=5), 1×107 exPBNK cells+0.2 mg/kg N-803 (n=5), 1×107 exPBNK cells+15 µg dinutuzimab +0.2 mg/kg N-803 (n=9), 15 µg dinutuzimab +0.2 mg/kg N-803 (n=5), or PBS (n=5) was intraperitoneally injected to each mouse. NK cells were administered once a week for 3 weeks and IgG, dinutuximab and N-803 were given twice a week for 6 weeks. The tumor size was measured with a caliper once a week and plotted as the mean±SEM for each group. The OS xenografted mice treated with exPBNK cells+dinutuximab + N-803 have significantly smaller tumor sizes than other groups. (D) Photons emitted from luciferase-expression OS cells were measured in regions of interest that encompassed the entire body and quantified using the living image software. Signal intensities (total flux) are shown at the time points plotted as mean±SEM. The OS xenografted mice treated with exPBNK cells+dinutuximab + N-803 have significantly lower bioluminescence signal than other groups. (E) After different treatments, OS xenografted mice were followed until death. The Kaplan-Meier survival curves for all groups were generated following therapy initiation using animal sacrifice as the terminal event. Comparison of survival between groups is shown. The combination of exPBNK cells+dinutuximab + N-803 significantly extended the survival of U2OS-Luc mice as compared with other groups. *p<0.05, **P<0.01, ***p<0.001, Dinut=dinutuximab. OS=U2OS cell line. The data were generated from the pooled two independent experiments. exPBNK, expanded peripheral blood natural killer cell; NK, natural killer; OS, osteosarcoma; PBS, phosphate-buffered saline.
We further confirmed the anti-tumor effects of N-803 combined with dinutuximab and exPBNK cells in human U2OS cells xenografted NSG mice. The U2OS xenografted mice treated with exPBNK cells+dinutuximab + N-803 had significantly smaller tumor sizes (figure 5C) and BLI signals than other groups (figure 5D), and significantly longer survival (figure 5E).
N-803 combined with dinutuximab and exPBNK cells significantly extended the survival of GBM and NB xenografted NSG mice
To confirm that the effect of combination therapy was not specific to OS disease, we investigated if the combination of N-803 with dinutuximab and exPBNK significantly enhances the overall survival of NSG mice with GBM or NB diseases. NSG mice were xenografted with tumor cell line M059K or SKNFI. We found that the combination of exPBNK cells+dinutuximab + N-803 (n=7) significantly extended the survival of M059K mice as compared with the control groups which were treated with PBS (n=4, p<0.001), exPBNK +IgG (n=5, p<0.001), exPBNK +N-803 (n=5, p<0.001), exPBNK +dinutuximab (n=4, p<0.001), and dinutuximab +N-803 (n=4, p<0.01) (figure 6A). Moreover, the combination of exPBNK cells+dinutuximab + N-803 (n=8) significantly extended the survival of SKNFI mice as compared with the control groups which were treated with PBS (n=6, p<0.001), exPBNK +IgG (n=5, p<0.01), exPBNK +N-803 (n=7, p<0.01), exPBNK +dinutuximab (n=7, p<0.05), and dinutuximab +N-803 (n=6, p<0.01) (figure 6B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The combination of exPBNK +N-803+dinutuximab significantly extended the survival of GBM or NB xenografted NSG mice. 4×106 of M059K-Luc (GBM) cells (A) or SKNFI-Luc (NB) (B) cells were subcutaneously injected in NSG mice on day 0. After confirming the tumor engraftment at day 7, 1×107 exPBNK cells+15 µg IgG, 1×107 exPBNK cells+15 µg dinutuzimab, 1×107 exPBNK cells+0.2 mg/kg N-803, 1×107 exPBNK cells +15 µg dinutuzimab +0.2 mg/kg N-803, 15 µg dinutuzimab +0.2 mg/kg N-803, or PBS was intraperitoneally injected to each mouse. NK cells were administered once a week for 3 weeks and IgG, dinutuximab and N-803 were given twice a week for 6 weeks. M059K-Luc (A) or SKNFI-Luc (B) xenografted mice were followed until death. The Kaplan-Meier survival curves for all groups were generated following therapy initiation using animal sacrifice as the terminal event. Comparison of survival between groups is shown. (A) The combination of exPBNK cells+dinutuximab + N-803 (n=7) significantly extended the survival of GBM mice as compared with the control groups which were treated with PBS (n=4, p<0.001), exPBNK +IgG (n=5, p<0.001), exPBNK +N-803 (n=5, p<0.001), exPBNK+ dinutuximab (n=4, p<0.001), and dinutuximab +N-803 (n=4, p<0.01). (B) The combination of exPBNK cells+dinutuximab + N-803 (n=8) significantly extended the survival of NB mice as compared with the control groups which were treated with PBS (n=6, p<0.001), exPBNK +IgG (n=5, p<0.01), exPBNK +N-803 (n=7, p<0.01), exPBNK +dinutuximab (n=7, p<0.05), and dinutuximab +N-803 (n=6, p<0.01). Dinut=dinutuximab. GBM=M059K cell line, NB=SKNFI cell line. The data for each of A and B were generated from the pooled two independent experiments. *<0.05, ** p<0.01, *** p<0.001, exPBNK, expanded peripheral blood natural killer cell; GBM, glioblastoma multiforme; NB, neuroblastoma; NK, natural killer; PBS, phosphate-buffered saline.
Discussion
Recently, there has been a significant increase in development of targeted cancer therapeutics, particularly against hematologic malignancies. However, the success in terms of developing novel therapeutics against solid tumors is still lagging. In this study, we demonstrated that combining dinutuximab with N-803 significantly enhances the cytotoxic potential of NK cells against NB, OS and GBM in vitro, and significantly improved the survival of NB, OS and GBM xenografted NSG mice.
N-803 has been shown to be a promising therapeutic agent in phase 1 clinical trials, resulting in a significant increase in number and function of NK cells, with an excellent safety profile.21 Consistent with those results, our data shows that N-803 significantly stimulated the proliferation of exPBNK cells (figure 1A,B). NK cells have been shown to exert their cytolytic effects using downstream signaling involving p38MAP kinase and JNK MAP kinase pathways.28 29 Using phosphoflow analysis, we showed that both IL-15 and N-803 significantly enhanced the proliferation of Stat3, AKT1 and p38MAP kinase (figure 1C), consistent with the increased proliferation of exPBNK cells. NK cells express a variety of activating and inhibitory receptors, and the net functionality of NK cells is a complex interplay of signals between activating and inhibitory receptors.30 We have previously shown that expression of activating NK cell receptor ligands (MIC A/B) on malignant cells mediates the improved cytotoxicity of NK cells by engaging with activating NK cells receptors.31 We have also shown in our previous studies that there is high expression of NKG2D on our ex vivo expanded PBNK cells.17 More recent studies have shown that decreased expression of the activating receptors NKp30, NKp46, NKG2D, and DNAM-1 on the peripheral NK cells was positively associated with tumor progression.32 We have shown that N-803 significantly enhanced the expression of NK activating receptors NKG2D, NKp30, NKp44 and NKp46 (figure 1D) and thereby enhancing the cytolytic potential of exPBNK cells. CD16 (FcγRIII) can trigger NK-mediated ADCC.33 The enhanced expression of CD16 on exPBNK cells by N-803 provides the rationale for the combinatorial therapy of exPBNK cells with N-803 and dinutuximab. Additionally, we observed that N-803 maintained the high expression levels of CD94, KIR2DL1 and KIR2DL2/3 on exPBNK cells (figure 1D) during coculture. CD94 forms a heterodimeric inhibitory receptor with NKG2A, and activating receptors with NKG2C, and E in humans.34 A recent study showed that the expression of CD94 on ex vivo-differentiated NK cells was associated with higher lytic potential, and higher ability to form immunological synapses with leukemic target cells.35 The human KIR are key regulators of the development, maturation, tolerance and activation of NK cells through a process termed as ‘education’, ‘licencing’, or ‘arming’.36 However, the specific roles of the enhanced CD94 and inhibitory KIRs by N-803 in NK cell function remains to be discovered.
Previous studies have demonstrated the efficacy of N-803 as an immuno-stimulatory molecule in its ability to further potentiate the immune effector cells either alone, or when combined with other therapeutic agents like monoclonal antibodies or immune checkpoint blockage agents.37 This further provided us with rationale of combining IL-15 superagonist with dinutuximab in an attempt to enhance the cytoxicity against GD2 positive solid tumors. We showed that the combining N-803 with dinutuximab had significantly higher in vitro cytotoxic potential against NB, OS and GBM cell lines as compared with N-803 and dinutuximab alone (figure 2A). NK cells exert their effector function by promoting the granule exocytosis pathway where perforin and granzyme B from the granules are released on conjugation with the target cells.38 However, it has been well published that NK cells undergo exhaustion and subsequently a reduction in granzyme B and perforin levels on serial contact with the target cells.39 Our data suggests that in large part the combination of N-803 and dinutuximab significantly enhanced the secretion both perforin (figure 2B) and IFN-γ (figure 2C) from exPBNK cells compared with all other groups against U2OS, M059K and SKNFI. These data suggest that increased cytotoxicity could in part be due to the ability of N-803 to overcome this NK cell ‘exhaustion and anergy’ and thereby providing a prolonged NK cell ADCC effect resulting in a more robust killing of tumor cells when combined with anti GD2 antibody in the presence of N-803.
It has now been well understood that the tumor microenvironment aids in the growth and survival of tumors in part by inhibiting the immunologic response.40 There has been a significant improvement in understanding of factors that can lead to a diminished efficacy of NK cell based targeted therapies against solid tumors. These factors include but are not limited to poor trafficking and infiltration into the tumor, increase in angiogenesis, downregulation of activating receptors on the effector cells and the presence of chronic immunosuppressive signals in the tumor microenvironment thereby resulting in inhibition of NK cell function and activity.14 41 Even though TRAIL induces apoptosis in some tumor cells and was considered for a potential use in anti-tumor therapy, several lines of evidence demonstrated that TRAIL induced the growth and proliferation of some tumor cells in vitro and promoted the proliferation and metastasis of tumor cells in mice.42 43 The PDGF factors and their receptors (PDGFRs) play important roles in oncogenesis, drug resistance, and are associated with clinical cancer recurrence.44 45
We showed that combining N-803 with dinutuximab significantly decreased the secretion of TRAIL and PDGF-BB (figures 3A and 4A), thereby overcoming some of the immunosuppressive effects of the tumor microenvironment and potentially preventing tumor relapse and metastasis. SCGF-β has been shown to be a predictor of responsiveness to therapy in certain solid tumors.46 We also showed that the combination of N-803 and dinutuximab significantly decreased the secretion of SCGF-β (figures 3A and 4A), partially contributing to the superior in vitro and in vivo anti-tumor efficacy of the combination therapy of exPBNK with N-803 and dinutuximab. RANTES is known as CC chemokine ligand 5. RANTES mediates the trafficking and homing of lymphoid cells such as T cells and NK cells by binding to G-protein-coupled receptors and induces the activation and proliferation of NK cells.47 48 However, RANTES in the tumor microenvironment also plays diverse roles to favor tumor survival and metastasis.49 50 Similarly, SDF-1α, known as CXC chemokine ligand-12, is another factor that is important for NK development and homing51 and but also has controversial functions in regulation of tumor growth, metastasis, and development of chemoresistance after binding to its receptor C-X-C chemokine receptor type 4 (CXCR4).52 We found that OS or GBM tumor cells significantly inhibited the secretion of RANTES and SDF-1α from exPBNK cells (p<0.001) (figures 3B and 4B). Further investigation is needed to unveil if the reduction of RANTES and/or SDF-1α creates pro- or anti-tumor microenvironment. Notably, increased secretion of MIG, IP-10 and MIP-1α and 1β likely resulted in improved trafficking of NK cells into the tumor tissues. In our in vivo animal model, the combination of dinutuximab with N-803 and exPBNK cells resulted in a superior survival benefit and a decreased tumor burden in an OS xenografted mouse model. We confirmed our findings in both a GBM and a NB xenografted mouse models that the combining N-803 with dinutuximab and exPBNK cells had superior in vivo antitumor efficacy.
Conclusions
In conclusion, these results demonstrate that there is significant improvement in the anti-tumor activity, both in vitro and in vivo, when N-803 is combined with dinutuximab as compared with either treatment group alone. Our preclinical data provides compelling evidence that the combination of N-803 and anti-GD2 monoclonal antibody dinutuximab with or without exPBNK would be a reasonable and potentially promising approach for designing future clinical studies against relapsed and refractory GD2 positive solid tumors.
Data availability statement
Data are available on reasonable request.
Ethics statements
Ethics approval
NSG mice were bred, treated, and maintained in the animal facility of New York Medical College with NYMC International Animal Care and Use Committee approved protocols. The animal experiments were conducted in accordance with the recommendations of the Guide for Care and Use of Laboratory Animals.
Acknowledgments
The authors would like to thank Erin Morris, BSN and Virginia Davenport, RN for their excellent assistance with the preparation of this manuscript. The authors would like to thank Dean A. Lee for providing the K562-mbIL21-41BBL cells and Hing Wong, Peter R. Rhode, Patrick Soon-Shiong, John H. Lee, and Jeffrey Safrit from ImmunityBio/Altor Bioscience for providing N-803, United Therapeutics for providing dinutuximab and Janet Ayello for her assistance with purchasing research reagents.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @DrPatSoonShiong
Presented at Presented in part at BMT Tandem meeting (2019), Houston, Texas and at Pediatric Blood & Marrow Transplant Consortium meeting (2019), New Orleans, LA.
Contributors YC and MSC conceived and designed the study; YC, GN and SJ developed the methodology, performed the analysis and interpreted the data; YC, GN, JMR and MSC wrote, reviewed and revised the manuscript; and PS-S, JTS and DL provided administrative, technical and material support. All authors approved the final manuscript for submission.
Funding The research for this study was supported by the grants from Pediatric Cancer Research Foundation (PI: MSC), New York Medical College School of Medicine Translational Science Institute Children Health Translational Research Award (PI: YC/JMR), Department of Defense W81XWH-16-PRCRP-TTSA #CA160461 (Partnering PI: MSC), and NIH 1U54 CA232561 01A1 (Sub-PI: MSC).
Competing interests MC serves as a consultant for Jazz, Omeros and Novartis and a Speakers Bureau for Jazz, Servier, Amgen, Sanofi and Sobi. DL reports personal fees and other from Kiadis Pharma, CytoSen Therapeutics, Courier Therapeutics, and Caribou Biosciences outside the submitted work; In addition, DL has a patent broadly related to NK cell therapy of cancer with royalties paid to Kiadis Pharma. PS-S is a majority shareholder of ImmunityBio, Inc. related to N-803. JTS is an employee of NantKwest, an affiliated company to ImmunityBio, Inc related to N-803. Other co-authors declare they have no conflict of interest.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.