Article Text

Abstract
Background Chimeric antigen receptor (CAR)-T cell immunotherapy has modified the concept of treatment in hematological malignancies. In comparison with pediatric patients, where responses are maintained over many years, older patients, such as those with non-Hodgkin’s lymphoma (NHL) and multiple myeloma (MM), present lower persistence of CAR-T cells that might be due to decreased fitness of T cells acquired with aging. Moreover, cord blood derived-NK cells (CB-NKs) and CAR-NK cells derived from CB-NK can be used ‘off-the-shelf’ as immune cells with antitumor properties for the treatment of cancer patients. However, to date, clinical studies have only demonstrated the safety of these therapies but not optimal efficacy. To confront the shortcomings of each therapy, we devised a novel approach consisting of simultaneous (CAR-)NK cell and CAR-T cell administration. In this setting, NK cells demonstrate an important immunoregulation of T cells that could be exploited to enhance the efficacy of CAR-T cells.
Methods A combinatorial treatment based on either CAR-T and CAR-NK cells or CB-NK and CAR-T cells in two models of NHL and MM was performed. Antitumor efficacy was analyzed in vitro and in vivo, and parameters related to early activation, exhaustion and senescence of T cells were analyzed.
Results We show that CAR-NK cells derived from CB-NK are only effective at high doses (high E:T ratio) and that their activity rapidly decreases over time in comparison with CAR-T cells. In comparison and to exploit the potential of ‘off-the-shelf’ CB-NK, we demonstrate that a low number of CB-NK in the CAR-T cell product promotes an early activation of CAR-T cells and their migration to MM cells leading to enhanced anti-MM efficacy. Moreover, cytokines related to CRS development were not increased, and importantly, CB-NK enhanced the fitness of both CARpos and CARneg T cells, promoting lower levels of exhaustion and senescence.
Conclusion This study demonstrates a relevant immunoregulatory role of CB-NK collaborating with CAR-T cells to enhance their antitumor activity. A novel and different approach to consider in CAR-T cell immunotherapy studies is presented here with the goal to enhance the efficacy of the treatment.
- adaptive immunity
- immunity
- innate
- immunotherapy
- killer cells
- natural
- receptors
- chimeric antigen
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All information is in the manuscript. All information is in the manuscript.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
In recent years, adoptive cell immunotherapy approaches, particularly administering chimeric antigen receptor (CAR)-modified T cells, have been established as new methods of treatment for patients with hematological malignancies.1 To date, most clinical studies in patients with B cell malignancies administer CAR-T cells in an autologous setting.2 3 However, there is a proportion of patients where the production of CAR-T cells results in an insufficient amount required for the treatment, and unfortunately, patients do not receive the therapy. For this type of patient, a universal source of immune cells, such as CAR-modified natural killer (NK) cells, are becoming an attractive option. However, whereas CAR-NK cells have demonstrated superiority over unmodified NK cells in preclinical studies4 5 and safety in clinical studies,6 their efficacy is still lower than that of CAR-T cells.1 7
Specifically, CAR-T cells directed to CD19 (CART-19) have achieved outstanding responses in acute lymphoblastic leukemia (ALL) and non-Hodgkin’s lymphoma (NHL) patients,3 8–10 and B-cell maturation antigen (BCMA) has emerged as the most promising target for CAR-T cells used to treat multiple myeloma (MM) patients.11–16 However, whereas pediatric ALL patients treated with CART-19 cells maintain durable responses that correlate with CAR-T cell persistence,3 NHL patients present poorer responses.10 Moreover, in MM patients, CART-BCMA cells do not persist long, and patients end up relapsing despite achieving initial complete responses.15 16 These findings indicate the need to improve CAR-T cell persistence and efficacy in NHL and MM patients to avoid relapses.
Cord blood-derived NK cells (CB-NKs) have been established as a source to obtain ‘off-the-shelf’ NK cells for the treatment of cancer patients.17 18 CAR-NK cells obtained from CB-NK have demonstrated improved antitumor activity compared with unmodified CB-NK5 and have also demonstrated safety in patients.6 19 We previously observed that when CB-NK come into contact with MM tumor cells, they secrete a variety of proinflammatory molecules that enhance the formation of tumor/immune cell clusters, bringing T lymphocytes and other immune cells into close contact and enhancing the antitumor activity of T lymphocytes.20 Therefore, we hypothesized that NK cells, either CB-NK or CAR-NK cells, would enhance the efficacy of CAR-T cells leading to more durable responses.
Here, we first confirmed that at low immune cell numbers, CAR-T cells are superior to CAR-NK cells. Then, to enhance the efficacy of CAR-T cells, we designed a combinatorial treatment based on CAR-T cells with either CAR-NK cells or CB-NK. Two CARs directed against CD19 (ARI-0001)2 and BCMA (ARI2h),14 which were previously developed by us and which have been administered to patients, were employed. We demonstrate that a combinatorial treatment for NHL with CART-19 and CARNK-19 cells does not improve the efficacy compared with the same total number of CART-19 cells alone, suggesting that there is a minimum number of CAR-T cells that is critical for the antitumor efficacy. However, a combinatorial treatment based on CAR-T cells with the addition of half the dose of CB-NK enhances the antitumor efficacy in a model of MM and ARI2h cells. This beneficial impact of CB-NK over ARI2h cells is initiated at incredibly early time points of tumor/immune cell contact where CB-NK facilitate earlier activation and enhanced migration of ARI2h cells to tumor cells. These events impact on the fitness of ARI2h cells in the long term by ameliorating exhaustion levels and the emergence of markers related to immunosenescence in ARI2h cells.
Methods
Donors of immune cells
CB-NK and peripheral blood T cells were obtained from the Banc de Sang i Teixits de Barcelona (BST) from either cord blood (CB) or peripheral blood from healthy donors after obtaining informed consent and being approved by the BST.
Tumor cell lines
Ramos, RPMI8226, U266 and K562 cell lines were purchased from American Tissue Culture Collection (Manassas, Virginia, USA). ARP1 cell line was kindly provided by Multiple Myeloma Research Center (Little Rock, Arkansas, USA). RPMI8226, K562 and ARP1 were cultured in RPMI with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Pen/Strep) and U266 with 15% FBS. K562-based artificial antigen-presenting cells expressing membrane-bound Interleukin (IL)-21 (K562-mb21-41BBL feeder cells) were cultured in RPMI with 10% FBS and 1% Pen/Strep. Tumor MM cell lines were modified to express GFP-FireFlyLuciferase (GFP-FFLuc) using the plasmids pLV-MSCV_Luc-T2A-GFP (kindly provided by Amer Najjar) coding for GFP-FFLuc, and pMD2.G and psPAX2 coding for VSV-G and gag/pol, respectively. Mycoplasma testing in cell lines was performed every 2 months.
Lentivirus production for CAR-T and CAR-NK cells
For CAR-T cells, either pCCL-EF1a-CAR19 or pCCL-EF1a-CARBCMA with packaging plasmids pMDLg-pRRE, pRSV-Rev and envelope plasmid VSV were used. For CAR-NK cells, pCCL-MSCV-CAR19 with pMDLg-pRRE, pRSV-Rev and envelope plasmid BaEV-TR (provided by Dr Els Verhoeyen) were used for production of virus particles. Production of virus particles to transduce T cells was done in HEK293T cells using the same protocol previously described by us.14 Production of virus for CAR-NK cells was done in HEK293T cells that were transfected with transfer vector pCCL-MCSV-CAR19 with pMDLg-pRRE, pRSV-Rev and BaEV-TR plasmid. JetPEI (PolyPlus Transfection) was used as a transfection reagent. Transfection was performed using NaCl and JetPei following the manufacturer’s protocol. Viral supernatants were collected at 48 hours and concentrated with Lenti-X Concentrator (ClonTech) following the supplier’s protocol and stored at −80°C until use.
CB-NK expansion
K562-mb21-41BBL feeder cells were used to expand untransduced NK cells. They were irradiated at 100 Gy and added at a feeder cell:CB-NK ratio of 2:1. CB-NK were left to expand for 14 days, with new feeder cells added on day 7.
T cells and CB-NK transduction
T cells were obtained from buffy coats by Ficoll and magnetic T cell depletion (Miltenyi Biotec). T cells were expanded in Click’s media (50% RPMI, 50% Click’s (Irvine Scientific), 5% human serum, 1% Pen/Strep), activated with Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific) and IL-2 (100 IU/mL) every other day. Experiments were performed after 8–10 days of T cell expansion. Viral transduction was performed after 48 hours of the expansion at a MOI of 10. NK cells were selected from CB units by Ficoll and magnetic NK cell depletion (Miltenyi Biotec). To produce CAR-NK (ARI3 cells), NK cells from a CB unit were expanded with NK MACS Medium (Miltenyi Biotec), 1% NK MACS Supplement, 5% human serum, IL-2 (500 IU/mL) and IL-15 (140 IU/mL). On day 5, CB-NK were transduced at a multiplicity of infection (MOI) of 5 with Vectofusin-1 (10 mg/mL) following Vectofusin-1 protocol (Miltenyi Biotec). Cells were washed 6–24 hours after transduction. On day 7, ARI3 cells were coexpanded with feeder cells adding IL2 (400 IU/mL) every other day for the next 7 days. Of note, throughout the manuscript, ‘ARI3 cells’ refers to CAR-NK cells and CB-NK refers to untransduced NK cells.
In vivo models
Immunodeficient NSG mice were purchased from The Jackson Laboratory. Mice used for experiments were of the same sex and 8–12 weeks old. Mice were irradiated on day 1 at a dose of 2 Gy in a biological radiator with two sources of 137 Cs (J.L. Shepherd, model MARK 1, 30 serial number 1199) and received itravenous tumor cells modified to express GFP-FFLuc on day 0. Different models of tumor burden were created for NHL depending on the dose of NHL cells administered. Thus, mice received 1×105, 3.5×105, or 5×105 NHL cells to create the low, medium, or high tumor burden models. Then, they received immune cells on day 7 (for NHL) or day 14 (for MM). The disease was followed weekly by bioluminescence as previously described.20 Bone marrow (BM) and spleen were harvested when mice were euthanized to analyze the presence of CAR-T cells and MM cells by flow cytometry.
Cytotoxicity assays
Luciferase killing assays were performed by coculturing target tumor cells expressing GFP-FFLuc with effector cells tested in each case in a white-walled 96-well plate. D-luciferin (20 mg/mL) was added 15 min prior to the bioluminescence reading in a Synergy HT Plate Reader (BioTek). The percentage of remaining tumor cells was calculated as (luminescence of sample/luminescence of tumor cells alone) ×100.
Cytokine production
Interferon Gamma (IFNγ), tumor necrosis factor alpha (TNFα) and IL-2 were quantified by ELISA (ELISA MAX Deluxe Set, Biolegend) following the manufacturer’s protocol. Luminex assays were conducted with ProcartaPlex multiplex immunoassay kit (Thermo Fisher Scientific).
Confocal microscopy
RPMI8226 cell line modified to express GFP was cocultured with CART cells stained with Cell Tracker Blue CMAC Dye (Thermo Fisher Scientific). Either CB-NK or non-transduced T cells (UT) were stained with CellTracker Deep Red Dye (Thermo Fisher Scientific) and added to the previously mentioned coculture. Images were acquired using a Leica SP5 microscope. 405, 488 and 633 lasers were used for excitation. For time lapse experiments, in vivo image acquisitions were performed every 30 s for 14 hours.
Challenges
ARP1-GFP-FFLuc (MM) cells were cocultured with CART cells at an effector:target ratio 0.5:1. In the condition with CB-NK, CB-NK were added at half the amount of CART cells, leaving a final ratio of CART:MM(:NK) 0.5:1(:0.25). The remaining tumor cells were assessed by fluorescent microscopy until no GFP was observed, and thus the challenge was over. T lymphocytes were then stained, and the phenotype was compared with that seen in unstimulated T cells (day 0) and CAR-T cells before exposure to tumor cells.
Flow cytometry
CAR expression was detected with either a recombinant BCMA-Fc protein (Enzo Life Sciences) or a recombinant CD19-Fc protein (Thermo Fisher Scientific) and an antihuman IgG Fc-BV-421 (Biolegend, clone: M1310G05). For T cell phenotyping, PD-1-APC (clone: J105), TIM-3-FITC (clone: F38-2E2), TIGIT-PerCP-Cy5.5 (clone: A15153G), LAG-3-PE (clone: 11C3C65), CXCR3-Alexa-Fluor 488 (Clone 1C6/CXCR3), CCR7-PerCP-Cy5.5 (clone: 150503), CD45RA-APC (clone: HI100), CD27-PE (clone: M-T271), CD28-FITC (Clone CD28.2), CD3-PE (clone SK7), CD4-APC-H7 (clone L200) and CD8-PE-Cy7 (clone: RPA-T8) antibodies were used. For NK cell phenotyping, CD16-Alexa488 (Clone 3G8), NKP30-PeCy7 (Clone P30-15), NKG2D-APC-Cy7 (Clone 1D11), NKG2A-PacificBlue (Clone S19004C), NKG2C-Pe (Clone S19005E), KIR2DL1-PE (Clone HP-DM1), KIR2DL2/L3-FITC (Clone DX27), KIR3DL1-BV421 (Clone DX9) and KIR2DL1/S1/S3/S5-PerCp Cy5.5 (Clone HP-MA4) were all from Biolegend. CD56-APC (Clone REA196, Miltenyi) was used for NK cells. Staining was performed in FACS buffer. For T cell determination in BM or spleen at in vivo end-point, mouse FcR blocking reagent (Miltenyi) was used. All experiments were read on a FACS Canto II (BD Biosciences) and analyzed with FlowJo software (Tree Star, Eugene, Oregon, USA).
Graphs and statistical analysis
Data were presented using GraphPad Prism software, and statistical analysis was performed using SPSS software V.20 using Student’s t-test to compare between two groups.
Results
CAR-NK cells demonstrate higher in vitro activity than CAR-T cells only at high E:T ratios, requiring IL2 to maintain efficacy at low E:T ratios, which can be provided by CAR-T cells
We first optimized a method to obtain a high number of CAR-NK cells directed against CD19 (ARI3 cells) starting from CB-NK. We used a CAR directed against CD19 with 4-1BB as costimulatory domain previously produced by our institution termed ARI-0001 (ARI1 from now on).21 Plasmids coding for ARI1 and ARI3 differed in the promoter (online supplemental figure 1A) as CB-NK presented higher transduction efficiencies using the MCSV promoter instead of the EF1a promoter. Another difference between ARI1 and ARI3 was the packaging plasmid that was used in the transduction. The use of a Vesicular Stomatitis Virus (VSV) packaging plasmid for virus production led to extremely low transduction efficiencies of CB-NK. Interestingly, Bari et al22 demonstrated that exchanging VSV for Baboon Envelope (BaEV) packaging plasmid improved efficiencies of NK cell transduction. Therefore, VSV and BaEV were compared confirming improved efficiency of CAR transduction with BaEV (online supplemental figure 1online supplemental figure 1B). Then, two different expansion methods were compared with BaEV (figure 1A). In the first method (A.1), CB-NK were expanded with IL2 and IL15 over 5 days. On day 5, CAR transduction was performed. On day 7, K562-mb21-41BBL feeder cells were added to CB-NK, and they were left to expand until day 14 with the addition of IL2 every other day. In the second method (A.2), CB-NK were expanded with IL2 and feeder cells for 5 days. On day 5, CAR transduction was performed. On day 7, CB-NK were left to expand until day 14 with the addition of fresh feeder cells and IL2 every other day. Method A.1 achieved a higher percentage of ARI3+ cells than method A.2 (figure 1B), and therefore, method A.1 was selected for all ARI3 cell productions.
Supplemental material

CAR-NK cells demonstrate higher in vitro activity than CAR-T cells only at high E:T ratios, requiring IL2 to maintain efficacy at low E:T ratios, which can be provided by CAR-T cells (see also online supplemental figure 1). (A) Diagram showing two different protocols (A.1 and A.2) used to obtain CAR-NK cells directed against CD19 (ARI3) from CB-NK. (B) Efficiencies of CB-NK transduction with both methods shown in figure part A. (C) Cytotoxicity assays of immune cells (untransduced T cells: UT, CAR-T cells against CD19 (ARI1), CB-NK and ARI3) against Ramos-NHL cell line performed at effector:target (E:T) ratio=10:1 at 24 hours and 48 hours. (D) Cytotoxicity assays of immune cells (UT, Ari1, CB-NK, ARI3 and ARI3+ IL2) against Ramos performed at different E:T ratios at 24 hours and 48 hours. (E) IFNγ and (F) TNF-α production of the cytotoxicity assays in (D). (G) Cytotoxicity assays at 24 and 48 hours against Ramos at different E:T ratios where immune cells are added either alone UT (1), ARI1 (1), or in combination of ARI1 and ARI3 cells, where the combination could have in comparison with ARI alone, half the dose of ARI1 and ARI3 (0.5+0.5) or the same dose of Ari1 and ARI3 cells (1+1). (H) IFNγ production of the cytotoxicity assays in (G). (I) Cytotoxicity assay versus Ramos cells over 7 days performed at 0.25:1 E:T ratio adding immune cells alone or the combination of ARI1+ARI3 (0.5+0.5) where the total number of immune cells is the same as in the other conditions. (J) IFNγ production of the cytotoxicity assays in (I). *P<0.05. **P<0.0001. Statistic in figure parts D, E and F is performed comparing to ARI3. Statistic in figure parts G–J is performed comparing ARI3 (1) or ARI1+ARI3 (0.5+0.5) to ARI1 (1). CAR, chimeric antigen receptor; CB-NK, cord blood-derived NK cells; NK, natural killer.
The anti-NHL activity of both CAR-T cells and CAR-NK cells directed against CD19 (ARI1 for CAR-T cells and ARI3 for CAR-NK cells: online supplemental figure 1A was compared in vitro. At higher effector:target (E:T) ratios, ARI3 cells presented higher antitumor activity than untransduced CB-NK or ARI1 cells (figure 1C). However, at low E:T ratios, ARI3 efficacy was significantly lower to that of ARI1 cells (figure 1D), being this difference even more pronounced at longer times (24 hours vs 48 hours). Lack of efficacy of NK cells at longer times might reflect their requirement for IL2, which is not present in these assays. Therefore, we confirmed that addition of exogenous IL2 improved the in vitro efficacy of ARI3 cells, especially at later time points (figure 1D). In terms of cytokine production, at higher E:T ratios, ARI3 cells produced a higher amount of IFNγ than CB-NK, with this production being increased further with the addition of IL2. However, production of IFNγ by ARI3 cells with and without IL2 was always lower than that of ARI1 cells (figure 1E). Regarding TNFα, this cytokine is rapidly produced at early time points and decays fast.14 While ARI3 cells produced higher TNFα levels than CB-NK, the highest TNFα production was detected for ARI1 cells (figure 1F).
ARI3 cells required IL2 at low E:T ratios to kill tumor cells, and interestingly, CAR-T cells produce high amounts of IL2. Moreover, we previously demonstrated that CB-NK can enhance the antitumor activity of untransduced (UT) T cells.20 Therefore, we hypothesized that ARI1 CAR-T cells would boost the activity of ARI3 CAR-NK cells, and vice versa, which would lead to an overall enhanced antitumor efficacy. Thus, combination of ARI1 and ARI3 cells, where the total number of immune cells was the same as ARI1 cells alone (0.5+0.5 vs 1), demonstrated increased anti-NHL activity (figure 1G) without impacting in higher IFNγ (figure 1H) and TNFα production (online supplemental figure 1C). Of note, when the total number of immune cells combining ARI1 and ARI3 was double that of ARI1 cells alone (1+1 vs 1), a higher anti-NHL activity and IFNγ production (figure 1G and H) was observed with hardly any changes for TNFα (online supplemental figure 1C). Last, long-term in vitro cytotoxicity assays performed with a low E:T ratio demonstrated superiority of this combinatorial treatment (ARI1+ARI3) versus ARI1 cells alone when the total number of immune cells was the same in both treatments (figure 1I). Of note, IFNγ levels were not increased (figure 1J).
CAR-NK cells do not maintain in vivo efficacy and enhance the migration of tumor cells to secondary lymphoid tissues in a model of NHL
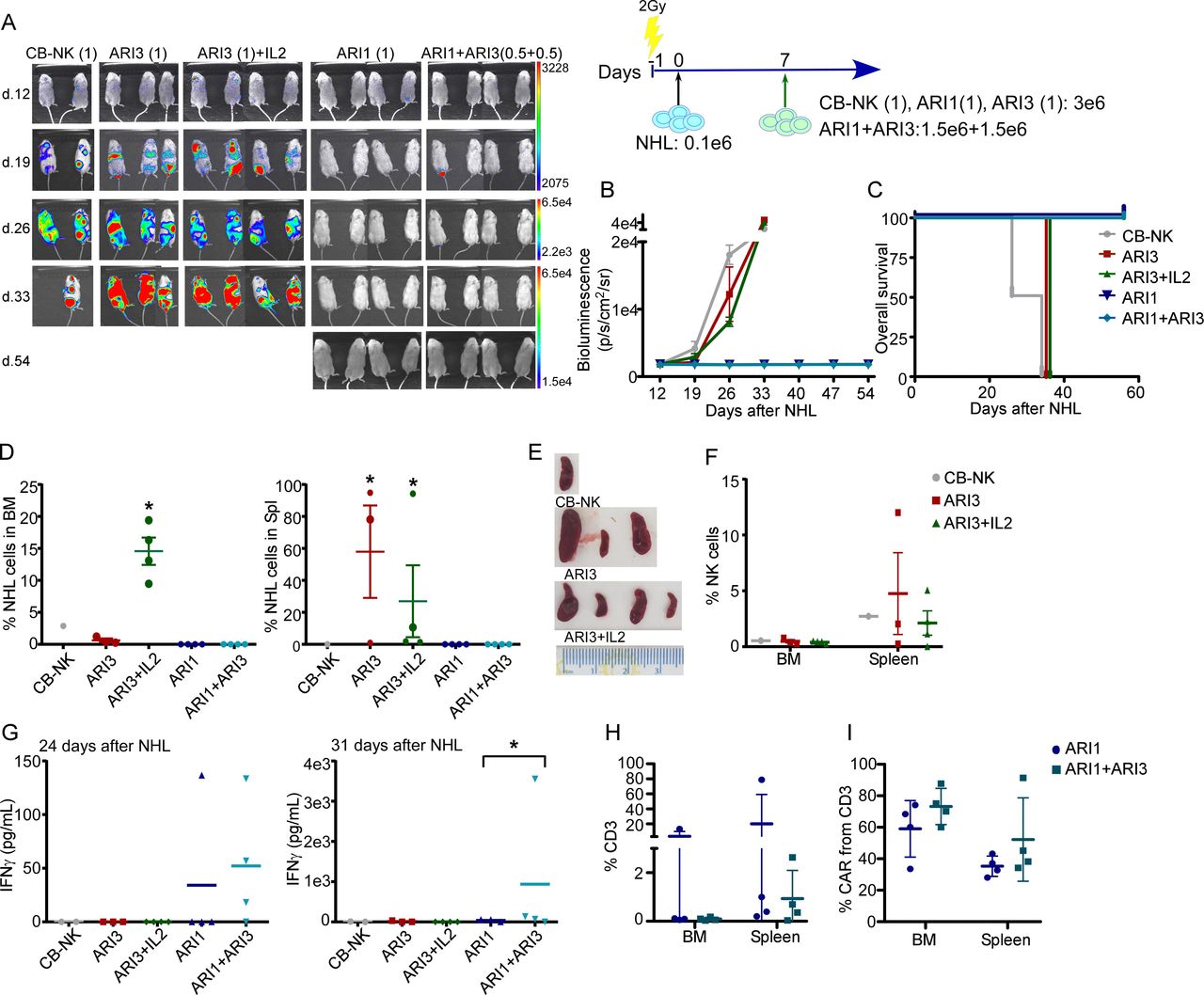
The impact of IL2 in the activity of ARI3 cells and the combinatorial treatment were further evaluated in vivo in different NHL tumor burden models. In a model of highly aggressive NHL, none of the immune cells administered (CB-NK, ARI1 or ARI3) were able to prevent disease progression at all (online supplemental figure 2A and 2B). Of interest, analysis of mice tissues demonstrated that ARI3 cell treatment maintained the bone marrow (BM) with the lowest levels of disease in comparison with untreated mice (online supplemental figure 2C). However, mice treated with either CB-NK or ARI3 cells presented a higher percentage of tumor cells in the spleen compared with both untreated and ARI1-treated animals (online supplemental figure 2C).
In a model of low tumor burden, treatment with either CB-NK or ARI3 cells alone was not able to stop disease progression (figure 2A–2C). ARI3 cells maintained the BM free of disease (figure 2D), but as previously observed in the high tumor burden model (online supplemental figure 2C), a high number of NHL cells were detected in the spleen (figure 2D). The addition of exogenous IL2 to ARI3 cells was highly detrimental by causing NHL progression, as likely due to the fact that IL2 also induces B cell proliferation,23 with this group showing a dramatic increase in the number of NHL cells in the BM compared with ARI3 cells without exogenous IL2 and a high number of NHL cells in the spleen (figure 2D). These findings were in agreement with the role of IL2 promoting the presence of B cells in the BM and also in the spleen but to a lesser degree.24 In all cases, the high number of NHL cells in the spleen correlated with splenomegaly (figure 2E). Moreover, NK cells were detected in the spleen in all groups treated with NK cells, so the presence of tumor cells in the spleen was not due to lack of NK cell migration (figure 2F). Treatment with either ARI1 cells alone or combined with ARI3 cells did not demonstrate differences in this model of low tumor burden (figure 2A–2D) as both treatments completely prevented NHL progression. However, higher IFNγ production was observed in the combinatorial treatment at 31 days. No increase was seen at 24 days (figure 2G). Finally, even though, in these two groups, there were low numbers of human-derived T cells in the BM and the spleen (figure 2H), the presence of CAR+ cells could be detected in both groups with no significant difference observed between groups (figure 2I).

CAR-NK cells do not maintain in vivo efficacy and enhance the migration of tumor cells to secondary lymphoid tissues in a model of NHL. (A) In vivo efficacy in a model of low tumor burden of NHL of ARI (1) and ARI3 (1) cells alone or combined where the total number of immune cells is the same (ARI1+ARI3 (0.5+0.5)). The exogenous addition of IL2 was also included (ARI3 (1)+IL2) and treatment with CB-NK as control. (B) Quantification of disease progression by weekly bioluminescence imaging and (C) overall survival of the different group of mice shown in figure part A. (D) Flow cytometry of bone marrow (BM) and spleen of mice at the end of the experiment showing the presence of NHL cells. (E) Spleen of mice treated with NK cells (CB-NK, ARI3 and ARI3+IL2). (F) Presence of NK cells in BM and spleen of mice treated with NK cells. (G) IFNγ levels detected in mice serum at the time points indicated in the different groups of mice. (H) Percentage of total T cells and (I) CAR-T cells in BM and spleen in mice treated with ARI1 or the combinatorial treatment based on ARI1+ARI3 (0.5+0.5) with the same total number of immune cells. (J) Ramos cell line was used as NHL cells. *P<0.05. Statistic shown in figure parts D and G is performed comparing to CB-NK. CAR, chimeric antigen receptor; CB-NK, cord blood-derived NK cells; NHL, non-Hodgkin’s lymphoma; NK, natural killer.
The number of CAR-T cells, but not CAR-NK cells, is critical in the prevention of NHL progression
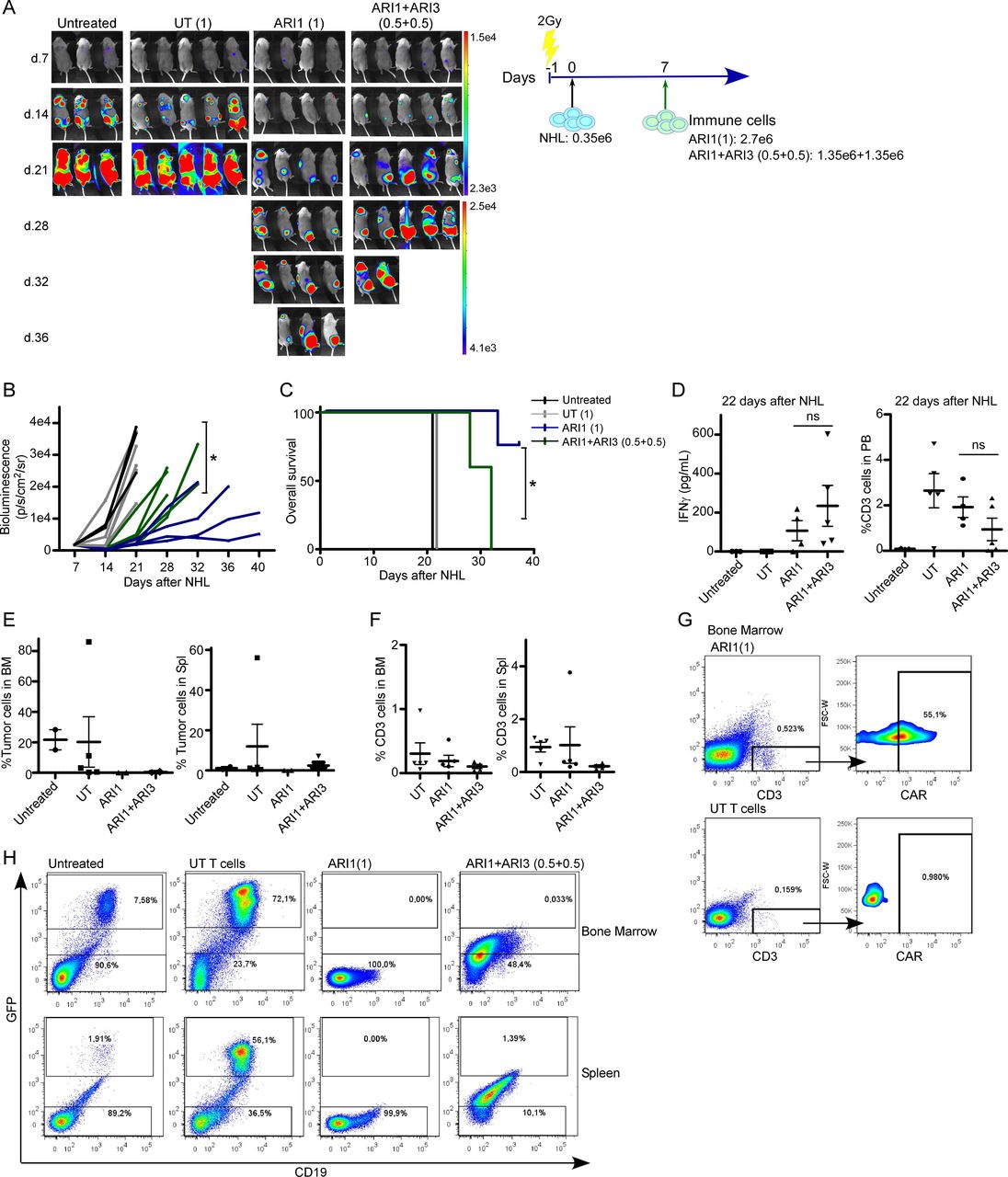
A third in vivo model of NHL with medium levels of tumor burden was performed to find out whether the combinatorial treatment based on ARI1+ARI3 was better than ARI1 cells alone. In this model of disease, the highest number of ARI1 cells was determinant to prevent disease progression, as groups treated with ARI1 cells alone retained the disease progression for longer time than the combinatorial treatment with half dose of ARI1 cells, leading to a higher survival of the mice (figure 3A–3C). In the peripheral blood (PB), 15 days after CAR treatment, a slight tendency for a higher production of IFNγ was detected for the combinatorial treatment, although it was not significant, and there were no differences in the number of T cells present in the PB, although it must be noted that the combinatorial treatment had half the dose of T cells (figure 3D). Analysis of mice tissues demonstrated that both treatments maintained the BM free of disease. In addition, tumor cells were not detected in the spleens of the ARI1 alone group, but a very low number of NHL cells was detected in the spleen of mice receiving the combinatorial treatment (figure 3E). In both groups, NK cells could not be detected, and the presence of T cells in both the BM and the spleen was extremely low (figure 3F), although CAR-T cells were still present (figure 3G). Of note, mice receiving the combinatorial treatment did not present a clear NHL population in the tissues. However, a population with low expression of GFP and CD19 (figure 3H), suggestive of a dying NHL cell population, was present, which might explain the higher bioluminescence signal detected in the combinatorial treatment (figure 3A and B).
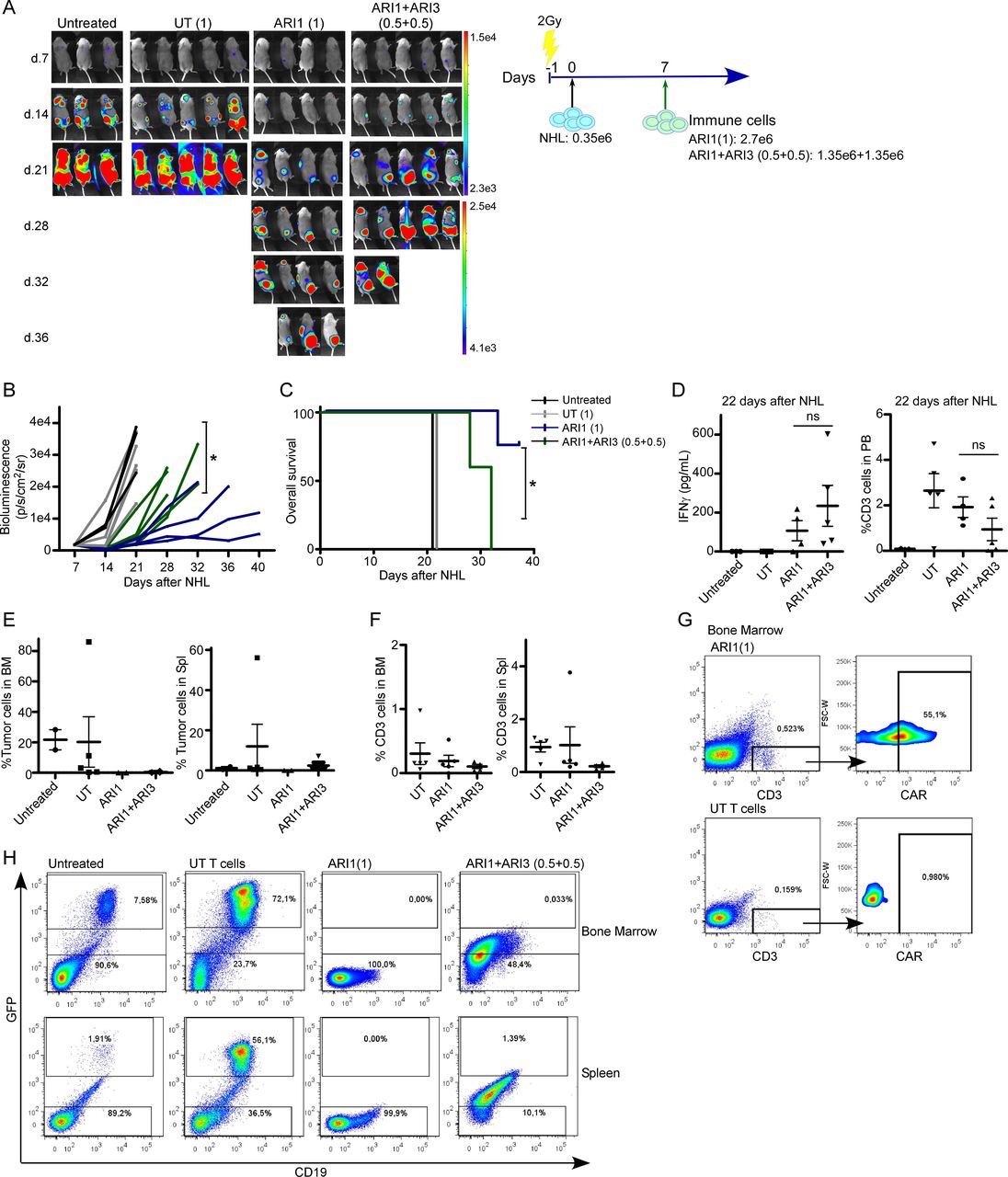
The number of CAR-T cells, but not CAR-NK cells, is critical in the prevention of NHL progression. (A) In vivo efficacy in a model of medium tumor burden of NHL of ARI (1) cells alone or combined with ARI3 cells where the total number of immune cells is the same (ARI1+ARI3 (0.5+0.5)). Treatment with untransduced T cells (UT) was added as control. (B) Quantification of disease progression by weekly bioluminescence imaging and (C) overall survival of the different group of mice shown in figure part A. (D) IFNγ levels detected in mice serum at the time points indicated in the different groups of mice. (E) Flow cytometry of bone marrow (BM) and spleen of mice at the end of the experiment showing the presence of NHL cells and (F) of T cells. (G) Flow plot showing the presence of CAR-T cells in BM in mice treated with ARI1 cells. (H) Representative flow plot showing presence of NHL cells based on GFP and CD19 expression in BM in spleen in the different groups of mice. For the group of UT T cells, the mouse with the highest number of GFP+ cells is shown. Ramos cell line was used as NHL cells. *P<0.05. CAR, chimeric antigen receptor; NHL, non-Hodgkin’s lymphoma; NK, natural killer; NS, not significant.
A small number of CB-NK improves the antitumor efficacy of CAR-T cells (ARI2h) in a model of MM
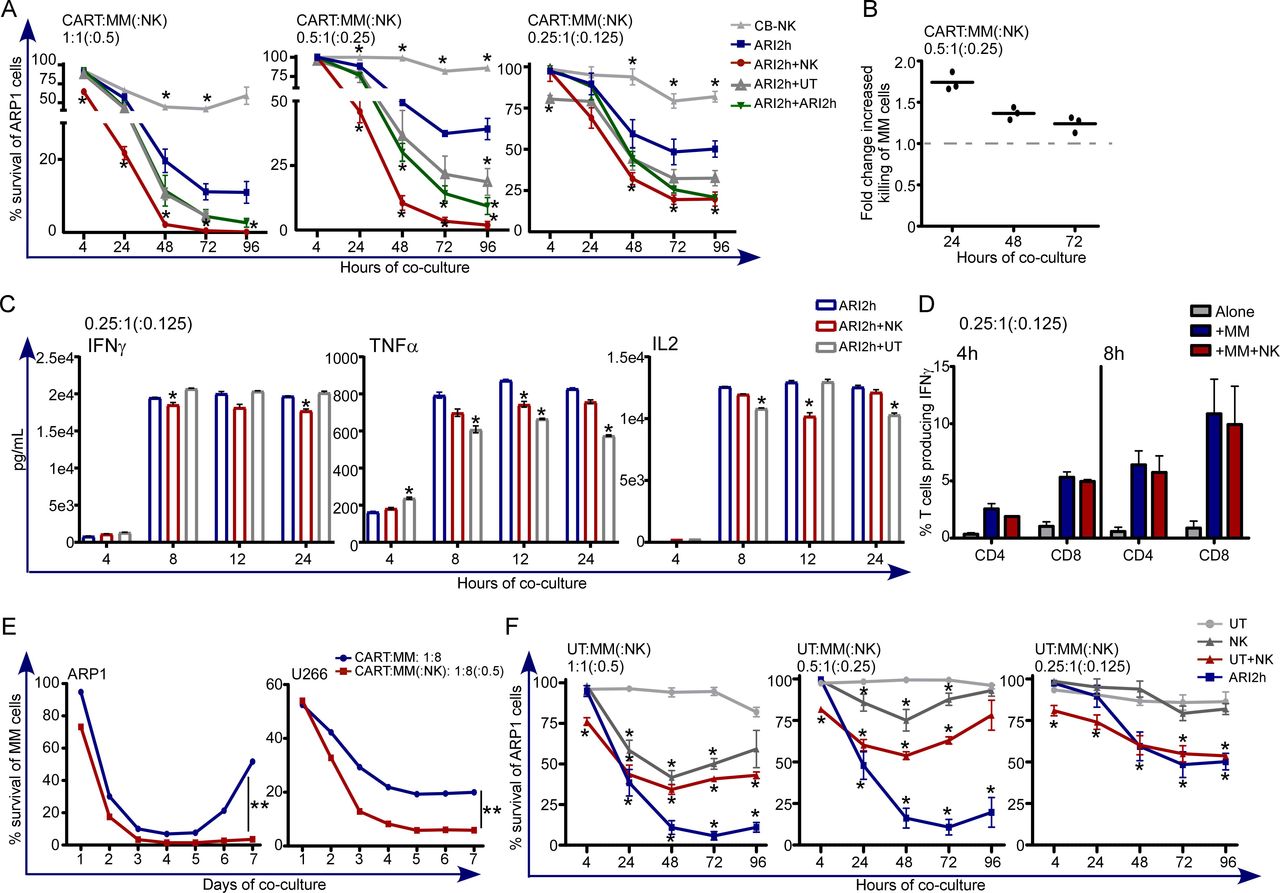
Our results determined that ARI3 cells presented much lower efficacy than ARI1 cells at low E:T ratios, which translated into low in vivo efficacy (figures 1 and 2). Moreover, the combinatorial treatment based on ARI1 and ARI3 cells showed that the number of ARI1 cells and the starting tumor cell burden was determinant to prevent disease progression (figure 3). Nevertheless, we previously determined that CB-NK are able to increase the antitumor activity of UT T cells by bringing UT T cells and MM cells into close proximity.20 Thus, we hypothesized that addition of CB-NK to CAR-T cells would increase the antitumor efficacy of CAR-T cells. Of note, patients receiving CAR-T cells receive a previous lymphodepleting treatment based on fludarabine and cyclophosphamide that removes NK cells. Moreover, NK cells represent around 10% of PB leukocytes, a much lower value than for T lymphocytes.25 Thus, we decided to design a treatment that includes NK cells, specifically with half the dose of CB-NK with respect to that of CAR-T cells. This hypothesis was tested in a model of MM and humanized CAR-T cells directed against BCMA (ARI2h cells). Addition of CB-NK to ARI2h cells increased the anti-MM in vitro efficacy. Of interest, this beneficial impact was enhanced when the E:T (ARI2h:MM(:NK)) ratios were lower (1:1(:0.5)) vs 0.5:1(:0.25)) in figure 4A. Moreover, UT T cells and ARI2h cells did not augment tumor killing to the same extent as with CB-NK when they were added at the same ratios, demonstrating that the highest beneficial effect occurred with the addition of CB-NK (figure 4A), especially at earlier time points (figure 4B). Of interest, IFNγ, TNFα and IL2 levels in the supernatant were not increased with the addition of CB-NK, being in some cases even slightly lower (figure 4C). Specifically, IFNγ production by either CD4+ or CD8+ T cells in these coculture assays did not increase when CB-NK were present (figure 4D). Furthermore, long-term cytotoxicity assays performed at a very low E:T ratio (1:8), confirmed the beneficial impact of CB-NK which even prevented the regrowth of MM cells (figure 4E). Of note, the pool of ARI2h cells that patients receive contains UT T cells. Therefore, we analyzed and confirmed that CB-NK also enhanced the anti-MM activity of UT T cells (figure 4F). Moreover, at low E:T ratios, the anti-MM activity of either UT or CB-NK alone was barely detectable and was much higher when both populations were combined (figure 4F).

A small number of CB-NK improves the in vitro antitumor efficacy of CAR-T cells (ARI2h) in a model of MM (see also online supplemental figure 3). (A) Cytotoxicity assays against ARP1 (MM cell line) adding as effectors immune cells alone (CB-NK and ARI2h) or ARI2h cells combined with half the dose of: CB-NK (ARI2h+CB-NK), UT T cells (ARI2h+UT) or ARI2h cells (ARI2h+ARI2 hour). Assays were performed at different E:T ratios. (B) Fold change increased killing with the addition of half the dose of CB-NK to ARI2h cells of cytotoxicity assay shown in figure part A at the E:T ratio of 0.5:1(:0.25) for CART:MM(:NK). (C) IFNγ, TNFα and IL2 production of the cytotoxicity assays in figure part A. (D) Flow cytometry analysis of IFNγ production by CD4+ and CD8+ T cells at the time-points shown adding ARI2h:MM(:NK) at (0.25:1(:0.125)) ratio. (E) Cytotoxicity assay versus ARP1 and U266 MM cells over 7 days performed adding ARI2h:MM(:NK) at (1:8(:0.5)) ratio. In parallel, the same condition in the absence of CB-NK was performed. (F) Cytotoxicity assays against ARP1 (MM cell line) adding as effectors immune cells untransduced T (UT T) cells combined with half the dose of CB-NK (UT+CB-NK). As controls, UT T cells, NK or ARI2h cells alone were compared. Assays were performed at different (E:T) ratios. *P<0.05 and **p<0.0001. Statistic in figure parts A and C is performed comparing to ARI2h. Statistic in figure part F is performed comparing UT T cells. CAR, chimeric antigen receptor; CB-NK, cord blood-derived NK cell; MM, multiple myeloma; NK, natural killer.
The beneficial impact of CB-NK over ARI2h cells was confirmed in an in vivo model for MM in terms of lower disease progression and higher survival (figure 5A–5C). Despite the beneficial impact obtained with CB-NK and ARI2h cells, mice treated only with CB-NK showed a high proportion of MM cells in the BM and the spleen (figure 5D). Moreover, IFNγ levels and total T cells in the PB of mice 28 days after treatment (figure 5E), and the presence of T cells in the BM and the spleen (figure 5F), did not differ between the ARI2h and ARI2+CB-NK groups.
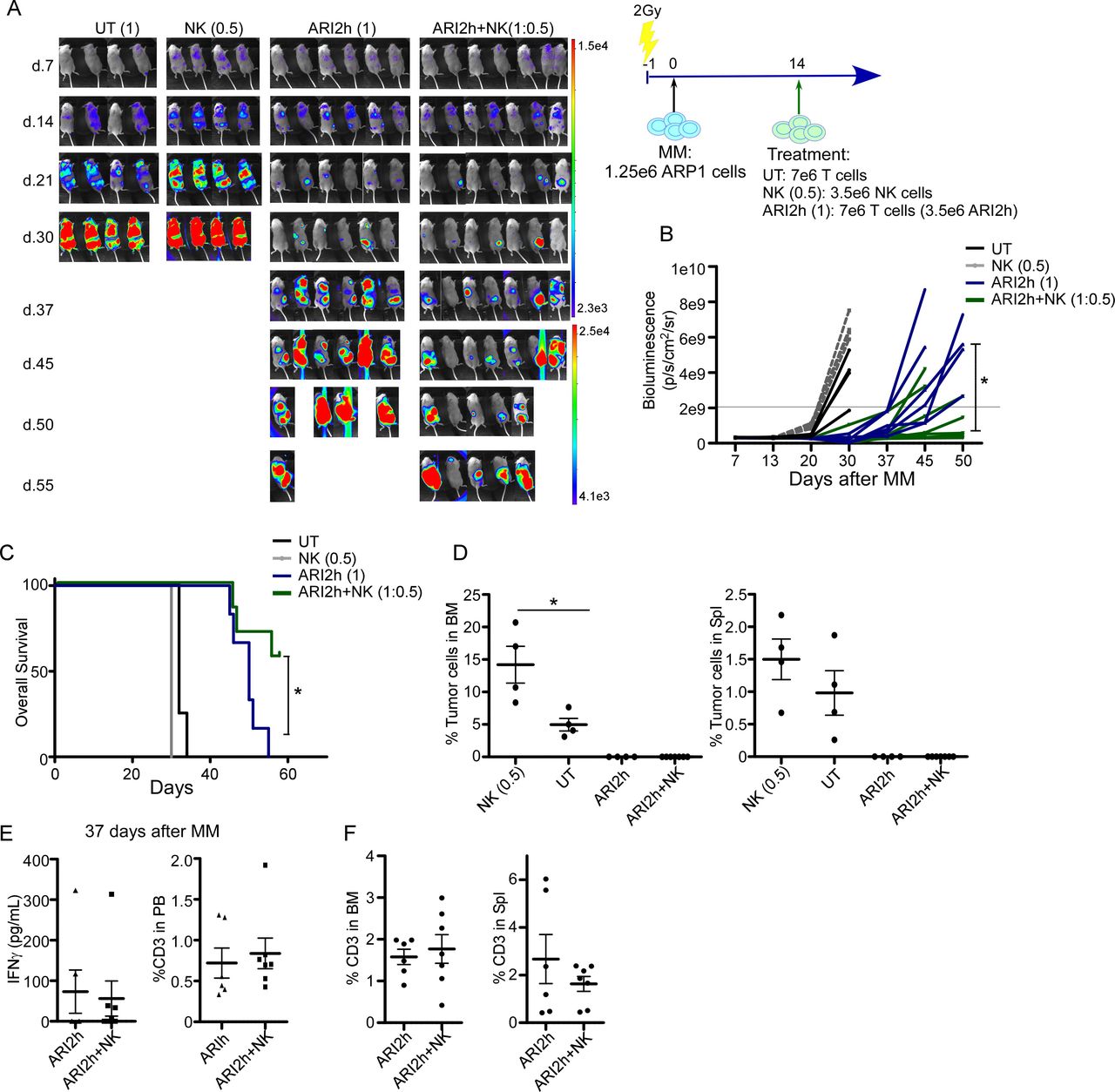
A small number of CB-NK improves the in vivo antitumor efficacy of CAR-T cells (ARI2h) in a model of MM. (A) In vivo efficacy in a model of MM (ARP1 cells) comparing AR2h (1) alone or with addition of half the dose of CB-NK (ARI2h+NK(1:0.5)). UT T cells (UT (1)) or CB-NK alone (NK (1)) were added as controls. (B) Quantification of disease progression by weekly bioluminescence imaging and (C) overall survival of the different group of mice shown in figure part A. (D) Flow cytometry of bone marrow (BM) and spleen of mice at the end of the experiment showing the presence of ARP1 MM cells. (E) IFNγ levels and CD3 T cells detected in mice serum and peripheral blood (PB) at the time point indicated in the different groups of mice. (F) Percentage of total T cells in BM and spleen in mice treated with ARI2h or ARI2h and half the dose of CB-NK (ARI2h+NK). *P<0.05. CAR, chimeric antigen receptor; CB-NK, cord blood-derived natural killer cell; MM, multiple myeloma; UT, untransduced.
CB-NK promotes the activation of ARI2h cells leading to enhanced migration of ARI2h cells to MM cells
NK cells, as part of the innate immune system, perform their activity at earlier time points than T cells,1 which was concordant with the highest impact of CB-NK over ARI2h cells observed at early time points (figure 4B). Therefore, we performed time-lapse in vivo imaging to visualize differences in the activity of ARI2h cells in the presence or absence of CB-NK over a period of 14 hours. As a control, UT T cells were added at the same number and in place of CB-NK. In vivo imaging showed that CB-NK accelerated the migration of ARI2h cells to MM cells, an effect that was not observed with UT T cells (online supplemental movies, figure 6A–6C). Specifically, CB-NK migrate incredibly quickly towards tumor cells (within the first 22 min in figure 6B) leading to enhanced formation of immune/tumor cell clusters. In turn, this promoted the migration of ARI2h cells to MM cells (figure 6C). Moreover, whereas CB-NK came into close proximity with tumor cells remarkably quickly, they started to distance from these clusters when tumor cells had already been eliminated (from 382 min onward in figure 6B). In parallel, movements of UT T cells added to ARI2h cells were hardly visible (online supplemental movies, figure 6B).
Supplementary video
Supplementary video

CB-NK promotes the activation of ARI2h cells leading to enhanced migration of ARI2h cells to MM cells (see also online supplemental movies 1 and 2). (A) Time lapse images over 14 hours of ARI2m cells cocultured with RPMI8226 MM cells modified to express green fluorescent protein and adding CB-NK at half the dose of ARI2h cells. In parallel, as control, UT T cells were added at half the dose of ARI2h cells. ARI2h and MM cells were added at 0.5:1 E:T ratio. ARI2h cells were stained with cell tracker CMAC and UT T cells or CB-NK with cell tracker Deep Red. (B) Analysis of the number of CB-NK or UT T cells and (C) Of ARI2h cells in the presence/absence of CB-NK in each field of view at the different time-points shown for experiment shown in figure part A). Data are calculated with four different time lapse images over 14 hours each. Movies with CB-NK derived from different CB units demonstrated the same pattern. (D) Percentage of CD69+ cells in the CD4+ and CD8+ ARI2h+ cells in the presence (ARI2h+NK) or absence (ARI2h) of CB-NK after coculturing ARP1 MM cells with ARI2h cells. (E) Representative plot of analysis shown in figure part D. (F) ARP1 MM cells were co-cultured with ARI2h cells for 6 hours at 0.5:1 (E:T) ratio in the presence or absence of half the dose of CB-NK. Supernatants were taken and analyzed using a ProcartaPlex multiplex immunoassay kit (n=4). Heat map shows the relative abundance of each molecule on a scale of blue (low) to red (high). (G) Molecules significantly differentially expressed in the two groups of heat map shown in figure part F. *P<0.05 and **p<0.0001. CB-NK, cord blood-derived NK cell; E:T, effector:target; MM, multiple myeloma; UT, untransduced.
The enhanced migration of CAR-T cells to MM cells due to the presence of CB-NK suggested a faster activation of CAR-T cells at earlier time points. Therefore, CD69 expression, a marker of T cell activation, was measured demonstrating a higher activation of CD4+ T cells in the presence of CB-NK (figure 6D and E). Furthermore, the secretion of cytokines in these cocultures was analyzed at 6 hours demonstrating a different secretome in the presence of CB-NK (figure 6F) and that cytokines involved in T cell migration, such as CCL3 and CCL5, were increased in the presence of CB-NK (figure 6G). Of interest, cytokines related to CRS development, such as TNFα, were not increased in the presence of CB-NK (figure 6F).
CB-NK improves the fitness of CARpos and CARneg T cells exposed to MM cells
Last, we analyzed how the impact of CB-NK on ARI2h cells observed at early time points (figure 6) would influence ARI2h cells exposed to MM cells in long-term challenge assays. These long-term assays were performed at low E:T ratios adding cells highly diluted so that the killing would take longer (figure 7A). These experiments were performed with six different donors for T cells and CB-NK from three different CB units. Of interest, the impact of CB-NK over the activity of T cells was not related to differences observed in the expression of some NK cell receptors (online supplemental figure 5). We observed that after encountering MM cells, the total number of CARpos and CARneg CD8+ cells was higher in the presence of CB-NK, suggesting either a higher expansion or a lower cell death (figure 7B and C). The transition of memory stages of T cells was analyzed after production of ARI2h cells and after challenging to MM cells. In the naïve/stem cell memory (SCM) compartment, a decrease in the proportion of naïve CARneg CD4 and CD8 T cells was observed during the expansion, with no changes observed in the SCM compartment (online supplemental figure 4A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CB-NK improves the fitness of CARpos and CARneg T cells exposed to MM cells (see also online supplemental figure 4 and 5). (A) T cells (D.0) were expanded and transduced to produce ARI2h cells (after Exp), which were cocultured in vitro for 6 days with ARP1 MM cells at 0.5:1 (E:T) ratio in the presence (Chall (NK)) or absence of CB-NK (Chall (no NK)). % of ARI2h cells was normalized at 40% for all assays. At those time points, the fitness of T cells was analyzed by flow cytometry (B–I). (B–C) Variation of the total number of CARpos (B) and CARneg (C) CD4+ and CD8+ cells after challenge to MM cells in the presence or absence of CB-NK. (D) Memory phenotype (CM: central memory; EM: effector memory; Eff: effector) of CD4+ and CD8+ ARI2h+ cells at the different time points shown in figure part A. (E) Analysis of exhaustion markers of CD4+ and CD8+ ARI2h+ and ARI2h- cells after challenge to Arp1 cells in the presence/absence of CB-NK. (F–G) % of CD27−CD28− cells in CD4+ and CD8+ ARI2h+ and ARI2h− cells at the different time points shown in figure part A. Figure part F is a representative plot of variation levels at the different time points, and figure part G is the comparative analysis versus the presence/absence of CB-NK. (H and I) Cell size and complexity of CD4+ and CD8+ ARI2h+ and ARI2h− cells at the different time points shown in figure part A. Plots shown at day 0 in figure parts D and F correspond to ARI2h− cells (before transduction). After Exp: after production of ARI2h cells. Experiments were performed with six donors of T cells and CB-NK from three CB units. Phenotype of CB units is shown in online supplemental figure 5. *P<0.05 and **p<0.0001. CB-NK, cord blood-derived NK cells; Chall, challenge of immune cells to MM cells; E:T, effector:target; MM, multiple myeloma; NK, natural killer.
In the central memory (CM) and effector memory (EM) compartments, at day 0, before production of ARI2h cells, there was a lower proportion of CM in comparison with EM T cells in both CARpos and CARneg T cells (figure 7D and online supplemental figure 4B). Of note, CM T cells present higher capacity for homing to secondary lymphoid organs, while EM T cells are more cytolytic and express integrins and chemokine receptors necessary for localization to peripheral tissues.26 The production/expansion of ARI2h cells induced an increase in CM and a decrease in EM for CARpos CD4 and CD8 T cells (figure 7D). For CARneg T cells, the expansion only caused an increase in the CM compartment (online supplemental figure 4B). After exposure to MM cells, the proportion of CM CARpos CD4 T cells decreased concomitant with an increase in EM CARpos cells (figure 7D). In the presence of CB-NK, these changes were more pronounced (figure 7D), suggesting a more differentiated phenotype and a faster capacity of CAR-T cells to migrate where MM cells are emerging. For CARpos CD8 T cells, exposure to MM cells caused an increase in the EM compartment only in the presence of CB-NK (figure 7D). Of interest, no changes were observed in the number of terminally differentiated effector ARI2h cells (figure 7D). However, an increase in effector cells was noticed for both CARneg CD4+ and CD8+ T cells that was more pronounced for CD8+ T cells in the presence of CB-NK (online supplemental figure 4B).
Moreover, the fitness of ARI2h cells after exposure to MM was analyzed by looking at exhaustion and immunosenescent parameters. The presence of CB-NK decreased the expression of exhaustion markers PD-1, TIM3 and LAG3 in both CARpos and CARneg T cells. Furthermore, TIGIT levels were also reduced in CARpos CD4+ T cells and both CARneg CD4+ and CD8+ T cells (figure 7E).
Loss of CD27 and CD28, known as markers related to the development of immunosenescence, specifically after chronic viral infection,27–29 were also improved in the presence of CB-NK. Specifically, exposure to MM cells induced an increase in the emergence of CD27−CD28− cells in CARpos T cells (figure 7F), a detrimental impact that was reduced in the presence of CB-NK for the CARpos CD8+ T cell population (figure 7G). Senescent cells are also characterized by presenting a higher cell size, an event that impairs cell proliferation.30 Importantly, production/expansion of ARI2h cells induced an increase in both cell size and complexity for all CARpos and CARneg T cells that continued to rise after exposure to MM cells (figure 7H). CB-NK avoided the increase in cell size and cell complexity of both CARpos and CARneg CD8+ and CD4+ T cells after exposure to MM cells (figure 7I) suggesting a delayed emergence of senescence in ARI2h cells due to the presence of CB-NK.
Discussion
CAR-T cell immunotherapy has modified the treatment of hematological malignancies. However, optimal clinical results fluctuate depending on the disease. Specifically, pediatric ALL patients treated with CART-19 cells maintain durable responses with the presence of CAR-T cells many years after administration,3 whereas older patients with conditions such as NHL10 and MM16 present poorer responses due to lack of persistence of CAR-T cells. This lack of persistence, among other factors, might be due to a decreased fitness of the immune cells acquired with aging.29 31 Here, we demonstrate that CB-NK can be combined with CAR-T cells, leading to enhanced in vivo antitumor efficacy without increasing parameters related to CRS in a model of MM. Importantly, CB-NK enhanced the fitness of CAR-T cells after in vitro exposure to MM cells promoting lower levels of exhaustion and senescence, suggesting the use of CB-NK as an approach to enhance the efficacy of CAR-T cell treatment.
A lot of strategies are being explored to enhance the efficacy of CAR-T cells without increasing associated CRS and neurotoxicity. These possibilities include the creation of complex constructs to enhance the persistence of CAR-T cells32 and also the use of universal allogeneic CAR-T cells that are derived from more ‘healthy’ T cells. In this regard, cord blood units can be used as a source to obtain ‘off-the-shelf’ immune cells including CB-NK and CAR-NK cells.1 6 17 33 Clinical studies indicate that NK cells are mostly effective as a consolidation therapy in AML patients,7 34–36 but their efficacy is much lower for other malignancies.7 However, CAR-NK cells derived from CB-NK have demonstrated safety in NHL patients. However, the durability of response or CAR NK persistence is unknown,6 37 suggesting that improvements either in the CAR construct or changes in clinical protocols might still be needed to ensure long-term responses. Here, we confirm that in comparison with CAR-T cells, CAR-NK cells are only effective at high doses (high E:T ratio), that their activity decreases rapidly over time, and that, in addition, they require IL2, which can be provided by T cells. In addition, the combination of CAR-NK and CAR-T cells demonstrated that a minimum number of CAR-T cells is required to achieve responses, an event confirmed in clinical studies.38 An ideal immune cell treatment should be able to maintain efficacy at the lowest dose possible and to persist long enough to avoid relapses. These results added to our previous findings demonstrating that proteins secreted by CB-NK enhance the anti-MM activity of UT T cells20 led us to hypothesize and confirm that CB-NK could be a tool to enhance the efficacy of CAR-T cells.
The choice of adding half the dose of CB-NK versus CAR-T cells was based on clinical studies in cancer where infusion of very high and repetitive doses of NK cells has not shown efficacy.39 Moreover, studies in microbial infections show that NK cells and T cells kill each other to avoid detrimental consequences of excessive inflammation. Thus, in cases of massive viral and bacterial infections, NK cells kill T cells to avoid a devastating inflammatory response by T cells that could lead to death.40–44 Similarly, regulatory T (T-reg) cells eliminate NK cells to avoid an exacerbated inflammatory response mediated by NK cells,45 suggesting the relevance of maintaining an optimal ratio between both populations. This intricate regulation occurs also in patients with cancer receiving NK cells, where responding patients had lower levels of T-reg cells than non-responding patients,46 and where T-reg depletion led to improved responses.35 However, another study demonstrated increased T-reg cells in responding patients47 indicating the complexity of these interactions. This complex regulation between both cell populations added to the fact that PB NK cells are present in much lower numbers than T cells led us to use only a half dose of CB-NK in comparison with CAR-T cells.
The innate antitumor activity of NK cells occurs at earlier time points than that of adaptive T cells, where NK cells are important in the regulation of the activity of adaptive T cells.48 We demonstrate that both CAR-NK cells and CB-NK initiate their antitumor activity earlier than CAR-T cells, which significantly, in the case of CB-NK, led to a faster activation of CAR-T cells and promotion of their migration to MM cells at early time points. Of note, prior to receiving CAR-T cells, patients undergo a lymphodepleting conditioning regimen15 16 to eliminate residual T cells and thus avoid a possible immunogenicity against murine components of the CAR.31 This conditioning regimen also depletes NK cells for at least 15 days,49 a time at which the performance of CAR-T cells is crucial. Our results show that adding a low number of CB-NK in the CAR product led to an enhancement of the efficacy of CAR-T cells after the first days of CAR-T cell treatment with no enhancement in the CRS and improved fitness of CAR-T cells. In addition, the immunoregulatory role of NK cells in avoiding detrimental inflammation by T cells, as observed in viral infections,40 could represent an advantage here to avoid severe CRS during the initial period after administering CAR-T cells.
Moreover, CB-NK enhanced the health of both CARneg and CARpos T cells, with lower expression of exhaustion and immunosenescence markers after exposure to MM cells. Exhausted or senescent T cells negatively impact the long-term persistence of CAR-T cells,50 51 which is critical to avoid relapses.3 15 16 T cell exhaustion is a common problem in cancer, which can be reversed with immunecheckpoints inhibitors. Specifically, MM patients receiving an autologous stem cell transplant develop exhausted CD4 and senescent CD8 T cells, with high expression of LAG3 and TIM3 associated with poor outcomes.52 Moreover, T cell immunoreceptor with Ig and ITIM domains (TIGIT) is another potential target of immune modulation in MM patients.53 Given the fact that clinical trials with immunocheckpoint inhibitors in MM were halted due to their high toxicity,54 the impact of CB-NK ameliorating exhaustion in T cells could represent a promising alternative. Moreover, MM patients are an elderly population with senescent T cells with lower proliferative capacity and impaired function.55 Here, CB-NK reversed parameters related to senescence in both CARneg and CARpos T cells, suggesting an interesting approach to improve immunosenescence in T cells in MM.
In summary, CB-NK and CAR-NK cells can be used as universal sources of immune cells for the treatment of cancer patients. However, their lower efficacy in comparison with CAR-T cells highlights the need to look for novel strategies to improve their therapeutic potential. Here, we present CB-NK as a novel approach to enhance the efficacy of CAR-T cells in a model of MM, importantly without enhancing parameters related to CRS development. Moreover, CB-NK prevent the emergence of exhausted and senescent T cells after exposure to MM cells, which suggests they could lead to enhanced persistence of CAR-T cells.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All information is in the manuscript. All information is in the manuscript.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the Ethics Committee of Hospital Clinic of Barcelona (2017/0438 and 2020/0048). All animal work was performed under approval of Ethical Committee of Animal Research (University of Barcelona, procedure 230/19). Mice were euthanized when they reached a score that determined the lack of mobility, lack of response to stimuli or weight loss as specified in our protocol approved by the Ethical Committee of Animal Research of the (procedure 230/19).
Acknowledgments
We would like to acknowle Multiple Myeloma Research Center (Little Rock, Arkansas) for providing the ARP1 cell line. We would also like to acknowledge Dean A Lee (The Ohio State University School of Medicine) for providing the feeder cells. We would like to acknowledge the facility of Confocal Fluorescence Microscopy at the University of Barcelona.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MB and LP-A contributed equally.
Contributors BM-A conceived and designed the study, LP-A performed in vitro and in vivo experiments related to chimeric antigen receptor (CAR)-natural killer (NK) cells and non-Hodgkin’s lymphoma (NHL). MB performed in vitro and in vivo experiments related to the combination of cord blood-derived NK (CB-NK) with CAR-T cells in multiple myeloma. LP-A, MB and BM-A analyzed data and wrote the manuscript. EV provided the BaEV plasmid. AN designed the scFv of ARI2 and provided plasmid coding for GFP-Ffluc. AMB performed the multiplex immunoassays, designed the flow cytometry panels and edited the manuscript. MJ provided the construct for ARI1 and provided funding. AU-I provided funding. All authors read and approved the manuscript.
Funding La Caixa Foundation (CP042702) and Institute of Health Carlos III (projects: PI17/01043, PI20/00991 and PI18/00775) provided funding for all studies.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.