Article Text

Abstract
Background Pancreatic cancer (PC) is a challenging diagnosis that is yet to benefit from the advancements in immuno-oncologic treatments. Irreversible electroporation (IRE), a non-thermal method of tumor ablation, is used in treatment of select patients with locally-advanced unresectable PC and has potentiated the effect of certain immunotherapies. Yeast-derived particulate β-glucan induces trained innate immunity and successfully reduces murine PC tumor burden. This study tests the hypothesis that IRE may augment β-glucan induced trained immunity in the treatment of PC.
Methods β-Glucan-trained pancreatic myeloid cells were evaluated ex vivo for trained responses and antitumor function after exposure to ablated and unablated tumor-conditioned media. β-Glucan and IRE combination therapy was tested in an orthotopic murine PC model in wild-type and Rag−/− mice. Tumor immune phenotypes were assessed by flow cytometry. Effect of oral β-glucan in the murine pancreas was evaluated and used in combination with IRE to treat PC. The peripheral blood of patients with PC taking oral β-glucan after IRE was evaluated by mass cytometry.
Results IRE-ablated tumor cells elicited a potent trained response ex vivo and augmented antitumor functionality. In vivo, β-glucan in combination with IRE reduced local and distant tumor burden prolonging survival in a murine orthotopic PC model. This combination augmented immune cell infiltration to the PC tumor microenvironment and potentiated the trained response from tumor-infiltrating myeloid cells. The antitumor effect of this dual therapy occurred independent of the adaptive immune response. Further, orally administered β-glucan was identified as an alternative route to induce trained immunity in the murine pancreas and prolonged PC survival in combination with IRE. β-Glucan in vitro treatment also induced trained immunity in peripheral blood monocytes obtained from patients with treatment-naïve PC. Finally, orally administered β-glucan was found to significantly alter the innate cell landscape within the peripheral blood of five patients with stage III locally-advanced PC who had undergone IRE.
Conclusions These data highlight a relevant and novel application of trained immunity within the setting of surgical ablation that may stand to benefit patients with PC.
- Tumor Microenvironment
- Immunity, Innate
- Phagocytosis
- Immunotherapy
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Recent evidence has demonstrated that inducing trained immunity by Bacillus-Calmette-Guérin and influenza vaccines, as well as natural compounds such as β-glucans, is capable of reducing tumor progression and controlling tumor metastasis. It is critical to understand how tumor cells elicit a trained response and whether the intensity or level of trained immunity is associated with therapeutic efficacy.
WHAT THIS STUDY ADDS
We demonstrated that irreversible electroporation (IRE)-ablated tumor cells elicited a potent trained response and augmented antitumor innate immunity. In vivo, β-glucan in combination with IRE reduced local and distant tumor burden, prolonging survival in a murine orthotopic pancreatic cancer (PC) model. Orally administered β-glucan in patients with stage III PC who had undergone IRE also induced a trained immunity phenotype in peripheral monocytes.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Based on our findings, it is suggested that inducing trained immunity in patients with PC may elicit antitumor innate immunity and augment the current immunotherapeutic modalities, such as immune checkpoint blockade therapy.
Introduction
Diagnosis and management of pancreatic cancer (PC), most commonly ductal adenocarcinoma, is challenging. Despite modest advancements in systemic chemotherapy, only 10.8% of patients survive to 5 years.1 2 Anatomic location and tumor biology often allow PC to remain symptomatically inconspicuous during early stages of disease. Therefore, many patients are diagnosed when local invasion or metastasis to distant organs has already occurred.3 Complete surgical resection offers the best chance for long-term survival, however, only 20% of patients are operative candidates at the time of diagnosis.4 5 Even with neoadjuvant chemotherapy followed by complete (R0) resection, the risk of recurrence is considerably high.6 Therefore, a critical need exists to develop and offer more efficacious treatment options within the setting of PC.
Immune checkpoint inhibitor (ICI) therapy has undoubtedly transformed many solid cancer treatment algorithms, however, the promises of ICI have failed to be translated to PC.7 This may be largely attributed to PC’s notoriously immunosuppressive tumor microenvironment (TME),8 which is a model for cancer immune evasion.9 PC is characterized by a highly fibrotic and desmoplastic stroma rich in immunosuppressive cells, particularly innate myeloid cells such as tumor-associated macrophages and myeloid-derived suppressor cells.10 11 Moreover, the PC TME promotes effector T-cell exclusion with low neoantigen abundance and tumor mutational burden, effectively eliminating proper antitumor adaptive responses creating both a structural and functional barrier to current ICI therapies.12
Irreversible electroporation (IRE)13 is an ablation technology that is currently approved for unresectable stage III locally advanced (LA) PC.14 IRE delivers targeted high voltage electric pulses across the tumor inducing an apoptotic cell death via creation of permanent cell membrane porosity. Compared with alternative ablation techniques, IRE is ‘non-thermal’ and elicits cell death without disruption of collagenous tissue structures.15 Recently, studies have demonstrated the ability of IRE to circumvent the immunosuppressive PC TME improving the efficacy of immunotherapy and delivery of chemotherapy.16 For example, IRE in combination with anti-programmed death protein-1 (anti-PD-1) significantly prolonged survival in a murine orthotopic PC model by alleviating tumorous hypoxia.17 IRE has also potentiated dendritic cell (DC) therapy previously shown to be ineffective in PC.18 IRE in conjunction with a toll-like receptor-7 agonist and anti-PD-1 eliminated untreated distant tumors.19 Target median survival in patients with LA or locally recurrent PC has also been exceeded by percutaneous IRE combined with chemotherapy as demonstrated in the phase II PANFIRE study.20 Although promising, there remain large gaps in knowledge regarding local and systemic immune effects of IRE.21 Taken together, independent applications of IRE or single agent immune based therapies are inadequate for the treatment of PC.
Contrary to the classic immune dichotomy, innate immune cells have been recently shown to possess a ‘de facto’ immunologic memory. This process, coined ‘trained immunity’, is defined as an enhanced cellular response (also known as the trained response) of initially trained innate cells following a secondary stimulus.22 23 Trained immunity agonists, such as β-glucan and Bacillus-Calmette-Guérin (BCG) vaccine, have previously been shown to exert antitumor activity.24 25 Intraperitoneal (IP) injected yeast-derived particulate β-glucan was recently reported to traffic to the murine pancreas and induce trained immunity phenotype leading to anti-PC immunity.26 Despite these encouraging findings, much of this data used prophylactic β-glucan prior to tumor challenge and all mice eventually succumbed to disease prompting further investigations. For example, how may the trained response be better activated or engaged in the setting of cancer? What level of trained response is beneficial for cancer therapy? What factors have the capacity to act as a secondary stimulus in the setting of β-glucan induced trained immunity? Our previous study showed that β-glucan trained pancreatic myeloid cells elicit a trained response on stimulation of tumor-derived damage-associated molecular patterns (DAMPs). Cell exposure to IRE is well known to increase synthesis and release of DAMPs and other danger signals.27
In this study, the hypothesis that IRE would enhance the anti-PC efficacy of β-glucan by inducing potent trained response is investigated. We demonstrate an effective and novel PC treatment combination whereby β-glucan-mediated trained immunity is augmented by IRE. This combination involves a pronounced and activated myeloid immune cell population within the PC TME. These findings suggest that the combination of trained immunity with IRE warrants further investigation by clinical trials.
Methods
Mice
Wild-type (WT) C57Bl/6 female and male mice, 6–8 weeks old, were purchased from Charles River Laboratories (Wilmington, Massachusetts, USA). B6.129S7-Ragtm1Mom/J (Rag−/−) mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). All mice were housed at the University of Louisville in a specific pathogen-free animal facility. All experiments were conducted in accordance with the relevant laws and institutional guidelines (IACUC#19471, 19536, 22115) provided by the Rodent Rearing Facility and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Louisville (Louisville, Kentucky, USA).
β-glucan preparation
Saccharomyces cerevisiae derived particulate β-glucan (Biothera), was dissolved in 1x PBS and sonicated at 20 pulses twice for 10 s each on ice using a Qsonica Q55-110 Q55 Sonicator (Cole-Parmer) before treatment or injection.
DTAF labeled β-glucan preparation and administration
5-((4,6-Dichlorotriazin-2-yl)amino) fluorescein hydrochloride (DTAF, 2 mg/mL, Sigma-Aldrich) was incubated with continuous shaking at room temperature (RT) for 8 hours in 20 mg/mL β-glucan in borate buffer (pH 10.8). The mixture was then washed and centrifuged in cold sterile endotoxin-free Dulbecco’s phosphate buffered saline (DPBS, Sigma-Aldrich) to remove any visible traces of DTAF in the supernatant. The DTAF-β-glucan was maintained and stored at 10 mg/L concentration in 4°C. Mice were given once daily 1 mg oral DTAF-β-glucan for 6 days or IP injected with 1 mg of DTAF-β-glucan 24 hours prior to euthanasia and harvest of the pancreas for trafficking studies.
Pancreas and tumor digestions/preparation of single-cell suspensions
Weighted and measured tumors or normal pancreases were mechanically minced and digested in a mixture of complete Roswell Park Memorial Institute (RPMI) 1640 and (1x) digestion buffer containing 300 U/mL collagenase I, 60 U/mL hyaluronidase, and 80 U/mL DNase (Sigma) at 37°C with 5% CO2 30 min with continuous rotation.The digestion reaction was stopped with cold complete RPMI 1640 then the cells were passed through a sterile nylon 40 µm basket filter. Undigested tissue was smashed through the filter using a sterile syringe stopper. Red blood cell lysis was performed by adding 0.5 mL of sterile ACK (Ammonium-Chloride-Potassium) buffer (Thermo Fisher Scientific).
Ex vivo restimulation assays
Single-cell suspensions from the pancreas were plated in a 24-well plate (2×106 cells/well) and stimulated with lipopolysaccharide (LPS) (10 ng/mL), recombinant MIF (rMIF), tumor-conditioned media, or IRE-conditioned media and incubated at 37°C with 5% CO2 for 4 hours in the presence of 1X brefeldin A (BioLegend). The cells were then harvested using a mini-cell scrapper (United Biosystems), pelleted, and stained for surface markers followed by intracellular tumor necrosis factor (TNF)-α staining.
In vitro ablations
Tumor cells were removed from culture with 0.25% trypsin, washed with complete Dulbecco’s Modified Eagle’s Medium, and resuspended in tumor-conditioned media at a concentration of 2×106 cells/mL. Cells were electroporated in 4 mm gap sterile electroporation cuvettes (Universal Medical) using the Safety Stand 630B adaptor for the BTX 830 square wave electroporation system, Harvard apparatus. The cells were subjected to electroporation at RT with the following parameters: Voltage 500–1000V, pulses 10–20, 0.1 ms duration, and 1 s pulse interval.
Phagocytosis assay
WT mice were IP injected with β-glucan, the pancreas was harvested 7 days later, and processed to single-cell suspensions. Positive selection using CD11b+ microbeads (Miltenyi Biotec) was performed using the autoMACS Pro Separator. Next, 0.75×106 of the resulting CD11b+ cells were washed with complete RPMI 1640 and incubated for 24 hours with either KPC tumor-conditioned media or IRE-conditioned media. Next, GFP+ KPC tumor cells were added to the activated pancreatic CD11b+ cells at a ratio of 4:1 and incubated for 1.5 hours at 37 °C. Samples were gently vortexed every 15 min. The reaction was stopped by adding 1 mL of cold PBS. Samples were incubated with Fc Block for 10 min at 4 °C, stained for viability, anti-CD11b (APC, BioLegend) for 30 min at 4 °C, and then analyzed using CyTEK. For analysis, after gating on live cells, CD11b+ cells that also co-expressed GFP were determined to be phagocytic.
Cytotoxicity assay
Pancreases from β-glucan IP treated mice were harvested and processed to single-cell suspensions. Positive selection using the autoMACS Pro Separator and CD11b+ Microbeads (Miltenyi Biotec) was performed to isolate myeloid cells. CD11b+ cells were then co-cultured with luciferase-expressing KPC pancreatic tumor cells or IRE ablated (500V, 20 pulses) luciferase-expressing KPC pancreatic tumor cells in a 96 well plate at 37°C for 24 hours. The plates were centrifuged at ambient temperature and 20 µL of the supernatant was mixed with 100 µL of the Luciferase Assay Reagent (Promega). Luciferase activity measured in the supernatant and whole cell lysates correlated with tumor cells that had been killed by the effector cells and was measured using a luminometer (Femtomaster FB 12, Zylux Corporation). Spontaneous luciferase signal from plated tumor cells was subtracted from the measurement of the supernatant. Luciferase values are represented as Relative Light Units.
In vivo orthotopic tumor model
Orthotopic pancreatic tumors were established by surgical implantation of 0.1×106 KPC cells into the pancreatic parenchyma of 6–8 weeks old female and male C57Bl/6 or Rag−/− mice. The KPC cell line was derived from the LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre (KPC) mouse model and purchased from Ximbio, as previously described.26 Briefly, mice were anesthetized with 2% inhaled isoflurane, given intramuscular buprenorphine analgesia (0.2 mg/kg), and supplemental oxygen. The abdomen was prepped and draped in sterile fashion. A 2 cm left upper quadrant laparotomy was performed. The tail of the pancreas was identified and eviscerated. A 30-gauge needle was inserted into the tail of the pancreas at its bifurcation. A 1:1 ratio of KPC tumor cells suspended in cold 1x PBS and Matrigel Matrix (Discovery Labware, Corning REF 35623) was infused into the pancreas using a 30-gauge needle creating a weal. The pancreas was placed back into anatomical position. The abdomen was closed in two layers using inner absorbable 5–0 Vicryl suture and skin closed with 4–0 Nylon suture (CP Medical). All mice were monitored for 48 hours following surgery and were administered buprenorphine for analgesia.
IRE and sham surgery
Fourteen days following tumor injection the mice were again anesthetized and prepped as described above. A separate 2 cm midline laparotomy was performed. The tumor was then identified within the tail of the pancreas, externalized, and measured using calipers. IRE was performed when the tumors reached 3–4 mm in maximum diameter using the BTX 830 square wave electroporation system, Harvard apparatus. Platinum tweezer trodes (BTX) were used to deliver 100 pulses at 1000 V/cm, 0.1 ms duration, and 0.1 s pulse interval. Sham/placebo surgery was performed for PBS control and β-glucan control groups, omitting only IRE.
ELISA
The supernatants of cultured and ablated KPC and S2013 were analyzed using murine and human MIF ELISA kits (R&D Systems). Supernatant from in vivo trained ex vivo stimulated CD11b+ cells were analyzed using murine TNF-α and interleukin (IL)-6 ELISA kits (BioLegend). To detect histone modification, histones were extracted from CD11b+ cells, which were positively selected from PBS, β-glucan, IRE, and β-glucan plus IRE treated KPC tumors using the Histone Extraction Kit (Active Motif, Catalong # 40028). Histone modifications were quantified using EpiQuik Total Histone H3, Acetyl histone H3K27, Tri-Methyl histone H3K4, and Tri-methyl histone H3K27 Quantification Kits (Colorimetric). Supernatants from in vitro trained human CD14+ cells were analyzed using human TNF-α ELISA kits (BioLegend). All assays were performed using the provided manufacturer instructions and all conditions were performed in duplicates.
qRT-PCR
RNA was extracted using TRIzol (Thermo Fisher) from CD11b+ cells isolated from PBS, β-glucan, IRE, and β-glucan plus IRE-treated KPC tumors using CD11b microbeads separation. RNA was then reverse transcribed to complementary DNA (cDNA) using iScript cDNA Synthesis Kit (Bio-Rad). Messenger RNA (mRNA) expression analysis was carried out using iQ SYBR Green Supermix and CFX Connect PCR Machine (Bio-Rad).
Surface staining for flow cytometry
Pancreas and pancreatic tumor single-cell suspensions were washed first with PBS and then Fc receptors were blocked for 10 min at 4°C. For pancreatic myeloid cells, single-cell suspensions were stained with viability dye-APC eFluor-780 (eBioscience), anti-CD45-PerCP-Cy5.5, anti-CD11b-PE-Cy7, anti-F4/80-FITC, anti-Ly6C-AlexaFluor 700, and anti-Ly6G-APC (BioLegend). For T cells, cells were stained with viability dye-APC eFluor 780, anti-CD3-PE-Cy5, anti-CD4-APC, anti-CD8-PerCp-Cy5.5, anti-CD69-AlexaFluor 700, anti-TIM-3-PE-Cy7, anti-LAG-3-PE-Dazzle 594, and anti-PD-1-APC (BioLegend). Cells were incubated at 4°C for 30 min, washed with cold PBS, filtered, and collected using CyTEK Aurora Northern lights 2 laser flow cytometer (CyTEKbio). All flow cytometry data were analyzed using FlowJo software (BD).
Intracellular staining
Mononuclear single-cell suspensions were stimulated in culture for 4–6 hours with LPS (10 ng/mL) or 1x PMA/I then stained for surface markers as previously described above after being washed with cold PBS. The cells were then fixed using 500 mL of fixation buffer (BioLegend) for 20 min at RT. Next, the cells were centrifuged at 1600 rpm for 5 min at 4°C, supernatants discarded, and then washed twice with 1x permeabilization buffer (BioLegend). Cells were then stained with anti-TNF-α-PE, anti-IFN-γ-PE, or anti-Granzyme B-PE (BioLegend) along with respective isotype controls and incubated at 4°C overnight. The samples were washed with 1 mL of 1x permeabilization buffer, filtered and suspended in 250 mL of 1x permeabilization buffer for acquisition by the CyTEK flow cytometer.
Histology
Mice followed for survival were humanely euthanized once showing signs of disease progression. The lungs were harvested and placed in 4% formalin followed by paraffin embedding. Tissues were cut in 5 mm sections and then stained with H&E.
Human subject study
Patients with pathologically confirmed and surgically unresectable stage III LA pancreatic adenocarcinoma were recruited after informed consent to undergo IRE followed by adjuvant oral β-glucan. All patients were provided a highly purified 500 mg β-glucan supplement (Wellmune, Immune Health Basics). Oral β-glucan administration was initiated at the time of post IRE hospital discharge and continued twice daily. Baseline blood was obtained preoperatively prior to IRE and at their 3-month postoperative office visit.
Isolation of human PBMC and CD14+ monocytes
Human blood was obtained via venipuncture after informed consent into sterile EDTA (ethylenediaminetetraacetic acid) collection tubes. Whole blood was mixed with RPMI 1640 in a 1:1 ratio and then gently layered over 5 mL of lymphocyte cell separation medium and then centrifuged at 2000 rpm for 20 min at RT. The resulting mononuclear cell layer was carefully removed and then washed in RPMI 1640. The cells were then stained for viability dye and anti-CD14 (FITC, BioLegend) and then sorted using the BD FACSAria.
In vitro training assay with β-glucan
Human CD14+ monocyte cells were cultured in complete RPMI 1640 at a concentration of 0.2×106 cells per 48 well plate in 500 µL total. The CD14+ cells were stimulated with 10 ng/mL of β-glucan for 24 hours at 37°C and 5% CO2. The cells were then washed and allowed to rest for 6 days. On day 7, the cells were stimulated with either LPS (10 ng/mL) or tumor-conditioned supernatant or IRE-treated supernatants for 24 hours. The supernatants were then collected for quantification of TNF-α using ELISA.
CyTOF mass cytometry sample preparation
Mass cytometry antibodies were either purchased from Fluidigm or were conjugated from commercially available purified antibodies to the appropriate metal isotope using the MaxPar X8 Polymer or MCP9 Polymer kits (Fluidigm). Peripheral blood from five patients with stage III LA-PDAC (pancreatic ductal adenocarcinoma) was acquired at baseline (prior to IRE or β-glucan) and 3 months post IRE while taking 1000 mg daily oral adjuvant β-glucan. Blood was processed into peripheral blood mononuclear cells (PBMCs) as described above and were kept frozen at –140°C prior to use. About 2×106 cells per sample were used. Cells were first stained for viability with 5 uM cisplatin (Fluidigm) in serum-free RPMI 1640 for 5 min at RT. Cells were then washed with RPMI 1640 containing 10% fetal bovine serum for 5 min at 300 × g. Cells were stained with the surface antibodies for 30 min at RT and washed twice with Maxpar Cell staining buffer (Fluidigm). The cytoplasmic/secreted antibody cocktail was then added and incubated with the cells for 30 min at RT. Following incubation, cells were washed with 1 mL of 1x Maxpar Perm-S buffer for 5 min at 800 × g and gently blotted to remove all liquid from the tube. In order to stain for nuclear antigens, cells were then suspended in 1 mL of 1x Maxpar nuclear antigen staining buffer for 30 min at RT. The nuclear antigen antibody cocktail was then added and incubated for 30 min at RT. Cells were washed twice for 5 min at 800 × g with 2 mL of Nuclear Antigen Staining Permeability buffer. Finally, cells were fixed with 1.6% formaldehyde for 10 min at RT, then incubated overnight in 125 nM of Intercalator-Iridium (Fluidigm) at 4 °C.
CyTOF data acquisition and analysis
Prior to acquisition, the cell samples were washed twice with Cell Staining Buffer (Fluidigm) and kept on ice. Directly prior to the acquisition, cells were suspended in a 1:9 solution of Cell Acquisition Solution: EQ 4 element calibration beads (Fluidigm). Using the CyTOF software, FCS files were normalized into .fcs files for data analysis. CyTOF data was analyzed using FlowJo, CytoBank software package, and the CyTOF workflow which includes a suite of packages available in R (r-project.org). For analysis conducted within the CyTOF workflow, FlowJo workspace files exported from flow Workspace and CytoML were used.
Statistical analysis
All results are presented as mean±SEM. Flow cytometry data was analyzed using FlowJo V.10.7.1 for Mac OSx. Statistical significance was determined by ordinary one-way analysis of variance with post hoc multiple comparison or by independent or paired samples t-test when appropriate using GraphPad Prism V.9.2 for Mac OSx. All p values of <0.05 were considered significant.
Results
IRE results in DAMP release from PC cells that elicits a potent trained response with enhanced phagocytosis and cytotoxicity
Enhanced TNF-α cytokine responses from β-glucan trained pancreatic CD11b+ myeloid cells after secondary exposure to tumor-conditioned media have been previously observed.26 Macrophage migration inhibitory factor (MIF) is an example of a DAMP released by PC cells capable of eliciting the trained response. However, it is unknown how increasing doses of MIF affect trained pancreatic CD11b+ TNF-α expression or if the level of trained response may be augmented. To address this, pancreatic myeloid cells from IP β-glucan trained mice were secondarily exposed to rMIF at increasing doses ex vivo. As demonstrated in figure 1A, rMIF increased β-glucan trained pancreatic CD11b+ myeloid cell TNF-α production in a dose-dependent manner. Prior studies have shown that IRE induces an immunogenic cell death. Further, exposure of PC cells to IRE releases DAMPs and other pro-inflammatory factors.17 18 We then compared MIF levels by ELISA from the tumor-conditioned media of cultured KPC cells to IRE-ablated KPC cells. After IRE, the concentration of MIF within the IRE-ablated supernatants increased nearly 80 times that of KPC-conditioned media alone (figure 1B). We next examined whether IRE-conditioned media may better serve as a secondary stimulus compared with tumor-conditioned media alone. Indeed, trained pancreatic CD11b+ myeloid cells released more TNF-α and IL-6 when exposed to ablated IRE-conditioned media compared with unablated KPC media alone (figure 1C). We then validated these findings by quantifying intracellular TNF-α using flow cytometry after short term culture (4 hours). Again, β-glucan-trained myeloid cells stimulated with supernatants derived from IRE-treated KPC cells increased intracellular TNF-α cytokine production compared with KPC media alone in both percent and mean fluorescent intensity (MFI) (figure 1D). This data further supports that soluble factors released from IRE-ablated PC cells can act as the secondary stimulus within the setting of trained immunity and that the specific levels of trained response can be augmented by increased doses of secondary stimulus.

IRE releases more MIF from PC tumor cells which elicits a potent trained response in β-glucan-trained pancreatic myeloid cells with enhanced phagocytosis and cytotoxicity. (A) Summarized pancreatic CD11b+TNF-α+ cells from untrained versus β-glucan trained mice on ex vivo restimulation with LPS or increasing doses of rMIF assessed by flow cytometry. n=3 per group. (B) Concentration of MIF (ng/mL) in supernatants after culture of 1×106 KPC cells for 24 hours or ablation of KPC cells as measured by ELISA, n=6 per group. (C) TNF-α and IL-6 (pg/mL) levels measured by ELISA from β-glucan trained CD11b+ pancreatic cells restimulated ex vivo with KPC-conditioned media or IRE-conditioned media for 24 hours. Representative flow cytometry contour plots and quantified percent and MFI of β-glucan trained CD11b+TNF-α+ cells restimulated with media control, KPC-conditioned media or IRE-conditioned media, n=3 per group. (E) β-glucan trained pancreatic CD11b+ cells were stimulated with media alone or KPC supernatant or IRE KPC supernatant and then cocultured with KPCGFP+ tumor cells. Representative flow plots and summarized data are shown. (F) Summarized results from cytotoxicity assay. CD11b+ cells from 7 day β-glucan trained mice were incubated at a ratio of 1:30 KPCLuc+ to CD11b+ cells for 24 hours. Data are representative of two or three independent experiments and presented as mean±SEM. ns, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. IL, interleukin; IRE, irreversible electroporation; LPS, lipopolysaccharide; MIF, migration inhibitory factor; PC, pancreatic cancer; rMIF, recombinant MIF; TNF, tumor necrosis factor.
Pancreatic CD11b+ myeloid cells also have an enhanced antitumor functional state after β-glucan training.26 We next sought to test whether β-glucan trained CD11b+ myeloid cells re-exposed to IRE-ablated media may demonstrate enhanced antitumor characteristics. Secondary ex vivo re-exposure of β-glucan-trained CD11b+ cells with IRE-conditioned media compared with KPC-conditioned media led to an increased phagocytosis of KPCGFP+ tumor cells in a dose-dependent manner as demonstrated by increased frequency and MFI of CD11b+GFP+ cells (figure 1E). Next, we co-cultured trained CD11b+ cells in the presence of luciferase expressing, IRE ablated or unablated, KPC cells and quantified cytotoxicity via resulting luciferase expression within the cell culture supernatants. Trained CD11b+ myeloid cells demonstrated an enhanced cytotoxicity via measured dead cell luminescence and percent cytotoxicity when co-cultured with IRE-treated KPC cells compared with untreated KPC cell controls (figure 1F). Taken together, these results suggest that IRE ablation results in increased release of DAMPs such as MIF in PC cells that induces a potent trained response and better promotes antitumor innate immune responses.
β-Glucan in combination with IRE reduces murine PC tumor burden and prolongs survival
β-Glucan has a specific tropism to the murine pancreas when injected IP stimulating a robust influx of trained myeloid cells that lead to reduced PC tumor burden with prolonged survival.26 IRE alone has been partially effective in treatment of solid tumors, positively altering the immunosuppressive PC stroma, and has been cooperative in combination with immunotherapies.28–30 Given these findings and our aforementioned ex vivo results, we hypothesized that the combination therapy of β-glucan and IRE would further reduce PC tumor burden compared with β-glucan or IRE alone and prolong overall survival. To test this, WT C57Bl/6 mice were challenged with 1×105 KPC cells on day 0 via orthotopic injection into the tail of the pancreas near its bifurcation. The tumors were allowed to progress for 7 days before mice were treated/trained with IP β-glucan. An additional 7 days were allowed for sufficient training to occur before mice underwent IRE or sham placebo surgery on day 14. Notably, β-glucan treated mice had reductions in maximum tumor diameter compared with PBS control treated mice at the time of IRE or sham surgery (online supplemental figure 1a). The mice then were monitored postoperatively for 10 days before humane euthanasia on study day 24 or were continually monitored for survival. On day 24, tumors were then grossly compared (figure 2A). Analysis of tumor burden first confirmed efficacy of IP β-glucan and IRE treatments as monotherapy. In comparison, combination therapy achieved a significant reduction in tumor burden (figure 2B) as demonstrated by measurements of tumor weight and maximum tumor diameter (figure 2C). To determine whether the combination therapy would have beneficial long-term implications for PC in this aggressive tumor model, we then followed these mice observing for survival. The data depicted in figure 2D demonstrate single IP treatment with β-glucan and IRE ablation alone significantly prolonged survival compared with PBS controls. The combination of IP β-glucan and IRE demonstrated an 81% increase in survival (median 70.5 days) compared with PBS control (median 39 days). Epigenetic and transcriptomic reprogramming are hallmarks of trained immunity, therefore we wanted to determine whether these tumors demonstrated such changes on the molecular level.31 We thus measured histone modifications in innate myeloid cells from each treatment group and found significantly more H3K27Ac, H3K4Me3, and H3K27Me3 expression in CD11b+ cells derived from β-Glucan plus IRE treated tumors (figure 2E.) We then performed RT-PCR analysis and found that the combination therapy down-regulated transcriptional expression of Arg-1 and IL-6 in CD11b+ cells (figure 2F), which have been found to drive immunosuppression in PC.32 33 β-Glucan treated mice were also found to have increased expression of TNF-α, iNOS (inducible nitric oxide), IL-1β, and IL-12 in CD11b+ cells (online supplemental figure 1b).These findings support our hypothesis, suggesting that IRE sustains the training effect of β-glucan ultimately to decrease PC progression and results in epigenetic and transcriptomic reprogramming in innate myeloid cells.
Supplemental material

β-Glucan in combination with IRE reduces PC tumor burden and prolongs survival. (A) Schematic representation of the experimental design. Murine KPC cells were orthotopically implanted into the pancreas at day 0. Mice were treated with one IP β-glucan injection (1 mg/mouse) or PBS control 7 days after tumor challenge. On day 14, sham surgery or IRE ablation was performed. (B) Representative image of orthotopic pancreatic KPC tumors on day 24 post tumor challenge from control and different treatment groups. (C) Tumor weight and maximum tumor diameter (right). PBS n=14, IRE n=15, β-glucan n=14, β-glucan+IRE n=15, respectively. (D) Overall survival. PBS n=5, IRE n=5, β-glucan n=7, β-glucan+IRE n=7, respectively. (E) Quantification of histone modification markers H3K27Ac, H3K4Me3, and H3K27Me3 compared with total H3 in CD11b+ cells measured by ELISA. (F) Arg-1 and IL-6 messenger RNA expression in CD11b+ cells measured by RT-PCR. PBS n=5, IRE n=5, β-glucan n=5, β-glucan+IRE n=5, respectively. Data are representative of two or three independent experiments and presented as mean±SEM. ns, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Arg-1, arginase 1; IL, interleukin; IP, intraperitoneal; IRE, irreversible electroporation; PBS, phosphate buffered saline; PC, pancreatic cancer; WT, wild-type.
β-Glucan and IRE increase total immune cell infiltration in the PC TME and IRE potentiates the trained innate response from myeloid cells early within PC tumors
Induction of peripheral trained immunity stimulates an increase of trained myeloid cells within the naïve murine pancreas. This effect is most prominent 7 days following single IP injection of β-glucan and wanes to normal naïve levels by 30 days post training. Additionally, IP injection of β-glucan has previously been given prophylactically, prior to tumor exposure.26 Therefore, there is a need to positively alter the PC TME in the setting of β-glucan training over time and to evaluate β-glucan given in a neoadjuvant setting. We next examined whether the combination of β-glucan and IRE modulated the intratumoral immune cell composition. Flow-cytometric analysis revealed comparable frequencies (gating strategy demonstrated in online supplemental figure 2a) of lymphocytes and myeloid cells including macrophages, neutrophils, monocytes, and dendritic cells at day 24 post tumor challenge (online supplemental figure 2b). However, absolute numbers of tumor infiltrating immune cells and myeloid cells per gram of tumor tissue were distinctively increased in the mice treated with combination therapy. These findings were consistent across all examined innate and adaptive cell populations (figure 3A–F). These data suggest that IRE allows for establishment of a prolonged influx of leukocytes to the PC TME in the setting of prior β-glucan exposure. Prior exposure to IP β-glucan increased CD11b+F4/80+ macrophage and CD11b+Ly6C+ monocyte TNF-α when re-exposed to LPS.26 Next, we wanted to evaluate whether the infiltrating innate cells exhibited a trained phenotype. To address this, tumors were harvested from the mice on day 24 post tumor challenge (figure 2A) and tumor single-cell suspensions were stimulated with LPS. As demonstrated in figure 3G–I, mice treated with single IP injection of β-glucan followed by IRE exhibited a trained immunity phenotype expressing significantly more TNF-α from all CD11b+ myeloid cells, CD11b+F4/80+ macrophages, and CD11b+Ly6C+ monocytes, as compared with PBS control, β-glucan and IRE alone groups. These data are consistent and supported by our prior findings in which exposure to β-glucan has demonstrated an influx of trained myeloid cells into the naïve (non-tumor bearing) murine pancreas.26 These data also suggest IRE is capable of potentiating the trained effect of β-glucan in vivo and support our hypothesis that IRE may enhance the anti-PC effect of β-glucan.

Combined β-glucan and IRE treatment increases total immune cell infiltration in the PC TME and IRE induces a trained innate response in monocytes and macrophages in early tumor progression. Absolute number of live CD45+ (A), CD11b+ (B), CD11b+F4/80+ macrophages (C), CD11b+Ly6G-Ly6C+ monocytes (D), CD11b+Ly6G+Ly6C- neutrophils (E), CD11b+CD11c+MHCII+ dendritic cells (F) per gram of tumor tissue as quantified by flow cytometry. (G) Absolute TNF-α from overall CD11b+ (H) CD11b+F4/80+ macrophages (I) and CD11b+Ly6C+ monocytes per gram of tumor tissue as quantified by flow cytometry. Data are representative of two or three independent experiments and presented as mean±SEM. ns, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. IRE, irreversible electroporation; PBS, phosphate buffered saline; PC, pancreatic cancer; TME, tumor microenvironment; TNF, tumor necrosis factor.
The combination of β-glucan and IRE reduces distant tumor burden late in disease progression
Despite local tumor control or complete tumor resection, many patients diagnosed with PC will experience distant progression.34 Therefore, we wanted to evaluate distant anatomical locations known to harbor PC metastasis to determine if our combination therapy impacted PC metastasis. To this end, we followed mice for survival and evaluated each respective group for lung metastases once control PBS mice demonstrated signs of advanced tumor burden. Strikingly, we observed no metastatic lung nodules in the mice that received dual therapy, whereas the control groups had significantly increased number and size of nodules (figure 4A,B). These data indicate that the combination therapy not only has local effect, but off target distant implications in reducing tumor burden as well. It is known that the immune influx to the naïve WT pancreas resulting from single dose IP β-glucan is transient, however it is unknown whether the trained response is sustained over time.26 To answer this question, we analyzed the myeloid cells derived from the primary tumors of each respective group once PBS controls reached described physiologic endpoints. Importantly, the trained TNF-α responses to stimulation with LPS from the CD11b+Ly6C+ monocytes and CD11b+F4/80+ macrophages appear to be lost over time (online supplemental figure 3). These data demonstrate that these aggressive PC tumors escape the trained response provided by single IP β-glucan treatment even with tumor ablation. However, the prior study used LPS as a secondary stimulus to evaluate trained response. We reasoned that the act of IRE itself may serve as the secondary stimulus engaging the trained myeloid cells to produce more TNF-α. To investigate this question, we then cultured single-cell suspensions from the tumors of these long duration survival mice in the absence of LPS to evaluate for unprovoked TNF-α production from the infiltrating myeloid cell population. We found that the CD11b+ myeloid cells obtained from tumors in mice who received combination therapy expressed an increase in frequency of TNF-α production to control treated mice, with a similar trend in MFI (figure 4C,D). This data implicates that IRE alone may serve as the secondary stimulus eliciting the trained response from β-glucan trained myeloid cells synergizing to control PC progression and metastasis.

The combination of β-glucan and IRE reduces distant tumor burden late in disease progression. (A) Representative H&E images of lungs harvested from PBS control (n=5), IRE (n=5), β-glucan (n=5), or β-glucan+IRE (n=5) at the time of PBS tumor endpoint. (B) Number of lung nodules per histologic section per group and average measured size (µm) of nodules (right). (C) Representative flow cytometry contour plots of CD11b+TNF-α from PBS control (n=4), IRE (n=4), β-glucan (n=4), or β-glucan+IRE (n=5) cultured in the presence of golgi plug without LPS stimulation. (D) Quantified flow cytometry data in percent CD11b+TNF-α and MFI (right). Data are representative of two independent experiments and presented as mean±SEM. ns, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. IRE, irreversible electroporation; LPS, lipopolysaccharide; MFI, mean fluorescent intensity; PBS, phosphate buffered saline; TNF, tumor necrosis factor.
The antitumor effect of β-glucan in combination with IRE occurs independent of the adaptive immune response
Adaptive immune cells are critical in antitumor immunity, however, the lack of evidence to support ICI in PC questions their roles in treating this particular solid tumor.35 Furthermore, the antitumor effect of β-glucan has previously been shown to occur independent of the adaptive response.26 36 In considering prior IRE studies, adaptive antitumor immunity has been provoked by ablation.29 37 38 In this study we did observe an increase in absolute number of CD4+ and CD8+ T cells (data not shown). To further investigate the T-cell phenotype within this novel therapy, we repeated our experimental design in figure 2A, and evaluated immune checkpoint molecules and activation markers on CD8+ and CD4+ T cells by flow cytometry. We found an overall decrease in PD-1 expression from CD8+ T cells (figure 5A) and increases in granzyme B expression (figure 5B) in β-glucan and IRE-treated mice, suggesting a more active effector CD8+ T cell phenotype. However, CD69 and TIM-3 expression was similar on CD8+ T cells among different groups (online supplemental figure 4a). Comparable frequencies of PD-1, granzyme B, CD69, and TIM-3, were also observed from CD4+ T cells (online supplemental figure 4b). Finally, β-glucan treatment decreased LAG-3 expression in both CD8+ and CD4+ T cells although the overall expression levels were low (online supplemental figure 4a,b). To definitively determine whether an adaptive immune response was involved in eradicating disease from the dual therapy, we used Rag−/− mice (lacking T and B cells) within our orthotopic KPC model (figure 5C). Treatment of Rag−/− mice with single dose IP β-glucan followed by IRE ablation similarly reduced tumor burden (figure 5D), as measured by reduction in tumor weight and maximum tumor diameters (figure 5E). We also observed a median survival of 107 days in the combination group, a 189% increase in survival from the PBS control (median 37 days). Additionally, three mice (50%) achieved recurrence-free survival (figure 5F). These data support a critical role for innate immune cells in the control and treatment of PC in the setting of treatment with β-glucan and IRE. Further, these antitumor innate immune responses occur without actively exhausting or suppressing adaptive T cells.

The antitumor effect of β-glucan and IRE occurs independent of the adaptive immune cells. (A) Representative flow cytometry contour plots of CD8+PD1+ T cells. Cells were first gated on viability and CD45+. (B) Quantified percent and MFI of CD8+PD-1+ T cells. (C) Representative flow cytometry contour plots of CD8+granzyme B+T cells. (D) Summarized percent and MFI of CD8+granzyme B+ expression. PBS n=5, β-glucan n=5, IRE n=4, β-glucan+IRE n=5, respectively. (E) Schematic representation of experimental design using Rag−/− mice. (F) Gross depiction of orthotopic tumors harvested from Rag−/− mice 24 days after tumor challenge. (G) Tumor weight and maximum tumor diameter of tumors depicted above. PBS n=5, IRE n=5 β-glucan n=5 β-glucan+IRE n=5, respectively. (H) Overall survival of Rag−/− KPC-bearing mice treated with different regimens. PBS n=5, IRE n=5 β-glucan n=5 β-glucan+IRE n=6, respectively. Data are presented as mean±SEM. ns, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. IRE, irreversible electroporation; MFI, mean fluorescent intensity; PBS, phosphate buffered saline; PD-1, programmed death protein-1.
Orally administered β-glucan traffics to the murine pancreas, increases myeloid cell frequency, induces trained immunity, and prolongs PC survival in combination with IRE
Thus far we have used IP delivered β-glucan to induce trained immunity within the murine PC TME. All previous studies using β-glucan have also employed the IP approach to induce trained immunity in various settings.26 36 39–41 However, this administration route of training poses several unique clinical and physiologic barriers considering obvious differences in the anatomy and localization between the human and mouse pancreas, limiting its translatability to humans.42 Therefore, we wanted to explore an alternative delivery method for induction of trained immunity by β-glucan. Until now, it has remained unknown whether orally delivered β-glucan may induce trained immunity in the pancreas. To answer this question, we first investigated the optimal dose of oral β-glucan which may induce trained immunity and determined six doses of 1 mg/mouse to produce peak trained response from pancreatic CD11b+ myeloid cells re-exposed to LPS (online supplemental figure 5). We also fed WT mice orally with DTAF labeled β-glucan for six consecutive days and evaluated for β-glucan trafficking within the pancreas on day 7. A single dose of IP DTAF-β-glucan was given 24 hours prior as a positive control for comparison. Oral β-glucan was found to traffic into the murine pancreas at low levels as quantified by the frequency and absolute numbers of CD11b+DTAF+ myeloid cells and it is less potent compared with IP injected β-glucan (figure 6A,B). Importantly, oral β-glucan also stimulated a distinct influx of CD11b+ myeloid cells in the pancreas compared with PBS control (figure 6C,D). In addition, these myeloid cells were also more responsive to secondary exposure to LPS expressing more TNF-α, indicating training had occurred by oral β-glucan (figure 6E,F). We also evaluated the trained response from other innate cell subtypes (online supplemental figure 6) and found the CD11b+CD11c+ cells to significantly increase TNF-α, implying an alternative underlying cellular mechanism compared with IP administered β-glucan. We next sought to determine if these trained cells would also respond to tumor-conditioned or IRE-conditioned media. Figure 6G exhibits representative data of CD11b+ myeloid cells derived from the pancreas of mice fed β-glucan for 6 days and then re-exposed to normal media, KPC conditioned media, or IRE-treated media. Myeloid cells cultured in the presence of the IRE-ablated media produced significantly more TNF-α in both frequency and MFI (figure 6H). These data support that the intensity of the trained response can be amplified and that IRE ablation augments the level of trained response. Next, we determined if orally administered β-glucan may also provide long-term tumor benefit in prolongation of survival and if its combination with IRE would further increase its hypothesized effect. WT mice were treated with daily neoadjuvant oral β-glucan beginning on day 7 post tumor implantation. This was also continued in a neoadjuvant manner for the combination group receiving IRE. After recovery from IRE or placebo surgery, oral β-glucan was again initiated in an adjuvant setting and continued daily for 4 weeks as mice were observed for survival (figure 6I). Importantly, mice who received oral β-glucan lived significantly longer than control PBS treated mice (median survival 52 vs 37 days, figure 6J). Moreover, improved overall survival was observed when oral β-glucan trained mice were treated in combination with IRE ablation (median survival 93 days) a 151% increase in survival compared with PBS control. Together, these data suggest that oral administration of β-glucan can also induce trained immunity in the murine pancreas, similarly prolonging PC survival, and works in treatment combination with IRE.

Orally administered β-glucan traffics to the murine pancreas, induces trained innate immunity, and prolongs survival in combination with IRE in PC-bearing mice. (A) WT mice were treated with one dose of IP DTAF-β-glucan or oral DTAF-β-glucan for 6 days. On day 7, mice were euthanized and pancreatic myeloid cells were assessed for β-glucan uptake by flow cytometry. Representative flow contour plots are shown. Cells were first gated on viable, CD45+CD11b+ cells. (B) Summarized per cent CD11b+DTAF+ cells and absolute number of CD11b+DTAF+ cells (right) are shown. PBS n=3, IP DTAF-β-glucan n=3, oral DTAF-β-glucan n=4 (C) Representative flow dot plots of viable, CD45+CD11b+ cells from the pancreas of PBS, IP β-glucan, or oral β-glucan treated mice. (D) Summarized per cent CD11b+cells and absolute number of CD11b+ cells (right) are shown. PBS n=4, IP β-glucan n=5, oral β-glucan n=5. (E) Representative flow contour plots of TNF-α producing CD11b+ cells from PBS, IP β-glucan, or oral β-glucan treated mice. (F) Quantified percent CD11b+TNF-α+ cells and MFI (right) are shown. PBS n=4, IP β-glucan n=5, oral β-glucan n=5 (G) Representative flow contour plots of TNF-α producing CD11b+ cells from the pancreas of mice fed six doses of oral β-glucan and exposed to media, KPC, or IRE conditioned media. (H) Summarized per cent and MFI of CD11b+TNF-α+ cells are shown. Media n=4, IP β-glucan n=4, oral β-glucan n=4 (I) Experimental scheme for oral β-glucan in combination with IRE for treatment of the orthotopic KPC model. C57Bl/6 mice were challenged with orthotopic KPC tumors on day 0 and allowed to recover for 1 week before beginning daily neoadjuvant oral β-glucan administration. After 1 week of neoadjuvant oral β-glucan, mice underwent IRE or sham surgery. Oral β-glucan was then continued daily for 4 weeks postoperatively. (J) Overall survival. PBS n=5, IRE n=5, β-glucan n=5, β-glucan+IRE n=5, respectively. Data are representative of two independent experiments and presented as mean±SEM. ns, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. DTAF, 5-((4,6-Dichlorotriazin-2-yl)amino) fluorescein hydrochloride; IP, intraperitoneal; IRE, irreversible electroporation; MFI, mean fluorescent intensity; PBS, phosphate buffered saline; PC, pancreatic cancer; SSC-H, Side Scatter-Height; TNF, tumor necrosis factor; WT, wild-type.
Effect of β-glucan on healthy donor and PC patient peripheral blood monocytes
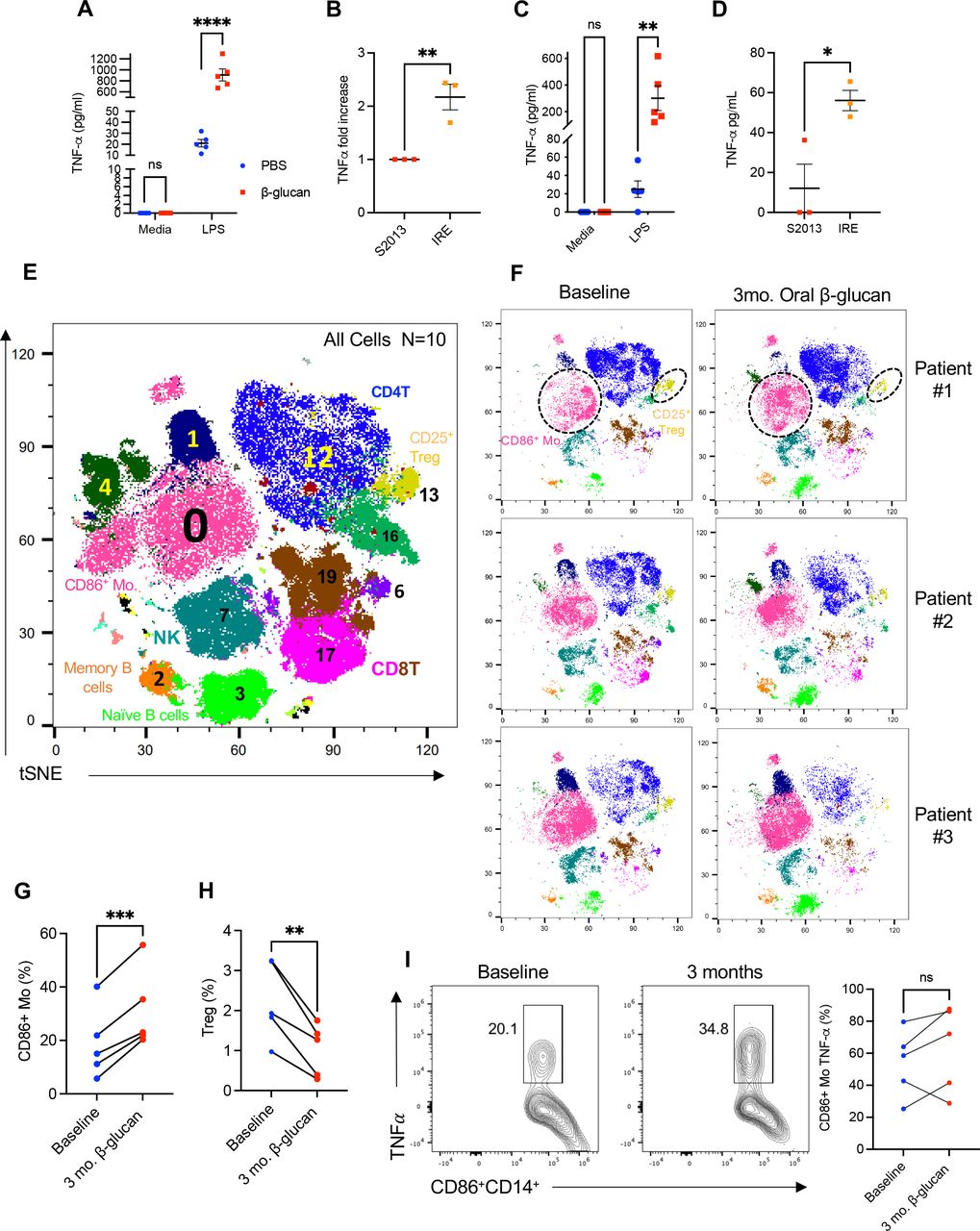
Induction of trained immunity via β-glucan has been previously demonstrated in healthy human myeloid cells, however it has yet to be evaluated in the context of PC or with IRE. As a validation of β-glucan training in human monocytes we repeated the in vitro training assay first described by Cheng et al.43 Healthy donor human CD14+ monocytes were sorted from the peripheral blood of five individual subjects, cultured in the presence of β-glucan for 24 hours, and then re-exposed to secondary stimulus (LPS) 7 days later. Consistent with prior findings, figure 7A, demonstrates that CD14+ monocytes trained with β-glucan showed increased TNF-α production on re-exposure to LPS. Next, we trained CD14+ monocytes with β-glucan, and then rechallenged them with either tumor-conditioned or IRE-ablated media from the human S2013 PC cell line. The trained CD14+ cells demonstrated significantly elevated levels of TNF-α after exposure to S2013 IRE-conditioned media (figure 7B). PC is known to cause a systemic immune cell exhaustion.44 Therefore, we wanted to determine if CD14+monocytes could be trained from the peripheral blood of patients with newly diagnosed treatment naïve PC. To test this, we repeated this experiment with CD14+ monocytes obtained from patients with PC. We observed that CD14+ monocytes derived from patients with treatment naïve PC were also trained by β-glucan as demonstrated by an increase in TNF-α production following LPS restimulation (figure 7C). Similarly, IRE-ablated tumor-conditioned media elicited more potent trained response compared with regular tumor culture supernatants as revealed by increased TNF-a production (figure 7D). Given these findings and our oral β-glucan data presented above, we next wanted to evaluate the potential trained immunity effect of oral β-glucan from peripheral blood of patients with stage III PC who were scheduled to receive IRE. To do so, baseline blood was collected from five patients prior to undergoing surgical IRE (baseline). After undergoing IRE and recovering to meet acceptable discharge standards the patients began taking 1000 mg oral β-glucan daily. Blood was then collected after 3 months of oral β-glucan therapy post IRE. To perform a comprehensive evaluation of the peripheral immune landscape, mass cytometry (CyTOF) was performed comparing baseline and 3-month samples. Twenty unique cell populations from the peripheral blood were identified and compared between the time points (figure 7E). Summary of cell surface markers and heatmap profile provided in online supplemental figure 7. Examination of t-SNE plots revealed there to be consistent observable differences in the peripheral immune landscape before and after oral β-glucan of all five patients (figure 7F). Notably, consistent increases in frequency of CD86+ monocytes, cluster 0, (figure 7G) were observed for all five patients whereas the frequency of regulatory T cells (Tregs), cluster 13, significantly decreased overtime (figure 7H). Encouragingly, four of five patients demonstrate heightened TNF-α production from CD86+ monocytes 3 months post IRE after daily oral β-glucan (figure 7I). Although preliminary, these data support the ability of oral β-glucan to positively alter the immune landscape of patients with advanced stage PC providing rationale for further clinical investigation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of β-glucan on healthy donor and PC patient peripheral blood mononuclear cells. (A) TNF-α production after in vitro β-glucan trained or untrained healthy donor CD14+ monocytes (n=5) obtained via cell sorting and restimulated with LPS measured by ELISA. (B) Fold change in TNF-α production by in vitro β-glucan-trained healthy donor CD14+ monocytes (n=3) on co-culture with S2013 or IRE-ablated media. (C) TNF-α production after in vitro β-glucan trained or untrained PC treatment naïve CD14+ monocytes (n=5) obtained via cell sorting and restimulated with LPS measured by ELISA. (D) TNF-α production by in vitro β-glucan-trained PC treatment naïve CD14+monocytes (n=3) on co-culture with S2013 or IRE-ablated media. (E) Representative t-SNE plot of the 10 experimental samples identifying 20 distinct cell clusters. (F) Individual t-SNE plots of PBMCs from three representative patients before (baseline) and after IRE and 3 months of oral β-glucan. (G) Quantified percent CD86+ monocytes and Tregs (H) at baseline and after IRE and 3 months of oral β-glucan. (I) Quantified percent of TNF-α production from CD86+ monocytes at baseline and 3 months post IRE and oral β-glucan. PC monocytes were restimulated with LPS (1 ng/mL). Data are presented as mean±SEM. ns, not significant; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. IRE, irreversible electroporation; LPS, lipopolysaccharide; NK, natural killer; Mo, monocytes; mo, months; PBMCs, peripheral blood mononuclear cells; PBS, phosphate buffered saline; PC, pancreatic cancer; TNF, tumor necrosis factor; Treg, regulatory T cell; t-SNE, t-distributed Stochastic Neighbor Embedding.
Discussion
Induction of trained immunity only recently emerged as a cancer therapeutic, expanding its relevance beyond host vaccination responses and protection against secondary infection.45 Mounting evidence exists supporting anticancer mechanisms underlying trained immunity.26 36 45 However, there is a dearth of information practically applying trained immunity to relevant tumor models and clinical treatment scenarios. Additionally, etiologies to provoke the trained response against carcinogenesis have not been reported, despite secondary stimuli being a critical component to engage the benefit of trained immunity. Our previous findings of the pancreas as a specific target of IP β-glucan trafficking and induction of peripheral trained immunity has meaningful implications in translating such immunotherapy to patients with PC.26 The immune modulatory potential of IRE in synergy with immunotherapy is becoming recognized in treatment of solid tumors.46 The release of tumor antigens, pro-inflammatory factors, and DAMPs by IRE provides a potential avenue to engage the trained response.19 Herein, we have described that factors such as MIF released from IRE in PC tumors serves as a secondary stimulus to induce a potent trained response in β-glucan-trained myeloid cells. Combined β-glucan-induced trained immunity with IRE therapy provided effective treatment against an aggressive orthotopic PC model. Importantly oral β-glucan, a more accessible route of delivery, induced trained immunity in the pancreas and altered the peripheral immune landscape in patients with advanced stage III PC after undergoing IRE. These findings provide encouraging results for the development of a novel combination therapy for treatment of PC, a disease traditionally refractory to available immunotherapies.
Heightened innate reaction to a secondary challenge is the hallmark of trained immunity, whereby host protection is inferred against an unrelated pathogen.23 Antitumor immune effects of trained immunity agonists have only recently been described in both prevention and treatment.47–49 However, no studies have discussed or demonstrated methods to provoke or augment the level of trained response. A unique finding from our study is that supernatant derived from IRE ablated cancer cells incited a more potent trained response from β-glucan trained myeloid cells compared with tumor-conditioned media alone. We first reported that tumor-conditioned media may specifically function as the secondary stimulus due to inherent release of DAMPs, such as MIF.26 However, this requires tumor progression or development to release soluble factors to act as the secondary stimulus to trained innate cells—an undesirable outcome particularly in the setting of an aggressive malignancy such as PC. Considering IRE has previously been reported to release DAMPs and other pro-inflammatory factors, we were prompted to posit this ablation modality as a more proactive method to reactivate the trained innate cells. Indeed, we found a substantial release of MIF from the cultured KPC cells by IRE compared with spontaneous release to the culture media. Further, re-exposure of β-glucan-trained pancreatic myeloid cells to IRE-ablated media increased their level of trained response compared with tumor-conditioned media alone. Aside from trained immunity, innate immune cells are well known to possess inherent anticancer function such as direct cell-mediated cytotoxicity and phagocytosis.50 Upregulation of these functions can be achieved via β-glucan alone yet more notably, we demonstrate these functions can be augmented via the action of IRE ablation. These data are the first to provide a novel and clinically available secondary stimulus capable of engaging and intensifying the level of trained response from myeloid cells.
The previously described decrease in PC tumor burden and prolongation in survival provided by β-glucan alone is encouraging yet modest. Further, the trained effect from increasing myeloid cells within the pancreas 7 days following single IP β-glucan injection declines overtime.26 These findings are characteristic of PC, whose immunosuppressive TME has easily resisted prior trials of mono and dual ICI.51 Therefore, we find it incumbent to provide combinatorial approaches that may prolong the antitumor trained immunity effect. Our findings that β-glucan and IRE independently reduce PC tumor burden is consistent with prior animal studies.17 19 26 Consequently, we found an enhanced therapeutic outcome when β-glucan and IRE were used sequentially. Our data also described changes in epigenetic and transcriptional profile in innate myeloid cells clearly supporting an augmented trained immune phenotype within β-glucan IRE treated tumors that is sustained over time. Immunologically, increases were observed in absolute number across all cell types suggesting that IRE allowed for a prolonged influx of leukocytes after β-glucan training. Our data finding prominent cytokine responses from monocytes and macrophages from the tumors of β-glucan trained mice also agrees with prior literature.52 53 However, this phenotype was lost overtime as tumor burden progressed suggesting PC tumors eventually escape the trained effect provided by single dose of IP β-glucan even in combination with IRE. More studies are needed to evaluate the exact duration of training to better understand the long-term benefit of trained immunity in the setting of cancer. Evaluation of long-term trained immunity in this combination therapy may best be addressed via cell subset deletion or adoptive transfer studies, which is a future direction of this work. Despite the loss of trained effect from monocytes and macrophages in mice with advanced disease, they demonstrated reduced metastatic lung nodules and increased TNF-α cytokine production from primary tumors without LPS re-stimulation. These data indicate IRE may act as the secondary stimulus to trained myeloid cells within the PC TME and the proposed combination therapy can be used to limit metastasis in the adjuvant setting.
IRE has provoked immunologic changes within the TME of subcutaneous and orthotopic PC tumor models. He et al, reported increased effector and memory T-cell frequency in mice treated with IRE, which subsequently rejected secondary tumor challenges.54 Clinical studies also have documented transient decreases in systemic Tregs after IRE, supporting combined use of IRE with immunotherapies to enhance T-cell activation.55 56 In our orthotopic model, increased absolute CD4+ and CD8+ T-cell numbers were observed in tumors following combination therapy, however no direct effect on cytokine production occurred. Additionally, these infiltrating CD8+ T cells in IRE plus β-glucan treated tumors expressed lower levels of PD-1 and LAG3 and higher levels of granzyme B indicating they are not exhausted rather than activated in this therapy. These findings are consistent with our prior findings that β-glucan cooperates with anti-PDL-1 Ab therapy to prolong survival in this KPC orthotopic model.26 These findings suggest a potential avenue to successfully implement ICI therapy in a PC model. In this study, a similar or even better therapeutic efficacy of β-glucan and IRE was observed in Rag−/− mice reducing tumor burden and prolonging survival. Although histologic analysis of the Rag−/− was not provided, we would expect these mice to have reduced lung metastatic burden as we observed long-term tumor-free survival. Further, the better therapeutic efficacy of this combination therapy in Rag−/− mice may imply critical roles of Tregs in this KPC model.57 Reduction of tumor burden in the absence of adaptive immunity questions the ability to provide a long- lasting antitumor protection. At present trained immunity is generally accepted to be shorter lived than classical adaptive immune cell memory, yet its duration is not well defined. Developing novel approaches such as this combination therapy to establish a ‘sustained’ trained immune response may hold the key for lasting innate immune memory.
This data agrees with our prior work where β-glucan significantly reduced tumor burden in NSG mice (lacking mature T, B, and natural killer (NK) cells) orthotopically implanted with KPC cells.26 While IRE is known to induce an immunogenic cell death releasing specific neoantigens that could contribute to T-cell activation, regulation of innate cell responses via release of DAMPs may be a greater consequence in the setting β-glucan training in PC. Regardless of T-cell activation/status, this data supports more effective antitumor therapy of trained immunity when paired with an active process of tumor killing such as IRE.
NK cells have been described in the context of trained immunity, particularly in the setting of murine and human cytomegalovirus (CMV) infection.58 On CMV reinfection, NK cells clonally expand and more rapidly degranulate providing enhanced immune protection to their host. These so-called ‘memory’ NK cells more closely resemble adaptive T-cell responses.22 59 Adoptive transfer of murine cytomegalovirus (MCMV) exposed NK cells increased survival to MCMV challenged mice. BCG vaccination in humans has also induced a long-lasting memory in NK cells, which demonstrate enhanced IFN-γ production even 1 year after vaccination.60 However, NK cells remain unimplicated in the setting of β-glucan training. β-Glucan trained mice induced lung interstitial macrophage training and reduced lung metastasis in NSG mice.49 Moreover, we have also previously observed myeloid cell training in the murine pancreas after monoclonal antibody depletion of NK cells as well as in NSG mice.26 Collectively, these data suggest that NK cells may not play an essential role in this combination therapy.
IP β-glucan holds promise in targeting PC via its unique trafficking and induction of trained immunity in the murine pancreas. Yet, there are substantial limitations to consider in translating this approach to patients. First, access and delivery of β-glucan to the peritoneal cavity is limited in humans. Direct peritoneal access is achieved at the time of diagnostic laparoscopy, definitive resection, or IRE in eligible patients, however timing for optimal administration of β-glucan remains in question. It is also likely that multiple doses would be needed or if subsequent doses would provide any further benefit. In addition, the murine pancreas resides in an intraperitoneal anatomical location as compared with retroperitoneal of the human pancreas.61 Influx of CD45+ immune cells to the pancreas also poses a theoretical threat of inflammation and subsequent pancreatitis. However, based on our prior data, WT mice treated with IP β-glucan have normal levels of serum amylase and histologic architecture of pancreatic acini and islet cells in comparison to PBS controls.26 Thus, we believe the phenomenon β-glucan trafficking to the pancreas to be safe, although more translational studies are needed to prove this point.
Oral administration of β-glucan offers many advantages over IP β-glucan injection such as ease of access and better patient compliance. Prior studies using oral β-glucan demonstrated effective anticancer properties.62 Human and animal studies have reported antiviral properties of oral β-glucan, yet no studies have used oral β-glucan as an agonist of trained immunity in the setting of cancer.63 64 We found that oral β-glucan traffics to the pancreas in small quantities inciting a phenotype of peripheral trained immunity. Oral β-glucans are known to directly interact with intestinal mucosa, to be taken up by M cells, and interact with resident macrophages and DCs being distributed throughout the general circulation.65 66 This may explain our finding that pancreatic CD11b+CD11c+ DCs to have increased cytokine production after oral β-glucan intake. However, the specific biodistribution of oral β-glucan, its overall tropism to the pancreas, and its effector cell targets remain to be better understood. Notably, systemic administration of IP β-glucan had more pronounced effects, which calls for more studies comparing antitumor immunity versus ease and safety of β-glucan administration routes in the PC setting. Myeloid cells isolated from pancreases of mice who received 7 days of oral β-glucan were also more responsive to IRE-ablated media. With this understanding, we also observed dramatic improvement in overall survival when oral β-glucan was given in combination with IRE. Together, these findings provide further credence to our above conclusions combining β-glucan with IRE in PC and call for further studies to address signaling pathways between IRE and trained myeloid cells.
Prior studies investigating induction of trained immunity in human subjects as a cancer therapeutic primarily focus on BCG in non-muscle invasive bladder cancer.67 68 While the anticancer effects and mechanisms of β-glucan continue to be appreciated, oral β-glucan is known to be well tolerated in humans and can augment innate immune cells similar to BCG.65 To date, few quality data exists evaluating use of β-glucan in patients with cancer. One study found oral β-glucan reduced adverse effects of mucositis from systemic chemotherapy in patients with colorectal cancer.69 In 23 patients with advanced stage breast cancer, oral β-glucan increased proliferation and activation of PBMCs measured by increased CD95 and CD45RA on CD14+ monocytes 15 days after treatment.70 Our previous study showed that oral β-glucan modulates peripheral monocyte composition and function in patients with human non-small cell lung carcinoma.71 The current study is the first report indicating monocytes obtained from patients with PC can be trained in vitro and to describe alternations in the peripheral immune phenotype in patients with PC after oral ingestion of β-glucan. Interestingly, we found increased frequency of CD86+CD11b+CD11c+ monocytes after 3 months of adjuvant oral β-glucan following IRE and decreased Treg frequency in five patients with stage III PC. In addition, CD86+ monocytes from four of five patients showed trained immunity phenotype. These data demonstrate that human monocytes from patients with PC can be trained through oral β-glucan, providing a potential avenue to modulate immunosuppressive myeloid cells within the PC TME. However, a more thorough assessment of the functional state of these cells, their influence on the TME, and their clinical effects and outcomes is needed.
In conclusion, these findings demonstrate the antitumor potential of yeast-derived β-glucan-induced trained immunity within the pancreas by two routes of administration whose effect can be augmented by IRE, providing a novel modality to augment trained response. This combination significantly reduced local and distant tumor burden in mice bearing orthotopic PC tumors, establishing solid proof of concept for future clinical trials. We also provide insight to the duration of trained effect demonstrated within the PC TME. This work builds on our previous findings first describing induction of peripheral trained immunity in the pancreas via β-glucan and is the first to describe changes within the peripheral immune landscape by oral β-glucan in a patient population with high-risk PC. Although our patient data are preliminary, these results provide optimism for future work investigating the combination of IRE and trained immunity, which we hypothesize will favorably alter the TME in human subjects.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by University of Louisville IRB#16.110. Participants gave informed consent to participate in the study before taking part.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @MattWoesteMD
Contributors MRW designed and performed all experiments, analyzed data, interpreted results, and wrote the manuscript. RS contributed to experimental design, analysis, and performed a critical review. AEG performed critical review. SL performed supporting experiments. DM-D contributed to experimental design and critical review. XH assisted in the preparation of materials and supporting experiments. CD performed supporting experiments, contributed to experimental design and critical review. HL and AP performed supporting experiments. RAM contributed materials and critical review. TH contributed acquisition of patient samples. MT and YL performed supporting experiments. KMM and RCGM provided materials and performed a critical review. JY directed experimental design and the overall study, interpreted data, supervised research, and assisted in writing and review of the manuscript. MRW and JY are responsible for the overall content as the guarantors.
Funding This work was partly supported by the NIH R01CA213990, R01AI128818, and the American Cancer Society Mission Boost Award MBGI-22-158-01-MBG (JY). The CyTOF was performed in the Functional Immunomics Core supported by the NIH P20GM135004.
Competing interests RCGM is an educational consultant for AngioDynamics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.