Article Text

Abstract
Background Harnessing the immune system to purposely recognize and destroy tumors represents a significant breakthrough in clinical oncology. Non-synonymous mutations (neoantigenic peptides) were identified as powerful cancer targets. This knowledge can be exploited for further improvements of active immunotherapies, including cancer vaccines, as T cells specific for neoantigens are not attenuated by immune tolerance mechanism and do not harm healthy tissues. The current study aimed at developing an optimized multitarget vaccine using short or long neoantigenic peptides utilizing virus-like particles (VLPs) as an efficient vaccine platform.
Methods Mutations of murine mammary carcinoma cells were identified by integrating mass spectrometry-based immunopeptidomics and whole exome sequencing. Neoantigenic peptides were synthesized and covalently linked to virus-like nanoparticles using a Cu-free click chemistry method for easy preparation of vaccines against mouse mammary carcinoma.
Results As compared with short peptides, vaccination with long peptides was superior in the generation of neoantigen-specific CD4+ and CD8+ T cells, which readily produced interferon gamma (IFN-γ) and tumor-necrosis factor α (TNF-α). The resulting anti-tumor effect was associated with favorable immune re-polarization in the tumor microenvironment through reduction of myeloid-derived suppressor cells. Vaccination with long neoantigenic peptides also decreased post-surgical tumor recurrence and metastases, and prolonged mouse survival, despite the tumor’s low mutational burden.
Conclusion Integrating mass spectrometry-based immunopeptidomics and whole exome sequencing is an efficient approach for identifying neoantigenic peptides. Our multitarget VLP-based vaccine shows a promising anti-tumor effect in an aggressive murine mammary carcinoma model. Future clinical application using this strategy is readily feasible and practical, as click chemistry coupling of personalized synthetic peptides to the nanoparticles can be done at the bedside directly before injection.
- immunotherapy
- vaccination
- breast neoplasms
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Driving the immune system to specifically recognize and destroy tumors represents a significant breakthrough in clinical oncology. Non-synonymous mutations were identified as powerful cancer targets. This knowledge can be exploited for further improvements of active and passive immunotherapies, including cancer vaccines. T cells specific for neoantigenic peptides are typically not attenuated by immune tolerance mechanism and do not harm healthy tissues.1 Recent clinical trials have shown that de novo immune responses can be induced by vaccination with neoantigens in melanoma and glioblastoma patients.2–5 The optimal peptide length for therapeutic cancer vaccines has been identified in preclinical and clinical studies. Short peptides of ∼8–11 amino acids (a.a.) are the typical CD8+ T cell epitopes, while long peptides of ∼15–32 a.a. contain both CD8+ and CD4+ T cell epitopes.6 7 Neoantigenic epitopes can be predicted based on their sequence, expression, proteasome cleavage sites, peptide abundance, major histocompatibility complex (MHC)-binding affinity and further physiochemical and structural features of peptide-MHC complexes. Long peptides are elongated at both ends with natural flanking residues that extend the repertoire of peptides binding to MHC-I to additional peptides binding MHC-II. Cytotoxic CD8+ T lymphocytes have been intensely characterized and identified as key players in anti-tumor immunity. Furthermore, CD4+ T cells have recently emerged as important contributors in enhancing the immune response against tumors.8 9
There exist many different vaccine platforms, based on mRNA, peptides/proteins, dendritic cells (DCs) or recombinant viruses.10–13 Oncolytic viruses have been developed since many years, leading to the approval of talimogene laherparepvec (T-VEC) to treat patients with metastatic melanoma.14 The success of this therapy is likely related to the immunogenicity of T-VEC, which is particularly useful when injected directly into tumors, thus reaching the site(s) where the microenvironment must become more supportive and less suppressive for T cell responses. The intratumoral injection route has also become promising for treating patients with virus-like particles (VLPs) that carry CpGs oligodeoxynucleotides but no antigen, whereby the rational is that tumor antigens are present in the tumors, making it unnecessary to be provided as vaccine. The quasicrystalline VLPs are multiprotein complexes with a highly defined repetitive surface geometry, mostly in the shape of an icosahedrons.15 Epitope repetitiveness serves as a potent pathogen-associated structural pattern (PASP) efficiently recognized by innate and adaptive immune cells.16 The outer surface of VLPs can be decorated with antigenic peptides. Inside of the VLPs, one can package innate immune stimulators such as toll-like receptor (TLR) ligands for efficient cellular activation.15 VLPs built with bacteriophage Qβ protein (Qβ-VLPs) are highly immunogenic and very versatile for the production of vaccine candidates with antigens and innate immune stimulators. In our earlier studies and clinical trials, we showed that such VLP vaccines induced strong tumor antigen-specific T cell responses in preclinical models and melanoma patients.17 18 Our Qβ (G10)-Melan-A vaccine was based on Qβ-VLPs packaged with G10, a TLR-9 ligand inducing a potent interferon (IFN) α response,19 and decorated with a long peptide derived from the Melan-AMart1 melanoma differentiation antigen. Chemical coupling of peptides to the Qβ-VLPs was performed using the (succinimidyl-6-((b-maleimidopropionamido)hexanoate) SMPH bifunctional cross-linker. Clinical trials showed that more than half of patients with stage II–IV melanoma generated strong tumor antigen-specific and in vivo functional CD8+ and CD4+ T cell responses.20 The tumors of some patients showed late loss of Melan-A expression, indicating that targeting of only a single antigen is problematic because of disease progression caused by antigen-loss variants. Indeed, preclinical models confirmed that targeting multiple antigens is superior in generating CD4+ and CD8+ T cell responses that can successfully infiltrate tumors, control tumor growth and avoid outgrowth of escape variants.21
The current study aimed at developing an optimized anti-tumor vaccine using short or long neoantigenic peptides. VLPs were packaged with a TLR-9 ligand and decorated with neoantigenic peptides discovered by peptidomic, genetic and transcriptomic techniques. Vaccines were composed of mixtures of four VLPs each with a different peptide, coupled by click chemistry permitting easy patient-individual vaccine preparation at the bedside. Our study provides proof of concept of an effective personalized therapeutic cancer vaccine that can readily be implemented for clinical use.
Materials and methods
Production and purification of Qβ-VLPs
The expression and production of the bacteriophage Qβ-VLPs were carried out as previously described.22–24
Cell lines and mice
4T1 mammary carcinoma cell line (ATCC CRL-2539) and EpH4-Ev wild-type mammary epithelial cell line (ATCC CRL-3063) were cultured in Gibco Dulbecco's Modified Eagle Medium (DMEM) medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. For EpH4-Ev cell line, 6 µg puromycin was added to the culture medium as per ATCC instructions. When cells were 80% confluent, medium was aspirated and cells were washed 3× with 1× phosphate-buffered saline (PBS) to remove excess medium. 1× trypsin was added and flasks were incubated at 37°C for 10 min. Cells were collected, resuspended in complete medium and kept on ice until use. Mycoplasma contamination was ruled out using Microsart AMP Mycoplasma Kit. Wild-type female Balb/cOlaHsd mice (8–12 weeks) were purchased from Harlan and kept in the animal facility at the University of Bern. 1×106 cells were inoculated subcutaneously (s.c.) in the flank region under isoflurane anesthesia. All animal procedures were conducted in accordance with the Swiss Animal Act (455.109.1—September 2008, 5) of University of Bern.
DNA sequencing and prediction of non-synonymous mutations
DNA was extracted using the commercially available Purelink Genomic DNA mini kit (Invitrogen) as per the manufacturer’s instructions. The purity and concentration of the DNA of both cell lines were measured using nanodrop. Whole exome sequencing (WES) was performed on an Illumina HiSeq 4000 machine using libraries prepared with a SureSelectXT Mouse All Exon capture kit (Agilent Technologies, Santa Clara, California, USA), to produce 150 paired end reads sufficient for a mean bait coverage of at least 150×. The sequence reads were aligned to the GRCm38.p6 mouse reference assembly using BWA-MEM V.0.7.17,25 and variants were predicted using the Genome Analysis Toolkit best practices protocol (GATK V.3.7). A set of reference variants for mouse was derived from data generated by the Wellcome Sanger Institute Mouse Genomes Project (available at ftp://ftp-mouse.sanger.ac.uk/REL-1807-SNPs_Indels/). Somatic mutations (SMs) were defined as variants present only in the 4T1 sample, while single nucleotide polymorphisms (SNPs) were defined as variants present in both the 4T1 and EpH4-Ev samples. The functional effect of the predicted SMs and SNPs was determined by annotating the variants with snpEff V.4.3T26 using the GRCm38.86 and mm10 databases.
Purification of MHC-I binding peptides
Anti-MHC-I monoclonal antibodies were purified from the supernatant of HIB 34.1.2 hybridoma cells (a gift from Angel Miguel Garcia-Lora, Hospital Universitario Virgen de las Nieves, Granada, Spain) using Protein A–Sepharose 4B beads (Invitrogen). Antibodies were cross-linked to Protein A–Sepharose 4B beads at a concentration of 5 mg of antibodies per 1 mL volume of beads at room temperature (RT) with dimethyl pimelimidate dihydrochloride (Sigma-Aldrich) in 0.2 M sodium borate buffer pH 9 (Sigma-Aldrich) at a final concentration of 20 mM for 30 min. 2.33×108 4T1 cells grown in culture or 8×108 grown in vivo in Balb/c mice per replicate were lysed in phosphate buffered saline containing 0.2 mM iodoacetamide (Sigma-Aldrich), 1 mM EDTA, 1:200 Protease Inhibitor Cocktail (Sigma-Aldrich), 0.5% sodium deoxycholate (Sigma-Aldrich), 1% octyl-β-D-glucopyranoside (Sigma-Alrich) and 1 mM phenylmethylsulfonylfluoride (Roche) at 4°C for 1 hour. Lysates were cleared by centrifugation at 4°C at 20 000 g for 50 min. MHC-I molecules were purified by incubating the cleared lysate with HIB antibodies cross-linked to Protein A–Sepharose 4B beads in affinity columns for 3 hours at 4°C. The affinity columns were washed first with 150 mM sodium chloride (NaCl) in 20 mM Tris-HCl pH 8, then with 400 mM NaCl in 20 mM Tris-HCl pH 8 and again with 150 mM NaCl in 20 mM Tris-HCl pH 8. The beads were washed with 20 mM Tris-HCl pH 8 and the MHC complexes were eluted by adding 1% trifluoroacetic acid (TFA) at RT. For the purification and concentration of MHC peptides, Sep-Pak tC18 96-well plates (Waters) were preconditioned with 80% acetonitrile (ACN) in 0.1% TFA and wahsed with 0.1% TFA. Elutions were loaded in the Sep-Pak tC18 96-well plates and the C18 wells were then washed with 0.1% TFA. The peptides were eluted with 28% ACN in 0.1% TFA. The peptide samples were dried using vacuum centrifugation (Thermo Fisher Scientific) and stored at −20°C.
Liquid chromatography and mass spectrometry
Peptides were separated on a 450 mm analytical column (8 µm tip, 75 µm inner diameter, PicoTipTMEmitter, New Objective) packed with ReproSil-Pur C18 (1.9 µm particles, 120 Å pore size, Dr Maisch GmbH) with an Easy-nLC 1200 (Thermo Fisher Scientific). The separation was performed at a flow rate of 250 nL/min by a gradient of 0.1% formic acid (FA) in 80% ACN (solvent B) and 0.1% FA in water (solvent A). With a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific), MS1 spectra were acquired from m/z=300–1650 with a resolution of 60,000 (m/z=200), an ion accumulation time of 80 ms and an auto gain control (AGC) was set to 3e6 ions. MS/MS spectra were acquired in a data-dependent manner on the 10 most abundant precursor ions with a resolution of 15,000 (m/z=200), an ion accumulation time of 120 ms and an isolation window of 1.2 m/z. The AGC was set to 2e5 ions. The dynamic exclusion was set to 20 s and a normalized collision energy of 27 was used.
Identification of neoantigenic peptides from MS immunopeptidomic data
Non-synonymous SMs and SNPs were incorporated into the headers of reference protein Fasta as previously described.27 Mouse GENCODE sequences and the associated gene annotations (V.M22; reference assembly V.GRCm38 from https://www.gencodegenes.org/mouse/releases.html) were scanned for the presence of 4T1-specific variants using genomic coordinates from the transcript annotations. Variants producing non-synonymous changes were identified and the corresponding a.a. changes were tagged with a unique ID and added to the header of the translated transcript sequence. We searched the immunopeptidomic MS data against the 4T1-specific customized reference database with MaxQuant V.1.5.9.4.28 The default settings were used except and in addition the following settings were included: variants were called from file, methionine oxidation and protein N-term acetylation were set as variable modifications and no fixed modification was set, enzyme specificity was set to ‘unspecific’ and peptide spectrum match false discovery rate (FDR) was set to 0.05 with no protein FDR. Binding affinity of the peptides to the MHC molecules was predicted by NetMHCpan V.4.1 software.29 Binding motif deconvolution of 9-mer MHC-I peptides was performed using the MixMHCp 2.130 31 with the default settings and deconvoluted motifs were manually assigned as H-2-Dd and H-2-Kd.
Verification of identified NeoAG
To verify the expression of the identified NeoAG, RNA from 4T1 mammary carcinoma cell line, 4T1 established tumor and EpH4-Ev wild-type mammary epithelial cell line were extracted using NucleoSpin RNA Kit and reverse transcribed using High-Capacity RNA-to-cDNA Kit as per the manufacturer’s instructions. Three primer pairs were designed of ~24 bp each, to amplify the mutated region leading to three amplicons of ~200 bp each. The PCR products were electrophoresed and after gel extraction, the amplicons with the expected size were purified using Zymoclean Gel DNA Recovery Kit. Amplicons were then sequenced using Sanger sequencing with the primers used for PCR amplification. The results confirmed the somatic missense mutations in 4T1 mammary carcinoma cell line and the established 4T1 tumors in Balb/c mice; however, these mutations were absent from the wild-type control mammary epithelial cell line.
Synthesis of neoantigenic peptides (short and long)
The identified NeoAG in 4T1 mammary carcinoma cell line were synthesized as short (15–16 a.a.) or long (32 a.a.) peptides using their flanking regions as listed in table 1.
Synthesized neoantigenic peptides (NeoAG) with flanking regions, additional lysine and an azide group (N3) at the C-terminus
An additional lysine (K) and an azide group (N3) were added at the C-terminus of the synthesized peptides to facilitate Cu-free click chemistry coupling. Neoantigenic short and long peptides are referred to as NeoAGS and NeoAGL, respectively. Neoantigenic MHC-I peptides (8 a.a. or 9 a.a. underlined), non-synonymous SMs (in bold).
Production of Qβ(1668)-NeoAGS and Qβ(1668)-NeoAGL vaccines
The naturally packaged single-stranded RNA (ssRNA) in Qβ-VLPs was digested as following: Qβ(ssRNA)-VLPs were washed 3× with 20 mM HEPES (Sigma-Aldrich) using Amicon-Ultra 0.5 mL tubes 100 kDa molecular weight cut-off (MWCO). RNAse A (Merck) was used to digest the ssRNA (1.2 mg/mL for each 3 mg/mL Qβ-VLPs) and incubated at 37°C for 3 hours in a shaker at 400 rpm. The digestion of the ssRNA was confirmed by 1% agarose gel stained with SYBR Safe dye for 30 min at 90 V. Qβ-VLPs were then re-packaged with B-type 1668 CpGs (5′′-TCC ATG ACG TTC CTG ATG CT-3′′) with phosphorothioate backbone purchased from Invitrogen. The re-packaging was confirmed by 1% agarose gel stained with SYBR Safe dye for 30 min at 90 V. The presence of Qβ(1668)-VLPs protein was detected by staining the 1% agarose gel with Coomassie Blue stain. Qβ(1668)-VLPs were then derivatized for 30 min at RT using dibenzocyclooctyne NHS ester (DBCO cross-linker) (Sigma-Aldrich) in a fivefold molar excess. Amicon-Ultra 0.5 mL tubes 100 kDa MWCO were used to remove excess DBCO cross-linker. The NeoAGS or NeoAGL peptides were synthesized by Pepscan BRESTO with the addition of lysine (K) and (N3) azide group at the C-terminus to facilitate their coupling to the derivatized Qβ(1668)-VLPs. The synthesized peptides were reconstituted in Dimethyl sulfoxide (DMSO) and added in a fourfold molar excess over Qβ(1668)-VLPs monomer. The vaccine was incubated 1 hour at RT and Amicon-Ultra 0.5 mL tubes 100 kDa MWCO were used to remove excess peptide. The coupling of the NeoAGS or NeoAGL to Qβ(1668)-VLPs was done separately for each neoantigenic peptide. The efficiency of the coupling was checked with SDS-PAGE (BIO-RAD).
Vaccination of mice
Balb/c mice were vaccinated after inoculation of 4T1 mammary carcinoma cell line as described in the Cell lines and mice section. Designed groups, vaccination doses and regimen were scheduled as described in the Results section. Briefly, we injected 1×106 4T1 s.c. in the flank as previous studies have shown that this method possesses an increased localization of the primary tumor, which makes it easier to be resected in surgery. Injecting 4T1 in mammary fat or orthotopically showed enhanced invasive growth pattern. The s.c. inoculation method of 4T1 results in metastases as per our primary data and as shown by other studies.32 Vaccination using s.c. injection (over the shoulders, into the loose skin over the neck) started 3 days after cell line inoculation as summarized in table 2. Mice were followed every 2 days to assess tumor volume and general health score. Three doses (Prime–Boost–Boost) of the vaccine (40 µg each dose) were administered over 17 days (please refer to the Efficient coupling of synthetic NeoAG peptides by Cu-free click chemistry to VLPs section). Tumors in the control group reached the ethically maximal tolerated size of 1 cm3 by day 17. For the experiment in the Long peptide vaccination decreases post-surgical tumor recurrence and lung metastases, and prolongs survival section, primary tumors were resected on day 14 from all groups and 2 vaccine doses were administered weekly. Mice weight was followed routinely and were euthanized when loosing 10%–15% of their original weight. Lungs were collected when mice were euthanized and lung weight was measured.
Treatment groups and vaccine doses
Analysis of blood cells
Blood was collected from mice’ tail vein on day 16 after inoculation of 4T1 mammary carcinoma. One hundred and fifty microliters of blood was collected in 500 µL 1× PBS containing heparin and kept on ice. Cells were centrifuged at 1200 rpm for 5 min and supernatant was aspirated. RBCs were lysed using 500 μL (Ammonium-Chloride-Potassium) ACK buffer (Sigma-Aldrich) on ice for 2–3 min. White blood cells were collected after 5 min of centrifugation at 1200 rpm. Supernatant was aspirated and cells were resuspended with 1× PBS containing 0.1% Bovine serum albumin (BSA) and again centrifuged. Pelleted cells were labeled with live/dead stain, anti-mouse CD16/CD32 (mouse BD Fc block) mAb clone 2.4G2 (BD Bioscience) for 10 min in the dark, centrifuged as described above and stained with PE anti-mouse CD8α mAb clone 53–6.7 (BD Bioscience), PE/Cyanine7 anti-mouse CD4 mAb clone RM4-5 (BioLegend), APC anti-mouse Ly6G mAb clone 1A8 (BioLegend), FITC anti-mouse Ly6C clone HK1.4 (BioLegend) and APC/Cyanine7 anti-mouse CD11b clone M/170 (BioLegend). Samples were read by FACSCaliber and analysis was done using GraphPad Prism V.8.4.2 (464). Gating for monocytic (CD11bHi Ly6CHi) and granulocytic (CD11bHi Ly6GHi) myeloid-derived suppressor cells (MDSCs) was carried out as illustrated in online supplemental figure 1.
Supplemental material
To assess the immunogenicity of the single NeoAG, mice were vaccinated on day 0 s.c. with 40 µg of Qβ(1668)-NeoAGL-1 or Qβ(1668)-NeoAGL-2 or Qβ(1668)-NeoAGL-3 or Qβ(1668)-NeoAGL-4 separately or with the multitarget vaccine Qβ(1668)-NeoAGL. Splenocytes were collected 7 days later for intracellular cytokine staining. Naïve mice and a control peptide (CP) were used.
Analysis of tumors
Tumors were measured every 3 days using a caliper using the formula V=(WxWxL)/2, V=final tumor volume in mm3, L=tumor length and W=tumor width. On day 17, mice were euthanised as tumors in the control group reached the size of 1 cm3. Tumors were collected in DMEM medium containing 10% FBS and 1% penicillin/streptomycin on ice. Tumor infiltrating lymphocytes (TILs) were collected as following: tumors were dissected into pieces and smashed using 70 µM cell strainer, cells were washed during the process using DMEM medium containing 10% FBS and 1% penicillin/streptomycin in 50 mL falcon tubes. Collected cells were added to 15 mL tubes containing 2 mL of 35% Percoll slowly. The tubes were centrifuged at 1800 rpm for 25 min at RT to isolate TILs. TILs were then resuspended in 200 µL PBS (0.1% BSA) and 100 µL was transferred to 96-well v-bottom plates and centrifuged at 1200 rpm for 5 min. Supernatant was discarded, and RBCs were lysed using ACK 500 µL ACK buffer on ice for 2–3 min. TILs were stained with live/dead, anti-mouse CD16/CD32 (mouse BD Fc block) mAb clone 2.4G2 for 10 min in the dark, centrifuged as described above and stained with PE anti-mouse CD8α mAb clone 53–6.7 and PE/Cyanine7 anti-mouse CD4 mAb clone RM4-5. In the second experiment, TILs were also stained with APC anti-mouse Ly6G mAb clone 1A8, FITC anti-mouse Ly6C clone HK1.4 and APC/Cyanine7 anti-mouse CD11b clone M/170. Plates were centrifuged at 1200 rpm for 5 min, supernatant was discarded, TILs were resuspended in PBS (0.1% BSA) and added to 5 mL round-bottom tubes with cell strainer to remove excess tumor debris. Samples were read by FACSCaliber and analysis was done using GraphPad Prism V.8.4.2 (464).
Intracellular cytokine staining
One hundred microliters of the TILs described in the Analysis of tumors section were transferred to sterile 96-well flat-bottom plates. TILs were additionally incubated with mouse IL-2 (mIL2-Ref: 11271164001-MERCK) 100 U/mL in DMEM medium containing 10% FBS and 1% penicillin/streptomycin at 37°C for 2 days. Cells were washed 3× with DMEM medium containing 10% FBS and 1% penicillin/streptomycin and a stimulation cocktail was added to 1 µg/mL of each NeoAG with Brefeldin and Monensin (1:1000) at 37°C for 6 hours. TILs were washed 3× with DMEM medium to remove the stimulation cocktail and then transferred to 96-well v-bottom plates for staining. TILs were stained with live/dead, anti-mouse CD16/CD32 (mouse BD Fc block) mAb clone 2.4G2 for 10 min in the dark, centrifuged as described above and stained with PE anti-mouse CD8α mAb clone 53–6.7 and PE/Cyanine7 anti-mouse CD4 mAb clone RM4-5. The plates were centrifuged at 1200 rpm for 5 min, supernatant was discarded and TILs were fixed using 100 µL of the fixation buffer (BD Cytofix) at 4°C for 15 min. The plates were again centrifuged at 1200 rpm for 5 min, supernatant was discarded and TILs were washed with 100 µl of 1× diluted permeabilization wash buffer (BioLegend) and centrifuged immediately at 1200 rpm for 5 min, supernatant was discarded. TILs were then stained with APC anti-mouse IFN-γ mAb clone XMG1.2 (MERCK) and PerCP-Cyanine5.5 anti-mouse TNF-α mAb clone MP6-XT22 (BioLegend). Plates were centrifuged at 1200 rpm for 5 min, supernatant was discarded, TILs were resuspended in PBS (0.1% BSA) and added to 5 mL round-bottom tubes with cell strainer to remove excess tumor debris. For splenocytes, 2×106 cells were used and a similar protocol was followed. The stimulation cocktail consists of 1 µg/mL of each NeoAG separately or combined. Samples were read by FACSCaliber and analysis was done using GraphPad Prism V.8.4.2 (464).
Immunohistochemistry
Formalin-fixed murine mammary carcinomas were assessed by immunohistochemistry (IHC) staining for CD4+ and CD8+ T cells using CD4 (clone 4SM95, rat, Thermo F. Scientific, 14‐9766) and CD8 (rat, Dianova, DIA‐808). For each tumor, one full cross-section was examined by a board-certified veterinary pathologist (SdB). The quantitative evaluation of peritumoral and intratumoral CD4+ and CD8+ T cells was done digitally using the software QuPath V.0.2.3. The number of CD4+ and CD8+ T cells was calculated per mm (tumor periphery) and per mm2 (intratumoral area).
In vivo depletion of CD8+ T cell
For depletion of CD8+ T cells, 200 µg anti-CD8mAb (BioXcell clone CD8β Lyt3.2) was administered intraperitoneally on day 1 and 100 µg every week after inoculation of 4T1 mammary carcinoma. Depletion efficiency was ~99% as determined by flow cytometry on day 3 and day 12. Flow cytometry was performed as explained in the Analysis of blood cells section.
Statistics
Data were presented as mean±SEM. Comparisons between two groups was performed by Student’s unpaired t test (two-tailed). Comparison between more than two groups was performed by one-way ANOVA or Bonferroni test. Area under curve (AUC) was used to measure tumor growth curves. Survival rate was analyzed by log-rank (Mantel-Cox) test. P values were ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05.
Results
NeoAG identification
The current study aimed at providing a proof of concept (POC) for the generation of a personalized VLP-based vaccine by incorporating tumor-specific NeoAG of mammary carcinoma. We have chosen the aggressive, highly metastatic and low mutational burden mammary carcinoma cell line (4T1).11 The wild-type mammary epithelial cell line, EpH4-Ev, was selected as germline reference cells. Both cell lines originate from Balb/c mice. NeoAG identification was performed by WES combined with immunopeptidomics (figure 1A). MHC-I binding peptides were eluted from 4T1 cells and were characterized by LC-MSMS. A 4T1-specific reference database that includes non-synonymous SMs was generated to match the MSMS data. In total, 2714 and 5077 MHC-I peptides were identified in the 4T1 mammary carcinoma cells grown in vitro and in vivo, respectively (online supplemental data 1). The peptides recapitulated the expected length distribution of MHC-I ligands, meaning the majority were 9 mers, and the 9-mer peptides were clustered into two groups, revealing the expected binding motifs of H-2-Dd and H-2-Kd (figure 1B–D). Despite the low mutational burden of 4T1, we identified four NeoAG by MSMS (figure 1E–H). The non-synonymous SMs were identified in Vrk3 (E391K), Thap3 (H75Y) and Sult2b1 (H299N) as shown in figure 1 and online supplemental table 1.
Supplemental material
Supplemental material
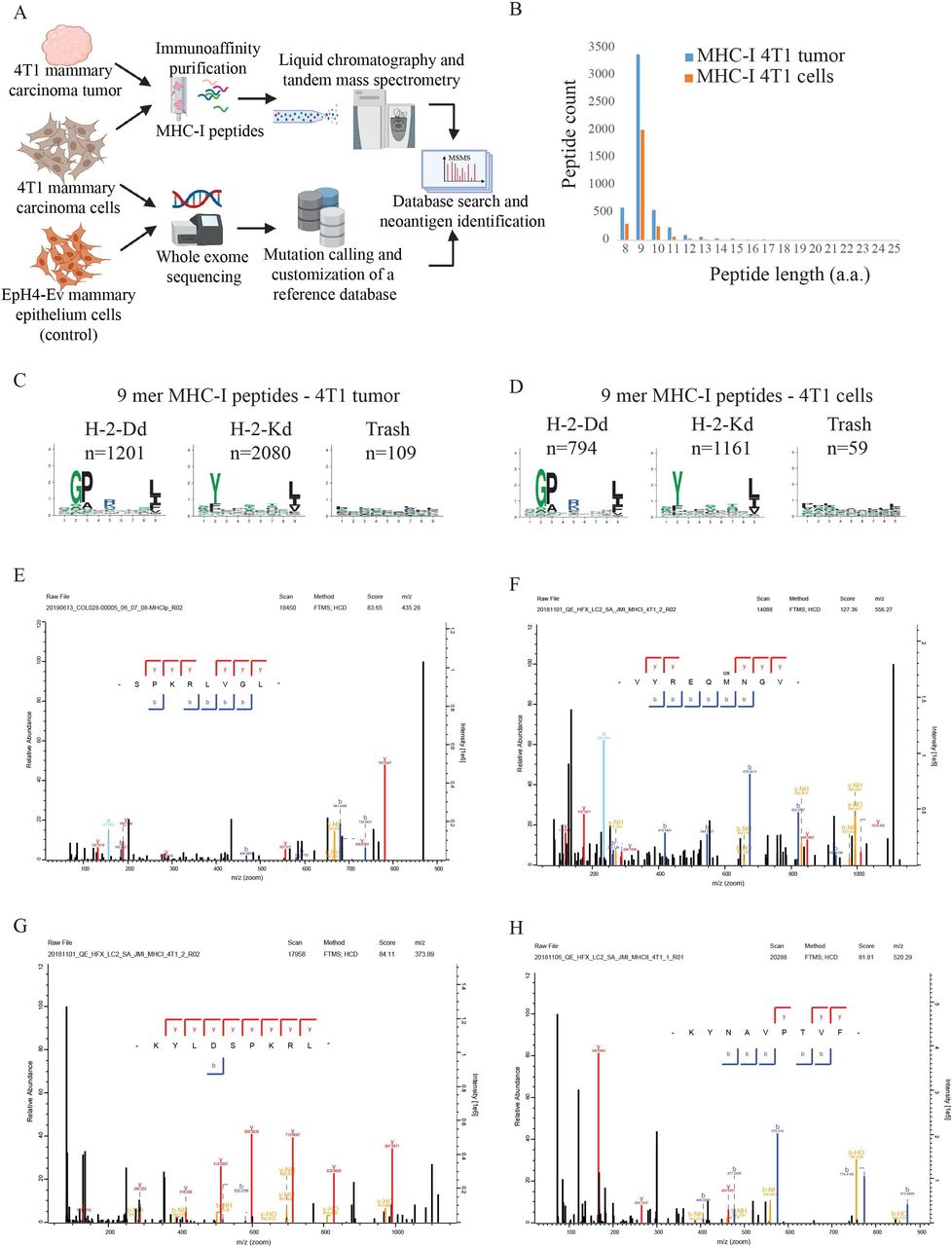
NeoAG identification. (A) Schematic representation of our approach for NeoAG identification. (B) Length distribution of MHC-I peptides identified by immunopeptidomics from 4T1 cells grown in vivo or in vitro. (C) Clustering of 9-mer MHC-I peptides identified in 4T1 cells grown in vivo and (D) in 4T1 cells grown in vitro. (E)–(H) Annotated MSMS spectra of the NeoAG identified by MaxQuant in 4T1 cells. MSMS spectra were exported from the MaxQuant viewer. NeoAG, neoantigens.
Verification of NeoAG sequence and expression
We performed RT-PCR to verify the sequence and expression of the NeoAG. RNA was extracted from the 4T1 mammary carcinoma cell line, from 4T1 tumor in vivo and from the EpH4-Ev wild-type mammary epithelial cell line as control. The results confirmed presence and expression of the non-synonymous SMs in 4T1 mammary carcinoma cell line as well as in 4T1 tumor in vivo in Balb/c mice. These mutations were absent in EpH4-Ev cells (online supplemental figure 2).
Supplemental material
Efficient coupling of synthetic NeoAG peptides by Cu-free click chemistry to VLPs
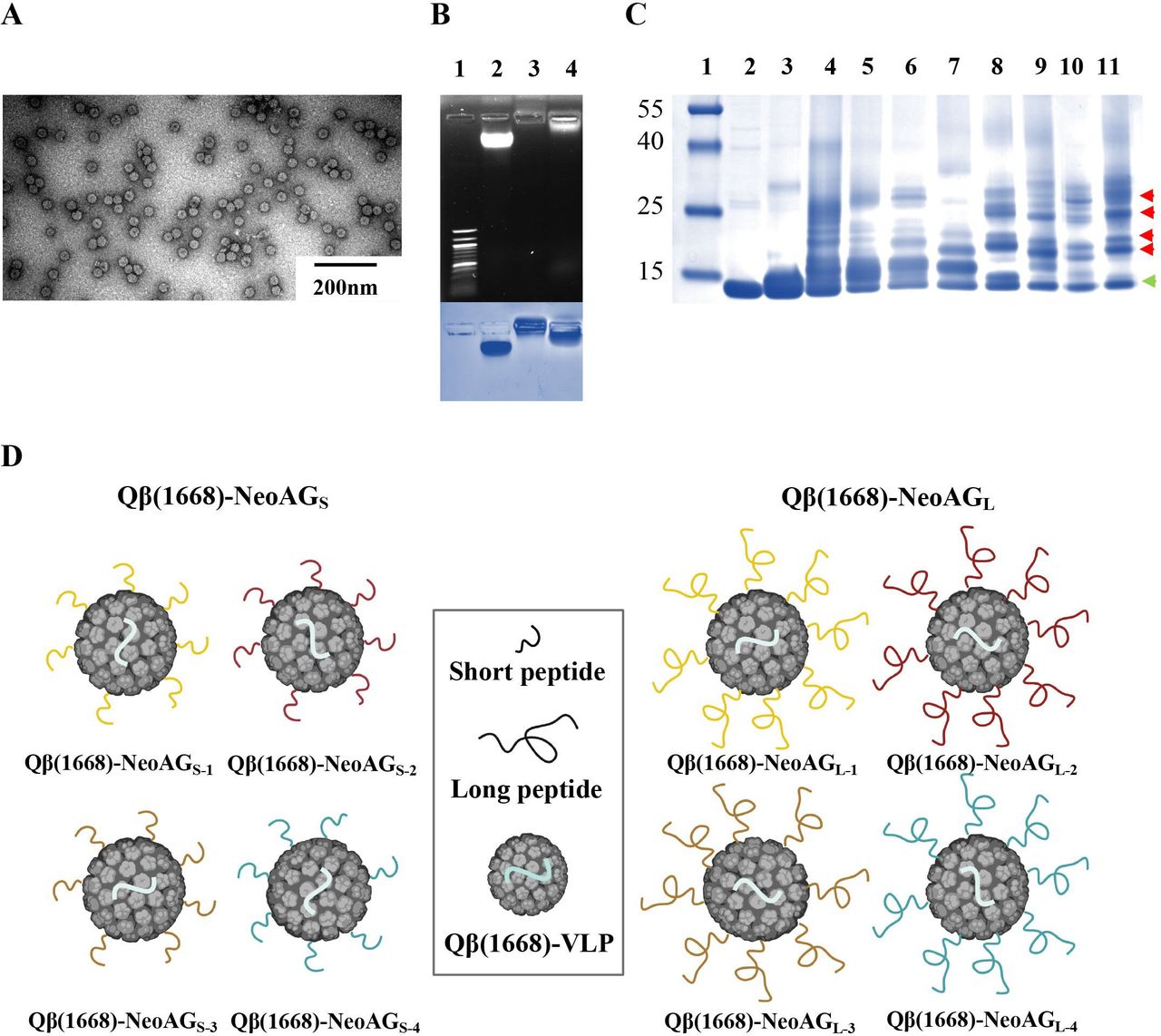
Qβ(ssRNA)-VLPs were expressed and purified as described previously.22 23 The integrity of Qβ(ssRNA)-VLPs was confirmed by electron microscopy (figure 2A). In a next step, Qβ(ssRNA)-VLPs were treated with RNaseA to digest the RNA that was naturally packaged during protein expression in Escherichia coli.15 33 The empty particles were then re-packaged with the B-type CpGs 1668, a TLR-9 ligand and potent activator of DCs, required for effective DC maturation and cross-priming. The re-packaging is based on direct diffusion of CpG into Qβ-VLPs via its natural pores followed by binding to arginine repeats inside the capsid which in the natural virus are binding the viral genome.34 The re-packaging process of Qβ-VLPs with the CpGs was confirmed in a 1% agarose gel stained with SYBR Safe dye (figure 2B, top), and the presence of the VLP’s capsid protein was assessed by staining the agarose gel with Coomassie Blue (figure 2B, bottom). In two previous and independent studies, we have shown that the bio-orthogonal Cu-free click chemistry is an efficient, safe (non-toxic) and fast method to chemically couple peptides of∼14 a.a. to Qβ-VLPs21 or to the plant-derived virus CuMVTT-VLPs.35 For several reasons, including ease of manufacturing and safety, this method is preferable over using the heterobifunctional cross-linker SMPH when considering the development of a personalized cancer vaccine (see also the Discussion section). Short (15–16 a.a.) or long (32 a.a.) neoantigenic peptides were synthesized with an additional lysine (K) and an azide group (N3) at the C-terminus (table 1) for coupling to Qβ(1668)-VLPs (table 2). The coupling efficiency was confirmed by SDS-PAGE followed by densitometric analysis. SDS-PAGE showed Qβ-VLP monomers (~14 kDa) as well as additional bands, indicating effective coupling of the synthesized NeoAG to Qβ(1668)-VLPs (figure 2C). Densitometric analysis of Qβ(1668) alone, Qβ(1668)-NeoAGS and Qβ(1668)-NeoAGL confirmed that the coupling was efficient (data not shown). Each NeoAG peptide (short or long) was coupled to Qβ(1668)-VLPs separately. To avoid possible outgrowth of any tumor escape variants, we prepared the final vaccines as multitarget NeoAG vaccines (figure 2D). The vaccine with the short peptides was termed Qβ(1668)-NeoAGS and the vaccine with the long peptides was termed Qβ(1668)-NeoAGL, each containing one of the neoantigenic peptides with the mutations identified in Vrk3 (E391K), Thap3 (H75Y) or Sult2b1 (H299N). The mutation in Vrk3 (E391K) was retrieved in two different MHC-I neoepitopes. The sequences and treatment doses are listed in table 2.

Efficient coupling of synthetic NeoAG peptides by Cu-free click chemistry to VLPs. (A) Electron microscopy imaging of the Qβ(ssRNA)-VLPs used for vaccine development. (B) (Top) 1% agarose gel stained with SYBR Safe dye, lane 1: DNA marker, lane 2: 10 µg of Qβ(ssRNA)-VLPs, lane 3: 10 µg of Qβ(empty)-VLPs digested with RNase A and lane 4: 10 µg of Qβ(1668)-VLPs re-packaged with B-type CpGs. (Bottom) Coomassie Blue staining of the agarose gel to confirm integrity of the VLPs, similar lanes as indicated on top. (C) SDS-PAGE stained with Coomassie Blue, lane 1: protein ladder, lane 2: 6 µg of Qβ(1668)-VLPs, lane 3: 6 µg of Qβ(1668)-VLPs derivatized with Cu-free click chemistry cross-linker (DBCO), lanes 4–7: 6 µg of Qβ(1668)-VLPs derivatized with Cu-free click chemistry cross-linker (DBCO) and coupled to NeoAG1 or NeoAG2 or NeoAG3 or NeoAG4 (15–16 a.a. short peptides), lanes 8–11: 6 µg of Qβ(1668)-VLPs derivatized with Cu-free click chemistry cross-linker (DBCO) and coupled to NeoAG1 or NeoAG2 or NeoAG3 or NeoAG4 (32 a.a. long peptides). The green arrow points to Qβ(1668)-VLP monomers and red arrows point to a peptide bound to Qβ monomer. (D) Cartoon illustrating the multitarget vaccines: Qβ(1668)-NeoAGS (left) and Qβ(1668)-NeoAGL (right). a.a., amino acids; NeoAG, neoantigens; ssRNA, single-stranded RNA; VLPs, virus-like particles.
Superior in vivo efficacy by the long peptide vaccine as opposed to the short peptide vaccine
We compared the in vivo anti-tumor efficacy of the short and long peptide vaccines. To this end, the aggressive 4T1 mammary carcinoma cell line was inoculated s.c. in Balb/c mice as described in the "cell lines and mice section". On day 3, day 7 and day 13, s.c. vaccinations were done with Qβ(1668)-NeoAGS, Qβ(1668)-NeoAGL or Qβ(1668) as control without peptides (figure 3A). Vaccination with the Qβ(1668)-NeoAGS significantly (p= 0.0091) hindered tumor progression as compared with the Qβ(1668) control. Interestingly, the long peptide vaccine Qβ(1668)-NeoAGL showed better enhanced anti-tumor efficacy (p<0.0001) (figure 3B–D). We also included a saline (PBS) control and no statistical difference was detected between mice that received saline or Qβ(1668) control (online supplemental figure 3). It is well known that infiltration of CD8+ T cells into the tumor correlates with better prognosis, which serves as an essential piece of evidence for effective immune responses in melanoma36 37 as well as breast cancer.38 Furthermore, it has been suggested that the majority of immunogenic NeoAG in personalized cancer vaccines activate CD4+ T cells.39 Accordingly, we determined the density of CD8+ and CD4+ T cells in each tumor. No significant differences were found for CD8+ (p=0.9759) or CD4+ (p=0.847) T cell infiltration between the control group and the group treated with the short peptide vaccine. Yet, a considerably increased CD8+ T cell infiltration was observed after long peptide vaccination (figure 3E,F). The infiltration of CD4+ T cell was also increased after long peptide vaccination (figure 3G,H). To study the systemic effects of the vaccinations, we characterized CD8+ and CD4+ T cells in peripheral blood 15 days post 4T1 inoculation. We found no significant differences between the groups (data not shown). We also assessed MDSCs characterized by CD11bHi Ly6CHi or CD11bHi Ly6GHi (figure 3I–K). Interestingly, the percentage of CD11bHi Ly6CHi was significantly decreased in the group treated with Qβ(1668)-NeoAGS when compared with the control group (p=0.026). The long peptide vaccine Qβ(1668)-NeoAGL was even more efficient in diminishing this population in comparison to the control group (p=0.0002).
Supplemental material

Superior in vivo efficacy by the long as opposed to the short peptide vaccine. (A) Treatment regimen using a prime and two boost vaccinations as summarized in table 2. (B) Tumor volume in mm3 measured on day 17 post 4T1 inoculation. Each dot represents an individual tumor. (C) Tumor growth curves of the treated groups. (D) Individual tumor growth curves of the treated groups. (E) and (G) Density of CD8+ or CD4+ T cells in tumors as determined by flow cytometry. The densities were determined by dividing the total number of CD8+ or CD4+ T cells in each tumor by the tumor volume in mm3, pre-gated on TILs. (F) and (H) Representative FACS plots showing the percentage of CD8+ or CD4+ or T cells in tumors, pre-gated on TILs. (I) and (J) Percentage of CD11bHi Ly6CHi or CD11bHi Ly6GHi cells in peripheral blood 15 days post 4T1 inoculation. (K) A representative FACS plot showing the percentage of CD11bHi Ly6CHi in peripheral blood 15 days post 4T1 inoculation, pre-gated on monocytes. Statistical analysis (mean±SEM) by one-way ANOVA (B), AUC and Student’s t-test (C) and Bonferroni test (E, G, I and J). (n=11), one representative of two similar experiments is shown. AUC, area under curve; NeoAG, neoantigens; TILs, tumor infiltrating lymphocytes.
In the tumor microenvironment, the long peptide vaccine reduced MDSCs and increased IFN-γ and TNF-α production by T cells
Based on its superiority, we focused on the long peptide vaccine Qβ(1668)-NeoAGL. To explore whether the induced immune response is mediated by a single NeoAG or the multiple ones, mice were vaccinated with Qβ(1668)-NeoAGL-1 or Qβ(1668)-NeoAGL-2 or Qβ(1668)-NeoAGL-3 or Qβ(1668)-NeoAGL-4 separately or with the multitarget vaccine Qβ(1668)-NeoAGL. Splenocytes of vaccinated mice were collected and stimulated with each corresponding NeoAG peptide, or with a combination of the four neoantigenic peptides for the group vaccinated with the multitarget vaccine. The results showed that vaccination with the multitarget vaccine induced significantly increased TNF-α production in CD8+ and CD4+ T cells in comparison to mice vaccinated with a single NeoAG (figure 4A,B). Significantly increased production of IFN-γ in CD8+ T cells but not in CD4+ T cells could also be detected in the group vaccinated with the multitarget vaccine (figure 4C,D).

In the tumor microenvironment, the long peptide vaccine reduced MDSCs and increased IFN-γ and TNF-α production by T cells. (A) and (B) Percentage of CD8+ TNF-α+ or CD4+ TNF-α+ cells in splenocytes from mice treated with vaccines with single long peptide 1,2,3 or 4, or all 4 long peptides (multitarget Qβ(1668)-NeoAGL vaccine, underlined). (C) and (D) Percentage of CD8+ IFN-γ+ or CD4+ IFN-γ+ T cells in splenocytes from the same mice. Naive and control peptide (CP) were used. (E) Tumor volume in mm3 measured on day 17 post 4T1 inoculation of control mice treated with Qβ(1668) or mice vaccinated with Qβ(1668)-NeoAGL. Each dot represents an individual tumor. (F) and (G) CD8+ or CD4+ T cell density in tumors as determined by flow cytometry and dividing the total number of CD4+ or CD8+ T cell in each tumor by the tumor volume in mm3. (H) and (I) Correlations between CD8+ or CD4+ T cells density in each tumor and its corresponding tumor volume in mm3. (J)–(L) Percentage of CD8+ IFN-γ+, CD8+ TNF-α+ and dual IFN-γ+, TNF-α+ CD8+ T cell in tumors. (N)–(P) Percentage of CD4+ IFN-γ+, CD4+ TNF-α+ and dual IFN-γ+, TNF-α+ CD4+ T cell in tumors. (M) and (Q) Representative FACS plots showing the percentage of dual IFN-γ+, TNF-α+ CD8+ or CD4+ T cell in tumors. (R) and (S) Percentage of CD11bHi Ly6CHi or CD11bHi Ly6GHi cells in tumors. Statistical analysis (mean±SEM) by Student’s t-test. (n=6), one representative of 2 similar experiments is shown. IFN, interferon; MDSCs, myeloid-derived suppressor cells; NeoAG, neoantigen.
In order to characterize the intratumoral immune response induced by vaccination, we compared tumors from control mice with those after vaccination with the long multitarget peptide vaccine Qβ(1668)-NeoAGL. The results confirmed that long peptide vaccination can significantly hinder 4T1 tumor growth (p=0.0022) when compared with the control group treated with Qβ(1668) (figure 4E). T cell density was assessed in each tumor by flow cytometry. We found significantly enhanced intratumoral infiltration of CD8+ and CD4+ T cells (p=0.0005 and p=0.0035, respectively) (figure 4F,G), inversely correlating with tumor volume (p=0.0089 and p=0.0079, respectively) (figure 4H,I). Furthermore, we determined the production of IFN-γ and TNF-α by CD8+ and CD4+ T cells after stimulation of T cells with a cocktail of the long peptides. Production of IFN-γ by specific CD8+ T cells was significantly increased (p=0.0022) (figure 4J). Despite a trend, the difference for TNF-α production was not significant, likely related to the high heterogeneity (figure 4K). When assessing specific CD8+ T cells producing both cytokines, a significant increase was found after long peptide vaccination as compared with control mice (p=0.0022) (figure 4L,M). The results also showed significant increase of IFN-γ or TNF-α production in specific CD4+ T cells (p=0.0022 and p=0.0303, respectively) (figure 4N,O) after long peptide vaccination compared with controls. This was also the case for the specific polyfunctional CD4+ T cells that simultaneously produced both cytokines (p=0.0022) (figure 4P,Q). Finally, we also studied the effect of vaccination on intratumoral monocytic and granulocytic myeloid cells. Interestingly, both intratumoral CD11bHi Ly6CHi and the CD11bHi Ly6GHi cells were significantly decreased after long peptide vaccination (figure 4R,S) indicating a decrease in myeloid-suppressor cells
Long peptide vaccination decreases post-surgical tumor recurrence and lung metastases, and prolongs survival
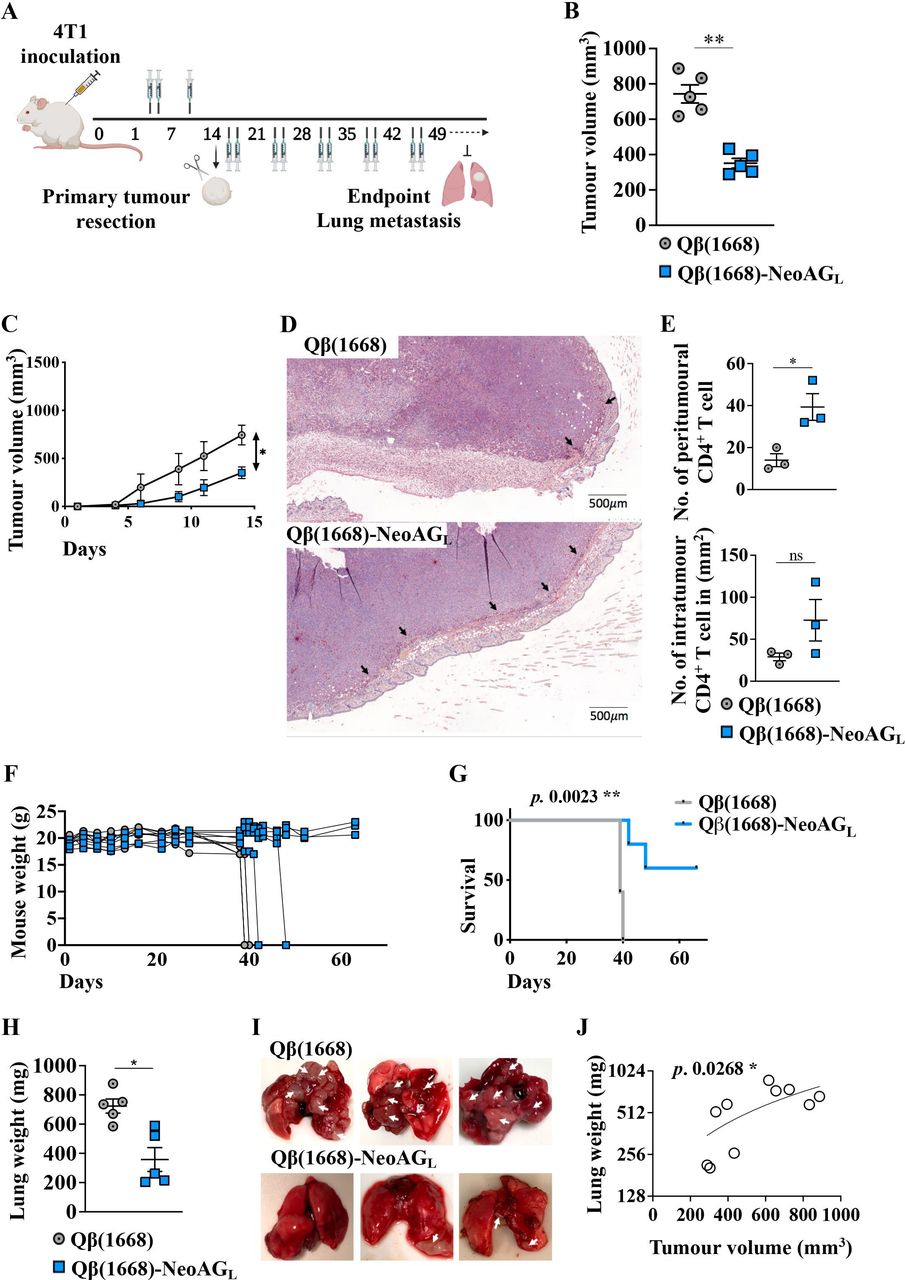
The observed efficacy of vaccination with Qβ(1668)-NeoAGL in vivo promoted us to address whether the vaccine may be useful in a clinically relevant perspective, namely, to prevent tumor progression after surgical resection of the primary tumor. To this end, 1 million 4T1 cells were inoculated in the left flank of the mice. Treatment with Qβ(1668) as a control or with the multitarget vaccine Qβ(1668)-NeoAGL was initiated 3 days later. Mice received 3 doses over 13 days and primary tumors were surgically resected on day 14, followed by 2 treatment doses weekly (figure 5A). Qβ(1668)-NeoAGL hindered 4T1 primary tumor progression as shown in figure 5B,C confirming our findings. By IHC, we investigated CD4+ and CD8+ T cell infiltration into the primary tumors (figure 5D). We found a significant increase (p=0.0229) in the numbers of peritumoral CD4+ T cells in the group vaccinated with Qβ(1668)-NeoAGL in comparison to the control group (figure 5E-top). The numbers of intratumoral CD4+ T cells were heterogeneous and showed a trend but no significant increase in the group vaccinated with Qβ(1668)-NeoAGL (figure 5E, bottom, and online supplemental figure 6). After surgical resection of primary tumors, the weight of mice in the control group remained stable for 10 days (until day 24 of primary tumor inoculation) but started to drop sharply thereafter when all the mice of the control group had to be sacrificed at days 39–40 after primary tumor inoculation. The mice vaccinated with the multitarget Qβ(1668)-NeoAGL showed much less weight loss and remained stable after day 40 (for the surviving mice) (figure 5F). However, the weight loss in some of the immunized mice required that 20% of the mice had to be euthanised on day 42 and 20% on day 48, whereas the remaining 60% of mice continued completely healthy and had stable weight until the end of the experiment on day 66 (figure 5G). We measured the weight of the lungs of euthanised mice to quantify lung metastases. The results reflect extensive 4T1 lung metastases in the control group, whereas long peptide vaccination resulted in significantly less lung metastases in the mice that were euthanised due to weight loss, and no lung metastases in the remaining 60% of the mice at the end of the experiment (p=0.0159) (figure 5H,I). Analyzing the volume of the primary tumor and lung weight revealed a significant correlation (p=0.0268) (figure 5J). To assess the role of CD8+ T cells in the effects of vaccination with Qβ(1668)-NeoAGL, we depleted CD8+ T cells. Three groups of mice were analyzed: a control group treated without tumor antigens (ie, Qβ(1668) alone), a group which received 3 doses of the multitarget Qβ(1668)-NeoAGL vaccine over 14 days, and a third group which received the same vaccination plus anti-CD8 monoclonal antibody. Efficient CD8+ T cells depletion of ~99% was confirmed in the third group by flow cytometry (online supplemental figure 5A). The effect of vaccination with Qβ(1668)-NeoAGL was significantly abrogated in the CD8+ T cell-depleted mice compared with non-depleted ones (p<0.0001) (online supplemental figure 5B). To further study the effect of CD8+ T cells depletion in the different groups, primary tumors were resected on day 14 and mice were followed for ~50 days without any additional vaccination. Our results showed reduced survival in the control group and the CD8+ T cell-depleted mice when compared with the vaccinated group (p=0.0024 and p=0.0008, respectively) (online supplemental figure 5C). Therefore, the beneficial anti-tumor effect of vaccination with Qβ(1668)-NeoAGL depends on CD8+ T cells.
Supplemental material
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Long peptide vaccination decreases post-surgical tumor recurrence and lung metastases, and prolongs survival. (A) Mice were inoculated with 1×106 4T1 cells on day 0 and vaccinated 3 times over 13 days. Primary tumors were surgically resected on day 14 under isoflurane anesthesia, followed by 2 vaccinations weekly until the end of the experiment. Mice were followed daily to monitor tumor recurrence, general health and mouse weight. (B) Tumor volume in mm3 measured on day 14 post 4T1 inoculation of control mice treated with Qβ(1668) or mice vaccinated with Qβ(1668)-NeoAGL. Each dot represents an individual tumor. (C) Tumor growth curves of the treated groups. (D) Representative immunohistochemistry (IHC) staining of CD4+ T cell in tumors of mice treated with Qβ(1668) as control or Qβ(1668)-NeoAGL. Arrows point to peritumoral CD4+ T cell (n=3). (E) Numbers of peritumoral (top) and intratumoral/mm2 (bottom) CD4+ T cell, respectively. (F) Mice weight (g) during the experiment. (G) Survival of mice in both groups; mice were euthanized when loosing 10%–15% of their original weight. (H) Lung weight (mg); mice were euthanized when loosing 10%–15% of their original weight and lungs were collected. (I) Representative photos of lungs from euthanized mice (control and Qβ(1668)-NeoAGL); arrows point to lung metastases. For the group treated with Qβ(1668)-NeoAGL, two lung samples are from the mice which were euthanized due to weight loss and one lung from a mouse that remained healthy and without metastases until the end of the experiment. (J) Correlation between primary tumor volume in mm3 and end-point lung weight (mg). Statistical analysis (mean±SEM) by Student’s t-test, AUC (C), log-rank test (H) and Ozone correlations (K). (n=5), one representative of 2 similar experiments is shown. IHC, AUC, area under curve; IHC, immunohistochemistry.
Discussion
In this study, we developed a universal approach for a personalized cancer vaccine. We found efficient tumor control through activation and functional competence of both CD8+ and CD4+ T cells, associated with reduction of suppressive myeloid cells both in circulation and within tumors. The vaccine was based on the successful identification of neoantigenic peptides using mass spectrometry-based immunopeptidomics combined with WES and wet-bench validation. The advantage of this experimental approach relies on the fact that it only identifies peptides that are indeed naturally presented by MHC molecules on the surface of tumor cells, thus avoiding the frequent epitope prediction failures by other techniques that do not rely on the physical identification of peptides. Indeed, our approach has been shown to enrich for NeoAG that are capable of controlling tumors in mice in vivo.40 To avoid possible outgrowth of tumor escape variants, we produced a multitarget vaccine with four neoantigenic peptides.
We synthesized short peptides corresponding to CD8+ T cell epitopes or long peptides, including both CD8+ and CD4+ T cell epitopes. Using the bio-orthogonal Cu-free click chemistry, peptides were coupled to VLPs consisting of recombinant bacteriophage Qβ protein and loaded with the B-type CpG 1668. The applied chemical coupling has several advantages as compared with other methods such as using SMPH linker.21 SMPH may react with internal cysteine within epitopes, causing their inactivation. Additionally, non-reacted SMPH on VLPs can be toxic and may complicate the Good manufacturing practice (GMP) process, in particular at bedside. Cu-free click chemistry is a non-toxic method, as the azide group added to the C-terminal end of the synthesized peptides is metabolically stable and lacks any reactivity with other biological functionalities in human cells. Cu-free chemistry has shown high selectivity in several studies, for example, for imaging, labeling and tracking cells in vivo.41 42 A key advantage of this method is that it can be applied at the bedside, enabling easy and rapid production of personalized vaccines for each patient directly where and when needed.
The short peptide vaccine significantly hindered tumor progression (p=0.0091) when compared with the antigen negative control vaccine. However, this vaccine did not enhance CD8+ nor CD4+ T cell infiltration into the tumors. Nevertheless, it significantly decreased (p=0.026) circulating MDSCs. The insufficient anti-tumor efficacy of the short peptide vaccine may be due to deficiency in activating CD4+ helper T cells that are often required to support the CD8+ T cells as shown in several studies.8 9 43 44 Indeed, vaccination with elongated peptides (by addition of natural flanking sequences) significantly increased the anti-tumor efficacy (p=0.0001). Additionally, long peptide vaccination elicited stronger T cell responses characterized by increased percentages of polyfunctional specific CD8+ and CD4+ T cells. Furthermore, long peptide vaccination enhanced the infiltration of CD4+ T cells into the tumors. It is known that CD4+ T cells can license DCs via CD40–CD40L interaction, which enhances antigen presentation to and activation of T cells.45 It may also be possible that CD4+ T cells themselves kill tumor cells.43 46 Probably more important is that tumor growth is inhibited by pro-inflammatory milieu generated by the effector cytokines, such as IFN-γ and TNF-α, which are not only released by CD8+ T cells but also by CD4+ T cells. However, despite the dependence on CD4+ T cells, the anti-tumor efficacy remained critically dependent on CD8+ T cells as shown by strongly reduced protection on in vivo depletion of CD8+ T cells.
There is general agreement that T cell vaccines must become more potent, particularly for the induction of CD8+ T cell responses, in order to increase the likelihood of clinical benefit. However, while CD4+ T cell responses are readily induced by the currently used cancer vaccines (eg, RNA, or free peptides mixed with adjuvants), CD8+ T cell responses induced by the majority of vaccines remain relatively scarce,4 5 particularly when vaccine antigens must undergo processing and cross-presentation by DCs which is an important but relatively inefficient process. Conversely, the requirement for cross-presentation has the advantage that only DCs present the vaccine’s MHC-I antigens to CD8+ T cells, favoring robust T cell priming and boosting, and avoiding tolerance induction.47
Immunosuppressive mechanisms are often responsible for the inefficiency of immunotherapy. Some of those mechanisms may be successfully hindered by therapies that appropriately activate T cell responses. Indeed, our long peptide vaccine led to significant reduction within tumors of granulocytic MDSCs known to exert negative effects on T cells, whereas appropriately activated T cells produce IFN-γ that counteracts MDSCs.48 These two cell populations may also be used as biomarkers for predicting treatment efficacy.
Surgical resection is usually the first step in the clinical management of cancer. Subsequently, residual microtumors as well as circulating cancer cells may cause potentially lethal recurrence.49 Immunotherapy is particularly necessary and potentially useful in patients who are at risk for subsequent disease recurrence and progression. Checkpoint blockade is often used in this setting. Alternatively, or in addition, personalized cancer vaccines may be considered, as they have the potential to activate the immune system more specifically and may thus have a favorable efficacy/toxicity profile, provided that they achieve sufficient anti-tumor effects for providing clinical benefit. This can be well addressed in the 4T1 tumor model, because these tumors can spontaneously metastasize from primary lesions to lymph nodes, lung, brain and bone, resembling triple-negative stage IV human breast cancer.50 After surgical resection of the primary tumors, we found that our multi-NeoAG vaccine led to reduction of tumor recurrence and metastases formation. Therefore, the use of our vaccine in this setting may be promising.
Vaccination alone may be insufficient to achieve satisfactory tumor control51 calling for combination therapies. For example, we and others have shown previously that combining a multitargeting vaccine with anti-CD25 mAb treatment to inhibit regulatory T cells significantly improved the anti-tumor efficacy in challenging mouse tumors.21 52 We envision that combining our new vaccine with anti-CD25 mAb, anti-OX40 mAb and/or checkpoint inhibitors may further improve therapy efficacy. The identification of patient-individual NeoAG remains challenging but is technically feasible.27 53 Together, our approach represents a further step making the vision of personalized cancer vaccines more realistic. Clinical development is encouraged because the vaccine induces efficient anti-tumor responses and is practically feasible as it can be formulated at bedside and may, therefore, be broadly applied.
Supplemental material
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study does not involve human participants.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Design of experiments, acquisition of data, interpretation and analysis of data: MOM, DES, SdB and MFB. Whole exome sequencing and mass spectrometry-based identification of neoantigens: MB-S, JM, HP, BJS and GC. Writing, revision and editing of manuscript: MOM, DES, MB-S and MFB. Technical, material and tool support: VPI, MV and SD. Study supervision: MOM and MFB. All authors read and approved the final manuscript.
Funding This work was supported by Qatar National Research Fund (PDRA4-0118-18002) and Swiss Cancer Research (KFS-4291-08-2017-R and KFS-5246-02-2021-R). The graphical abstract, Figure 1A, Figure 3A and Figure 5A were generated with BioRender.com.
Competing interests MM, DES and MFB are shareholders of DeepVax GmbH, involved in the development of cancer immunotherapy.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.