Article Text

Abstract
Background Cancer-associated neoantigens (neoAg) derived from tumor genomic sequencing and predictive algorithms for mutated peptides are a promising basis for therapeutic vaccines under investigation. Although these are generally designed to bind major histocompatibility complex class I and induce CD8 cytolytic T lymphocyte (CTL) activity, results from preclinical and clinical studies demonstrate that the majority of neoAg vaccines efficiently induce CD4 T helper (Th) responses but not CTL. Despite this, these vaccines have demonstrated clinical efficacy. Therefore, understanding the mechanisms of CD4 + T cell-mediated tumor protection is critical to optimizing this immunotherapeutic strategy.
Methods We investigated this phenomenon in the mineral oil-induced plasmacytoma (MOPC).315.BM (MOPC315) mouse model of multiple myeloma, a malignancy of plasma cells. MOPC315 cells express in their lambda chain a unique tumor-specific neoAg, an idiotypic (Id) peptide. We generated a vaccine formulated with this Id peptide fused to a heat shock protein HSC70 binding (HSB) motif co-delivered with poly (I:C). The immunogenicity of the Id-vaccine was measured in splenocytes by ELISpot. Mice were challenged with MOPC315 cells and antitumor immunity was assessed by co-incubating splenocytes and bone marrow mononuclear cells derived from vaccinated mice and controls, with the Id antigen and irradiated MOPC315 cells. The frequency of activated CD4 and CD8 T cells and their phenotype were characterized by flow cytometry.
Results Id-vaccine efficiently induced antigen-specific CD4 Th activity and antitumor immunity, protecting mice from MOPC315 tumor growth. CD4 cytolytic activity was not detected under these conditions. Polyfunctional CD8 T cells homed to the bone marrow microenvironment of protected mice and preferentially expanded only when restimulated ex vivo with both Id peptide and MOPC315 cells. Protective activity was abrogated by depletion of either CD4 or CD8 lymphocytes.
Conclusion These results demonstrate that Id-HSB +poly (I:C) vaccine protects against MOPC315 growth by priming Id-specific CD4 Th cells that confer protection against tumor but are not directly cytotoxic. These data indicate that activation of CD8 CTL against MOPC315-associated antigens not present in the vaccine is one of the major mechanisms of tumor immunity.
- antigens
- tumor-associated
- carbohydrate
- CD4-positive T-lymphocytes
- immunogenicity
- vaccine
- tumor microenvironment
- CD8-positive T-lymphocytes
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- antigens
- tumor-associated
- carbohydrate
- CD4-positive T-lymphocytes
- immunogenicity
- vaccine
- tumor microenvironment
- CD8-positive T-lymphocytes
Introduction
Immunologic therapy for cancer has pursued the hypothesis that effective tumor antigen (Ag)-specific CD8 (cytotoxic) T cell activity is necessary to kill cancer cells and confer clinical benefit. Therefore, the majority of therapeutic strategies are designed to prime or boost such T cells, promote cellular or molecular mechanisms such as proinflammatory cytokines and co-stimulatory molecule agonists to enhance their activity, or to block inhibitory factors such as immune checkpoints that impair antitumor cytotoxic T cells. The efficacy of immune checkpoint inhibitors (anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and anti-programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1)), bi-specific engineered T cell engaging agents (eg, blinatumomab CD19×CD3), and chimeric antigen receptor T cells1 2 supports this overall concept. Therapeutic cancer vaccines are another promising strategy, which deliver cancer-associated or-specific Ags to prime/boost CD8 T cells. Several different classes of tumor-associated Ags have been investigated, including cancer-associated proteins such as survivin,3 MUC14 or cancer-testis Ags5 in many tumor types. A novel class of tumor-specific targets, mutational neoantigens (neoAg) has emerged as potential targets in tumors that feature high mutational burdens.6 These were first identified in animal models and clinical trials of idiotype-based vaccines in non-Hodgkin’s lymphoma,7–12 a truly tumor-specific neoAg due to the combinatorial and mutational biology that produces a unique idiotype. This principle was broadly demonstrated in clinical trials of CIs, as translational studies demonstrated that expansion of neoAg-specific T cells correlated with clinical response,13 14 which in turn spawned investigation into formulating ‘personalized’ neoAg vaccines for clinical application. These neoAgs are derived from novel bioinformatic prediction methods applied to tumor exome sequencing and gene expression data to identify expressed mutations predicted to be immunogenic based on criteria such as binding affinity or similarity to infectious disease Ags.15
Although these strategies were designed to stimulate CD8 cytolytic T lymphocyte (CTL), clinical trials and laboratory models have repeatedly demonstrated that tumor Ag-specific CD4 (helper) T cells are capable of conferring protective antitumor immunity, even in the absence of CD8 activity directed against the same Ags. In a clinical trial of neoAg vaccines in melanoma, the majority of neoAg peptides stimulated CD4 T cells.16 Vaccine-induced or adoptively transferred tumor Ag-specific CD4 T cells have demonstrated clinical responses, even though they did not exhibit direct cytolytic activity against tumor cells.17 In one such study, vaccine-elicited NY-ESO-1-specific CD4 T cells were correlated with clinical benefit in melanoma.18 Interestingly, there was evidence of epitope spreading to other melanoma-associated Ags such as MelanA-MART1 and GP100, but the CD4/CD8 phenotype of these responses was not reported. These findings suggest that while tumor Ag-specific CD4 T cells can orchestrate protective immunity, the cellular and molecular mechanisms as well as the nature of the Ags they target remain unclear. Thus, understanding this phenomenon and how it results in protective immunity is critical to the development of next-generation cancer immunotherapy.
We investigated the role of neoAg-specific CD4 T cells in an animal model of multiple myeloma (MM), a cancer of plasma cells that is the second most common hematological malignancy. The only reported curative treatment for this disease is allogeneic hematopoietic stem cell transplantation (allo-SCT), demonstrating that immunologic therapy in the form of the graft-versus-tumor effect can eradicate MM cells and prevent relapse.19 However, allo-SCT has very high procedure-associated mortality and the majority of patients with myeloma are ineligible for this procedure due to age and medical limitations. Therefore, we investigated a neoAg vaccine strategy as a means to induce protective anti-MM immunity without the morbidity and mortality associated with allo-SCT. Mineral oil-induced plasmacytoma (MOPC).315.BM cells (MOPC315) are malignant plasma cells derived from the immune competent BALB/c background and mimic many of the features of the human disease.20 MOPC315 cells produce a monoclonal IgA which is a unique MOPC315-associated Ag due to somatic mutations in the third hypervariable region of the lambda2 L chain, resulting in a neoAg called an idiotypic (Id) peptide. This peptide, pId315, is presented by the major histocompatibility complex (MHC) class II molecule I–Ed to Id-specific CD4+ T cells.10 Transgenic mice whose T cells expressed a pId315:I-Ed-specific T cell receptor (TCR) were protected against a challenge with MOPC315 cells.17 21
Here, we used a novel pId315 vaccine formulation that efficiently induced Id-specific CD4 T cells and conferred protection against MOPC315 tumor growth. In this setting, no pId315 specific CD8 T cell or B cells responses were detected. Ex vivo restimulation of splenocytes and bone marrow mononuclear cells (BMMC) with Id alone only showed CD4 T cell activity. However, restimulation with MOPC315 cells demonstrated CD8 T cell activation, and this was amplified by stimulation with both Id peptide and MOPC315 cells. Moreover, depletion of CD8 T cells after Id vaccination resulted in loss of protection. Furthermore, immunophenotyping of BMMC from protected and unprotected test animals revealed increased populations of effector/memory CD4 and CD8 T cells. Taken together, these results show that neoAg-specific CD4 T cells confer protective immunity through activation of CD8 T cells against tumor-associated Ags not expressed by the vaccine.
Material and methods
Mice and cell lines
Eight to ten week-old female BALB/c mice were purchased from Charles River laboratories and used for immunizations and tumor challenge. Mice were immunized with 100 µg of two different Id peptides either two times or three times at days 0, 14 and 21 in combination with 50 µg of poly (I:C) (InvivoGen). One peptide (Id22) included the 11-mer idiotypic minimal epitope corresponding to residues 91–101 of the λ2315 L chain of M315 myeloma protein produced by MOPC315 cells, linked to a heat shock protein 70 binding motif (HSB). The second peptide (Id27) included the same 11-mer epitope flanked by the wild-type sequence of λ2315. Both peptides were provided by Agenus.
MOPC.315.BM cell line was kindly provided by Dr B Bogen (University of Oslo) in 2015. A20 lymphoma cell line was kindly provided by Dr J Brody (Icahn School of Medicine at Mount Sinai).
For CD8 + and CD4+T cell depletion, we injected mice three consecutive days before tumor challenge with 100 µg of anti-mouse CD8β (clone 53–5.8) or anti-mouse CD4 (GK1.5) and their isotype controls rat IgG1 and rat IgG2b, respectively (Bio X Cell). Depletion was verified in mice peripheral blood mononuclear cells (PBMCs) before tumor challenge and 2 and 3 weeks after depletion.
Tumor challenge
Bone marrow (BM) tumors were generated by injecting intravenously 100,000 MOPC.315.BM cells in the tail vein. Tumors were monitored by measuring weekly levels of M315 protein in the blood by ELISA as previously described.22 23 Mice were checked every other day and daily at week 3 after tumor challenge. They were euthanized when they developed hind limb paralysis or after 80 days of survival. Splenocytes and BMMC were harvested and cryopreserved.
Interferon-γ ELISpot
ELISpot has been performed following the manufacturer’s instructions (BioLegend). Briefly, 96 wells plates (Millipore) were coated with 10 µg/mL of anti-mouse interferon-γ (IFN-γ) (BioLegend, AN18) overnight at 4°C. Splenocytes were harvested 7 days after the last immunization and plated at 4×105 and 2×105 cells/well with Id22, Id27 peptide or their wild type (WT) counterpart peptide at 5 µg/mL. Medium alone and Concanavalin A (Sigma) were used as negative and positive control, respectively. After 24 hours of incubation at 37°C, plates were washed and incubated for 1 hour at room temperature with biotinylated anti-mouse IFN-γ antibody (BioLegend, R4-6A2), then washed and incubated for 1 hour at room temperature with streptavidin-AP. After extensive washing, 100 µl/well of the substrate (NBT/BCIP, KPL) was added to measure spot development. Spots were read using an ImmunoSpot CTL reader and calculated per million splenocytes.
CD4 and CD8 T cells were separated using Dynabeads Untouched Mouse T Cells kit (Invitrogen) following manufacturer’s instructions.
Flow cytometry
Antibodies were purchased from BioLegend and Thermo Fischer. We used BioLegend antibodies for regular flow cytometry assays analyzed on the BD LSRII CD3ε (145–2 C11), CD4 (GK1.5), CD8α (53–6.7), CD138 (281-2), I-A/I-E (M5/114.15.2), IFN-γ (XMG1.2), TNFα (MP6-XT22) and CD16/32 (93).
For intracellular staining, BMMC or splenocytes were incubated with Id22 peptide, irradiated MOPC cells or both. Brefeldin A (BioLegend) was used for cytokine accumulation (4–5 hour) and cell stimulation cocktail (eBiosciences) as positive control. After surface staining, Cytofix/Cytoperm (BD BioSciences) was used to fix and permeabilize the cells and LIVE/DEAD fixable aqua dye for viability (Invitrogen).
For spectral flow cytometry, the following panel (table 1) was used:
Antibodies for spectral flow cytometry
Cytotoxicity assay
Splenocytes and BMMC were harvested and stimulated with 5 µg/mL of Id22 peptide for 7 days. Recombinant mouse interleukin-2 was added every 2–3 days. CD4 and CD8 T cells were separated using Dynabeads Untouched Mouse T Cells kit (Invitrogen) following manufacturer’s instructions and diluted to several E:T cell ratios in 96-well U-bottom culture plates in RPMI 1640 medium (Thermofisher) supplemented with 5% fetal bovine serum (FBS) (Sigma-Aldrich), 100 U/mL penicillin, and 100 U/mL streptomycin (Thermofischer). MOPC cells were added to effector cells and incubated for 4 hours. Supernatants were recovered and specific lysis was assayed with the CytoTox 96 lactate dehydrogenase (LDH) release kit (Promega, Madison, Wisconsin, USA) according to the manufacturer’s instructions.
Statistics
Data are presented as means±SEM. All statistical comparisons were performed using Prism V.8.0 Software (GraphPad Software, La Jolla, California, USA). Log rank (Mantel-Cox) test was used for survival in tumor challenge experiments and one-way or two-way analysis of variance with Tukey’s tests for multiple comparisons.
Results
Idiotypic peptide vaccine elicits specific CD4 T cell responses that protect from tumors
The MOPC315.BM cell line, derived from the MOPC315,24 mimics human multiple myeloma disease in that cells injected intravenous into immunosufficient BALB/c mice home to the BM and cause osteolytic disease.20 MOPC315.BM cells secrete a mutated IgA myeloma protein called M315. The M315 λ2 L chain expresses three somatic mutations (Y94S95T96→ F94R95N96) that create an immunogenic neoAg comprizing residue 91–101 of the CDR3 region.10 This neoAg, also called an Id peptide, is presented by the MHC class II molecule I-Ed to Id-specific CD4 T cells. In TCR-transgenic mice, Lauritzen et al showed that recognition of pId315:I-Ed by CD4 + T cells in TCR-transgenic mice results in protection against MHC-IINEG MOPC315 cells by an indirect mechanism that involves tumor infiltrating macrophages and that does not require CD8 + T cells or B cells.23 25
Here, we use a vaccination strategy where two different modified Id-peptides were synthesized for immunization of mice. The first is a 27-mer peptide (Id27) containing the 11-mer minimal neoAg epitope pId315 flanked by the wild type λ2 sequence. The second is a 22-mer (Id22) peptide where the 11-mer epitope is fused to an HSC70-binding motif (HSB) via a 4-amino acid linker (figure 1A). We immunized BALB/c mice either two times or three times subcutaneously with either Id22 or Id27 in combination with poly (I:C) adjuvant. Seven days after the last immunization, we quantified IFN-γ-secreting cells in the spleen by ELISpot assay. We observed that on three immunizations with Id22 peptide, the number of IFN-γ-secreting cells induced was double compared with only two immunizations; and was four times higher than three immunizations with Id27 peptide (figure 1B,C). Notably, these immune responses were mainly CD4 T cell responses as demonstrated by assays of separated CD4 and CD8 T cells.
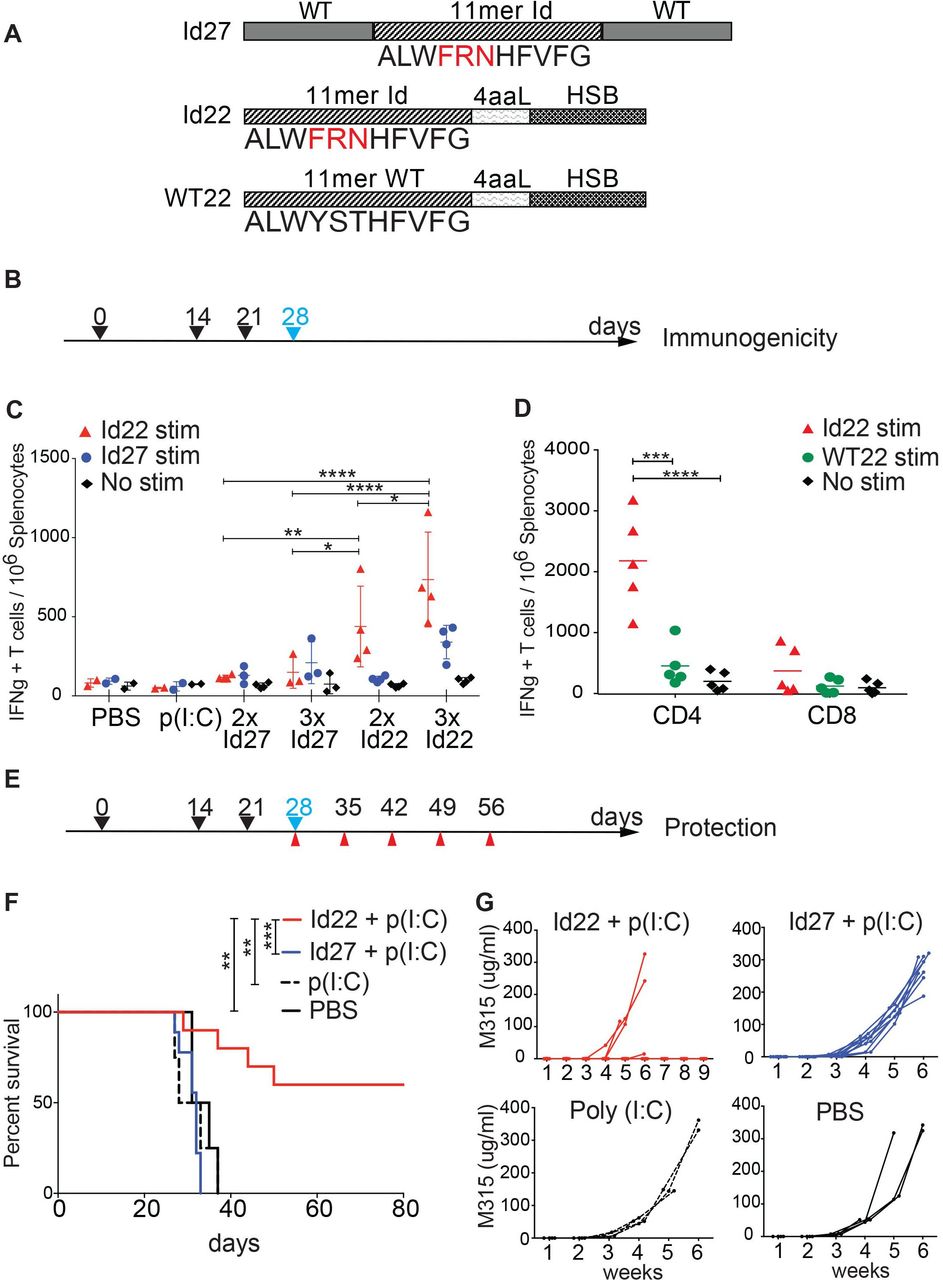
Idiotype peptide vaccine elicits protective antitumor CD4 T cell responses. (A) Peptides design for immunization. Two different idiotype 2315 peptides were synthesized to be used as vaccines in combination with the poly (I:C) adjuvant. Id27 consists of the 11-mer Id minimal epitope with three somatic point mutations (in red) flanked by the WT sequence. Id22 is composed of the 11-mer Id connected by a four amino acids linker (4aaL) to an Hsc70 binding motif (HSB). A third peptide, WT22, is composed of the 11 aa WT sequence linked to HSB and was used as a control for T cell restimulation. (B) Timeline schedule for immunogenicity experiment showing immunization days (black arrows) and spleens collection day (blue arrow). (C) Immunogenicity of the idiotype peptides. Groups of mice were immunized subcutaneously two times (2 x) at days 0 and 14 or three times (3 x) at days 0, 14 and 21 with 100 µg of Id22 (n=4) or Id27 (n=3) in combination with 50 µg of poly (I:C). Control mice were immunized with poly (I:C) (n=2) or PBS (n=2). Splenocytes were harvested on day 28 to measure IFN-γ production by ELISpot. Cells were restimulated with Id22, Id27 peptide or no stimuli. (D) five mice were immunized three times at days 0, 14 and 21 with 100 µg of Id22 in combination with 50 µg of poly (I:C). Splenocytes were harvested at day 28. CD4 and CD8 T cells were separated using magnetic beads and IFN-γ responses were measured by ELISpot using Id22 or its wild type counterpart WT22 for stimulation. (E) Timeline schedule for tumor protection experiment showing immunization (black arrows), MOPC.315.BM tumor challenge (blue) and blood collection (red) days. (F) Kaplan-Meier survival curve of vaccinated mice challenged with MOPC.315.BM. Four groups of mice were immunized three times with Id22 (n=10), Id27 (n=10), Poly (I:C) (n=4), or PBS (n=4) as controls and MOPC315.BM tumor cells were injected at day 28. (G) Sera were collected every 7 days for measurement of M315 protein levels in the blood by ELISA. Data were analyzed using two-way analysis of variance and Mantel-Cox test. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. HSB, heat shock binding; Id, idiotypic; IFN, interferon; MOPC, mineral oil-induced plasmacytoma; PBS, phosphate-buffered saline; WT, wild type.
The addition of the HSC70 binding motif (HSB) significantly enhanced immunogenicity of the peptide vaccine. The CD4 response was specific for the minimal Id-peptide pId315 since the wild-type counterpart WT22 (WT 11-mer linked to the HSC70 binding motif, figure 1A) failed to elicit any CD4 T cell responses (figure 1D).
We then examined the efficacy of Id22 or Id27 in protecting mice against tumor growth in a prophylactic model. We immunized mice using the three dose schedule, challenged them with 105 MOPC.315.BM cells by intravenous injection 1 week after the last immunization, and monitored tumor progression by measuring weekly levels of M315 protein in the serum (figure 1E–G). Sixty per cent (6/10) of mice immunized with Id22 peptide exhibited significant protection from tumor growth up to 80 days of observation. In contrast, all mice vaccinated with Id27 peptide and control animals that received either poly (I:C) alone or phosphate-buffered saline demonstrated increasing M-protein and morbidity/mortality during the observation period. These results show that the Id22 +poly (I:C) vaccine induced Ag-specific CD4 T cell activity and conferred protective immunity against tumor challenge.
Id22-specific CD4 T cells do not directly kill MOPC315 cells
Id22-specific CD4 T cells could mediate tumor protection by 1. Direct cytotoxic activity against tumor cells, 2. Indirect activation of natural killer (NK) and/or myeloid cells, or 3. Cross-priming tumor-specific cytotoxic CD8 T cell activity. To determine if Id22-specific CD4 T cells could directly eliminate tumor cells in our system, we isolated CD4 T cells from splenocytes of Id22 vaccinated mice and co-cultured them with MOPC.315.BM cells at different E:T ratios in cytotoxicity assays. No direct killing of tumor cells was observed (figure 2A).

Id22-specific CD4 T cell do not directly kill MOPC cells. (A) CD4 T cells were isolated from Id22 vaccinated mice and co-cultured with MOPC.315.BM at different effector:target (E:T) ratios. The percentage of cytotoxic activity was measured using LDH release in vitro assay. (B) MHC class II expression on MOPC cells in vivo. Bone marrow mononuclear cells (BMMC) were harvested from unvaccinated mice (No vax +Chall.), or Id22-vaccinated mice, three immunizations with Id22 +poly (I:C), unprotected from tumor growth (Id vax +Chall., unprotected) when tumors reached their endpoint at day 37 after tumor challenge. BMMC were harvested from Id vax +Chall., protected mice at the end of the experiment at 80 days after tumor challenge. (C) MOPC cells (red) and normal plasma cells (blue) were stained using anti-mouse CD138, CD4 (to identify MOPC cells) and MHC-II antibodies. Id, idiotypic; LDH, lactate dehydrogenase; MHC, major histocompatibility complex; MOPC, mineral oil-induced plasmacytoma.
MOPC.315.BM cells do not express MHC class II at baseline, drastically reducing the likelihood that CD4 T cells can directly recognize them. However, it has been reported in some cases that MHC class II can be upregulated in vivo in response to IFN-γ.26 We assessed the expression of MHC class II in vivo by staining MOPC.315.BM cells from BM aspirates of Id22 vaccinated mice or controls that were challenged with MOPC.315.BM cells and selected test animals that were either protected from tumor growth or not. MOPC.315.BM cells were identified by flow cytometry, appearing as CD4+, CD138 +cells in the CD3-negative fraction of BMMC,27 whereas normal plasma cells were CD4− CD138+. Protected mice eliminated MOPC315 cells from their BM (fig, 2B, red quadrants) in comparison with unprotected or unvaccinated mice, while normal plasma cells were unaffected (figure 2B, blue quadrants). MOPC315 cells did not express MHC class II in vivo in any of the conditions (figure 2C red histograms and online supplemental figure 2). The number of MOPC cells in PRO mice was too low to assess expression of MHC-II, however, MOPC cells in UNP mice who were vaccinated in the same way than PRO mice did not express MHC-II in vivo. Therefore,it is likely that MOPC315 cells were not directly killed by Id-specific CD4 T cells under these conditions. This is similar to the findings of Tveita et al28 in which Id-specific CD4 T cells could mediate rejection of I-Ed knocked out MOPC cells, supporting an indirect mechanism in the absence of MHC-II.
Supplemental material
Id22 vaccine elicits tumor-specific CD8 T cell activation in the BM
We next investigated the activity of spleen and BM CD4 and CD8 T cells in this model. We harvested splenocytes and BMMC from Id22-immunized mice and restimulated them in vitro for 8 days by co-culture with Id22 peptide in the presence of irradiated MOPC.315.BM cells (iMOPC), then measured IFN-γ and tumor necrosis factor (TNF)α secretion by intracellular cytokine staining. BMMC were collected after mice reached endpoint (hind limb paralysis) for challenged only (CHA) or unprotected (UNP) mice, 7 days after the last immunization for vaccinated only mice (IDV), and 80 days after tumor challenge for protected mice (PRO). Figure 3A shows an example of CD4 and CD8 T cells activation in the spleen in comparison with the BM of protected mice (PRO). We observed the presence of polyfunctional IFN-γ+TNFα+CD4 T cells in the spleen as expected. However, polyfunctional CD8 T cells are present only in the BM but not in the spleen suggesting a recruitment of CD8 T cells to the tumor microenvironment (TME). Figure 3B shows accumulative data from vaccinated and protected mice (PRO), vaccinated only mice (IDV) and unvaccinated and challenged mice (CHA). The spleens of CHA mice were infiltrated by tumor cells when mice reached their endpoint and contained very few T cells, and therefore could not be used for comparison. Contemporaneous assessment of BM T cell populations was precluded due to the morbidity of tumor burden in the unprotected groups, therefore we cannot definitively rule out similarities in CD8 + T cell trafficking. However, the absence of tumor-specific CD8 T cells in the BM of IDV mice suggests that these polyfunctional CD8 T cells only migrate to the BM in the presence of tumor cells. Next, we compared T cell activation in three groups of mice after restimulation with Id22 peptide and iMOPC or Id22 alone. We found that in unvaccinated mice, CD4 and CD8 T cells did not produce proinflammatory cytokines in response to tumor restimulation; in vaccinated only mice, CD4 T cells were activated in response to Id22 stimulation and in vaccinated and protected mice, activation of CD4 T cells was similar to vaccinated only mice. Interestingly, CD8 T cells activation significantly increased when BMMC were stimulated with both Id22 and iMOPC but not when BMMC were stimulated with Id22 peptide alone (figure 3C). When we compared restimulation with both Id22 and iMOPC, Id22, iMOPC, irradiated A20 (iA20) alone or Id22 and iA20 (a BALB/c lymphoma cell line included as a B-lineage control) in PRO mice, the frequency of polyfunctional CD8 T cells in the BM was increased only after stimulation with both Id22 peptide and iMOPC but not after stimulation with iMOPC or iA20 cells alone (figure 3D). Polyfunctional CD8 T cells frequency was higher in Id22 +iA20 stimulated cells in comparison with iA20 alone and this could be explained by either (a) the low number of samples (n=2) for this condition that did not achieve statistical significance or (b) MOPC and A20 cells might share common Ags since they are both B-lineage cells in the BALB/c background. These results indicate that CD8 T cells recognized MHC class I-restricted, tumor-associated Ags that were not the immunized Ag. Moreover, these CD8 T cells are most likely memory cells because they were still present in the BM of protected mice 80 days after tumor challenge.
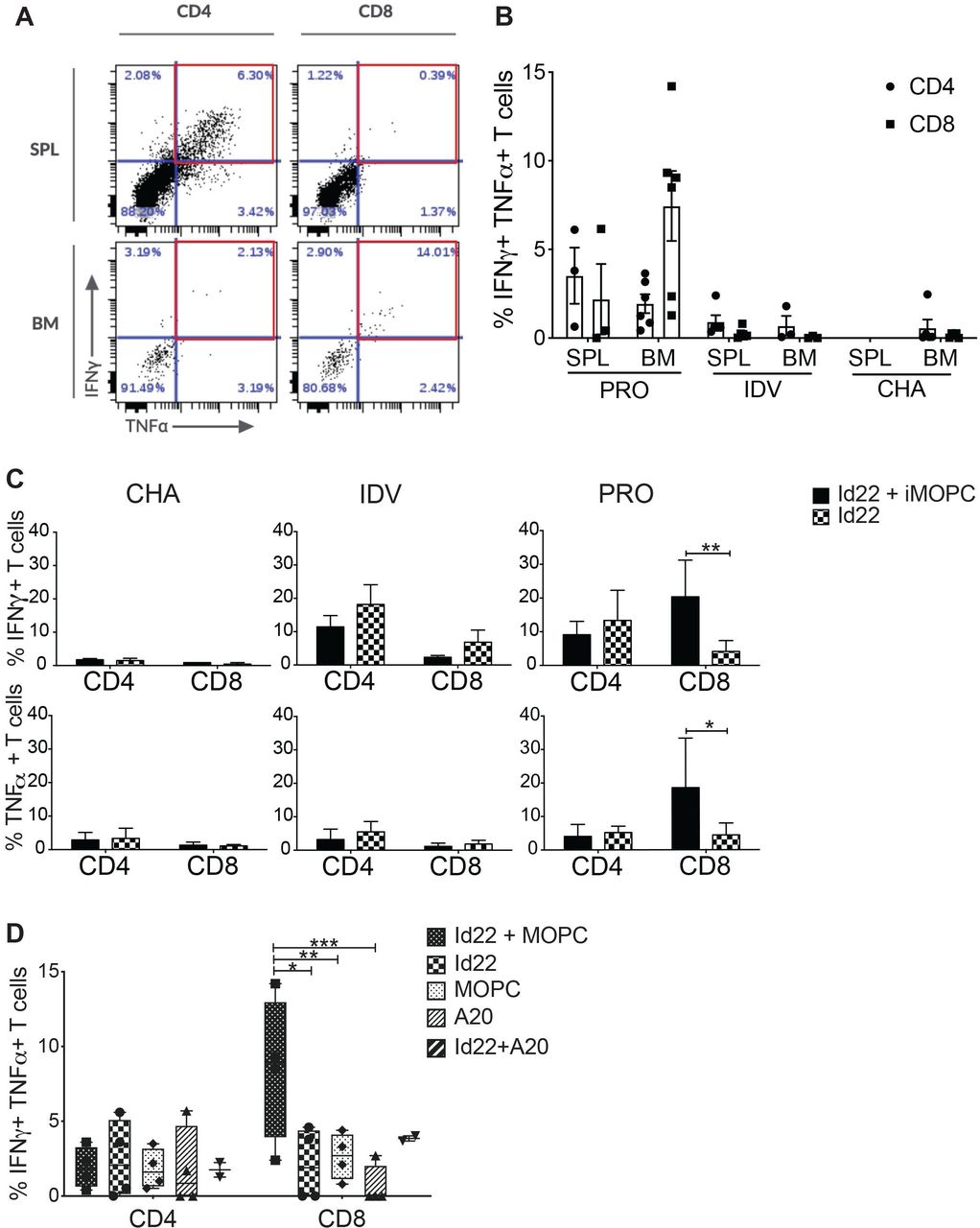
Id22 vaccine elicits tumor-specific CD8 T cell activation in the BM. CD4 and CD8 T cell activation was assessed by measuring IFN-γ and TNFα production by intracellular cytokine staining (ICS) after 8 days of in vitro restimulation. Splenocytes and BMMC were harvested from mice that were vaccinated and protected from MOPC tumor challenge (PRO) and restimulated with Id22 peptide and irradiated MOPC cells (iMOPC). (A) Flow cytometry plots of CD4 and CD8 T cell activation in the spleen (SPL) and bone marrow (BM) of a vaccinated and protected mouse. Red squares depict polyfunctional (IFN-γ+, TNFα+) T cells. (B) Average percentages of IFN-γ+, TNFα+CD4 or CD8 T cells in the spleen or BM of protected mice (PRO,n=3SPL/6BM), Id22 vaccinated only mice (IDV, n=5SPL/3BM)) and challenged only mice (CHA, n=5 BM) after in vitro restimulation. (Some samples did not have sufficient live lymphocytes to measure their activation after 8 days of in vitro restimulation) (C) T cell activation was assessed as previously described in the BM of mice either challenged with MOPC cells only (CHA), vaccinated with the Id22 peptide only (IDV) or Id22 vaccinated and challenged with MOPC cells (PRO) after restimulation with Id22 peptide and irradiated MOPC cells (iMOPC) or Id22 peptide alone. (D) Average percentages of polyfunctional T cells in BMMC of PRO mice after restimulation with either Id22 and iMOPC cells, Id22 alone, iMOPC alone, irradiated A20 cells (negative control), or Id22 and irradiated A20 cells. Statistics were analyzed using two-way analysis of variance and Mantel-Cox tests. **p<0.01; *p<0.05. ***p<0.001. Results are representative of three independent experiments. BMMC, bone marrow mononuclear cells; Id, idiotypic; IDV, immunization for vaccinated; IFN, interferon; MOPC, mineral oil-induced plasmacytoma; TNF, tumor necrosis factor.
Altogether, these results suggest that both the Id22-specific CD4 helper activity and MHC class I-restricted CD8 cytotoxic activity contributed to antitumor immunity.
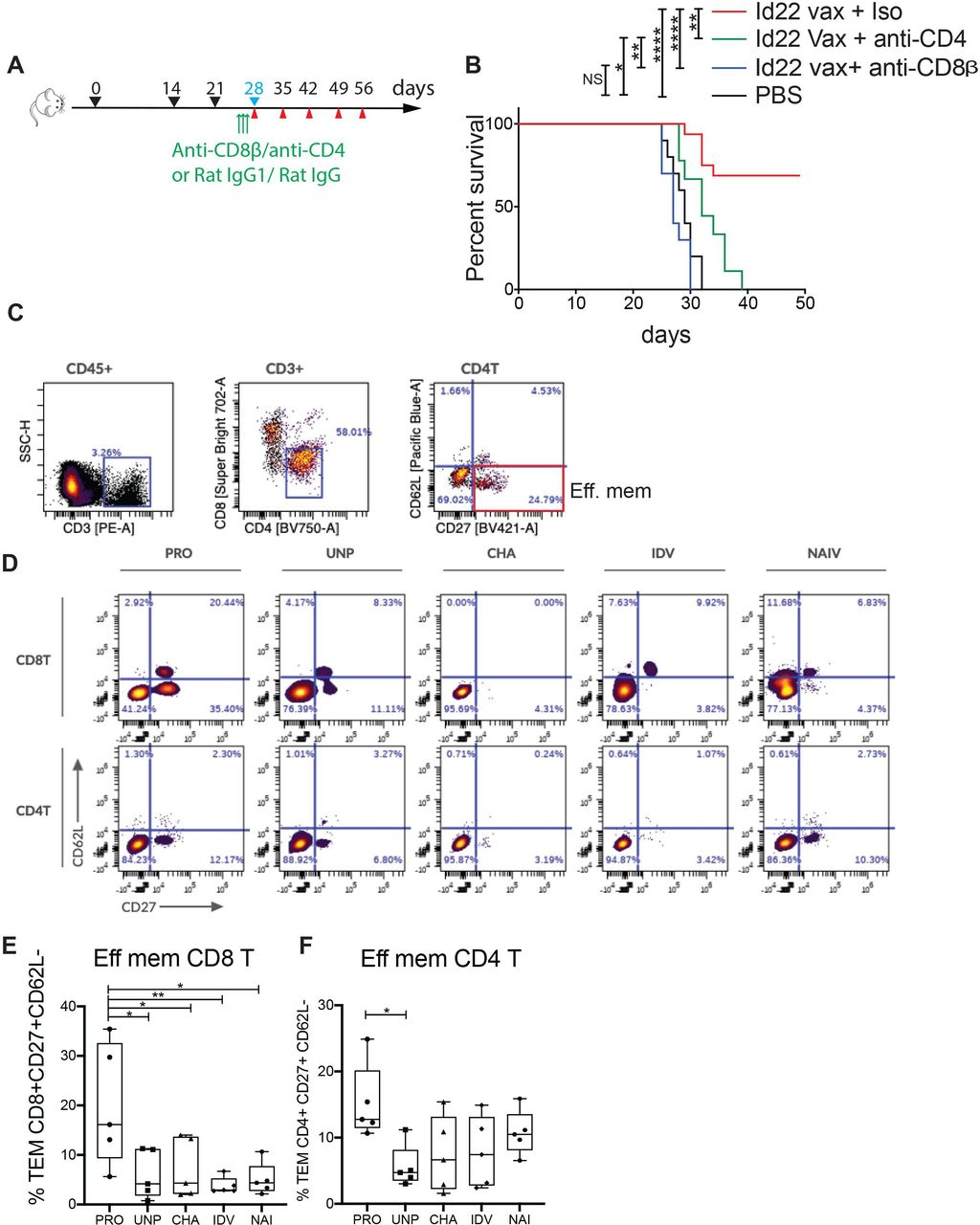
To verify that both CD4 or CD8 T cells were required for tumor rejection, we immunized mice with Id22 vaccine as previously described and depleted CD4 or CD8 T cells for three consecutive days before tumor challenge by intraperitoneal injection of anti-CD4 or anti-CD8β antibody. Control groups received Id22 vaccine and isotype controls. Depletion was verified by flow cytometry of PBMCs at day 0 before tumor challenge and day 7 after tumor challenge (online supplemental figure 1). In the absence of either CD4 or CD8 T cells, mice were no longer able to stop tumor growth, comparable to unvaccinated mice, whereas 70% of non-depleted vaccinated mice were protected against tumor growth (figure 4A,B). This finding, coupled with the observed lack of cytolytic activity of CD4 T cells (figure 2A), indicated that tumor Ag-specific CD8 T cells mediated tumor killing.
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD8 tumor protection and memory are mediated by Id22-specific CD4 T cells. (A) Timeline schedule for CD4 or CD8 T cell depletion after vaccination and before tumor challenge showing immunization days (black arrows), MOPC.315.BM tumor challenge (blue), blood collection (red) and injection of depleting antibodies (green). (B) Kaplan-Meier survival curve of vaccinated and control mice after MOPC challenge. Two groups of 10 mice were immunized subcutaneously with 100 µg of Id22 peptide in combination with 50 µg of poly (I:C). They were injected three times intrapertioneally with 100 µg of anti-CD4 or anti-CD8β depleting antibodies 3 days before MOPC challenge at days 25, 26 and 27. A third group of mice (n=16) was vaccinated the same way and injected with rat IgG1 and IgG2a isotype controls. Five PBS-injected mice were used as vaccination controls. (C) Representative gating strategy: BMMC from mice were restimulated for 3 days with Id22 peptide and iMOPC cells, then immunophenotyping was performed by multicolor flow cytometry. PRO, Id22 vaccinated and protected from tumor growth, UNP, vaccinated and unprotected, CHA, non-vaccinated and challenged with MOPC cells, IDV, vaccinated only, and NAI, naïve mice. CD4 and CD8 effector memory T cells (TEM) were selected using CD62L and CD27 antibodies (CD27 +CD62L−). (D) Representative plots of CD4 + and CD8+ TEM cells in the BM of one mouse per group. (C) Frequency of CD4 + TEM (E) and CD8 + TEM (F) cells in the BM of five mice per group. Statistics were analyzed using Mantel-Cox test for survival and one-way analysis of variance for multiple comparisons. **p<0.01; *p<0.05. ***p<0.001, ****p<0.0001. BMMC, bone marrow mononuclear cells; iMOPC, irradiated MOPC; MOPC, mineral oil-induced plasmacytoma; PBS, phosphate-buffered saline; TME, tumor microenvironment.
In order to investigate the differences in the immune environment of tumor protected mice in comparison with the other groups of mice and the implication of subsets of lymphoid and myeloid cells in tumor protection, we performed high dimensional immunophenotyping, by spectral multicolor flow cytometry, of the BM cellular immune compartment in the different groups of test animals compared with controls (figure 4C,D). Protected mice had a significantly higher proportion of effector memory CD8 T cells (CD27+, CD62L–) compared with all other groups (figure 4E), supporting the interpretation that these cells were the proximal mediators of tumor killing. We observed similar levels of CD4 effector memory T cells in all groups, with the only significant difference in comparison between protected and unprotected mice that were vaccinated and tumor challenged (figure 4F). We also compared the frequency of NK cells, macrophages and regulatory T cells between groups but we did not see any differences (online supplemental figure 3).
Supplemental material
Together, these data show that vaccine induced, Id-specific CD4 T cell activity confer protection against MOPC.315.BM tumor growth, through cooperation of CD4 and CD8 T cells most likely by cross-priming effector memory CD8 T cells against non-Id, tumor-associated Ags that home to the TME (BM).
Discussion
We report here that a novel neoAg Id-HSB fusion vaccine elicited protective CD4 T cell immunity in the MOPC315 model by activation of CD8 T cells against non-immunized, tumor-associated Ags. We observed very low to no Id-specific CD8 T cells after vaccination, consistent with Bogen et al’s findings that the minimal epitope binds to MHC class II molecules.29 The activation of Id-specific CD4 T cells is similar to previous neoAg studies where peptides predicted to bind to MHC-I molecules preferentially or exclusively activate CD4 T cells.16 30 In this model, Id-specific CD8 T cells do not seem to play a major role in tumor protection. This may reflect either tolerance due to the close sequence of the Id-peptide to self-Ag or T cell exhaustion due to long exposure to high Ag levels secreted by MOPC tumor cells. In the TME (BM), we observed expansion of polyfunctional CD8 T cells only when restimulated with MOPC315 cells and Id peptide in vitro, and selective depletion experiments showed that both CD4 and CD8 T cells were required for tumor protection in vivo. It was clear that direct CD4 cytotoxic T cell activity was not a significant factor, as CD4 T cells did not demonstrate lytic activity in vitro and the target MOPC315 cells did not express MHC class II in vitro or in vivo. NeoAg-specific CD4 T cells have been shown to confer protection against tumor growth even in the absence of CD8 T cells directed against the same Ag.30 Our study supports a model in which neoAg-specific CD4 T cells protect against myeloma growth by (1) CD4-CD8 cooperation, most likely through cross-priming of CD8 T cells against distinct tumor-associated Ags and (2) promoting migration of effector memory CD8 T cells to the TME.
Although Id vaccination is a clinically proven strategy against B cell malignancies such as non-Hodgkin’s and follicular lymphoma,12 31 this approach failed to induce protective immunity against MM.32 We showed here that fusion of a HSC70 binding motif (HSB) to the idiotype λ2315 peptide sequence was sufficient to confer tumor protection when delivered with the toll like receptor 3 (TLR3) ligand poly (I:C). These results support Ag uptake and internalization as important factors that determine Ag presentation and efficacy. HSP70 protein is known to preferentially chaperone peptides to MHC-I presentation pathways,33 but it can also chaperone external Ags through the MHC class II pathway.34 This can play a significant role in recruiting Ag-specific CD4 T helper cells to mediate dendritic cells (DCs) licensing and amplify cross-priming of CD8 T cells. Our results suggest that HSB binds to endogenous HSP70 protein, which may account (in part or whole) for the efficient priming of CD4 T helper activity observed. In addition, TLR ligands such as poly (I:C) promote maturation of APC such as DCs, which then prime CD4 T cells through contact-dependent and soluble factors, resulting in cross-priming and homing of tumor-specific CD8 T cells to the tumor. This is supported by the presented model, since CD8 depletion completely revoked tumor protection even in the presence of Id-specific CD4 T cells. In our system, the protection appears to be primarily T cell-mediated, however, we do not rule out a supporting role of other antitumor mechanisms including cytokines, NK cells and/or macrophages, similar to observations in the Id TCR-transgenic model.17 28 Further investigations are needed to elucidate the full complement of mechanisms by which the HSB fusion and TLR ligand co-delivery improve vaccine efficacy.
Epitope spreading, the phenomenon of T cell responses against non-target Ags that emerge after a tumor Ag-specific immunotherapy, was observed in patients with different malignancies following neoAg vaccination35 and in tumor-specific CD4 T cell adoptive transfer trials.18 36 However, the CD4/CD8 distribution of these T cell responses was not described and may reflect a similar mechanism to the model we propose. CD4 T cell help is essential for recruitment and cytolytic function of CD8 T cells37 and long-term maintenance of CD8 memory cells.38 Here, we observed an increased frequency of memory CD4 and CD8 T cells only in the BM in vaccinated mice that resisted tumor growth, confirming the role of CD4 T cell help in recruiting memory cells to the TME. Thus, elucidating the Ags recognized by cross-primed CD8 T cells may contribute to optimized vaccines composed of CD4 and CD8 Ags with appropriate fusion to HSB and co-delivered adjuvant/immunostimulatory components.
Our data highlight the impact of tumor vaccine formulation on the efficacy of the vaccine to induce elimination of tumor cells as well as the importance of CD4 T cell help in the recruitment, activation and maintenance of memory CD8 T cells in the TME. Therefore, understanding the mechanisms of cross-priming and Ag spreading may lead to better prediction of Ag presentation and selection of peptides for vaccines to generate potent and durable antitumor immune responses.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Animal experiments were approved by the Institutional Animal Care and Use Committee of Mount Sinai. ARRIVE1 reporting guidelines were used.
Acknowledgments
We would like to thank the flow cytometry core at the Icahn School of Medicine at Mount Sinai for their help with the spectral flow cytometer.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @joccastle
Contributors SB, HC, DL and JC designed the project. SB designed, performed experiments, and analyzed the data. RR-S and DG performed experiments. BB provided MOPC315.BM cell line and DL provided the Id peptides. Manuscript was written by SB and revised by HC, BB, DL and JC. All authors reviewed the manuscript. SB is guarantor of the work.
Funding This work was funded by Agenus, Lexington, Massachusetts, USA, and by the Mount Sinai Multiple Myeloma research fund.
Competing interests DL is an employee of Agenus and JC was an employee of Agenus at the time the research was being conducted. All other authors do not have relevant competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.