Article Text

Statistics from Altmetric.com
Our discussion on the targetability of neoantigens in diffuse midline glioma (DMG) is important since (1) DMG as a pediatric high-grade glioma is a devastating disease and a major cause of childhood cancer deaths and (2) adoptive T cell therapy has been shown to be very efficient when targeting ubiquitously expressed CD19 as a B cell lineage-antigen, lacking however bulk evidence about the clinical feasibility of the approach for targeting single amino acid mutations. Since the K27M mutation in H3.3 is a tumor driver in DMG,1 such an approach would be extraordinarily effective and precise, as exclusively cancer cells will be targeted by T cells. Nevertheless, effective tumor-killing without causing dose-limiting pathology in normal somatic tissues will only be possible when the respective epitope carrying the mutation is effectively processed and presented in the cancer cells. In this important aspect our data in Immisch et al2 and the Chheda et al data3 differ substantially. We cannot detect naturally processed H3.3K27M epitope in HLA-A*02:01, neither by MS-based immunopeptidomics nor by T cell coculture using modified T cells expressing a TCR generated with our mouse platform2 or the TCR published by the Okada group3 as a control. Therefore, we are afraid that false hope is given to children and parents participating for example in clinical trials already set up (NCT02960230) or to be started in the future (NCT04749641, NCT05478837), that altogether aim to target the H3.3K27M26-35 epitope in HLA-A*02:01-positive patients. Consequently, the statement in the letter to the editor by Chheda et al that the endogenous presentation of the H3.3K27M epitope in HLA-A*02:01 may not be robust is an understatement. Treatment-induced cell stress, such as ionizing radiation, may rather suppress H3.3 expression4 and negatively impact the epitope-generating proteasome activity.5
We can only speculate on the discrepancies between the data of Chheda et al,3 figure 1 in the letter to the editor and Immisch et al,2 but varying expression levels of mutant H3.3, dysfunctional antigen processing machinery (APM) or downmodulation of HLA-A*02:01 in (diffuse intrinsic pontine glioma, DIPG) tumors as well as insufficient expression of the H3.3K27M26-35-specific TCRs in peripheral blood mononuclear cells (PBMCs) can be excluded:
Using T cells genetically modified with either our 27633 TCR or the 1H5 TCR, no recognition of DIPG cell lines SF8628-A2 and SF7761-A2 cells harboring a heterozygous mutation of H3.3 (figure 2A and online supplemental figure 1D in Immisch et al, respectively),2 supposedly similar to DIPG17 cells, could be demonstrated. In four additional tumor cell lines (one of which was the same U87MG cells as used by Chheda et al3) we even did not achieve T cell recognition when the mutant H3.3 was overexpressed 15–20-fold (online supplemental figures 1B and 2B in Immisch et al).2 More strikingly, using the H3.3 mutant-specific antibody RM192 in western blotting, we detected mutant H3.3 naturally expressed from the heterozygous mutant allele in midline glioma cell lines (figure 2B in Immisch et al)2 as well as overexpressed in tumor cell lines (figure 2B and online supplemental figure 2A in Immisch et al).2 Mutant H3.3K27M protein could also be detected by immunohistochemistry in a series of midline gliomas.6 There is no doubt that the sensitivity to detect mutant H3.3 by H3.3K27M26-35-specific T cells would be by far higher than that of immunodetection in western blotting or histology by a mutant H3.3-specific antibody, if the respective T cell epitope would be naturally processed and presented.
Since we accompanied our T cell recognition assays with cell lines overexpressing CDK4R24L and found recognition by the respective CDK4-specific TCR14/352 in all cases, we can exclude any deficiencies in APM or HLA-A*02:01 expression that could have caused the lack of neoantigen presentation to T cells. In addition, we can exclude that lack of recognition is caused by differential epitope trimming or destruction due to specific ERAP1 allotypes (online supplemental table 1 in Immisch et al).2
In the majority of our T cell assays, we used TCR chimeras consisting of human variable and mouse constant region, allowing preferred pairing of the introduced TCRs. Already this configuration achieves similar peptide reactivity and multimer-binding (figure 1C and online supplemental figure 4D in Immisch et al, respectively)2 in comparison to the siRNA-knockdown modifications as used in Chheda et al (their figures 1Dand 3C, respectively).3 In addition, when endogenous epitope processing is circumvented by expressing a triple H3.3K27M26-35 epitope interconnected with the preferred proteasomal cleavage site AAY, intracellularly generated H3.3K27M26-35 epitope is recognized by our configuration of recombinant TCR expression (figure 2A in Immisch et al).2 With CRISPR/Cas9 knockout of the endogenous human TCR, we excluded any formation of chimeric non-functional TCRs (online supplemental figure 4 in Immisch et al).2 Still no recognition of H3.3K27M+ cell lines was observed although again peptide recognition and multimer binding were comparable to the data as shown in Chheda et al.3 Since CRISPR/Cas9 knockout is a complete knockout of endogenous TCRs whereas siRNA-knockdown only interferes with endogenous TCR expression, we consider it highly unlikely this as a reason for the complete lack of recognition. The in vitro cytotoxicity assay shown in figure 1A of the letter to the editor is difficult to judge as peptide-loaded cancer cells have not been included in the experiment—it seems that H3.3K27M cells do not grow in the presence of 1H5 TCR-modified T cells rather than that they are completely eradicated as we show for CDK4R24L targeted or H3.3K27M peptide loaded tumor cells (figure 3 in Immisch et al).2 In order to rule out cross-reactivity of 1H5 TCR-modified T cells to wildtype H3.3, TNFα or IL-2 release (figure 1B in the letter to the editor and Chheda et al3) may not be sensitive enough, as we show that IFNγ release from the same cells we measured for TNFα/IL-2 may differ by a factor of 9 or 3, respectively (online supplemental figure 2C in Immisch et al).2
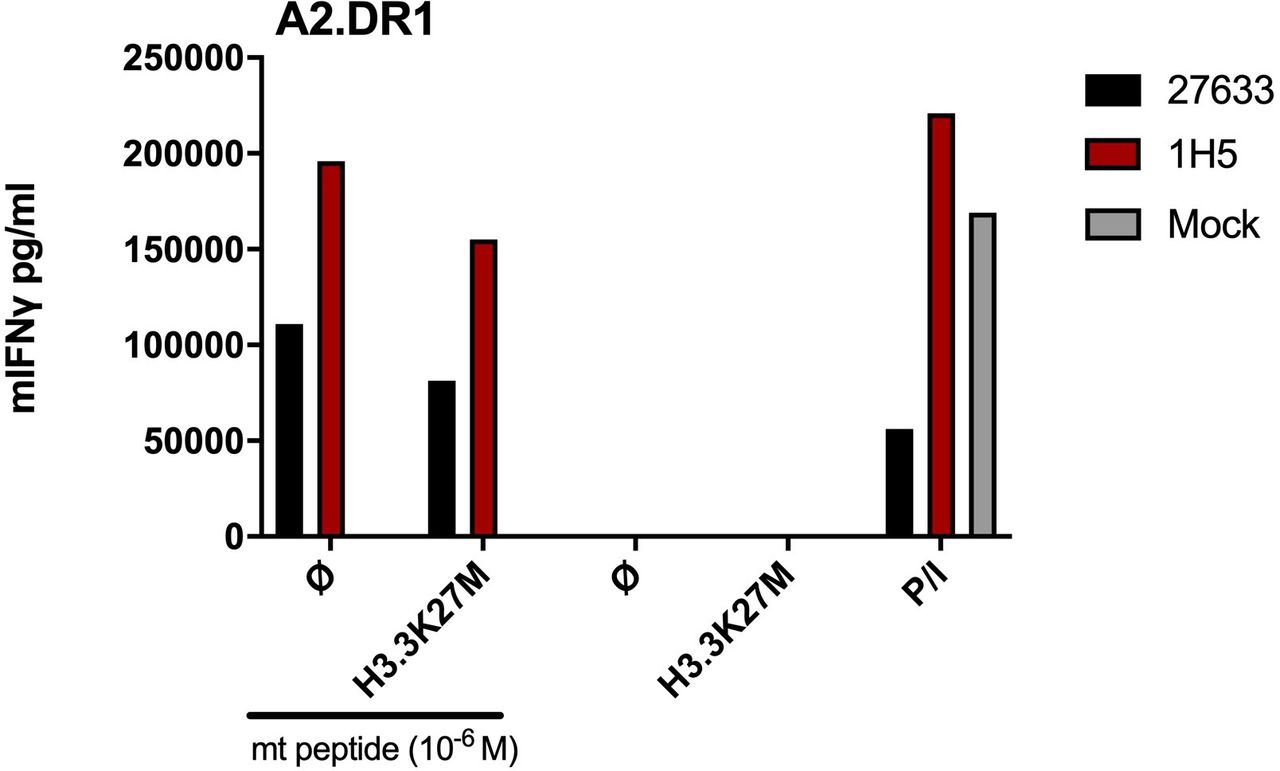
Concerning the data by the Platten group,7 brought up as supporting observations, two aspects need to be considered: (1) when immunizing with the H3.3K27M26-35 epitope, H3.3K27M-specific T cells can presumably be generated since this epitope is a good binder for HLA-A*02:01, as also shown by the Sette group by direct binding assays.3 Such amplification of H3.3K27M peptide-specific CD8+ T cells upon vaccination with the minimal H3.3K27M epitope (that does not need to be endogenously processed) has also been shown in the PNOC007 clinical trial (NCT02960230) and is comparable with our mouse experiments for obtaining the H3.3K27M TCR 27633 (figure 1A in Immisch et al),2 but lacks to prove recognition of tumor cells naturally expressing H3.3K27M. (2) When mice were immunized with a 27mer H3.3K27M14-40 peptide,7 unconventional ‘super-bulged’ peptide–HLA-I presentation or trimmings, for example by ERAP1/28, cannot be excluded and in vivo reactivity of CD8+ or CD4+ T cells cannot be distinguished. In the course of our experiments analyzing 27 633 and 1H5 TCRs for recognition of H3.3K27M+ tumor cells,2 we already included the H3.3K27M-transduced A2.DR1 sarcoma cell line used in Ochs et al.7 As with the human H3.3K27M+ tumor cell lines, we also found no CD8+ T cell reactivity against this H3.3K27M+ sarcoma cell line, unless the cells were pulsed with H3.3K27M26-35 peptide (figure 1).

{kind=link}
H3.3K27M TCR-transduced T cells fail to recognize A2.DR1 mouse sarcoma cell line overexpressing the mutant H3.3 histone. Levels of IFNγ secretion of 27633- and 1H5-TCR transduced T cells obtained from SV40TCR-I mice (Jackson Laboratories 005236, TCR-I). TCR-I mice are transgenic for a T-cell αβ receptor (irrelevant in this), which recognizes the H-2Db–restricted SV40-Tag epitope I. Parental A2.DR1 sarcoma cells were lentivirally transduced to express H3.3K27M full length cDNA (A2.DR1-H3.3K27M).7 A total of 104 transduced CD8+ T cells were cocultured at a 1:1 E:T ratio. P/I represents maximum IFNγ secretion from mouse T cells stimulated with PMA/Ionomycin.
The data for detection of H3.3K27M-specific T cells in HLA-A*02:01+ H3.3K27M+ DIPG patients do not speak for spontaneous induction of H3.3K27M-specific T cells as Chheda et al (their figure 1A)3 and Ochs et al (their figure 4B)7 both use in vitro stimulation with H3.3K27M26-35 peptide in the presence of hIL-2/IL-7/IL-15 for 5d or hIL-2 for 9d prior to ELISPOT analysis or multimer staining, respectively. Interestingly, Ochs et al7 rather demonstrate specific H3.3K27M epitope co-localisation with HLA-DR in H3.3K27M-mutant human glioma tissue by in situ proximity ligation assay (figure 4A and online supplemental figure 3D in Ochs et al),7 which further supports involvement of CD4+ T cells in H3.3K27M recognition. It should be noted here that within the vaccine H3.3K27M25-35 trial the Okada group reported progressive upregulation of HLA-DR in several myeloid-derived populations (doi:10.1093/neuonc/noz036.138), which similarly may have allowed a CD4+ T cell driven anti-H3.3K27M response. Recently, a MultIceNTER Phase I Peptide VaCcine Trial for the Treatment of H3K27M-Mutated Gliomas (INTERCEPT H3) started recruiting (NCT04808245) and exploits a long peptide targeting H3K27M. Here, treatment is not restricted to HLA-A*02:01+ patients but allows the discovery of patient-individual TCRs binding H3K27M presented by other MHCI but also on MHCII molecules.
Apart from the cytotoxicity assays, we also repeated MS-based immunopeptidomics. Our mass spectrometric analysis in the same U87MG cell line as used in Chheda et al,3 was controlled not only by loading cells with the heavy synthetic counterparts of mutant H3.3 and CDK4 epitopes, but also by detecting the naturally processed/presented CDK4R24L epitope in U87MG cells that overexpressed the respective cDNA (figure 2E in Immisch et al).2 We used four replicates and could only detect the mutant CDK4R24L but not H3.3K27M epitope on both, MS1 and MS2 level. Only for CDK4R24L overexpressing cells we could also demonstrate recognition by CDK4-specific TCR14/35-engineered T cells, thus directly confirming our MS data (figure 2D in Immisch et al).2 In Chheda et al,3 the mass spectrometric identification of a peak that may be indicative for HLA class I presented mutated H3.3K27M26-35 peptide appears only to be achieved in a single replicate on MS1 but not MS2 level (figure 2, unlabeled peptide version in Chheda et al)3 and therefore rather not exclude that this HLA presentation may happen than it can be taken as definitive proof.
Unfortunately, the new data in the letter to the editor do not resolve the differences between our results and those from the Okada group, but we meanwhile received support for our data also from other labs. We are convinced that the HLA-A*02:01 restricted H3.3K27M26-35 peptide is not an effective target for cancer immunotherapy with peptide vaccination or T cell therapy due to the lack of respective antigen density at the cell surface. Since DMG patients with H3.3K27M mutation are a vulnerable cohort desperately in need of new therapies future studies to identify immunogenic targets around this mutation should be pursued. Generally, we here claim the cautionary note that in silico predicted (neo)antigens, based on gene sequence or peptide binding analysis, should first be validated by testing for presence of the expected HLA ligands on the relevant cells, either by T cell assays or by mass spectrometry, before an extensive (and expensive) development program towards clinical use is started.
Ethics statements
Patient consent for publication
Footnotes
Contributors GW designed study and response to the letter to the editor, LI, GP and OP conducted experiments. LI, GP, PM, TB and GW analyzed data and wrote the response to the letter to the editor.
Funding Funding was provided by grants from the German Research Foundation (SFB-TR36), the Berlin Institute of Health (CRG-1), Deutsche Krebshilfe (111 546 and 70113456), DKTK joint funding (NEO-ATT), European Union (ERC Advanced Grant 882963) and the Helmholtz-Gemeinschaft, Zukunftsthema ‘Immunology and Inflammation’ (ZT-0027).
Competing interests None declared.
Provenance and peer review Commissioned; internally peer reviewed.
Linked Articles
- Immune cell therapies and immune cell engineering
- Letter